

Abstract
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease that selectively affects optic nerves and spinal cord. It is considered a severe variant of multiple sclerosis (MS), and frequently is misdiagnosed as MS, but prognosis and optimal treatments differ. A serum immunoglobulin G autoantibody (NMO-IgG) serves as a specific marker for NMO. Here we show that NMO-IgG binds selectively to the aquaporin-4 water channel, a component of the dystroglycan protein complex located in astrocytic foot processes at the blood-brain barrier. NMO may represent the first example of a novel class of autoimmune channelopathy.
Inflammatory demyelinating diseases of the central nervous system (CNS) are recognized to be immune-mediated, but no disease-specific microbial antigen or autoantigen has been identified to date. Neuromyelitis optica (NMO; formerly known as Devic's disease) accounts for approximately one third of the cases of multiple sclerosis (MS) that are encountered in Asia, where it is known as optic-spinal MS (1). The characteristic immunopathology of NMO is restricted to spinal cord and optic nerves, and affects gray and white matter. IgG, IgM, and products of complement activation are deposited in a vasculocentric pattern, which suggests a pathogenic role for autoantibody (2). Blood vessels within demyelinating lesions are distinctively thickened and hyalinized, disproportionate to the patient's age (3). Active lesions exhibit swelling, infiltrating macrophages, activated microglia, demyelination, axonal loss, prominent necrosis, and variable perivascular inflammation, with evidence of eosinophils and products of their exocytosis (2, 3). Chronic lesions are characterized by gliosis, cystic degeneration, cavitation, and atrophy.
Typically, NMO has a worse outcome than MS, with frequent and early relapses (4, 5). Vision and ambulation are impaired within 5 yr of its onset in 50% of patients, and 20% succumb to respiratory failure from cervical myelitis (5). Plasmapheresis has been reported to improve the neurologic outcome for patients who have NMO with severe longitudinally extensive myelitis of recent onset (6). This observation further supports an autoantibody-mediated pathogenesis for NMO. We recently described an IgG specific for NMO in the serum of 73% of patients who had NMO, and in 58% of patients who had the Asian optic-spinal form of MS. Patients who had classical (western) MS—for which no biomarker is recognized—were uniformly seronegative (7). Thus, seropositivity for NMO-IgG allows early diagnostic distinction between patients who have NMO and those who have MS. This distinction is important prognostically and therapeutically because optimal treatments differ for NMO (immunosuppression; reference 8) and MS (immunomodulation with β-IFN or glatiramer acetate; reference 9). NMO-IgG binds to the abluminal face of microvessels, pia, subpia, and Virchow-Robin sheath in sections of normal mouse CNS tissues. Its partial colocalization with laminin (7) is consistent with the autoantigen being a component of the glia limitans at the blood–brain barrier (BBB; reference 10). In this report we show that NMO-IgG binds to the aquaporin-4 (AQP4) water channel.
RESULTS AND DISCUSSION
NMO antigen is in CNS and non-CNS tissues, and colocalizes with AQP4
To determine whether the NMO antigen is restricted to the CNS, we tested NMO-IgG–positive patients' sera by indirect immunofluorescence on sections of normal mouse liver, kidney, and stomach tissues. In contrast to the characteristic intense staining of pial and microvascular elements in the brain, NMO-IgG did not bind to any vascular or visceral autonomic neural elements in stomach, kidney, or liver (e.g., myenteric and submucosal plexi, or sympathetic nerves accompanying arterioles). However, NMO-IgG bound prominently to distal urine-collecting tubules in the renal medulla and to basolateral membranes of epithelial cells deep in the gastric mucosa (Fig. 1 A); the immunoreactive gastric mucosal cells were identified immunohistochemically as parietal cells (not depicted). The distribution of NMO-immunoreactivity in CNS, kidney, and gastric mucosa suggested the water channel protein, AQP4, as a candidate antigen (11). By use of dual immunostaining with AQP4-specific rabbit IgG, confocal microscopy demonstrated that the antigen to which NMO patients' IgG binds colocalizes with AQP4 in all of these tissues (Fig. 1 A).
