

Abstract
Psoriasis is one of the most common T cell–mediated autoimmune diseases in humans. Although a role for the innate immune system in driving the autoimmune T cell cascade has been proposed, its nature remains elusive. We show that plasmacytoid predendritic cells (PDCs), the natural interferon (IFN)-α–producing cells, infiltrate the skin of psoriatic patients and become activated to produce IFN-α early during disease formation. In a xenograft model of human psoriasis, we demonstrate that blocking IFN-α signaling or inhibiting the ability of PDCs to produce IFN-α prevented the T cell–dependent development of psoriasis. Furthermore, IFN-α reconstitution experiments demonstrated that PDC-derived IFN-α is essential to drive the development of psoriasis in vivo. These findings uncover a novel innate immune pathway for triggering a common human autoimmune disease and suggest that PDCs and PDC-derived IFN-α represent potential early targets for the treatment of psoriasis.
Psoriasis is the most common autoimmune disease of the human skin, affecting ∼2% of the population worldwide (1). Similar to Crohn's disease and rheumatoid arthritis, psoriasis results from an overt self-perpetuating activation of autoimmune T cells (2–4). Through the secretion of Th1 cytokines, these T cells contribute to the epidermal hyperproliferation in genetically predisposed individuals. The initial onset of the lesions is commonly followed by chronic relapses of the disease triggered by infections, mechanical stress, and drugs (5). Although it is still unclear how these environmental factors drive the pathogenic T cell cascade, it has been suggested that innate immune pathways may provide the missing link (6, 7).
Plasmacytoid pre-DCs (PDCs) are a rare cell population in the peripheral blood and secondary lymphoid organs characterized by plasma cell–like morphology and a unique surface phenotype (8). PDCs represent key effectors in innate antiviral immunity because of their unique capacity to secrete large amounts of IFN-α in response to viruses (9, 10). Upon viral stimulation, PDCs differentiate into DCs (11, 12) and/or induce an IFN-α–dependent maturation of bystander myeloid DCs with the ability to drive Th1 responses (13), thus providing a unique link between innate and adaptive antiviral immunity. During homeostasis, PDCs are encountered exclusively in the blood and lymphoid organs; however, viral infection leads to an active recruitment of PDCs from the blood into peripheral sites of infection (14). Recent studies have shown that PDCs may also accumulate in peripheral tissues during certain noninfectious inflammatory disorders (15–18), including psoriasis (17, 19), although a functional relevance has not been demonstrated.
There are three scientific observations that suggest a role for IFN-α in psoriasis. First, psoriatic skin lesions demonstrate an activated IFN-α signaling pathway (20–23). Second, continuous excessive IFN-α/β signaling in IFN regulatory factor (IRF)-2−/− mice causes an inflammatory skin disease resembling psoriasis (24). Finally, treatment of psoriasis patients with recombinant IFN-α for unrelated conditions (e.g., viral infections or tumors) can exacerbate psoriasis (25–28). We therefore hypothesized that IFN-α produced by PDCs may contribute to the pathogenesis of psoriasis.
We show that PDCs infiltrate the normal-appearing skin of psoriatic patients and become activated to produce IFN-α early during the development psoriatic skin lesions. Furthermore, we demonstrate that PDC-derived IFN-α is essential in driving the local activation and expansion of pathogenic T cells leading to the development of psoriatic skin lesions. Thus, activation of PDCs to produce IFN-α in the skin of psoriatic patients represents a key innate immune pathway to initiate the autoimmune T cell cascade leading to psoriasis.
Results
PDC accumulation in the skin of psoriasis patients
To assess the presence of PDCs in psoriatic skin, we performed immunohistochemistry and confocal laser scan microscopy using an antibody specific for human blood PDCs (anti–BDCA-2; reference 29). High numbers of PDCs were found throughout the T cell–rich infiltrate in the dermis of primary psoriatic plaque lesions (Fig. 1, a and b). In contrast, PDCs were completely absent in the normal skin of healthy donors or the inflamed skin of atopic dermatitis patients (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050500/DC1), as previously reported (18). We confirmed the specificity of BDCA-2 for PDCs in psoriatic skin by flow cytometry analysis of dermal single cell suspensions, showing that BDCA-2+ PDCs expressed high levels of CD123 (Fig. 1 c), were positive for MHC class II and CD4, lacked both lineage markers (CD3, CD14, CD20, and CD56) and myeloid markers (CD11b and CD11c), and displayed characteristic plasmacytoid morphology (reference 30; Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050500/DC1). Interestingly, we observed an increased frequency of PDCs in both plaque lesions (mean, 8.6%; range, 3–14% of total dermal mononuclear cells) and the nearby uninvolved (normal appearing) skin of psoriatic patients (mean, 3.1%; range, 1–5.6%) compared with the skin of healthy individuals (consistently <0.03%; Fig. 1 c). In addition, we found a decrease of PDCs in the peripheral blood of psoriasis patients (mean, 0.18%; range, 0.08–0.26%) compared with the peripheral blood of healthy individuals (mean, 0.32%; range, 0.18–0.65%; Fig. 1, c and d), suggesting that the accumulation of PDCs in psoriatic skin resulted from an active redistribution of PDCs from the blood into the skin, similar to a phenomenon observed in systemic lupus erythematosus (SLE; reference 31).
Figure 1.
