

Abstract
In the central nervous system (CNS), the transcription factor nuclear factor (NF)-κB is a key regulator of inflammation and secondary injury processes. After trauma or disease, the expression of NF-κB–dependent genes is highly activated, leading to both protective and detrimental effects on CNS recovery. We demonstrate that selective inactivation of astroglial NF-κB in transgenic mice expressing a dominant negative (dn) form of the inhibitor of κBα under the control of an astrocyte-specific promoter (glial fibrillary acidic protein [GFAP]–dn mice) leads to a dramatic improvement in functional recovery 8 wk after contusive spinal cord injury (SCI). Histologically, GFAP mice exhibit reduced lesion volume and substantially increased white matter preservation. In parallel, they show reduced expression of proinflammatory chemokines and cytokines, such as CXCL10, CCL2, and transforming growth factor–β2, and of chondroitin sulfate proteoglycans participating in the formation of the glial scar. We conclude that selective inhibition of NF-κB signaling in astrocytes results in protective effects after SCI and propose the NF-κB pathway as a possible new target for the development of therapeutic strategies for the treatment of SCI.
In spinal cord injury (SCI) progression, the first phase of damage, dependent on mechanical destruction of the nervous tissue, is followed by a secondary phase caused by severe local disturbance of the blood supply and a massive release of proinflammatory mediators and neurotoxins from invading and resident cells (1). Although inflammation is the physiological process by which vascularized tissues respond to injury, an overactivation of this process can lead to additional damage to the spinal cord, preventing neural recovery and regeneration and, ultimately, resulting in worsening of the clinical outcome (2). A prominent source of inflammatory mediators is astroglia, which, in response to injury, undergoes a profound activation known as reactive astrogliosis, whose biological importance is still a matter of debate (3). Even though reactive astrocytes release factors essential for neuronal survival and wound healing (4, 5), they are also responsible for the production of molecules detrimental to functional recovery and for creating an environment unfavorable to nerve regeneration (6).
Many of the processes occurring in reactive astrocytes as a consequence of central nervous system (CNS) damage are regulated by NF-κB (7). In brain injury and SCI models, NF-κB is highly activated (8–11) and the expression of NF-κB–dependent genes is up-regulated (12, 13), indicating a critical function of this factor in CNS pathophysiology. NF-κB activation in astrocytes induces both detrimental and protective effects on CNS recovery. As for the detrimental function, NF-κB activation promotes the transition of astrocytes to a substrate nonpermissive to neurite outgrowth (14). Inhibition of NF-κB down-regulates the glial response to excitotoxic brain injury, resulting in neuroprotection (15). As for the protective function, NF-κB activation in astrocytes leads to the synthesis of neurotrophins (nerve growth factor and brain-derived neurotrophic factor) essential for neuronal survival (16). After brain injury, NF-κB–dependent expression of calbindin exerts a cytoprotective effect preventing astrocyte apoptosis (17). Collectively, these reports point to a complex function of NF-κB in astroglial cells, whose biological importance still needs to be fully clarified.
To address the contribution of astroglial NF-κB to CNS pathophysiology, we generated a transgenic (TG) mouse line in which NF-κB was selectively inactivated in astrocytes by targeting the overexpression of a dominant negative (dn) form of the inhibitor of κBα (IκBα) to astrocytes by means of the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter. Taking advantage of this model, we provide evidence that inactivation of NF-κB in astrocytes has beneficial implications in SCI that result in the reduction of lesion volume, increased white matter preservation, reduction in proteoglycan expression, modulation of the inflammatory response, and dramatically improved functional recovery after contusive injury.
Results
Generation and characterization of GFAP-IκBα-dn mice
Unlike whole animal knockouts of p65 and p50 that display either lethal (p65) or severe (p50) immunological complications (18, 19), GFAP-IκBα-dn mice survive normally to adulthood and do not exhibit any apparent abnormal sensorimotor phenotype. Transgene expression, measured by RT-PCR, was detected in the CNS (both brain and spinal cord) and in the peripheral nervous system (sciatic nerve; Fig. 1 A). This latter finding is justified by the well-documented expression of GFAP in nonmyelinating Schwann cells (20). No ectopic expression was detected in the heart, lung, kidney, liver, or spleen (Fig. 1 A). To functionally confirm the ability of truncated IκBα to prevent NF-κB activation, a series of electrophoretic mobility shift assays (EMSAs) was performed on nuclear extracts from astrocytes isolated from WT and GFAP-IκBα-dn mice (Fig. 1, B and C). WT astrocytes stimulated with TNF-α exhibited a strong induction of NF-κB DNA binding activity, whereas no induction was detected in TG astrocytes (Fig. 1 B). To confirm the cell specificity of transgene expression, similar EMSA experiments were performed on neurons from GFAP-IκBα-dn mice and WT littermates (Fig. 1 C). As expected, when stimulated with TNF-α, GFAP-IκBα-dn neurons showed an increase in NF-κB DNA binding activity comparable to WT neurons, demonstrating the complete functionality of the NF-κB pathway in this cell type (Fig. 1 C). To identify the molecular composition of the NF-κB dimers, supershift experiments were performed on astrocyte cultures from WT mice (Fig. 1 D). Preincubation of extracts from TNF-α–treated cultures with anti-p65 and -p50 antibodies induced a supershift or an attenuation of the specific electrophoretic band, respectively, identifying the p65–p50 dimer as the main NF-κB complex recruited after TNF-α treatment in our model.
Figure 1.

Biochemical characterization of GFAP-IκBα-dn mice. (A) RT-PCR analysis of GFAP-IκBα-dn (IκBα-dn) expression in the brain (Br), spinal cord (SC), sciatic nerve (SN), heart (Ht), kidney (Kd), lung (Ln), liver (Lv), and spleen (Sp). β-Actin was amplified as a control. Samples lacking RT (−RT) were amplified as controls for genomic DNA contamination. Genomic DNA from a TG animal and water were used as positive (+) and negative (−) controls for the PCR reaction, respectively. (B) Inhibition of NF-κB DNA binding activity exclusively in astrocytes from GFAP-IκBα-dn (IκBα-dn) mice. Cultures were treated with TNF-α (+, 10 ng/ml for 30 min) or medium alone (−). (C) Induction of NF-κB DNA binding activity exclusively in neurons from GFAP-IκBα-dn (IκBα-dn) mice. Cultures were treated with TNF-α (+, 10 ng/ml for 30 min) or medium alone (−). (D) NF-κB supershift in TNF-α–stimulated WT astrocytes. Nuclear extracts from WT astrocytes, treated with TNF-α (+, 10 ng/ml for 30 min) or medium alone (−) were preincubated with anti-p65 or -p50 antibodies. The supershifted p65–p50 dimer is detected in the top left (ss). Binding specificity to the NF-κB consensus sequence was demonstrated by displacement of the 32P-labeled NF-κB oligonucleotide in the presence of 100-fold excess cold NF-κB and by a lack of displacement in the presence of 100-fold excess cold AP-1.
To investigate possible phenotypic abnormalities resulting from the genetic manipulation, a series of behavioral tests was conducted. Locomotor behavior in a novel environment was assessed with the open field and grid walk tests. In both tasks, male and female GFAP-IκBα-dn mice performed equally to their WT counterparts (Fig. S1, A and B, available at http://www.jem.org/cgi/content/full/jem.20041918/DC1), indicating the absence of gross locomotor deficits.
Sensorimotor function was further evaluated with the following tests: vibrissae-elicited and visually elicited reflexive placing of forelimbs, wire test, tactile/proprioceptive hindlimb placing, grip strength, and balance beam assessments. In all tests, both male and female GFAP-IκBα-dn mice performed equally to WT mice (unpublished data). In addition, WT and GFAP-IκBα-dn mice performed comparably well in the visual cliff test, indicating the absence of visual acuity deficits (unpublished data). Finally, when tested with the elevated plus maze and the light/dark transition test, GFAP-IκBα-dn mice exhibited levels of anxiety similar to WT littermates (unpublished data). Together, these data indicate the absence of gross behavioral abnormalities related to the expression of the transgene, validating our experimental model as suitable for CNS injury studies.