Figure 1.
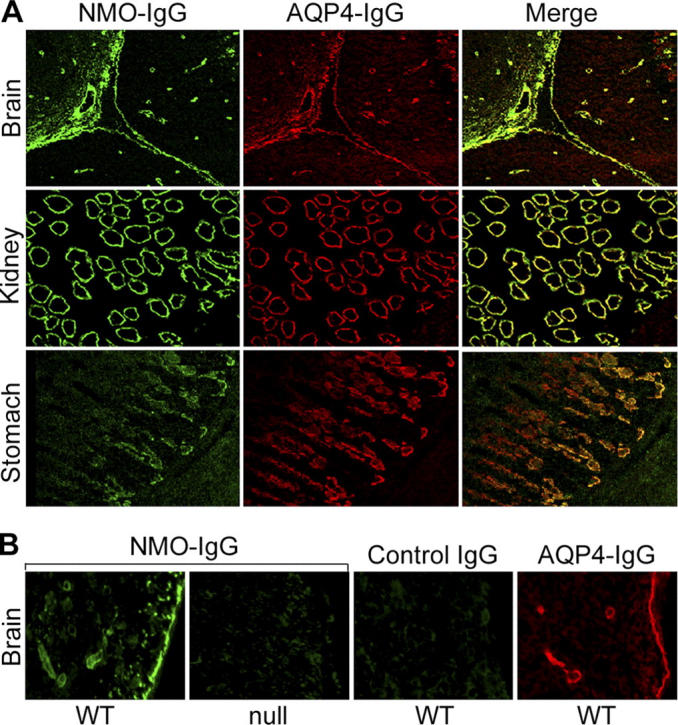
Immunofluorescence reveals NMO-IgG colocalization with AQP4 in mouse tissues. (A) Brain: Virchow-Robin space (pial–astrocyte interface) at the junction of two folia in mouse cerebellar cortex and midbrain. NMO-antigen (green, fluorescein-conjugated anti-human IgG), AQP4 water channel protein (red, rhodamine-conjugated anti–rabbit IgG); merged images yellow. Kidney: colocalization of NMO and AQP4 antigens in distal collecting tubules of medulla. Stomach: basolateral membranes of epithelial cells in deep gastric mucosa. (B) WT mouse brain binds NMO-IgG (panel 1) in a pattern that is indistinguishable from its binding of AQP4-specific IgG (panel 4); pia, subpia, and microvessels are stained. However, AQP4 knockout mouse brain (null) did not bind NMO-IgG (panel 2), and the serum of a control patient who had neuropsychiatric disease did not bind to WT brain (panel 3).
NMO-IgG does not bind to CNS tissue from AQP4-null mice
Next, we investigated whether IgG in any of six coded patients' sera bound to frozen sections of brain tissue obtained from transgenic AQP4 null mice (11). Three were NMO-IgG positive sera and three were control sera from patients who had neuropsychiatric diagnoses (age- and sex-matched to the NMO patients). IgG in the three NMO patients' sera bound to microvessels, pia, and subpia in the wild-type brain tissue in a pattern that was indistinguishable from that yielded by rabbit anti-AQP4–IgG (Fig. 1 B). However, neither human serum IgG nor the rabbit anti-AQP4–IgG bound detectably to AQP4-null mouse brain tissue.
NMO-IgG binds selectively to membranes of AQP4-transfected cells
We tested the same six patients' coded sera, in blinded conditions, for IgG binding to a stably transfected human embryonic kidney cell line (HEK-293) expressing a transgene encoding full-length AQP4. No patient's IgG bound to the HEK-293 cells transfected with plasmid encoding GFP without AQP4 (Fig. 2). However, IgG in the sera of all three patients who had NMO stained the periphery of AQP4-transfected cells intensely in a plasma membrane pattern (corresponding to that seen with rabbit anti-AQP4–IgG), where it colocalized with GFP. The distribution of GFP-AQP4 that did not colocalize with NMO-IgG was compatible with endoplasmic reticulum or cytoplasmic transport packages, presumably inaccessible to NMO-IgG in nonpermeabilized cells. IgG in the three controls' sera did not bind to the GFP-AQP4–transfected cells.