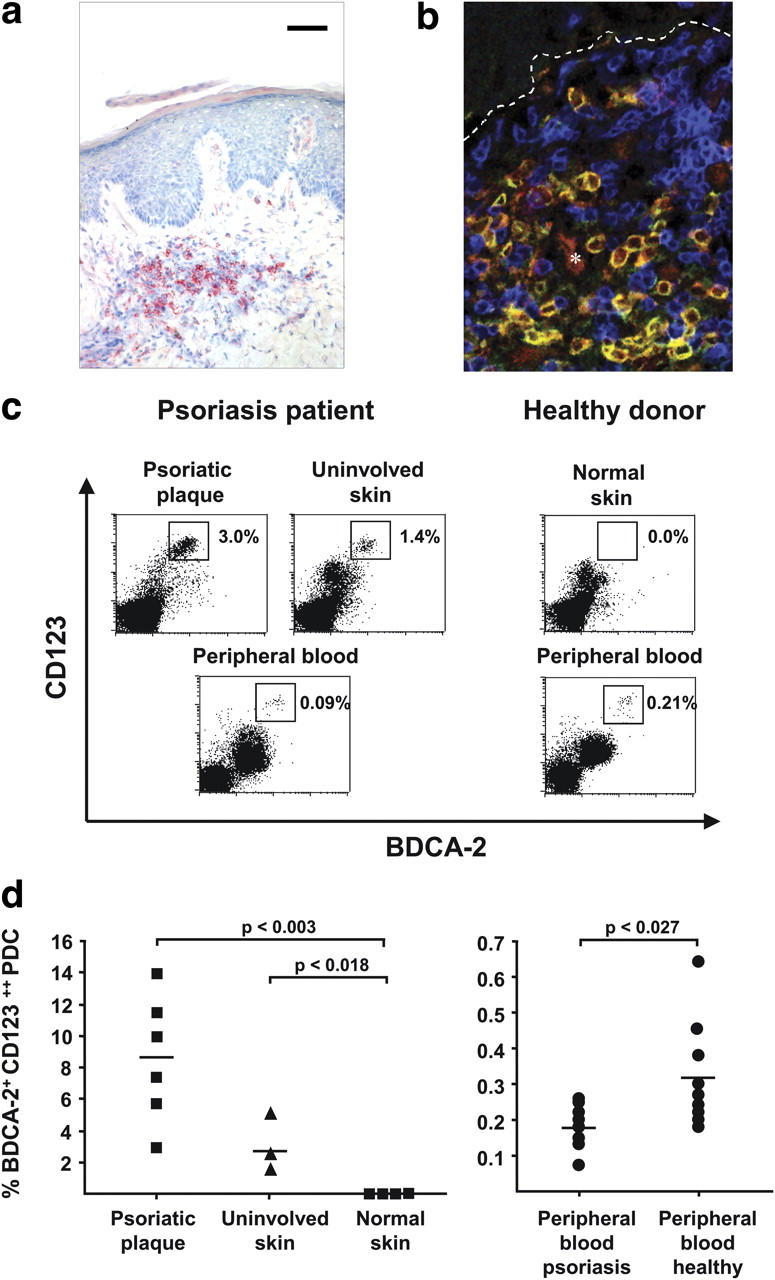
PDC infiltration in the skin of psoriasis patients. (a) Immunohistochemical analysis of BDCA-2 expression in psoriatic plaque lesions. A representative staining (n = 8) at 400× is shown. Bar, 40 μm. (b) CLSM of psoriatic plaque lesions stained for BDCA-2 (green), CD123 (red), and CD3 (blue). A representative analysis shows PDCs costaining for BDCA-2 and CD123 (yellow), T cells staining for CD3 (blue), and CD123 single-positive endothelial cells (*, red). Dashed lines indicate the border between the epidermis (top) and dermis (bottom). (c and d) Flow cytometric quantification of PDCs in dermal single cell suspensions isolated from psoriatic plaque lesions, uninvolved skin, normal skin, and peripheral blood of psoriatic patients and healthy individuals according to their BDCA-2+ CD123high phenotype. Representative flow cytometry analyses are shown in panel c and the percentages of BDCA-2+ CD123high PDCs are indicated in the plots. The results of multiple donors are plotted in panel d. Horizontal bars represent the mean.
PDCs are activated in psoriatic skin lesions
Given that PDCs are present in the peripheral blood, the uninvolved skin, and the plaque lesions of psoriatic patients, we sought to investigate the activation profile of PDCs in these three compartments by examining their expression levels of costimulatory molecules CD80 and CD86 and maturation marker CD83. Peripheral blood PDCs of psoriatic patients exhibited a typical resting phenotype of PDCs, lacking the expression of CD80 and CD83 and expressing low levels of CD86 (Fig. 2). A similar resting phenotype was observed in PDCs accumulating in the uninvolved skin of psoriasis patients (Fig. 2). In contrast, PDCs in psoriatic plaque lesions showed expression of CD80 and CD83 and enhanced levels of CD86 when compared with PDCs in uninvolved skin and peripheral blood (Fig. 2). These data indicate that PDCs are activated locally in psoriatic skin lesions.
Figure 2.

Local activation of PDCs in psoriatic plaque lesions. Flow cytometry analysis of CD86, CD80, and CD83 expression by BDCA-2+ CD123+ PDCs in dermal single cell suspensions of plaque lesions, uninvolved skin, and peripheral blood derived from the same psoriasis patient. Open histograms represent staining of DC activation markers; closed histograms represent isotype control. Data are representative of three independent experiments. Differences in the percentages of CD86high, CD80+, and CD83+ expression on PDCs between plaque lesions and uninvolved skin (P = 0.007, P = 0.02, and P = 0.005, respectively) and between plaque lesions and peripheral blood (P = 0.006, P = 0.02, and P = 0.005, respectively) were significant.