Inactivation of astroglial NF-κB has no effect on spinal cord architecture, neuronal number, or constitutive cell death
Standard cresyl violet histopathology and immunohistochemistry with the astrocyte-specific marker GFAP and neuron-specific Nissl staining did not reveal any gross morphological anomalies in the architecture of the naive spinal cord in GFAP-IκBα-dn mice (Fig. S2, A–D, available at http://www.jem.org/cgi/content/full/jem.20041918/DC1). To rule out any subtle abnormality undetectable by standard qualitative histology, we stereologically counted the number of neurons in naive WT and GFAP-IκBα-dn spinal cords based on NeuN immunostaining. No difference was detected between the two genotypes (neuronal number ± SEM: WT, 370,479 ± 12,948; GFAP, 377,895 ± 20,308), suggesting that inactivating NF-κB in astrocytes does not lead to developmental anomalies in the neuronal population (Fig. S2 E).
Based on the well-documented implication of NF-κB in regulating both pro- and antiapoptotic mechanisms (21), we further investigated whether GFAP-IκBα-dn mice displayed a different pattern of cell death when compared with WT mice. TdT-mediated dUTP-biotin nick-end labeling (TUNEL) staining of the naive spinal cord revealed the absence of constitutive cell death (absence of TUNEL+ cells) in both WT and GFAP-IκBα-dn mice (Fig. S2 F), suggesting that, at least in the spinal cord, astroglial NF-κB is not a critical regulator of apoptotic/necrotic programs under nonpathologic conditions.
Activation of NF-κB induced by SCI is prevented in GFAP-IκBα-dn mice
The role of astroglial NF-κB in SCI pathophysiology was evaluated in a model of moderate traumatic injury. We initially demonstrated that activation of NF-κB occurred after injury in WT mice (Fig. 2). NF-κB activation was evaluated by Western blot with an antibody that specifically recognizes an epitope overlapping the nuclear localization signal of the p65 subunit and, thus, selectively binds to the activated form of p65 (p65*). Western blot analysis indicated increased NF-κB activation as early as 1 h after SCI that persisted for up to 3 d. These results parallel our previous findings in a similar contusion model in the rat (8). Activated p65 was detected as a doublet of bands migrating at an apparent molecular mass of 65 and 67 kD, respectively, with the heavier band possibly representing a hyperphosphorylated form of p65. Indeed, it has been reported that phosphorylation enhances the transactivation potential of p65, which is required for optimal NF-κB activation (22). Remarkably, SCI-induced NF-κB activation was almost completely prevented in GFAP-IκBα-dn mice, where only a mild increase in activated p65 could be detected at 1 and 3 d.
Figure 2.

Time course of NF-κB activation after SCI in WT and GFAP-IκBα-dn mice. NF-κB activation was evaluated by Western blot analysis with an antibody that specifically recognizes the activated form of p65 (p65*). The arrowhead indicates p65* migrating at exactly 65 kD. 20 μg of proteins/sample were loaded, and blots were probed for β-actin as a control. A representative blot is shown (n = 3). nv, naive.
Inactivation of astroglial NF-κB improves functional recovery after SCI
To assess whether the blockade of NF-κB had any effect on functional recovery after SCI, locomotor performance in the open field was recorded weekly for up to 8 wk after injury and scored with the Basso mouse scale (BMS; reference 23). Compared with WT animals, GFAP-IκBα-dn mice exhibited a dramatically improved functional recovery that was statistically significant as early as 1 d after injury (P < 0.04; Fig. 3 A). Although recovery reached a plateau 2 wk after SCI in WT mice (final BMS ± SEM: 3.3 ± 0.2), GFAP-IκBα-dn mice showed a progressive gain of function until 6 wk after injury (final BMS ± SEM: 6.3 ± 0.4). Histological analysis of the injured cords 8 wk after SCI showed an apparently smaller lesion in GFAP-IκBα-dn mice compared with WT (Fig. 3 B). Intense GFAP immunoreactivity was detected in both genotypes around the lesion and in the surrounding white matter, indicating the formation of a glial scar. At the site of impact, WT and TG mice displayed an equivalent degree of damage (Fig. 3 B, cross sections). As typical of spinal cord lesions in mice, no cavitation or vacuolization of the tissue was observed. Rather, the lesion area was filled with cells packed at high density in both genotypes (Fig. 3 B, blue).
Figure 3.
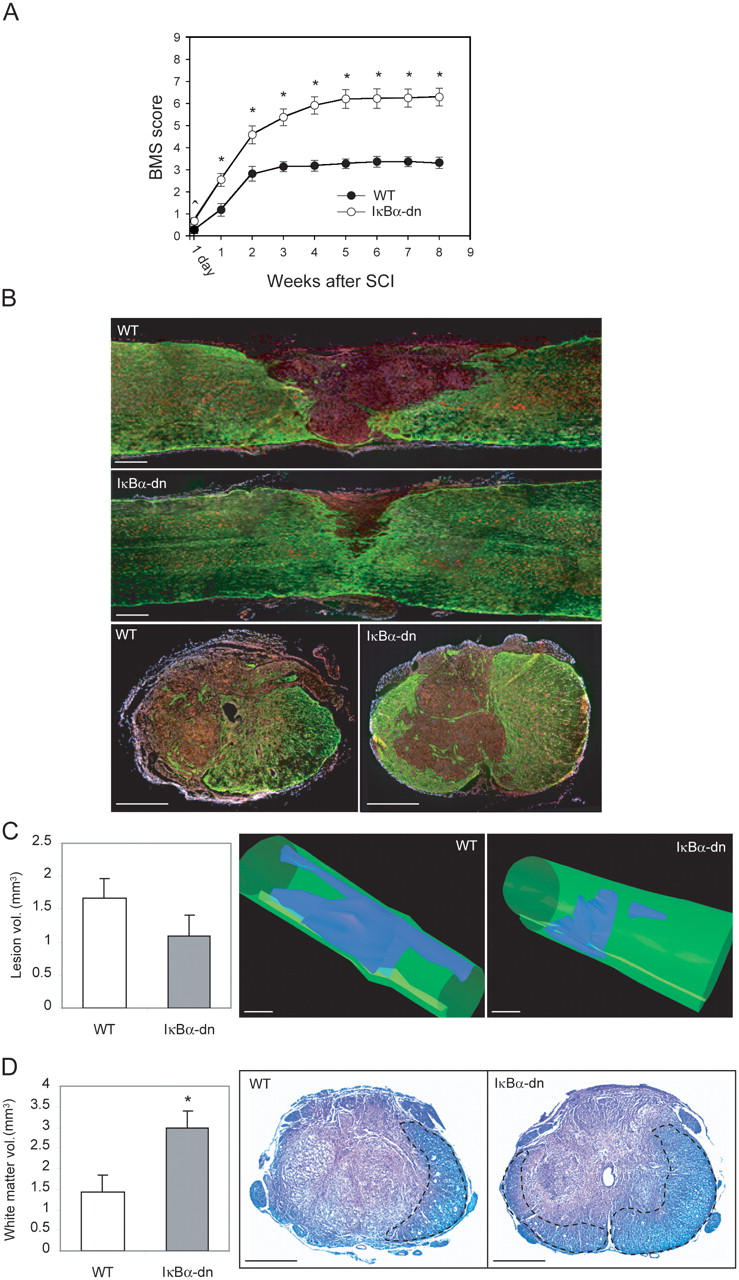
Behavioral and histological analysis of WT and GFAP-IκBα-dn mice 8 wk after SCI. (A) Evaluation of hindlimb locomotor function. WT (n = 12) and GFAP-IκBα-dn (IκBα-dn; n = 12) mice were tested 1 d after SCI and weekly thereafter for 8 wk. Motor behavior was scored under blind conditions with the BMS. ^, P < 0.04; *, P < 0.001 versus WT, as determined by one-way ANOVA and the Tukey test. (B) GFAP labeling (green), fluorescent Nissl (red), and DAPI (blue) staining of spinal cord sections 8 wk after SCI. (C) Lesion volume assessment of the two genotypes (n = 5). (right) A three-dimensional reconstruction of the lesion. (D) White matter sparing. Volume of myelinated white matter was measured in WT (n = 5) and GFAP-IκBα-dn (n = 5) mice on Luxol-stained sections. Contoured areas with darker Luxol staining delineate intact white matter. *, P < 0.02, as determined by the Student's t test. Morphometric analyses shown in C and D were conducted under blind conditions. Bars, 450 μm.