Figure 2.

NMO-IgG binds selectively to AQP4-transfected cells. NMO-IgG (red, Alexa Fluor 594–conjugated anti–human IgG) binds to the plasma membrane of cultured human embryonic kidney (HEK-293) cells transfected with AQP4 (green, [GFP]-AQP4), but not to the plasma membrane of control HEK-293 cells transfected with plasmid encoding GFP only. Nucleus is blue (stained with Hoescht). Overlap staining is yellow in the merged images. Note: GFP produced in control transfected cells is distributed throughout the nucleus and cytoplasm; GFP fused to AQP4 is membrane-bound. Cytoplasmic GFP-AQP4 (presumably in endoplasmic reticulum and transport packages) is not accessible to NMO-IgG in nonpermeabilized cells. Bars, 10 μm.
NMO-IgG binds to the AQP4 water channel component of the dystroglycan complex
The selective binding of NMO-IgG to substrates expressing AQP4, and its lack of reactivity with vector-transfected cells and AQP4-null mouse brain, strongly implicate AQP4 as the autoantigen of NMO-IgG. However, in astrocytic endfeet, the distribution of AQP4 is dependent on its association with elements of the dystroglycan complex at the BBB (12–14). Therefore, it is conceivable that the target of NMO-IgG is one of these associated proteins. To investigate this possibility, we tested lysates of the AQP4 and control transfected cell lines by immunoprecipitation. Despite endogenous expression of α-syntrophin, β-dystroglycan, and dystrophin (Dp71) in HEK-293 cells (Fig. 3 A), only GFP-AQP4 was precipitated from lysates by serum IgG from patients who had NMO (and by rabbit anti-AQP4–IgG; Fig. 3 B). The immunoprecipitates did not contain α-syntrophin, β-dystroglycan, Dp71, or free GFP (Fig. 3 B). Control human IgG did not precipitate GFP-AQP4 or any of the dystroglycan-complex proteins.
Figure 3.

NMO-IgG immunoprecipitates GFP-AQP4 but not related dystroglycan complex proteins. (A) Lysates of HEK-293 cells transfected with GFP-AQP4 or GFP vector, and nontransfected cells were separated by SDS PAGE and probed with IgG's specific for GFP, α-syntrophin, β-dystroglycan, or dystrophin (Dp71). Expression of α-syntrophin, β-dystroglycan, or dystrophin (Dp71) did not differ appreciably in any cell lysate. (B) Immune complexes were captured on protein G-agarose after incubating lysates of HEK-293 cells (stably transfected with GFP-vector or GFP-AQP4) with pooled serum from patients who had NMO, pooled serum from control patients who had neuropsychiatric disease, or a rabbit AQP4-specific IgG. The complexes were analyzed by Western blot for GFP-AQP4 and for dystroglycan complex proteins (present in both lysates). NMO-IgG and AQP4-IgG captured GFP-AQP4. Control patients' IgG did not capture GFP-AQP4. No IgG captured α-syntrophin, β-dystroglycan, or Dp71 (dystrophin). The cell lysate lane represents 1.5% of the total input used for immunoprecipitations. IgG from individual patients who had NMO yielded similar results.
Our results identify a water channel protein as the first defined autoantigen pertinent to an inflammatory demyelinating disorder of the human CNS. AQP4 is an integral protein of astrocytic plasma membranes (15–18), and is highly concentrated in foot process domains facing microvessels where it interacts with dystrophin-associated proteins (10, 17). The AQP4 channel is mercurial-insensitive, and is the predominant water channel in the CNS. It has a pathophysiologic role in brain edema formation following water intoxication (14, 19, 20) or focal cerebral ischemia (21). Brain edema occurring in oncologic contexts is attributed to the up-regulation of AQP4 in high-grade astrocytomas and in reactive astrocytes related to cerebral adenocarcinoma metastases (21, 22).