IFN-α is expressed early and transiently during the development of psoriasis
Because PDCs have the unique ability to produce large amounts of IFN-α on activation, we sought to analyze IFN-α expression in psoriatic skin. Initial investigations did not reveal substantial up-regulation of IFN-α mRNA in primary psoriatic plaque lesions compared with the normal skin of healthy individuals. However, psoriatic plaque lesions, but not uninvolved skin or normal skin, consistently demonstrated a significantly increased expression of IRF-7, an IFN-α–inducible gene (P < 0.0001; references 32 and 33; Fig. 3 a), and the presence of MxA protein, a marker for IFN-α activity (references 34 and 35; Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050500/DC1). Given the presence of an IFN-α signature in the absence of detectable levels of the IFN-α cytokine, we questioned whether IFN-α had been produced earlier during the development of the psoriatic plaque lesion. To analyze the early stages of psoriasis development, we took advantage of a xenograft model of human psoriasis developed in our laboratory in which the uninvolved (prepsoriatic) skin of psoriasis patients spontaneously converts into a full-fledged psoriatic skin lesion on transplantion onto AGR−/− mice within 35 d (36). This model system depends on the activation and proliferation of autoimmune T cells derived from the engrafted prepsoriatic skin (36) and allows temporal analyses of psoriasis development. Human IFN-α mRNA expression levels in skin grafts increased as early as day 7 after transplantation and reached a peak at day 14 before rapidly declining (Fig. 3 b, top left), whereas its signature, detected by increased IRF-7 expression levels, persisted throughout the 35-d development of the psoriatic plaque (Fig. 3 b, top right). It is noteworthy that skin grafts of control mice transplanted with the normal skin of healthy donors did not develop an IFN-α signature within 35 d (unpublished data). The early induction of IFN-α in engrafted prepsoriatic skin was paralleled by the activation and expansion of pathogenic T cells (Fig. 3 b, bottom). In contrast, the psoriatic phenotype, defined by the typical epidermal hyperplasia involving a thickening of the epidermis (acanthosis), as well as the elongation of the epidermal rete ridges (reflected in the papillomatosis index), showed delayed kinetics starting at day 21 after transplantation and reaching complete development at day 35 (Fig. 3 b, bottom). These data indicate that IFN-α expression is an early and transient event during the development of the psoriatic phenotype and precedes a completely activated epidermal compartment. During this early wave of IFN-α secretion, MxA expression was confined to cells of the dermal compartment and involvement of cells of the epidermal compartment occurred at later time points (after day 14 of engraftment; unpublished data), indicating that IFN-α originates in cells of the dermal compartment.
Figure 3.

Quantification of IFN-α and IRF-7 mRNA levels in primary plaque lesions and developing psoriasis. (a) Normalized IFN-α and IRF-7 mRNA expression (fg/25 ng of total mRNA) in primary psoriatic plaque lesions compared with normal skin and measured by quantitative real-time RT-PCR. The increased IRF-7 mRNA expression levels in psoriatic skin lesions (n = 24, mean = 32.09) compared with normal skin (n = 11, mean = 5.026) were significant (P < 0.0001, unpaired Student's t test). (b, top) Kinetics of normalized human IFN-α (gene copies per picogram of human mRNA) and IRF-7 (fg/10 pg of human mRNA) expression during psoriasis development in the AGR−/− xenograft model. (b, bottom) Human CD3+ T cells (open circles) and papillomatosis index (closed circles) during the development of psoriasis in the AGR−/− model. The arrow indicates the onset of a psoriatic phenotype defined by the typical epidermal hyperplasia involving both an increase in papillomatosis and acanthosis (not depicted) indices. Data represent the mean of three independent experiments.
PDCs are the principal IFN-α–producing cells in developing psoriasis
To identify the cellular source of IFN-α, we sampled the margin zone of spreading psoriatic plaque lesions as a surrogate for early developmental disease stages in psoriasis patients (37). Intracellular staining of dermal single cell suspensions demonstrated that IFN-α protein was expressed in developing psoriatic skin lesions and was confined to BDCA-2+ PDCs (Fig. 4). In contrast, IFN-α was not detectable in PDCs or other cells derived from uninvolved skin (Fig. 4) or peripheral blood (unpublished data) of the same psoriasis patient. These data indicate that PDCs represent the principal IFN-α–producing cell in developing psoriasis.
Figure 4.

IFN-α production in developing psoriatic lesions is mediated by PDCs. Flow cytometric analysis of surface BDCA-2 and intracellular IFN-α2 expression in dermal single cell suspensions derived from developing psoriatic lesions (across the margin zone of advancing psoriatic plaques) and uninvolved skin. In developing psoriatic lesions, intracellular IFN-α expression was detected among BDCA-2+ PDCs but completely absent in dermal cells lacking BDCA-2, a cell fraction which mainly consists of T cells and myeloid APCs. Data are representative of three independent experiments. Percentages of positive cells are indicated in each quadrant. Differences in the percentages of PDCs positively stained for IFN-α in comparison with matched isotype control, as well as differences in the percentages of IFN-α–expressing BDCA-2+ PDCs compared with non-PDCs, were significant (P < 0.003, unpaired Student's t test).
Blocking of IFN-α/β signaling inhibits the development of psoriasis
To further investigate the importance of PDC-derived IFN-α in the development of psoriasis, we sought to block IFN-α signaling in vivo during the spontaneous conversion of uninvolved skin into psoriatic skin lesions in the AGR−/− xenograft model perviously described (36). Transplanted mice were treated with either neutralizing antibodies to IFN-α/β receptor (38) or an isotype-matched control antibody, and histological analyses of grafts were performed at day 35. Whereas skin grafts from mice receiving the isotype-matched control antibodies developed into full-fledged psoriatic lesions, with T cell numbers and epidermal hyperplasia reaching similar levels to those of primary psoriatic plaques of the graft donor, treatment with neutralizing anti–IFN-α/β receptor antibody completely inhibited both the activation and expansion of pathogenic T cells (Fig. 5 a) and the development of the psoriatic phenotype (Fig. 5 b and Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20050500/DC1). These data indicate a requirement for IFN-α/β signaling for the T cell–dependent development of psoriasis.
Figure 5.

IFN-α/β signaling is required for the development of psoriatic skin lesions. Human CD3+ T cell count (a) and papillomatosis index in skin grafts before transplantation (uninvolved prepsoriatic skin at day 0) and 35 d after transplantation subsequent to the administration of isotype-matched control Ab or neutralizing anti–IFN-α/β receptor mAb (b). Values of primary psoriatic plaques derived from the graft donors are provided in comparison. Error bars in panel a represent one SD. Dots in panel b represent independently grafted mice or patient samples. p-values calculated by the unpaired Student's t test are indicated.