Inactivation of astroglial NF-κB increases white matter sparing after SCI
To confirm the qualitative observations described, a stereological quantification of lesion volume and white matter sparing was performed. Reduced lesion volume was observed in GFAP-IκBα-dn mice (Fig. 3 C), although values did not reach statistical significance. At the lesion epicenter where tissue disruption was comparable in the two genotypes, no difference was observed between WT and TG mice in the total number of spared neurons (neuronal number ± SEM: WT, 302,026 ± 61,740; GFAP-IκBα-dn, 251,842 ± 17,419). Importantly, stereological quantitation of white matter on Luxol-stained serial sections (Fig. 3 D) revealed more than double the amount of intact white matter (Fig. 3 D, contoured areas) in GFAP-IκBα-dn mice compared with WT mice (mm3 ± SEM: 3.00 ± 0.35 vs. 1.44 ± 0.34, respectively). Coimmunolabeling of tissues with myelin basic protein (MBP; red) and neuronal class III β-tubulin (TuJ1; green) demonstrated colocalization of intact myelin with neuronal fibers bridging the lesion area in GFAP-IκBα-dn mice (Fig. 4, C–F), indicating the presence of preserved, and possibly functional, axons. In all the WT sections tested, we were unable to colocalize MBP labeling with TuJ1 within the lesion core (Fig. 4, A and B), indicating that axonal fibers running through the lesion area are nonmyelinated and, thus, functionally compromised.
Figure 4.
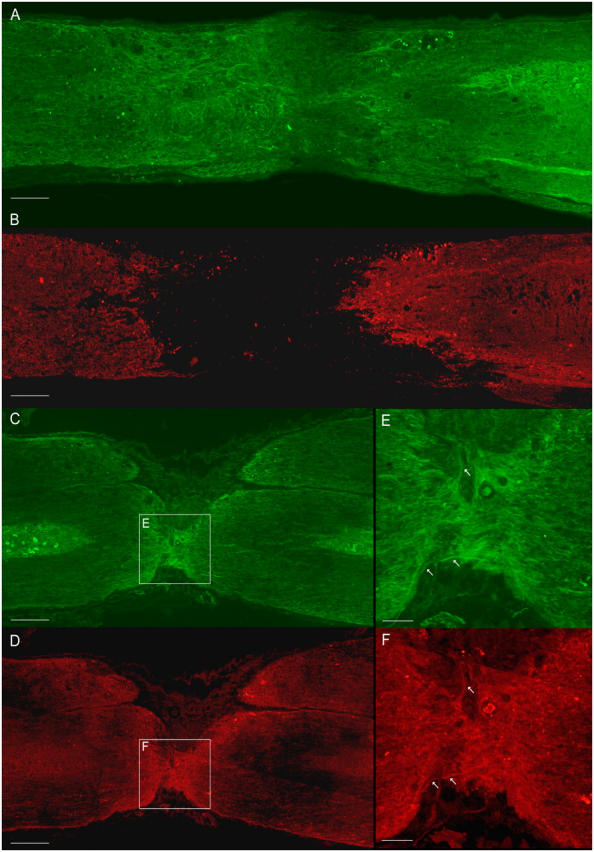
Immunostaining for MBP and TuJ1 in WT and GFAP-IκBα-dn spinal cords 8 wk after SCI. WT (A and B) and GFAP-IκBα-dn (C and D) sections of the spinal cord were double labeled for MBP (red; B, D, and F) and TuJ1 (green; A, C, and E). White arrows in E and F show colocalization of MBP with TuJ1 in myelinated axons. Bars: (A–D) 180 μm; (E and F) 50 μm.
Inhibition of astroglial NF-κB results in reduced expression of neurocan and phosphacan after SCI
The expression of chondroitin sulfate proteoglycans (CSPGs), which are essential constituents of the glial scar, was evaluated by immunohistochemistry in WT and GFAP-IκBα-dn mice 8 wk after injury. Both neurocan and phosphacan were highly expressed within the lesion core and in the white matter in WT mice (Fig. 5, A and C), whereas greatly reduced immunoreactivity was observed in TG mice (Fig. 5, B and D). In both genotypes, neurocan and phosphacan were detected on GFAP+ processes at the lesion edge and in the white matter and were colocalized with the microglia/macrophage marker OX-42 in the lesion core and in the white matter (unpublished data). To begin elucidating the mechanisms at the basis of the decreased CSPG expression in TG mice, we examined the expression of TGF-β and decorin, molecules that are known to be important regulators of such proteoglycans. We first evaluated the expression of the three TGF-β isoforms by RNase protection assay (RPA; Fig. 6 A). No differences were detected for TGF-β1 and TGF-β3 in response to injury and between WT and TG mice. TGF-β2 expression, however, was significantly induced 6 h after SCI in WT mice, and this effect was completely abrogated in GFAP-IκBα-dn mice (P < 0.05; Fig. 6 A). This result is particularly important considering the role of TGF-β2 in stimulating the expression of CSPGs (24), and it correlates with the reduced deposition of neurocan and phosphacan at later times after injury (Fig. 5).
Figure 5.
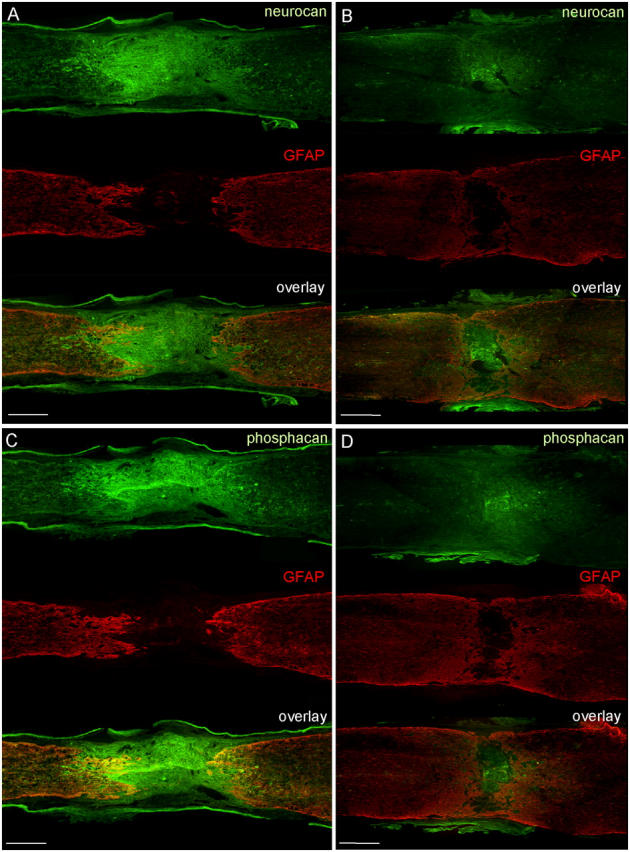
Neurocan and phosphacan immunohistochemistry 8 wk after SCI. WT (A and C) and GFAP-IκBα-dn (B and D) sections of the spinal cord were double labeled for GFAP (red) and either neurocan (green; A and B) or phosphacan (green; C and D). Bars, 450 μm.
Figure 6.