The present study is the first to implicate AQP4 in the pathogenesis of any autoimmune disorder. NMO may represent the first example of a novel class of autoimmune channelopathies. Unlike MS, the cerebrospinal fluid in patients who have NMO lacks evidence of intrathecal synthesis of IgG, such as oligoclonal bands (4). IgG produced in peripheral lymphoid tissues has limited access to the extracapillary space in the brain because of the endothelial tight junctions that constitute the BBB. However, this barrier is not absolute (23, 24). If AQP4-specific IgG is the proximal cause of NMO, we envisage two potential mechanisms for the pathogenicity of the small amount of IgG that penetrates this barrier: (a) initiation of an inflammatory immune response against astrocytic AQP4 protein by direct binding of antigen-specific IgG molecules to the outer face of the water channel; the homotetrameric structure of this protein would enable potent activation of complement by appropriate IgG classes; and (b) in the absence of initial complement activation, inflammation and demyelination may be a consequence of IgG-mediated dysregulation of tissue water homeostasis. We speculate that endothelial leakage accompanying inflammatory events initiated by the binding of NMO-IgG to the extracellular face of AQP4 accounts for the abundance of IgM in immune complexes deposited around penetrating vessels in lesions of autopsied spinal cord from patients who had NMO (2). Vascular leakage would allow secondary binding of IgM rheumatoid factor (an extremely potent activator of the complement cascade) to astrocytic endfeet displaying NMO-IgG; low affinity interactions of other IgMs entering the lesion site would amplify the inflammatory process (Eisen, H.N., personal communication). Numerous autoantibodies (nonorgan-specific and organ-specific) are a frequent serologic finding in patients who have NMO (4). However, unlike NMO-IgG, no other autoantibody is restricted to patients who have NMO. Lucchinetti et al. classified four MS lesion subtypes immunohistochemically (25). The most common (“pattern 2” MS lesion) is characterized by deposits of complement and immunoglobulins. However, immune complexes in “pattern 2” MS lesions are located at sites of active myelin destruction and not in the prominent perivascular distribution that characterizes NMO lesions.
The wide distribution of AQP4 does not explain the restricted immunopathology of patients who have NMO. Renal abnormalities have not been reported in NMO; however AQP4-null mice have only minimal perturbation of renal function, which reflects the small and noncritical nephron segment that expresses AQP4 (11). Although NMO-IgG binds to rodent brain tissues, inflammatory lesions in the CNS of patients characteristically are restricted to optic nerves and spinal cord. This incongruity may relate to potential species differences in the distribution of AQP4 in the CNS of humans and mice, or differences in accessibility of AQP4 epitopes to IgG in vivo and in vitro. Regional differences in the spatial distribution or molecular orientation of AQP4 epitopes on astrocytic endfeet at sites other than optic nerve and spinal cord might preclude complement activation (26) or intermolecular cross-linking (27). Now that a putative target autoantigen has been identified for NMO, and human IgG of high titer is available, direct investigation of potential pathogenic autoimmune mechanisms is feasible in laboratory animals by active immunization and by passive transfer of selected patients' autoantibodies (28, 29).
MATERIALS AND METHODS
These studies were approved by the Institutional Review Board of Mayo Clinic, Rochester, Minnesota, and the Institutional Animal Care and Use Committees of Mayo Clinic and the University of California, San Francisco.
Antibodies.
We selected sera from 13 adult patients (9 were pooled) in whom NMO-IgG was detected at 1:3,840 dilution or greater by indirect immunofluorescence on mouse CNS tissues (7), and from 3 healthy age-matched subjects, 2 patients who had classical MS, 1 patient who had idiopathic myelopathy, and 3 patients who had miscellaneous neuropsychiatric disorders. We pooled sera from 50 additional patients who had miscellaneous disorders. Affinity-purified rabbit IgG specific for rat AQP4 (C-terminal residues 249–323) was purchased from Sigma-Aldrich; normal rabbit serum was obtained from a New Zealand white rabbit. Mouse monoclonal IgGs were purchased from: Affinity BioReagents, Inc. (anti-α-syntrophin); Santa Cruz Biotechnology, Inc. (anti-GFP); Novocastra Laboratories Ltd. (anti-β-dystroglycan); and Sigma-Aldrich (anti-dystrophin). Goat anti–human IgG (fluorescein-conjugated and rhodamine-conjugated) and rhodamine-conjugated anti–rabbit IgG were purchased from Southern Biotechnology Associates, Inc. Alexa Fluor 594–conjugated goat anti–human IgG was purchased from Molecular Probes. Horseradish peroxidase conjugated anti–mouse IgG was purchased from Southern Biotechnology Associates, Inc.