PDC-derived IFN-α is essential for the development of psoriasis
Because we have demonstrated that PDCs are a principal source of IFN-α in developing psoriatic lesions, we next sought to determine whether psoriasis development was mediated by PDC-derived IFN-α. For this purpose, we took advantage of the anti–BDCA-2 monoclonal antibody that specifically binds to human PDCs and suppresses their ability to secrete IFN-α (29). Given that human cells derived from the transplanted skin do not recirculate in the AGR−/− mice (36), in vivo treatment with anti–BDCA-2 selectively targets human PDCs present in engrafted prepsoriatic skin. Intravenous injection of anti–BDCA-2 antibody led to a >90% reduction of IFN-α expression in engrafted skin at day 13 after transplantation (Fig. 6 a), confirming that PDCs represented the principal IFN-α–producing cells in the engrafted skin. Anti–BDCA-2 antibody injections completely inhibited the activation and expansion of pathogenic T cells and the development of a psoriatic phenotype (Fig. 6, b and c; and Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20050500/DC1). To test whether IFN-α was sufficient to reverse the anti–BDCA-2–mediated inhibition of psoriasis development, we administered recombinant human IFN-α during anti–BDCA-2 antibody treatment. In contrast to the anti–BDCA-2 treatment alone, the addition of IFN-α induced a strong activation and expansion of pathogenic T cells and the development of a full-fledged psoriatic plaque lesion (Fig. 6, b–d), demonstrating that PDC-derived IFN-α was not only necessary but also sufficient to drive psoriasis development from prepsoriatic skin. In contrast, IFN-α was not able to induce T cell expansion and psoriasis development in normal skin transplanted onto AGR mice, suggesting that factors already present in normal-appearing prepsoriatic skin are a prerequisite for the IFN-α–mediated induction of the pathogenic T cell cascade leading to psoriasis (Fig. 6, b and c).
Figure 6.

PDC-derived IFN-α is necessary and sufficient to drive the development of psoriatic skin lesions. (a) Normalized human IFN-α mRNA expression in skin grafts harvested 13 d after transplantation subsequent to the administration of isotype-control Ab or anti–BDCA-2 mAb. The mean plus one SD of two independent experiments is given. Human CD3+ T cell count (b) and papillomatosis index (c) in skin grafts before transplantation (uninvolved skin at day 0) and 35 d after transplantation subsequent to the administration of isotype-matched control Ab, anti–BDCA-2 mAb, or anti–BDCA-2 plus recombinant human IFN-α. Dots represent independently grafted mice. In panel b, the mean of these experiments plus one SD is given. p-values were calculated using the unpaired Student's t test. As a control, normal skin (from healthy donors) was transplanted with or without systemic injections of recombinant human IFN-α. (d) Histologic analysis of the skin grafts harvested at day 35 after transplantation. The development of a psoriatic phenotype is inhibited by the administration of anti–BDCA-2 and fully restored by the addition of recombinant human IFN-α. The dashed line indicates the border between the epidermis and the dermis.
Discussion
Psoriasis is a Th1 cell–mediated autoimmune disease affecting the skin of genetically predisposed individuals. In recent years, there has been growing interest in the role of innate immunity to explain the interplay between environmental triggers and the exacerbation of the autoimmune T cell cascade leading to psoriasis. We uncovered a key innate immune pathway for triggering psoriasis. We show that PDCs accumulate in the skin of psoriasis patients and become activated to produce IFN-α early during disease development. Furthermore, we demonstrate that, through the production of IFN-α, PDCs drive the activation and expansion of autoimmune T cells in prepsoriatic skin, leading to the development of psoriasis. Thus, our findings revise the current immunopathogenic understanding of psoriasis by identifying a key proximal event based on the presence of PDCs in psoriatic skin and their innate activation to produce IFN-α.
PDCs represent a unique cell type in antiviral immunity because of their unique ability to secrete large amounts of IFN-α on viral stimulation through toll-like receptor (TLR)-7 and TLR-9 (39, 40). PDCs have also been found in the inflamed tissue of autoimmune diseases, including lupus erythematosus (15), rheumatoid arthritis (41), and psoriasis (17, 19), although a functional relevance had not been demonstrated. We provide the first direct evidence that PDCs play a key role in the elicitation of a common autoimmune disease. We show that PDCs are increased in the normal-appearing prepsoriatic skin of psoriasis patients compared with the skin of healthy controls and demonstrate that this accumulation represents a conditioning factor for future flares of the disease. Through their ability to secrete IFN-α in response to innate activation signals, PDCs in prepsoriatic skin determine the onset of local autoimmune inflammation leading to disease formation. Because injury to the skin is a well known elicitation factor for psoriasis (42), PDC activation may result from the release of skin-derived products on infectious or mechanical stress (simulated in the AGR−/− model by the transplantation procedure). In psoriatic skin, potential activation signals may include pathogen- or self-derived single-stranded RNA, recently identified as potent IFN-α inducers in PDCs through TLR-7 (43). Accordingly, we have observed that psoriasis can be exacerbated by topical application of imiquimod, a synthetic TLR-7 agonist (19). Furthermore, it has been recently demonstrated that TLR-mediated inflammation of the peripheral target organ is a prerequisite for the conversion of T cell autoreactivity into overt autoimmune disease (44).
Our study defines IFN-α as the molecular mediator of PDC function in eliciting psoriasis. PDC-derived IFN-α may drive the “quiescent” autoimmune T cells in prepsoriatic skin into activated pathogenic effectors through the induction of myeloid DC activation/maturation. An unabated activation of myeloid DCs through IFN-α with the consequent activation of autoimmune T cells has been recognized as a key pathogenic event in SLE (13, 45). In line with these findings, we have previously shown that myeloid DCs in psoriatic skin are also activated and have the ability to stimulate autoimmune T cells (46). The presence of high levels of lesional IFN-α might favor cross-presentation of sequestered tissue-specific autoantigens by myeloid DCs (47). PDC-derived IFN-α may also enhance the survival and expansion of T cells through the induction of IL-15 (48) or may act directly on T cells by promoting their expression of T-bet and IL12-Rβ2 (49), thus potentiating the Th1 cell bias of pathogenic T cells in psoriasis.
Our data provides the direct evidence that IFN-α is a master cytokine in psoriasis development. In contrast to TNF-α, a validated therapeutic target for psoriasis expressed throughout psoriatic inflammation (36, 50–52), production of IFN-α is a tightly regulated, transient event occurring early during the development of psoriasis. Furthermore, in contrast to the broad expression of TNF-α by a variety of cells, including myeloid DCs, T cells, and keratinocytes, the production of IFN-α in psoriatic skin seems to be confined to dermal PDCs. This may reflect the specialization of PDCs to recognize distinct TLR ligands (39, 40), as well as their extraordinary ability to produce large amounts of IFN-α, because of a prolonged endosomal retention of TLR ligands with consequent sustained MyD88/IRF-7 signaling (53). We therefore propose a spatial and temporal view of psoriasis development in which PDC-derived IFN-α represents an early upstream event preceding autoimmune inflammation and the development of psoriasis.