Expression of TGF-β and decorin in WT and GFAP-IκBα-dn mice after SCI. (A) Gene expression of TGF-β1, -β2, and -β3 6 h and 1 d after SCI was measured by RPA. Semiquantitative measurements were obtained by normalizing to GAPDH and L32. Results are expressed as the percentage of WT 6 h after SCI and represent the mean ± SEM (n = 4). *, P < 0.05, as determined by one-way ANOVA and the Tukey test. (B) Decorin expression 3 d and 1 wk after SCI was measured by real-time RT-PCR and normalized to β-actin. Results are expressed as the percentage of WT 3 d after SCI and represent the mean ± SEM (n = 3). Samples were also analyzed by agarose gel electrophoresis. −, negative control for the PCR.
The expression of decorin, a small dermatan/GSPG that is considered a natural antagonist to scar formation (25), was quantified by real-time PCR (Fig. 6 B). No differences were detected between WT and GFAP-IκBα-dn mice at 3 d and 1 wk after injury.
Inactivation of astroglial NF-κB results in reduced expression of the chemokines CXCL10 and CCL2 after SCI
To investigate the molecular mechanisms relevant to the improved functional outcome in GFAP-IκBα-dn mice, we evaluated the expression of NF-κB–dependent chemokines known to be up-regulated after SCI as critical modulators of the secondary injury response (26). mRNA levels of such molecules were measured by RPA 6 h and 1 d after SCI. Regulated on activation, normal T cell expressed and secreted (RANTES) expression was up-regulated at 6 h and still elevated at 1 d, with no noticeable difference between WT and TG mice (Fig. 7 A). On the contrary, statistically significant differences in the expression of CXCL10 (or IP10) and CCL2 (or MCP-1) were observed (P < 0.05). CXCL10 expression was greatly increased in WT mice 1 d after SCI, and this effect was inhibited in GFAP-IκBα-dn mice (Fig. 7 A). Similarly, SCI-induced CCL2 up-regulation was reduced in GFAP-IκBα-dn mice compared with the WT (Fig. 7 A).
Figure 7.

Chemokine expression in WT and GFAP-IκBα-dn mice after SCI. (A) Gene expression of RANTES, CXCL10, and CCL2 6 h and 1 d after SCI was measured by RPA. Semiquantitative measurements were obtained by normalizing to GADPDH and L32. Results are expressed as the percentage of WT 6 h after SCI and represent the mean ± SEM (n = 4). *, P < 0.05 as determined by one-way ANOVA and the Tukey test. (B–D) Colocalization of CXCL10 immunostaining (green) with GFAP (red) in WT (B) and GFAP-IkBa-dn mice (C) 3 d after SCI. Bars, 20 μm.
To further characterize the source of CXCL10 and CCL2, we performed immunohistochemical studies 3 d after SCI. CXCL10 (Fig. 7, green) was widely expressed throughout the cord in the two genotypes and immunoreactivity perfectly colocalized with GFAP (Fig. 7, B and C, red), indicating that astrocytes represent the sole source of the molecule at this phase of injury. Identical results were obtained for CCL2 (unpublished data). These data, in combination with the RPA data (Fig. 7 A), confirmed that NF-κB acts as a direct regulator of CXCL10 and CCL2 expression. Furthermore, they provide evidence that inhibiting NF-κB in astrocytes effectively reduces the inflammatory response in the early phase of injury because both CXCL10 and CCL2 are important chemotactic factors involved in the development of inflammation by driving the infiltration of leukocytes into the damaged spinal cord.
CXCL10 and CCL2 expression also temporally correlated with the appearance of OX-42+ monocytes/macrophages at the lesion site in both WT (Fig. S3, A and B, available at http://www.jem.org/cgi/content/full/jem.20041918/DC1) and TG (Fig. S3, C and D) mice 3 d after SCI. OX-42+ cells were also very numerous outside of the lesion area and displayed the typical stellate morphology of activated microglia (unpublished data). Immunostaining for the T cell–specific marker CD3 revealed the absence of T lymphocytes within the lesion 3 d after injury (unpublished data), which is in agreement with a previous study indicating that, in the mouse, T cell infiltration occurs at later times after injury (2 wk; reference 27).
Inactivation of astroglial NF-κB results in increased expression of IL-6 after SCI
Finally, we evaluated the expression of two classic NF-κB-dependent cytokines, TNF-α and IL-6, because of their documented proinflammatory function and up-regulation after SCI (12, 28). TNF-α was up-regulated at comparable levels in WT and GFAP-IκBα-dn mice (Fig. 8). Expression peaked at 6 h and declined by 1 d. Conversely, at 6 h after injury, IL-6 expression was significantly higher in GFAP-IκBα-dn mice compared with the WT (P < 0.05; Fig. 8). This represents a rather unexpected result because NF-κB is a major transcriptional activator of IL-6 expression.
Figure 8.

Expression of TNF-α and IL-6 in WT and GFAP-IκBα-dn mice after SCI. Gene expression of TNF-α and IL-6 6 h and 1 d after SCI was measured by RPA. Semiquantitative measurements were obtained by normalizing to GADPDH and L32. Results are expressed as the percentage of WT 6 h after SCI and represent the mean ± SEM (n = 4). *, P < 0.05, as determined by one-way ANOVA and the Tukey test.
Discussion
We provide evidence that the NF-κB signaling pathway operating in astrocytes is a major contributor to the pathological events occurring after SCI. By generating a TG model in which NF-κB is specifically inactivated in astrocytes, we demonstrated that, under nonpathologic conditions, astroglial NF-κB is not a critical regulator of spinal cord development and function. Indeed, TG mice display normal locomotor behavior and retain complete integrity of spinal cord architecture. Furthermore, we demonstrated the absence of constitutive cell death in GFAP-IκBα-dn mice (similar to WT mice), indicating that, under physiologic conditions, the NF-κB pathway operating in astrocytes is not involved in the regulation of apoptotic mechanisms in the spinal cord. According to our data, astrocytes do not require the presence of functional NF-κB for their viability under normal conditions, unlike hepatocytes and B lymphocytes in which NF-κB acts as a physiologic survival factor preventing apoptotic cell death (29, 30). In addition, we demonstrated that inhibition of NF-κB in astrocytes does not have any indirect effect on the viability of other CNS cell types (e.g., neurons) under physiologic conditions.
After contusive SCI, NF-κB is rapidly activated in WT mice (Fig. 2), with a temporal profile that mirrors that observed in the rat (8). In contrast, NF-κB activation is almost completely prevented in GFAP-IκBα-dn mice. This result has two important implications. First, it is a further confirmation that the transgene is highly functional in blocking NF-κB. Second, at least at early times after injury, it suggests that astrocytes are the major cell type in which the “bulk” of NF-κB activation occurs.
From a behavioral point of view, GFAP-IκBα-dn mice exhibited, over time, a dramatic functional improvement compared with WT mice. At the end of an 8-wk period of post-injury testing, TG mice locomotor capability was substantially ameliorated with respect to WT, as indicated by an approximately three-point increase in the BMS score. This translated into the ability of GFAP-IκBα-dn mice to perform coordinated movements and plantar stepping. WT mice, on the other hand, not only lacked coordination, but were often unable to support their weight when moving. Histologically, GFAP-IκBα-dn mice exhibited a statistically significant increase in white matter preservation. This likely accounts for their improved functional outcome caused by a higher number of spared axons extending through the intact white matter, the presence of which was demonstrated in double-labeling experiments showing colocalization of MBP with the neuronal-specific marker TuJ1 (Fig. 4). We also cannot exclude the possibility that a certain degree of regeneration might occur in GFAP-IκBα-dn mice, allowing for reinnervation of peripheral muscles and, consequently, better locomotion. In support of this speculation, a recent study by de Freitas et al. (14) demonstrated that the capacity of heat-stressed (reactive) astrocytes to inhibit neurite growth is dependent on NF-κB activation. In our TG model, the nonpermissive properties of astrocytes may be abrogated by the constitutive inactivation of the NF-κB pathway, resulting in an increased ability of damaged neurons to regenerate their axons. To specifically test this hypothesis, axonal tracing experiments are currently ongoing. Morphological analysis of the injured spinal cords in WT mice revealed the presence of a more elongated lesion than in GFAP-IκBα-dn mice (Fig. 3, B and C), suggesting that astroglial NF-κB may be involved in the spreading of the damage to the areas surrounding the site of injury.