Fusion constructs and stable cell lines.
AQP4 was amplified from a Quick Clone cDNA library (adult human cerebellum, BD Biosciences) and cloned into pEGFP-C2 (CLONTECH Laboratories, Inc). HEK-293 cells were stably transfected with pEGFP-C2 with and without AQP4 using Fugene 6 transfection reagent according to the manufacturer's protocol (Roche). Stable cell lines were selected using 0.8 μg/ml geneticin (GIBCO BRL). Expression of the GFP and GFP-AQP4 in the stable cell lines was visualized in Western blot analyses using anti-GFP–IgG.
Cells and tissues.
We used three different substrates for immunostaining: (a) frozen sections of adult mouse cerebellum, midbrain, liver, kidney, and stomach; (b) frozen sections of cerebellum from adult transgenic AQP4-null mice and wild-type control mice (11); and (c) human embryonic kidney (HEK-293) cells transfected with plasmids encoding GFP-AQP4 or Vector-GFP.
Immunofluorescence.
Cells and tissues were fixed in 10% formalin for 4 min, washed in PBS, and blocked for 1 h in PBS containing 10% goat serum. After incubation with primary antibodies for 40 min (tissues) or 1 h (cell lines), the substrates were washed three times in PBS and incubated with anti–human or anti–rabbit IgG for 30 min, washed in PBS, stained with the DNA dye Hoescht at 1 μg/ml, and mounted in Prolong antifade mounting media (Molecular Probes). Images were captured using a Zeiss confocal microscope.
Immunoprecipitations and Western blot.
HEK-293 cells that were stably transfected with pEGFP-C2 or GFP-AQP4 were lysed in buffer containing: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 1% NP-40. Lysates were clarified by centrifugation at 3700 rpm for 8 min to remove cellular debris, and divided into three aliquots. A pool of high-titered sera from nine patients who had NMO (20 μL/ml), and subsequently from individual patients, was added to one aliquot; pooled sera from 50 control patients was added to a second aliquot; and rabbit anti-AQP4–IgG (Sigma-Aldrich) was added to a third aliquot (2 μL/ml). Lysates were incubated with antibodies overnight at 4°C. Protein G-agarose (Zymed Laboratories) was added to each lysate (30 μl/ml). After incubating for 1 h at 4°C, beads were pelleted and washed three times in 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, and 0.1% NP-40. Beads were resuspended in standard 2X SDS Laemmli sample buffer containing β-mercaptoethanol (20 μl/ml), to release immune complexes.
Subsequent steps were at room temperature. Immune complexes were electrophoresed in 4–15% gradient polyacrylamide gels and transferred electrophoretically to nitrocellulose paper. The nitrocellulose paper was blocked in buffer (20 mM Tris, pH 7.6, 137 mM sodium chloride, 0.1% Tween 20) containing 10% powdered milk, and exposed for 1 h to a panel of IgG probes specific for: α-syntrophin (1:1,000); GFP (1:1,000); β-dystroglycan (1:100), and Dp71 dystrophin (1:1,000). Blots were washed three times for 5 min in 20 mM Tris, pH 7.6, 137 mM sodium chloride, 0.1% Tween-20 and then incubated for 30 min with horseradish peroxidase–labeled goat anti–mouse IgG or goat anti–rabbit IgG. After washing, bound IgG was detected autoradiographically by enhanced chemiluminescence (GE Healthcare).
Transgenic AQP4 –/– mice.
Transgenic AQP4-null mice were generated and characterized as described previously (11). These mice totally lack AQP4 protein.
Acknowledgments
This study was supported by the Mayo Foundation.