Increasing evidence indicates that IFN-α plays a pivotal role in the pathogenesis of other autoimmune disorders such as SLE, insulin-dependent diabetes mellitus (IDDM), and rheumatoid arthritis (for review see reference 54). As for psoriasis, IFN-α treatment for unrelated conditions can induce or exacerbate these autoimmune diseases (55–57). Evidence for a pathogenic role of IFN-α in SLE was provided by the finding that SLE patients have increased serum levels of IFN-α that coincide with exacerbations of the disease (58, 59). In addition, large numbers of PDCs are found in the skin lesions of SLE and may be activated to produce IFN-α by immune complexes consisting of anti–double-stranded DNA antibodies and DNA derived from apoptotic cells (60). Patients with IDDM demonstrate increased levels of IFN-α in the serum and in the pancreas (61). In addition, convincing evidence for a role of IFN-α in the pathogenesis of IDDM has been obtained from murine studies (44, 62). IFN-α signature has also been detected in the synovium of rheumatoid arthritis patients and has been correlated with the infiltration and activation of PDCs (63). Thus, the organ-specific accumulation of PDCs and their innate activation to produce IFN-α may represent a common proximal pathway that drives the efferent arm of immune responses leading to exacerbation of the underlying autoimmune diseases in susceptible individuals.
In conclusion, this study identifies PDCs and PDC-derived IFN-α as being important upstream initiators of psoriasis development. Given the side effects and limitations of current antipsoriatic therapies, including TNF-α blockers for long term disease control (64), we propose that new strategies targeting PDCs and PDC-derived IFN-α should be considered both for prevention and early therapeutic intervention in psoriasis and, potentially, other related autoimmune diseases.
MaterialS and Methods
Tissue samples, immunohistochemistry, and confocal laser scanning microscopy (CLSM) analysis.
Human psoriasis studies were approved by the Institutional Review Board of the University Hospital of Zurich. Biopsies from patients with stable plaque-type psoriasis were taken after informed consent was obtained and diagnosis of psoriasis was histologically confirmed. Cryopreserved skin specimens were fixed in acetone, subsequently stained with an excess of primary Ab, including anti–human BDCA-2 mAb (Miltenyi Biotec), anti–human MxA (a gift of J. Pavlovic, University of Zurich, Zurich, Switzerland), or IgG isotype control, and the signals were amplified by sequential incubations with rabbit anti–mouse IgG xenoantibodies and alkaline phosphatase–anti-alkaline phosphatase complexes as previously described (36). The immunoreaction was visualized with a developing solution containing neufuchsin (DakoCytomation) and counterstained with 1% hematoxylin. For immunofluorescence analysis, acetone-fixed cryopreserved specimens were stained with unlabeled anti–BDCA-2 mAb followed by FITC-labeled anti–mouse IgG mAb (DakoCytomation), anti-CD123 PE (BD Biosciences), and anti-CD3 APCs (BD Biosciences) for 1 h each. Immunofluorescence was analyzed by CLSM (DM IRB/E; Leica).
Isolation of dermal cells and flow cytometry analysis.
Skin keratome biopsies (20 × 20 × 0.4 mm) of plaque psoriasis lesions and uninvolved skin (normal-appearing skin at a distance of ≥0.5 cm from the lesion) were taken from the buttocks or upper thigh of patients with confirmed plaque-type psoriasis as described previously (46). Patients did not receive topical or systemic therapy for at least 4 wk before the study. For analysis of developing psoriasis lesions, biopsies were taken across the margin zone (immediately inside and outside of the clinical edge) of spreading psoriatic lesions as defined clinically (37). Dermal sheets were separated from epidermal sheets, cut into small pieces (1–5 mm), and carefully enzymatically digested to yield single cell suspensions as previously described (46). Three-color staining was performed by using anti–BDCA-2 FITC, CD123 APCs (Miltenyi Biotech), and PE-labeled CD3, CD4, CD11b, CD11c, CD14, CD20, CD56, CD80, CD86, CD83, and HLA-DR (all obtained from Becton Dickinson) or their corresponding isotype controls. For intracellular IFN-α detection, cells were first surface stained with anti–BDCA-2 FITC, followed by permeabilization and incubation with PE-labeled anti–human IFN-α2 mAb (Chromaprobe Inc.) or IgG2b isotype control. Cells were analyzed using a flow cytometer (FACSCalibur; Becton Dickinson) and data were processed using CellQuestPro (Becton Dickinson).
Real-time quantitative PCR.
Total RNA from homogenized skin specimens was extracted and reverse transcribed as previously described (36). Complementary DNA was quantitatively analyzed for the expression of IFN-α and IRF-7 transcripts by real-time PCR using primers designed against most human IFN-α sequences and against human IRF-7 (left, 5′-TCCCCACGCTATACCATCTACCT-3′; right, 5′-ACAGCCAGGGTTCCAGCTT-3′; Applied Biosystems). 18S ribosomal RNA was used for normalization. In the AGR model, IFN-α was quantified in transplanted skin by using a primer kit recognizing most human IFN-α genes and that did not recognize its mouse counterpart (Search-LC). Human GAPDH mRNA levels were quantified using human-specific primers (left, 5′-ATTGCCCTCAACGACCACTTTG-3′; right, 5′-TTGATGGTACATGAAAGGTGAGG-3′) and used for normalization as previously described (36).
Animals and transplantation procedure.
Animal studies were approved by the Kantonale Veterinaersamt of Zurich. AGR129 mice, deficient in type I (A) and type II (G) IFN receptors, in addition to being RAG-2−/−, were kept pathogen free throughout the study. Keratomes of uninvolved prepsoriatic or normal skin (a gift of S. Baldi, University Hospital of Zurich, Zurich Switzerland) as controls were transplanted to the back of mice using an absorbable tissue seal as previously described (36). 35 d after engraftment, transplanted skin was removed and snap frozen for histological or mRNA expression analysis. CD3+ T cell counts, acanthosis, and papillomatosis index were determined histologically as previously described (36). CD3+ T cell values represent the mean cell count of three random fields assessed at 400× by two independent investigators. The indicated papillomatosis and acanthosis values represent the mean of 10 random areas of each sample.