To investigate the molecular mechanisms at the basis of the functional improvement in GFAP-IκBα-dn mice, we evaluated the expression of molecules known to be secreted by astrocytes and participate in the formation of the glial scar, namely the CSPGs neurocan and phosphacan, at chronic time points (8 wk after SCI). CSPGs represent a class of extracellular matrix molecules highly expressed within the glial scar and inhibitory to axonal regeneration (31, 32). Strategies aimed at degrading CSPGs attenuate their inhibitory activity and promote functional recovery after injury (33, 34). In our model of SCI, we demonstrated that expression of neurocan and phosphacan is highly attenuated in GFAP-IκBα-dn mice compared with the WT (Fig. 5). This may result in reduced glial scarring and, therefore, establish a more favorable environment to neuronal regeneration. In addition, our data indicate that NF-κB plays a role in inducing neurocan and phosphacan production in astrocytes. NF-κB may accomplish this function either by directly activating the transcription of the two genes or by indirectly inducing the expression of molecules responsible for proteoglycan synthesis. Among these molecules, a possible candidate is TGF-β, a potent fibrogenic factor implicated in scar formation (35, 36), whose expression is regulated by NF-κB (37, 38). Within the TGF-β family, TGF-β2 expression has been correlated with the deposition of scar tissue after SCI (24). We demonstrate that early expression of TGF-β2 induced by SCI is significantly reduced in GFAP-IκBα-dn mice (P < 0.05; Fig. 6 A). This represents an important finding and allows us to speculate that inhibition of astroglial NF-κB, by blocking the production of TGF-β2, may remove a prominent stimulating factor for the synthesis of scar-forming CSPGs and, therefore, help explain the recovery of function observed in TG mice. In addition, because it has been recently demonstrated that decorin reduces the expression of neurocan and phosphacan and promote axon growth across SCIs (39), we quantified decorin gene expression in search of a possible correlation with the reduced CSPGs observed in GFAP-IκBα-dn mice. Expression of decorin, a small dermatan/CSPG acting as a natural inhibitor to scar formation (25) by blocking the activity of TGF-β (40) and by antagonizing the epidermal growth factor receptor (41), did not change between WT and TG mice at early times after SCI, suggesting that decorin does not participate in the antifibrotic effect observed. This result also indicates that NF-κB does not play a role in the transcriptional regulation of mouse decorin (at least under these experimental conditions), as it does for TGF-β.
Another interesting finding is that early SCI-induced up-regulation of CXCL10 and CCL2 is considerably reduced in GFAP-IκBα-dn mice. Because both CXCL10 and CCL2 are involved in blood cell chemotaxis, a reduction in their expression could translate into a reduced infiltration of leukocytes into the lesion area and contribute to containing the inflammatory response after injury. In support of this hypothesis, recent studies demonstrated that antibody-mediated neutralization of CXCL10 enhances tissue sparing, reduces T cell infiltration, and improves functional recovery after SCI (42, 43). This evidence, combined with our results indicating the expression of CXCL10 and CCL2 in astrocytes 3 d after injury (Fig. 7, B and C), suggests that astroglial NF-κB is an important factor in driving the production of CXCL10 and CCL2 after SCI and that blocking this pathway has a beneficial effect on the functional outcome. The fact that GFAP-IκBα-dn mice display a reduced expression, but not a complete abrogation, of these chemokines 1 d after SCI indicates that CXCL10 and CCL2 are likely regulated by other transcription factors in addition to NF-κB. Furthermore, we cannot exclude the possibility that the induction of CXCL10 and CCL2 detected in both genotypes at these early times after injury is caused by the contribution of cell types other than astrocytes, particularly endothelial cells, which are known sources of CXCL10 and CCL2 (44–46).
Gene expression experiments produced rather unexpected results with respect to TNF-α and IL-6, genes that are classically regulated by NF-κB. First, no difference was detected in TNF-α expression between WT and TG mice after SCI. This suggests that the early production of TNF-α occurs in cell types other than astrocytes, most likely neurons and microglia. Second, after SCI, GFAP-IκBα-dn mice exhibited an up-regulation of IL-6 expression considerably higher than that of WT mice, indicating that astroglial NF-κB is not only not required for IL-6 expression at this early stage, but may also even represent an inhibitory signal. As for the role of IL-6, an increasing body of evidence has been accumulating on IL-6 neuroprotective function in CNS injury (47) and against glutamate excitotoxicity (48, 49). A recent study by Cafferty et al. in IL-6 knockout mice (50) determined that IL-6 is required for conditioning injury-induced regeneration of dorsal column axons after SCI. Based on these data, we can speculate that an increased IL-6 expression in GFAP-IκBα-dn mice early after SCI may activate protective mechanisms leading to increased neuronal survival and/or regeneration.
In conclusion, our results provide the first direct evidence that NF-κB–dependent processes initiated in astrocytes are responsible for the development of damage after SCI and that inhibition of such processes by gene-targeted inactivation of astroglial NF-κB leads to dramatic functional improvement after SCI. This, in a field where very little pharmacological tools are available, identifies a potential new therapeutic target for the development of treatments for SCI. Because NF-κB is a ubiquitous molecule essential for several vital functions, including inhibition of apoptotic cell death (51), we are well aware that inactivating NF-κB could result in deleterious side effects. However, by limiting the treatment within a relatively narrow window in the early stages of injury when the “burst” of NF-κB activation (and therefore the escalation of the inflammatory response) occurs, such effects are likely to be prevented or at least contained. Furthermore, the feasibility and efficacy of treatments targeting the inhibition of NF-κB (i.e., proteasome inhibitors; references 52 and 53) have been documented in various animal models, and several clinical trials with these molecules are now ongoing for cancer and inflammation-based pathologies (i.e., ischemia-reperfusion injury, rheumatoid arthritis, and psoriasis). In this scenario, our studies provide clinical evidence that such treatments could potentially be applied to the treatment of SCI.
MATERIALS AND METHODS
Generation of GFAP-IκBα-dn mice
GFAP-IκBα-dn mice were generated at the Transgenic Core Facility of the University of Miami. The original pGfa2Lac-1 plasmid containing a 2.2-kb 5′ flanking sequence of the human GFAP promoter (gfa2) upstream of the LacZ gene and a segment of the mouse protamine-1 gene (mP1) as a polyadenylation site was a gift of M. Brenner (University of Alabama-Birmingham, Birmingham, AL; reference 54). The cDNA encoding for a truncated form of the human IκBα gene (IκBα-dn; amino acids 37–317), excised from the pCMV4-IκBα-dn vector provided by D.W. Ballard (Vanderbilt University School of Medicine, Nashville, TN; references 54 and 55), was cloned downstream of the gfa2 promoter. The resulting GFAP-IκBα-dn-mP1 expression cassette was gel purified, microinjected into 1-d-old C57BL/6 × SJL embryos, and transplanted into pseudopregnant C57BL/6 × SJL mice. Transgenic founders were backcrossed to isogenicity (8–10 generations) onto WT C57BL/6 mice. All mice used in our experiments were 2–4 mo old and were obtained by breeding heterozygous GFAP-IκBα-dn males with WT females. WT littermates from the same breeding group were used as controls. All animals were housed in a 12-h light/dark cycle in a virus/antigen-free facility with controlled temperature and humidity and were provided with water and food ad libitum.
Traumatic SCI
Surgeries were performed at the Animal and Surgical Core Facility of the Miami Project to Cure Paralysis according to protocols approved by the Institutional Animal Care and Use Committee of the University of Miami. Contusion injury was induced with the Electromagnetic Spinal Cord Injury Device (developed at Ohio State University) adapted to the mouse (56). In brief, adult female mice (3–4 mo old, 20–24 g in weight) were laminectomized between vertebrae T8 and T10 and lowered at a predetermined impact force of 2,000 dynes (moderate injury). After surgery, animals were housed separately and treated with topical antibiotics and subcutaneous Ringer's solution to prevent fluid loss. Manual bladder expression was performed twice a day. 50–80 mg of prophylactic antibiotic gentocin was administered daily to prevent urinary tract infections.