V.A. Lennon and T.J. Kryzer are named inventors on a patent application filed by Mayo Foundation for Medical Education and Research that relates to the neuromyelitis optica antigen and its application for the detection of NMO autoantibody. The authors have no other potential conflicting financial interests.
References
- 1.Misu, T., K. Fujihara, I. Nakashima, I. Miyazawa, N. Okita, S. Takase, and Y. Itoyama. 2002. Pure optic-spinal form of multiple sclerosis in Japan. Brain. 125:2460–2468. [DOI] [PubMed] [Google Scholar]
- 2.Lucchinetti, C.F., R.N. Mandler, D. McGavern, W. Bruck, G. Gleich, R.M. Ransohoff, C. Trebst, B. Weinshenker, D. Wingerchuk, J.E. Parisi, and H. Lassman. 2002. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain. 125:1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mandler, R.N., L.E. Davis, D.R. Jeffery, and M.K. Kornfeld. 1993. Devic's neuromyelitis optica: a clinicopathological study of 8 patients. Ann. Neurol. 34:162–168. [DOI] [PubMed] [Google Scholar]
- 4.Wingerchuk, D.M., W.F. Hogancamp, P.C. O'Brien, and B.G. Weinshenker. 1999. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. 53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 5.Wingerchuk, D.M., and B.G. Weinshenker. 2003. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology. 60:848–853. [DOI] [PubMed] [Google Scholar]
- 6.Keegan, M., A.A. Pineda, R.L. McClelland, C.H. Darby, M. Rodriguez, and B.G. Weinshenker. 2002. Plasma exchange for severe attacks of CNS demyelination: predictors of response. Neurology. 58:143–146. [DOI] [PubMed] [Google Scholar]
- 7.Lennon, V.A., D.M. Wingerchuk, T.J. Kryzer, S.J. Pittock, C.F. Lucchinetti, K. Fujihara, I. Nakashima, and B.G. Weinshenker. 2004. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 8.Mandler, R.N., W. Ahmed, and J.E. Dencoff. 1998. Devic's neuromyelitis optica: a prospective study of seven patients treated with prednisone and azathioprine. Neurology. 51:1219–1220. [DOI] [PubMed] [Google Scholar]
- 9.Goodin, D.S., E.M. Frohman, G.P. Garmany Jr., J. Halper, W.H. Likosky, F.D. Lublin, D.H. Silberberg, W.H. Stuart, and S. van den Noort. 2002. Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology. 58:169–178. [DOI] [PubMed] [Google Scholar]
- 10.Moore, S.A., F. Saito, J. Chen, D.E. Michele, M.D. Henry, A. Messing, R.D. Cohn, S.E. Ross-Barta, S. Westra, R.A. Williamson, et al. 2002. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 418:422–425. [DOI] [PubMed] [Google Scholar]
- 11.Ma, T., B. Yang, A. Gillespie, E.J. Carlson, C.J. Epstein, and A.S. Verkman. 1997. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J. Clin. Invest. 100:957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amiry-Moghaddam, M., R. Xue, F.M. Haug, J.D. Neely, A. Bhardwaj, P. Agre, M.E. Adams, S.C. Froehner, S. More, and O.P. Otterson. 2004. Alpha syntrophin deletion removes the perivascular but not the endothelial pool of aquaporin-4 at the blood-brain barrier and delays the development of brain edema in an experimental model of acute hyponatremia. FASEB J. 18:542–544. [DOI] [PubMed] [Google Scholar]
- 13.Frigeri, A., G.P. Nicchia, B. Nico, F. Quondamatteo, R. Herken, L. Roncali, and M. Svelto. 2001. Aquaporin-4 deficiency in skeletal muscle and brain of dystrophic mdx mice. FASEB J. 15:90–98. [DOI] [PubMed] [Google Scholar]