Neutralization studies.
Dosage and schedule of antibody administration were deduced based on previous data with anti–human mAbs against other cell surface molecules (36) and administered as follows: (a) i.v. injection of 30 μg neutralizing anti–human IFN-α/β Receptor Chain 2 (CD118) mAb (clone MMHAR-2; PBL Biomedical Laboratories) twice weekly for 35 d, starting at day 0 after transplantation; and (b) i.v. injection of 30 μg anti–BDCA-2 mAb (reference 29; provided by J. Schmitz, Miltenyi Biotec, Bergisch-Gladbach, Germany) twice weekly for 35 d, starting at day 0 after transplantation. For IFN-α reconstitution experiments, 30,000 IU recombinant human IFN-α2a (Roferon A; Roche) were administered systemically by s.c. injections three times a week for 35 d. Dosage corresponds to the therapeutic dose of 8 Mio IU used in humans and was deduced by an allometric approach as previously described (36).
Online supplemental material.
Fig. S1 shows absent expression of BDCA-2 in normal skin and atopic dermatitis skin. Fig. S2 shows plasmacytoid morphology and the classical phenotype of PDCs in psoriatic lesions. Fig. S3 shows IFN-α activity in psoriatic plaque lesions but not the uninvolved skin of psoriatic patients, normal skin of healthy individuals, or skin lesions of atopic dermatitis patients. Figs. S4 and S5 show induction and inhibition of psoriasis development in the AGR model as measured by the epidermal acanthosis index. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050500/DC1.
Acknowledgments
We would like to thank J. Schmitz for providing anti–human BDCA-2 mAb, S. Baldi for the specimen of normal skin, T. Baechi for helping in performing confocal microscopy, and the excellent technical assistance by A. Perez, C. Dudli, and G. Tonel.
This work was funded by a grant from the Swiss National Science Foundation (32-100833/1) to M. Gilliet and F.O. Nestle.
The authors have no conflicting financial interests.
Abbreviations used: CLSM, confocal laser scanning microscopy; IDDM, insulin- dependent diabetes mellitus; IRF, IFN regulatory factor; PDC, plasmacytoid pre-DC; SLE, systemic lupus erythematosus; TLR, toll-like receptor.
M. Gilliet's present address is Dept. of Immunology, M.D. Anderson Cancer Center, Houston, TX 77030.
References
- 1.Lebwohl, M. 2003. Psoriasis. Lancet. 361:1197–1204. [DOI] [PubMed] [Google Scholar]
- 2.Lew, W., A.M. Bowcock, and J.G. Krueger. 2004. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and ‘Type 1’ inflammatory gene expression. Trends Immunol. 25:295–305. [DOI] [PubMed] [Google Scholar]
- 3.Adorini, L., and F. Sinigaglia. 1997. Pathogenesis and immunotherapy of autoimmune diseases. Immunol. Today. 18:209–211. [DOI] [PubMed] [Google Scholar]
- 4.Davidson, A., and B. Diamond. 2001. Autoimmune diseases. N. Engl. J. Med. 345:340–350. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths, C.E., and J.J. Voorhees. 1996. Psoriasis, T cells and autoimmunity. J. R. Soc. Med. 89:315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nickoloff, B.J., and F.O. Nestle. 2004. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J. Clin. Invest. 113:1664–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowes, M.A., W. Lew, and J.G. Krueger. 2004. Current concepts in the immunopathogenesis of psoriasis. Dermatol. Clin. 22:349–369. [DOI] [PubMed] [Google Scholar]
- 8.Liu, Y.J. 2005. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 23:275–306. [DOI] [PubMed] [Google Scholar]
- 9.Siegal, F.P., N. Kadowaki, M. Shodell, P.A. Fitzgerald-Bocarsly, K. Shah, S. Ho, S. Antonenko, and Y.J. Liu. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science. 284:1835–1837. [DOI] [PubMed] [Google Scholar]
- 10.Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919–923. [DOI] [PubMed] [Google Scholar]
- 11.Kadowaki, N., S. Antonenko, J.Y. Lau, and Y.J. Liu. 2000. Natural interferon α/β–producing cells link innate and adaptive immunity. J. Exp. Med. 192:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fonteneau, J.F., M. Gilliet, M. Larsson, I. Dasilva, C. Munz, Y.J. Liu, and N. Bhardwaj. 2003. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 101:3520–3526. [DOI] [PubMed] [Google Scholar]
- 13.Blanco, P., A.K. Palucka, M. Gill, V. Pascual, and J. Banchereau. 2001. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 294:1540–1543. [DOI] [PubMed] [Google Scholar]
- 14.Vanbervliet, B., N. Bendriss-Vermare, C. Massacrier, B. Homey, O. de Bouteiller, F. Briere, G. Trinchieri, and C. Caux. 2003. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell–derived factor 1 (SDF-1)/CXCL12. J. Exp. Med. 198:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farkas, L., K. Beiske, F. Lund-Johansen, P. Brandtzaeg, and F.L. Jahnsen. 2001. Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am. J. Pathol. 159:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jahnsen, F.L., F. Lund-Johansen, J.F. Dunne, L. Farkas, R. Haye, and P. Brandtzaeg. 2000. Experimentally induced recruitment of plasmacytoid (CD123high) dendritic cells in human nasal allergy. J. Immunol. 165:4062–4068. [DOI] [PubMed] [Google Scholar]