Assessment of functional recovery after SCI
Recovery of function after SCI was determined by scoring the locomotor hindlimb performance in the open field with the BMS (23), a 0–9 rating system based on a modification of the BBB scale (57) and specifically designed for the mouse. Under blind conditions, a team of two investigators evaluated the mice over a 4-min time period 1 d after SCI and weekly thereafter. Before surgery, animals were acclimated to the open field and to handling to prevent fear and/or stress behaviors that could bias the locomotor assessment.
Histopathology and immunohistochemistry
For paraffin histopathology, animals were perfused with FAM (37% formaldehyde, acetic acid, and methanol, 1:1:8 by volume) fixative. Tissue segments containing the lesion area (3 mm on each side of the lesion) were paraffin embedded and cut into 15-μm-thick sections. Sections were stained with cresyl violet for general morphology and tissue architecture and Luxol fast blue for myelinated white matter. Neurons were labeled with an antibody against the specific marker NeuN (1:500; Chemicon) and visualized with 3-3′-diaminobenzidine staining using the Elite ABC kit (Vector Laboratories) according to manufacturer's instructions. For immunofluorescent staining, animals were perfused with 4% paraformaldehyde in 0.1 M PBS. Tissues were cryoprotected in 0.1 M PBS + 20% sucrose and cryostat cut into 15-μm-thick sections. Sections were incubated overnight at 4°C with antibodies against GFAP (1:1,000; DakoCytomation), OX-42 (1:100; Serotec), MBP (1:500; Chemicon), TuJ1 (Covance), CXCL10, and CCL2 (both 1:500; R&D Systems), followed by secondary species-specific fluorescent antibodies (Alexa Fluor 488 and 594, 1:500; Molecular Probes) for 1 h at room temperature.
Morphometric analysis
Neuronal cell count.
Neurons were identified by NeuN immunostaining. Sets of 10 sections/animal were selected according to unbiased sampling criteria (58), and the total number of neurons was stereologically quantified with StereoInvestigator (MicroBrightField, Inc.).
Lesion volume.
Lesion volume was determined as previously described (12). Sets of 10 cross sections were selected according to unbiased sampling criteria (58) and were stained with cresyl violet, and the volume of damaged tissue was measured on a three-dimensional reconstruction with Neurolucida (MicroBrightField, Inc.).
White matter sparing.
Sets of serial cross sections were stained with Luxol fast blue. Intact white matter was traced as previously described (59), and the volume was calculated on a three-dimensional reconstruction with Neurolucida (MicroBrightField, Inc.).
All morphometric analyses were performed by a single investigator under blind conditions.
Primary astrocyte and neuronal cultures
Primary astrocytes were prepared according to the protocol described by Bolego et al. (60), with minor modifications. In brief, the brains of 7-d-old pups were dissected and placed in HBSS without Ca/Mg and supplemented with 0.6% glucose. Tissue was incubated for 30 min at 37°C in 0.125% trypsin + 10 μg/ml DNase I and triturated. Cells were collected, resuspended in medium 199 + 100 IU/ml penicillin/streptomycin and 10% FBS, and plated onto poly–l-lysine–coated dishes. 2 d before use, cells were switched to a chemically defined serum-free medium consisting of 199 + N2.
Primary cortical neurons were obtained with the following procedure. Cortices of E15 embryos were dissected in L15 medium supplemented with 200 IU/ml penicillin/streptomycin. Tissues were incubated in 0.25% trypsin/ethylenediaminetetraacetate for 5 min at 37°C. Cells were collected, resuspended in Eagle's basal medium + 10% FBS, 2 mM glutamine, 100 IU/ml penicillin/streptomycin, 25 mM KCl, and plated onto poly–l-lysine–coated dishes. 1 d before use, neurons were switched to a serum-free medium consisting of Eagle's basal medium + N2. All media and reagents for tissue cultures were obtained from GIBCO BRL.
Total RNA isolation and RT-PCR
Total RNA was extracted with TRIzol (Invitrogen) according to the manufacturer's instructions. 2 μg RNA were reverse transcribed with 200 U/sample SuperScript II (Invitrogen) and 90 ng/sample of random hexamers (Promega). The GFAP-IκBα-dn transgene was amplified from 0.1-μg aliquots of cDNA in a standard PCR buffer (50 mM KCl, 1.5 mM MgCl2, and 10 mM Tris-HCl, pH 8.3) containing 10 pmol of forward GFAP (5′-TTC ATA AAG CCC TCG CAT CC-3′) and reverse IκBα (5′-ACA GCC AGC TCC CAG AAG TG-3′) primers and 0.5 U/sample of AmpliTaq DNA polymerase (Applied Biosystems). Mouse β-actin was amplified as a control for the PCR reaction. Samples lacking reverse transcriptase were amplified as a control for genomic DNA contamination.
Real-time PCR
Real-time PCR was performed with the Rotor-Gene 3000 (Corbett Research) on cDNA samples obtained as described in the Total RNA…RT-PCR section using the intercalating dye SYBR green (Molecular Probes) in the presence of primer pairs specific for either decorin (forward, 5′-ATT CGC ATC TCA GAC ACC AAC ATA ACT-3′; reverse, 5′-GAC TGC CAT TCT CCA TAA CGG TGA T-3′) or β-actin (forward, 5′-ATG GTG GGA ATG GGT CAG A-3′; reverse, 5′-CAC GCA GCT CAT TGT AGA AGG-3′)
EMSA
After treatments, cells were collected for whole cell extract (neurons) or nuclear extract (astrocytes) preparation. Whole cell extracts were obtained by scraping cells in ice-cold lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM ethylenediaminetetraacetate, and 1% Triton X-100, pH 7.5) supplemented with protease inhibitors (Complete; Roche). After incubation on ice for 30 min, suspensions were spun at 21,000 g for 20 min and supernatants (whole cell extracts) were collected. Nuclear extracts were prepared according to Dignam et al. (61). EMSAs were performed as previously described (8).
RPA
RPA experiments were performed with the Multi-Probe Ribonuclease Protection Assay RiboQuant (BD Biosciences). Total RNA was isolated from spinal cord segments consisting of 2 mm of tissue rostral and caudal to the lesion epicenter. Sham-operated (laminectomy alone) WT and GFAP-IκBα-dn mice were used as controls. 2-μg RNA aliquots from each sample were hybridized to an α-32P-labeled multiprobe template set (mCK-2b and mCK-3b mouse cytokine sets, 3.95 × 105 cpm/μl; mCK-5c mouse chemokine set, 3.6 × 105 cpm/μl) according to the manufacturer's instructions. RNase-protected probes were resolved on 4.75% polyacrylamide gels and visualized by autoradiography. Autoradiograms were quantified with Quantity One (Bio-Rad Laboratories). Samples were normalized against L32 and GAPDH.
Western blot for activated p65
For the detection of activated p65 in spinal cord extracts, proteins were resolved by SDS-PAGE on 10% gels, transferred to nitrocellulose membranes, and blocked overnight in 5% nonfat milk. Membranes were probed with an antibody recognizing the activated form of p65 (1:1,000; Chemicon), followed by horseradish peroxidase–conjugated secondary antibody. Proteins were visualized with a chemiluminescent kit (ECL; GE Healthcare). Blots were also probed for β-actin as a loading control.
Statistical analysis
Statistical analysis was performed with a one-way analysis of variance (ANOVA) followed by the Tukey test for multiple comparisons. In the case of single comparisons, the Student's t test was applied. P ≤ 0.05 was considered statistically significant.
Online supplemental material
Fig. S1 shows the behavioral characterization of GFAP-IκBα-dn mice. Fig. S2 depicts the histological analysis of naive WT and GFAP-IκBα-dn spinal cords. Fig. S3 shows OX-42 immunostaining in WT and GFAP-IκBα-dn mice 3 d after SCI. Additional protocols are provided as online materials and methods. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20041918/DC1.
Acknowledgments
We thank Dr. A. Marcillo and the Animal and Surgical Core Facility of the Miami Project to Cure Paralysis, where all the surgical procedures were performed.