- 14.Vajda, Z., M. Pedersen, E.-M. Füchtbauer, K. Wertz, H. Stødkilde-Jørgensen, E. Sulyok, T. Dóczi, J.D. Neely, P. Agre, J. Frøkiaer, and S. Nielsen. 2002. Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc. Natl. Acad. Sci. USA. 99:13131–13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung, J.S., R.V. Bhat, G.M. Preston, W.B. Guggino, J.M. Baraban, and P. Agre. 1994. Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA. 91:13052–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nico, B., A. Frigeri, G.P. Nicchia, F. Quondamatteo, R. Herken, M. Errede, D. Ribatti, M. Svelto, and L. Roncali. 2001. Role of aquaporin-4 water channel in the development and integrity of the blood-brain barrier. J. Cell Sci. 114:1297–1307. [DOI] [PubMed] [Google Scholar]
- 17.Amiry-Moghaddam, M., D.S. Frydenlund, and O.P. Ottersen. 2004. Anchoring of aquaporin-4 in brain: molecular mechanisms and implications for the physiology and pathophysiology of water transport. Neuroscience. 129:999–1010. [DOI] [PubMed] [Google Scholar]
- 18.Hasegawa, H., T. Ma, W. Skach, M. Matthay, and A.S. Verkman. 1994. Molecular cloning of a mercurial insensitive water channel expressed in selected water transporting tissues. J. Biol. Chem. 269:5497–5500. [PubMed] [Google Scholar]
- 19.Manley, G.T., M. Fujimura, T. Ma, N. Noshita, F. Filiz, A.W. Bollen, P. Chan, and A.S. Verkman. 2000. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxification and ischemic stroke. Nat. Med. 6:159–163. [DOI] [PubMed] [Google Scholar]
- 20.Agre, P., and D. Kozono. 2003. Aquaporin water channels: molecular mechanisms for human diseases. FEBS Lett. 555:72–78. [DOI] [PubMed] [Google Scholar]
- 21.Saadoun, S., M.C. Papadopoulos, D.C. Davies, S. Krishna, and B.A. Bell. 2002. Aquaporin-4 expression is increased in oedematous human brain tumours. J. Neurol. Neurosurg. Psychiatry. 72:262–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papadopoulos, M.C., S. Krishna, G.T. Manley, and A.S. Verkman. 2004. Aquaporin-4 facilitates the reabsorption of excess fluid in vasogenic brain edema. FASEB J. 18:1291–1293. [DOI] [PubMed] [Google Scholar]
- 23.Brimijoin, S., P. Hammond, M. Balm, and V. Lennon. 1991. Experimental autoimmunity to brain acetylcholinesterase. Cholinesterases: Structure, Function, Mechanism, Genetics, and Cell Biology. J Massoulié, F. Bacou, E. Barnard, A. Chatonnet, B. Doctor, D.M. Quinn, editors. American Chemical Society, Washington DC. 332–335.
- 24.Poduslo, J.F., and G.L. Curran. 2001. Amyloid β peptide as a vaccine for Alzheimer's disease involves receptor-mediated transport at the blood-brain barrier. Neuroreport. 12:3197–3200. [DOI] [PubMed] [Google Scholar]
- 25.Lucchinetti, C., W. Brück, J. Parisi, B. Scheithauer, M. Rodriguez, and H. Lassmann. 2000. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47:707–717. [DOI] [PubMed] [Google Scholar]
- 26.Lennon, V.A., E.H. Lambert, and G.E. Griesmann. 1984. Membrane array of acetylcholine receptors determines complement-dependent mononuclear phagocytosis in experimental myasthenia gravis. Fed. Proc. 43:1764. [Google Scholar]
- 27.Lennon, V.A. 1978. Myasthenia gravis: a prototype immunopharmacological disease. The Menarini Series on Immunopathology. P.A. Miescher, editor. Schwabe & Co., Basel. 178–198.
- 28.Lennon, V.A., L.G. Ermilov, J.H. Szurszewski, and S. Vernino. 2003. Immunization with neuronal nicotinic acetylcholine receptor induces neurological autoimmune disease. J. Clin. Invest. 111:907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vernino, S., L.G. Ermilov, L. Sha, J.H. Szurszewski, P.A. Low, and V.A. Lennon. 2004. Passive transfer of autoimmune autonomic neuropathy to mice. J. Neurosci. 24:7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]