- 17.Wollenberg, A., M. Wagner, S. Gunther, A. Towarowski, E. Tuma, M. Moderer, S. Rothenfusser, S. Wetzel, S. Endres, and G. Hartmann. 2002. Plasmacytoid dendritic cells: a new cutaneous dendritic cell subset with distinct role in inflammatory skin diseases. J. Invest. Dermatol. 119:1096–1102. [DOI] [PubMed] [Google Scholar]
- 18.Bangert, C., J. Friedl, G. Stary, G. Stingl, and T. Kopp. 2003. Immunopathologic features of allergic contact dermatitis in humans: participation of plasmacytoid dendritic cells in the pathogenesis of the disease? J. Invest. Dermatol. 121:1409–1418. [DOI] [PubMed] [Google Scholar]
- 19.Gilliet, M., C. Conrad, M. Geiges, A. Cozzio, W. Thurlimann, G. Burg, F.O. Nestle, and R. Dummer. 2004. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch. Dermatol. 140:1490–1495. [DOI] [PubMed] [Google Scholar]
- 20.van der Fits, L., L.I. van der Wel, J.D. Laman, E.P. Prens, and M.C. Verschuren. 2004. In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-alpha sensitivity is unaltered. J. Invest. Dermatol. 122:51–60. [DOI] [PubMed] [Google Scholar]
- 21.Schmid, P., P. Itin, D. Cox, G.K. McMaster, and M.A. Horisberger. 1994. The type I interferon system is locally activated in psoriatic lesions. J. Interferon Res. 14:229–234. [DOI] [PubMed] [Google Scholar]
- 22.Fah, J., J. Pavlovic, and G. Burg. 1995. Expression of MxA protein in inflammatory dermatoses. J. Histochem. Cytochem. 43:47–52. [DOI] [PubMed] [Google Scholar]
- 23.Suomela, S., L. Cao, A. Bowcock, and U. Saarialho-Kere. 2004. Interferon alpha-inducible protein 27 (IFI27) is upregulated in psoriatic skin and certain epithelial cancers. J. Invest. Dermatol. 122:717–721. [DOI] [PubMed] [Google Scholar]
- 24.Hida, S., K. Ogasawara, K. Sato, M. Abe, H. Takayanagi, T. Yokochi, T. Sato, S. Hirose, T. Shirai, S. Taki, and T. Taniguchi. 2000. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity. 13:643–655. [DOI] [PubMed] [Google Scholar]
- 25.Funk, J., T. Langeland, E. Schrumpf, and L.E. Hanssen. 1991. Psoriasis induced by interferon-alpha. Br. J. Dermatol. 125:463–465. [DOI] [PubMed] [Google Scholar]
- 26.Pauluzzi, P., F. Kokelj, V. Perkan, G. Pozzato, and M. Moretti. 1993. Psoriasis exacerbation induced by interferon-alpha. Report of two cases. Acta Derm. Venereol. 73:395. [DOI] [PubMed] [Google Scholar]
- 27.Downs, A.M., and M.G. Dunnill. 2000. Exacerbation of psoriasis by interferon-alpha therapy for hepatitis C. Clin. Exp. Dermatol. 25:351–352. [DOI] [PubMed] [Google Scholar]
- 28.Ketikoglou, I., S. Karatapanis, I. Elefsiniotis, G. Kafiri, and A. Moulakakis. 2005. Extensive psoriasis induced by pegylated interferon alpha-2b treatment for chronic hepatitis B. Eur. J. Dermatol. 15:107–109. [PubMed] [Google Scholar]
- 29.Dzionek, A., Y. Sohma, J. Nagafune, M. Cella, M. Colonna, F. Facchetti, G. Gunther, I. Johnston, A. Lanzavecchia, T. Nagasaka, et al. 2001. BDCA-2, a novel plasmacytoid dendritic cell–specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon α/β induction. J. Exp. Med. 194:1823–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grouard, G., M.C. Rissoan, L. Filgueira, I. Durand, J. Banchereau, and Y.J. Liu. 1997. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J. Exp. Med. 185:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cederblad, B., S. Blomberg, H. Vallin, A. Perers, G.V. Alm, and L. Ronnblom. 1998. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-alpha-producing cells. J. Autoimmun. 11:465–470. [DOI] [PubMed] [Google Scholar]
- 32.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya, M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 13:539–548. [DOI] [PubMed] [Google Scholar]
- 33.Kawai, T., S. Sato, K.J. Ishii, C. Coban, H. Hemmi, M. Yamamoto, K. Terai, M. Matsuda, J. Inoue, S. Uematsu, et al. 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5:1061–1068. [DOI] [PubMed] [Google Scholar]
- 34.Ronni, T., K. Melen, A. Malygin, and I. Julkunen. 1993. Control of IFN-inducible MxA gene expression in human cells. J. Immunol. 150:1715–1726. [PubMed] [Google Scholar]
- 35.Roers, A., H.K. Hochkeppel, M.A. Horisberger, A. Hovanessian, and O. Haller. 1994. MxA gene expression after live virus vaccination: a sensitive marker for endogenous type I interferon. J. Infect. Dis. 169:807–813. [DOI] [PubMed] [Google Scholar]
- 36.Boyman, O., H.P. Hefti, C. Conrad, B.J. Nickoloff, M. Suter, and F.O. Nestle. 2004. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-α. J. Exp. Med. 199:731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vissers, W.H., C.H. Arndtz, L. Muys, P.E. Van Erp, E.M. De Jong, and P.C. Van De Kerkhof. 2004. Memory effector (CD45RO+) and cytotoxic (CD8+) T cells appear early in the margin zone of spreading psoriatic lesions in contrast to cells expressing natural killer receptors, which appear late. Br. J. Dermatol. 150:852–859. [DOI] [PubMed] [Google Scholar]
- 38.Colamonici, O.R., and P. Domanski. 1993. Identification of a novel subunit of the type I interferon receptor localized to human chromosome 21. J. Biol. Chem. 268:10895–10899. [PubMed] [Google Scholar]
- 39.Kadowaki, N., S. Ho, S. Antonenko, R.W. Malefyt, R.A. Kastelein, F. Bazan, and Y.J. Liu. 2001. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194:863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jarrossay, D., G. Napolitani, M. Colonna, F. Sallusto, and A. Lanzavecchia. 2001. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 31:3388–3393. [DOI] [PubMed] [Google Scholar]