This work was supported by National Institutes of Health grants NS37130 and NS051709 (both to J.R. Bethea) and by the Miami Project to Cure Paralysis.
The authors have no conflicting financial interests.
Abbreviations used: ANOVA, analysis of variance; CNS, central nervous system; CSPG, chondroitin sulfate proteoglycan; dn, dominant negative; EMSA, electrophoretic mobility shift assay; GFAP, glial fibrillary acidic protein; IκBα, inhibitor of κBα; MBP, myelin basic protein; RPA, RNase protection assay; SCI, spinal cord injury; TG, transgenic; TuJ1, neuronal class III β-tubulin.
R. Brambilla and V. Bracchi-Ricard contributed equally to this work.
W.-H. Hu's present address is Dept. of Physiology, Medical College of Virginia, Virginia Commonwealth University, Richmond, VA 23298.
References
- 1.Schwab, M.E. 2002. Repairing the injured spinal cord. Science. 295:1029–1031. [DOI] [PubMed] [Google Scholar]
- 2.Popovich, P.G., and T.B. Jones. 2003. Manipulating neuroinflammatory reactions in the injured spinal cord: back to basics. Trends Pharmacol. Sci. 24:13–17. [DOI] [PubMed] [Google Scholar]
- 3.Ridet, J.L., S.K. Malhotra, A. Privat, and F.H. Gage. 1997. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 20:570–577. [DOI] [PubMed] [Google Scholar]
- 4.Liberto, C.M., P.J. Albrecht, L.M. Herx, V.W. Yong, and S.W. Levison. 2004. Pro-regenerative properties of cytokine-activated astrocytes. J. Neurochem. 89:1092–1100. [DOI] [PubMed] [Google Scholar]
- 5.Faulkner, J.R., J.E. Herrmann, M.J. Woo, K.E. Tansey, N.B. Doan, and M.V. Sofroniew. 2004. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24:2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menet, V., M. Prieto, A. Privat, and M. Gimenez y Ribotta. 2003. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc. Natl. Acad. Sci. USA. 100:8999–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Neill, L.A., and C. Kaltschmidt. 1997. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 20:252–258. [DOI] [PubMed] [Google Scholar]
- 8.Bethea, J.R., M. Castro, R.W. Keane, T.T. Lee, W.D. Dietrich, and R.P. Yezierski. 1998. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J. Neurosci. 18:3251–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider, A., A. Martin-Villalba, F. Weih, J. Vogel, T. Wirth, and M. Schwaninger. 1999. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 5:554–559. [DOI] [PubMed] [Google Scholar]
- 10.Nomoto, Y., M. Yamamoto, T. Fukushima, H. Kimura, K. Ohshima, and M. Tomonaga. 2001. Expression of nuclear factor kappaB and tumor necrosis factor alpha in the mouse brain after experimental thermal ablation injury. Neurosurgery. 48:158–166. [DOI] [PubMed] [Google Scholar]
- 11.Beni, S.M., R. Kohen, R.J. Reiter, D.X. Tan, and E. Shohami. 2004. Melatonin-induced neuroprotection after closed head injury is associated with increased brain antioxidants and attenuated late-phase activation of NF-kappaB and AP-1. FASEB J. 18:149–151. [DOI] [PubMed] [Google Scholar]
- 12.Bethea, J.R., H. Nagashima, M.C. Acosta, C. Briceno, F. Gomez, A.E. Marcillo, K. Loor, J. Green, and W.D. Dietrich. 1999. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J. Neurotrauma. 16:851–863. [DOI] [PubMed] [Google Scholar]
- 13.Siren, A.L., R. McCarron, L. Wang, P. Garcia-Pinto, C. Ruetzler, D. Martin, and J.M. Hallenbeck. 2001. Proinflammatory cytokine expression contributes to brain injury provoked by chronic monocyte activation. Mol. Med. 7:219–229. [PMC free article] [PubMed] [Google Scholar]
- 14.de Freitas, M.S., T.C. Spohr, A.B. Benedito, M.S. Caetano, B. Margulis, U.G. Lopes, and V. Moura-Neto. 2002. Neurite outgrowth is impaired on HSP70-positive astrocytes through a mechanism that requires NF-kappaB activation. Brain Res. 958:359–370. [DOI] [PubMed] [Google Scholar]
- 15.Acarin, L., B. Gonzalez, and B. Castellano. 2001. Triflusal posttreatment inhibits glial nuclear factor-kappaB, downregulates the glial response, and is neuroprotective in an excitotoxic injury model in postnatal brain. Stroke. 32:2394–2402. [DOI] [PubMed] [Google Scholar]
- 16.Zaheer, A., M.A. Yorek, and R. Lim. 2001. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochem. Res. 26:1293–1299. [DOI] [PubMed] [Google Scholar]
- 17.Mattson, M.P., B. Cheng, S.A. Baldwin, V.L. Smith-Swintosky, J. Keller, J.W. Geddes, S.W. Scheff, and S. Christakos. 1995. Brain injury and tumor necrosis factors induce calbindin D-28k in astrocytes: evidence for a cytoprotective response. J. Neurosci. Res. 42:357–370. [DOI] [PubMed] [Google Scholar]
- 18.Beg, A.A., W.C. Sha, R.T. Bronson, S. Ghosh, and D. Baltimore. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 376:167–170. [DOI] [PubMed] [Google Scholar]
- 19.Sha, W.C., H.C. Liou, E.I. Tuomanen, and D. Baltimore. 1995. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 80:321–330. [DOI] [PubMed] [Google Scholar]
- 20.Jessen, K.R., L. Morgan, H.J. Stewart, and R. Mirsky. 1990. Three markers of adult non-myelin-forming Schwann cells, 217c(Ran-1), A5E3 and GFAP: development and regulation by neuron-Schwann cell interactions. Development. 109:91–103. [DOI] [PubMed] [Google Scholar]
- 21.Kucharczak, J., M.J. Simmons, Y. Fan, and C. Gelinas. 2003. To be, or not to be: NF-kappaB is the answer–role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 22:8961–8982. [DOI] [PubMed] [Google Scholar]
- 22.Viatour, P., M.P. Merville, V. Bours, and A. Chariot. 2005. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem. Sci. 30:43–52. [DOI] [PubMed] [Google Scholar]
- 23.Basso, D.M., and L.F. Fisher. 2003. The Basso mouse scale for locomotion is a more sensitive indicator of recovery than the BBB scale in mice with spinal cord injury. J. Rehab. Research Dev. 40:26. [Google Scholar]
- 24.Lagord, C., M. Berry, and A. Logan. 2002. Expression of TGFbeta2 but not TGFbeta1 correlates with the deposition of scar tissue in the lesioned spinal cord. Mol. Cell. Neurosci. 20:69–92. [DOI] [PubMed] [Google Scholar]
- 25.Iozzo, R.V. 1999. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J. Biol. Chem. 274:18843–18846. [DOI] [PubMed] [Google Scholar]
- 26.McTigue, D.M., M. Tani, K. Krivacic, A. Chernosky, G.S. Kelner, D. Maciejewski, R. Maki, R.M. Ransohoff, and B.T. Stokes. 1998. Selective chemokine mRNA accumulation in the rat spinal cord after contusion injury. J. Neurosci. Res. 53:368–376. [DOI] [PubMed] [Google Scholar]
- 27.Sroga, J.M., T.B. Jones, K.A. Kigerl, V.M. McGaughy, and P.G. Popovich. 2003. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J. Comp. Neurol. 462:223–240. [DOI] [PubMed] [Google Scholar]
- 28.Wang, C.X., B. Nuttin, H. Heremans, R. Dom, and J. Gybels. 1996. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J. Neuroimmunol. 69:151–156. [DOI] [PubMed] [Google Scholar]
- 29.Beg, A.A., and D. Baltimore. 1996. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 274:782–784. [DOI] [PubMed] [Google Scholar]