- 41.Van Krinks, C.H., M.K. Matyszak, and J.S. Gaston. 2004. Characterization of plasmacytoid dendritic cells in inflammatory arthritis synovial fluid. Rheumatology (Oxford). 43:453–460. [DOI] [PubMed] [Google Scholar]
- 42.Kupper, T.S., and R.C. Fuhlbrigge. 2004. Immune surveillance in the skin: mechanisms and clinical consequences. Nat. Rev. Immunol. 4:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diebold, S.S., T. Kaisho, H. Hemmi, S. Akira, and E.S.C. Reis. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 44.Lang, K.S., M. Recher, T. Junt, A.A. Navarini, N.L. Harris, S. Freigang, B. Odermatt, C. Conrad, L.M. Ittner, S. Bauer, et al. 2005. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat. Med. 11:138–145. [DOI] [PubMed] [Google Scholar]
- 45.Banchereau, J., V. Pascual, and A.K. Palucka. 2004. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 20:539–550. [DOI] [PubMed] [Google Scholar]
- 46.Nestle, F.O., L.A. Turka, and B.J. Nickoloff. 1994. Characterization of dermal dendritic cells in psoriasis. Autostimulation of T lymphocytes and induction of Th1 type cytokines. J. Clin. Invest. 94:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Bon, A., N. Etchart, C. Rossmann, M. Ashton, S. Hou, D. Gewert, P. Borrow, and D.F. Tough. 2003. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 4:1009–1015. [DOI] [PubMed] [Google Scholar]
- 48.Zhang, X., S. Sun, I. Hwang, D.F. Tough, and J. Sprent. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 8:591–599. [DOI] [PubMed] [Google Scholar]
- 49.Hibbert, L., S. Pflanz, R. De Waal Malefyt, and R.A. Kastelein. 2003. IL-27 and IFN-alpha signal via Stat1 and Stat3 and induce T-Bet and IL-12Rbeta2 in naive T cells. J. Interferon Cytokine Res. 23:513–522. [DOI] [PubMed] [Google Scholar]
- 50.Leonardi, C.L., J.L. Powers, R.T. Matheson, B.S. Goffe, R. Zitnik, A. Wang, and A.B. Gottlieb. 2003. Etanercept as monotherapy in patients with psoriasis. N. Engl. J. Med. 349:2014–2022. [DOI] [PubMed] [Google Scholar]
- 51.Chaudhari, U., P. Romano, L.D. Mulcahy, L.T. Dooley, D.G. Baker, and A.B. Gottlieb. 2001. Efficacy and safety of infliximab monotherapy for plaque-type psoriasis: a randomised trial. Lancet. 357:1842–1847. [DOI] [PubMed] [Google Scholar]
- 52.Schottelius, A.J., L.L. Moldawer, C.A. Dinarello, K. Asadullah, W. Sterry, and C.K. Edwards III. 2004. Biology of tumor necrosis factor-alpha: implications for psoriasis. Exp. Dermatol. 13:193–222. [DOI] [PubMed] [Google Scholar]
- 53.Honda, K., Y. Ohba, H. Yanai, H. Negishi, T. Mizutani, A. Takaoka, C. Taya, and T. Taniguchi. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 434:1035–1040. [DOI] [PubMed] [Google Scholar]
- 54.Theofilopoulos, A.N., R. Baccala, B. Beutler, and D.H. Kono. 2005. Type I interferons in immunity and autoimmunity. Annu. Rev. Immunol. 23:307–335. [DOI] [PubMed] [Google Scholar]
- 55.Gota, C., and L. Calabrese. 2003. Induction of clinical autoimmune disease by therapeutic interferon-alpha. Autoimmunity. 36:511–518. [DOI] [PubMed] [Google Scholar]
- 56.Guerci, A.P., B. Guerci, C. Levy-Marchal, J. Ongagna, O. Ziegler, H. Candiloros, O. Guerci, and P. Drouin. 1994. Onset of insulin-dependent diabetes mellitus after interferon-alpha therapy for hairy cell leukaemia. Lancet. 343:1167–1168. [DOI] [PubMed] [Google Scholar]
- 57.Passos de Souza, E., P.T. Evangelista Segundo, F.F. Jose, D. Lemaire, and M. Santiago. 2001. Rheumatoid arthritis induced by alpha-interferon therapy. Clin. Rheumatol. 20:297–299. [DOI] [PubMed] [Google Scholar]
- 58.Bengtsson, A.A., G. Sturfelt, L. Truedsson, J. Blomberg, G. Alm, H. Vallin, and L. Ronnblom. 2000. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 9:664–671. [DOI] [PubMed] [Google Scholar]
- 59.Bennett, L., A.K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, and V. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vallin, H., A. Perers, G.V. Alm, and L. Ronnblom. 1999. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J. Immunol. 163:6306–6313. [PubMed] [Google Scholar]
- 61.Huang, X., J. Yuang, A. Goddard, A. Foulis, R.F. James, A. Lernmark, R. Pujol-Borrell, A. Rabinovitch, N. Somoza, and T.A. Stewart. 1995. Interferon expression in the pancreases of patients with type I diabetes. Diabetes. 44:658–664. [DOI] [PubMed] [Google Scholar]
- 62.Stewart, T.A., B. Hultgren, X. Huang, S. Pitts-Meek, J. Hully, and N.J. MacLachlan. 1993. Induction of type I diabetes by interferon-alpha in transgenic mice. Science. 260:1942–1946. [DOI] [PubMed] [Google Scholar]
- 63.Lande, R., E. Giacomini, B. Serafini, B. Rosicarelli, G.D. Sebastiani, G. Minisola, U. Tarantino, V. Riccieri, G. Valesini, and E.M. Coccia. 2004. Characterization and recruitment of plasmacytoid dendritic cells in synovial fluid and tissue of patients with chronic inflammatory arthritis. J. Immunol. 173:2815–2824. [DOI] [PubMed] [Google Scholar]
- 64.Sterry, W., J. Barker, W.H. Boehncke, J.D. Bos, S. Chimenti, E. Christophers, M. De La Brassinne, C. Ferrandiz, C. Griffiths, A. Katsambas, et al. 2004. Biological therapies in the systemic management of psoriasis: International Consensus Conference. Br. J. Dermatol. 151:3–17. [DOI] [PubMed]