- 30.Grumont, R.J., I.J. Rourke, L.A. O'Reilly, A. Strasser, K. Miyake, W. Sha, and S. Gerondakis. 1998. B lymphocytes differentially use the Rel and nuclear factor κB1 (NF-κB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J. Exp. Med. 187:663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKeon, R.J., M.J. Jurynec, and C.R. Buck. 1999. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J. Neurosci. 19:10778–10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asher, R.A., D.A. Morgenstern, P.S. Fidler, K.H. Adcock, A. Oohira, J.E. Braistead, J.M. Levine, R.U. Margolis, J.H. Rogers, and J.W. Fawcett. 2000. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J. Neurosci. 20:2427–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradbury, E.J., L.D. Moon, R.J. Popat, V.R. King, G.S. Bennett, P.N. Patel, J.W. Fawcett, and S.B. McMahon. 2002. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 416:636–640. [DOI] [PubMed] [Google Scholar]
- 34.Grimpe, B., and J. Silver. 2004. A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J. Neurosci. 24:1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westergren-Thorsson, G., A. Schmidtchen, B. Sarnstrand, L.A. Fransson, and A. Malmstrom. 1992. Transforming growth factor-beta induces selective increase of proteoglycan production and changes in the copolymeric structure of dermatan sulphate in human skin fibroblasts. Eur. J. Biochem. 205:277–286. [DOI] [PubMed] [Google Scholar]
- 36.Romaris, M., A. Bassols, and G. David. 1995. Effect of transforming growth factor-beta 1 and basic fibroblast growth factor on the expression of cell surface proteoglycans in human lung fibroblasts. Enhanced glycanation and fibronectin-binding of CD44 proteoglycan, and down-regulation of glypican. Biochem. J. 310:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rameshwar, P., R. Narayanan, J. Qian, T.N. Denny, C. Colon, and P. Gascon. 2000. NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: an inflammatory response beyond the realm of homeostasis. J. Immunol. 165:2271–2277. [DOI] [PubMed] [Google Scholar]
- 38.Strauch, E.D., J. Yamaguchi, B.L. Bass, and J.Y. Wang. 2003. Bile salts regulate intestinal epithelial cell migration by nuclear factor-kappa B-induced expression of transforming growth factor-beta. J. Am. Coll. Surg. 197:974–984. [DOI] [PubMed] [Google Scholar]
- 39.Davies, J.E., X. Tang, J.W. Denning, S.J. Archibald, and S.J. Davies. 2004. Decorin suppresses neurocan, brevican, phosphacan and NG2 expression and promotes axon growth across adult rat spinal cord injuries. Eur. J. Neurosci. 19:1226–1242. [DOI] [PubMed] [Google Scholar]
- 40.Stander, M., U. Naumann, W. Wick, and M. Weller. 1999. Transforming growth factor-beta and p-21: multiple molecular targets of decorin-mediated suppression of neoplastic growth. Cell Tissue Res. 296:221–227. [DOI] [PubMed] [Google Scholar]
- 41.Santra, M., C.C. Reed, and R.V. Iozzo. 2002. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J. Biol. Chem. 277:35671–35681. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez, R., J. Glaser, M.T. Liu, T.E. Lane, and H.S. Keirstead. 2003. Reducing inflammation decreases secondary degeneration and functional deficit after spinal cord injury. Exp. Neurol. 184:456–463. [DOI] [PubMed] [Google Scholar]
- 43.Glaser, J., R. Gonzalez, V.M. Perreau, C.W. Cotman, and H.S. Keirstead. 2004. Neutralization of the chemokine CXCL10 enhances tissue sparing and angiogenesis following spinal cord injury. J. Neurosci. Res. 77:701–708. [DOI] [PubMed] [Google Scholar]
- 44.Mancardi, S., E. Vecile, N. Dusetti, E. Calvo, G. Stanta, O.R. Burrone, and A. Dobrina. 2003. Evidence of CXC, CC and C chemokine production by lymphatic endothelial cells. Immunology. 108:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piali, L., C. Weber, G. LaRosa, C.R. Mackay, T.A. Springer, I. Clark-Lewis, and B. Moser. 1998. The chemokine receptor CXCR3 mediates rapid and shear-resistant adhesion-induction of effector T lymphocytes by the chemokines IP10 and Mig. Eur. J. Immunol. 28:961–972. [DOI] [PubMed] [Google Scholar]
- 46.Feng, L., C. Matsumoto, A. Schwartz, A.M. Schmidt, D.M. Stern, and J. Pile-Spellman. 2005. Chronic vascular inflammation in patients with type 2 diabetes: endothelial biopsy and RT-PCR analysis. Diabetes Care. 28:379–384. [DOI] [PubMed] [Google Scholar]
- 47.Loddick, S.A., A.V. Turnbull, and N.J. Rothwell. 1998. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 18:176–179. [DOI] [PubMed] [Google Scholar]
- 48.Carlson, N.G., W.A. Wieggel, J. Chen, A. Bacchi, S.W. Rogers, and L.C. Gahring. 1999. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J. Immunol. 163:3963–3968. [PubMed] [Google Scholar]
- 49.Thorns, V., G.F. Walter, and F. Licastro. 2002. Effects of IL6 and IL1beta on aFGF expression and excitotoxicity in NT2N cells. J. Neuroimmunol. 127:22–29. [DOI] [PubMed] [Google Scholar]
- 50.Cafferty, W.B., N.J. Gardiner, P. Das, J. Qiu, S.B. McMahon, and S.W. Thompson. 2004. Conditioning injury-induced spinal axon regeneration fails in interleukin-6 knock-out mice. J. Neurosci. 24:4432–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barkett, M., and T.D. Gilmore. 1999. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 18:6910–6924. [DOI] [PubMed] [Google Scholar]
- 52.Rajkumar, S.V., P.G. Richardson, T. Hideshima, and K.C. Anderson. 2005. Proteasome inhibition as a novel therapeutic target in human cancer. J. Clin. Oncol. 23:630–639. [DOI] [PubMed] [Google Scholar]
- 53.Elliott, P.J., T.M. Zollner, and W.H. Boehncke. 2003. Proteasome inhibition: a new anti-inflammatory strategy. J. Mol. Med. 81:235–245. [DOI] [PubMed] [Google Scholar]
- 54.Brenner, M., W.C. Kisseberth, Y. Su, F. Besnard, and A. Messing. 1994. GFAP promoter directs astrocyte-specific expression in transgenic mice. J. Neurosci. 14:1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brockman, J.A., D.C. Scherer, T.A. McKinsey, S.M. Hall, X. Qi, W.Y. Lee, and D.W. Ballard. 1995. Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol. Cell. Biol. 15:2809–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jakeman, L.B., Z. Guan, P. Wei, R. Ponnappan, R. Dzwonczyk, P.G. Popovich, and B.T. Stokes. 2000. Traumatic spinal cord injury produced by controlled contusion in mouse. J. Neurotrauma. 17:299–319. [DOI] [PubMed] [Google Scholar]
- 57.Basso, D.M., M.S. Beattie, and J.C. Bresnahan. 1995. A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma. 12:1–21. [DOI] [PubMed] [Google Scholar]
- 58.Gundersen, H.J., P. Bagger, T.F. Bendtsen, S.M. Evans, L. Korbo, N. Marcussen, A. Moller, K. Nielsen, J.R. Nyengaard, B. Pakkenberg, et al. 1988. The new stereological tools: dissector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 96:857–881. [DOI] [PubMed] [Google Scholar]
- 59.Noble, L.J., and J.R. Wrathall. 1985. Spinal cord contusion in the rat: morphometric analyses of alterations in the spinal cord. Exp. Neurol. 88:135–149. [DOI] [PubMed] [Google Scholar]
- 60.Bolego, C., S. Ceruti, R. Brambilla, L. Puglisi, F. Cattabeni, G. Burnstock, and M.P. Abbracchio. 1997. Characterization of the signalling pathways involved in ATP and basic fibroblast growth factor-induced astrogliosis. Br. J. Pharmacol. 121:1692–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dignam, J.D., R.M. Lebovitz, and R.G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]