

Abstract
MHC molecules associated with autoimmunity possess known structural features that limit the repertoire of peptides that they can present. Such limitation gives a selective advantage to TCRs that rely on interaction with the MHC itself, rather than with the peptide residues. At the same time, negative selection is impaired because of the lack of negatively selecting peptide ligands. The combination of these factors may predispose to autoimmunity. We found that mice with an MHC class II–peptide repertoire reduced to a single complex demonstrated various autoimmune reactions. Transgenic mice bearing a TCR (MM14.4) cloned from such a mouse developed severe autoimmune dermatitis. Although MM14.4 originated from a CD4+ T cell, dermatitis was mediated by CD8+ T cells. It was established that MM14.4+ is a highly promiscuous TCR with dual MHC class I/MHC class II restriction. Furthermore, mice with a limited MHC–peptide repertoire selected elevated numbers of TCRs with dual MHC class I/MHC class II restriction, a likely source of autoreactivity. Our findings may help to explain the link between MHC class I responses that are involved in major autoimmune diseases and the well-established genetic linkage of these diseases with MHC class II.
Genetic predisposition to autoimmune disorders, such as type 1 diabetes and rheumatoid arthritis, is associated with the MHC (1, 2), although the nature of this association is not entirely clear. In addition, most of the established linkages are to MHC class II, whereas MHC class I–reactive CD8+ T cells play a critical role in the development of organ-specific diseases, such as type 1 diabetes. Structural studies of MHC class II molecules associated with autoimmunity revealed that these molecules have specific features that introduce a bias in the repertoire of peptides that can be bound (3–5). For example, human and mouse MHC class II molecules associated with type 1 diabetes have an identical amino acid substitution (Aspβ57>Ser) in the β chain that leads to a change in the overall properties of class II molecules (6). Resolution of the structure of I-Ag7 molecules (4, 5) revealed that this MHC class II protein preferentially, although not exclusively, binds peptides with acidic P9 residues. Similarly, HLA-DR4 (DRA*0101, DRB1*0401), associated with a high frequency of rheumatoid arthritis, possesses a Lys at position β71, which also leads to a bias in peptide repertoire; peptides with negatively charged amino acids in position p4 bind DR4 preferentially (3, 7).
We reasoned that limitation of peptide diversity should influence negative selection of T cells—which is peptide-specific—and their positive selection. During positive selection, a limited peptide repertoire should provide an advantage to TCRs that are less dependent on interactions with specific peptides, but which rely on recognition of MHC molecules per se, with possible assistance from the coreceptor proteins. This unusual TCR repertoire containing TCRs with autoreactive potential, in turn, may be supported by the impairment of negative selection.
To test this hypothesis, we took advantage of a transgenic mouse strain in which MHC class II/peptide complex diversity is diminished to a single variety: the I-Ab complex with a peptide derived from the Eα protein (AbEp; reference 8). Mice expressing AbEp were shown to select a significant proportion of “autoreactive” T cells activated by WT Ab molecules (8). Here, we describe various indicators of autoimmunity in AbEp mice. Moreover, when TCR genes cloned from an Ab-reactive CD4+ T cell (MM14.4) isolated from an AbEp mouse were expressed as transgenes, the TCR-transgenic mice developed autoimmunity—an inflammatory skin disease. Surprisingly, dermatitis was caused by MM14.4+CD8+ T cells, which suggested that these T cells carried a TCR with dual MHC class I/MHC class II restriction. Further examination confirmed that conclusion and revealed that AbEp mice select elevated numbers of dual–MHC-restricted TCRs (dr-TCRs) that may contribute to a potential pool of autoreactive T lymphocytes.
RESULTS
Mice with biased MHC–peptide repertoire develop autoimmune reactions
To determine whether animals with limited MHC-peptide repertoire are prone to autoimmunity, we histologically examined multiple organs from AbEp mice (Fig. 1). Although no obvious changes were found in young (<3 mo) mice (unpublished data), mononuclear infiltrates in multiple organs were detected in older animals (>6 mo). ∼30% of mice have shown lymphocytic infiltrates in the pancreatic islets and around blood vessels (perivasculitis; Fig. 1 A). In addition, sialoadenitis, thyroiditis, and interstitial nephritis were observed. This latter group of lesions is not unusual in aging mice of common mouse strains. AbEp mice have been derived by a complicated breeding scheme that included several genetic backgrounds (8). They underwent continuous inbreeding in our facility, and are considered inbred. They are congenic to mice lacking MHC class II Ab molecules and the invariant chain (Ii), termed C2Δ mice (8). Thus, AbEp mice could have lesions seen in other common strains. However, infiltration of the pancreatic islets is not common at all, except in NOD mice that are prone to spontaneous type 1 diabetes. We found that the infiltrates contained CD4- and CD8-positive T cells (Fig. 1 B) which indicates that limited peptide repertoire presented to the TCRs by MHC class II molecules also may lead, in some manner, to activation of autoimmune CD8+ T cells.
Figure 1.

Mice with extremely limited and partially limited MHC peptide repertoires are prone to autoimmunity. (A) Histologic examination of organs from AbEp mice >6-mo-old revealed mononuclear infiltrates in different organs. (B) Lymphoid infiltrates around a large peritoneal blood vessel (top left), and a pancreatic islet (right and bottom left). Hematoxylin-eosin staining (top), anti–CD4-FITC (bottom left), and anti–CD8-FITC (bottom right). LN, lymph node. (C) ANAs in the sera of >5-mo-old AbEp mice expressing Ii. Negative control B6 serum scored “0,” serum from B6lpr/lpr mouse scored “10,” while a serum from a representative AbEp,Ii+ mouse scored “5” on a relative scale. (D) Distribution of ANA presence and titers in AbEp,Ii+ mice and control mice. All data are for sera dilution of 1:50.
We also searched for signs of generalized autoimmunity, such as generation of antinuclear antibodies (ANAs), but did not detect them. However, that can be explained by lack of expression of the Ii in AbEp mice, which makes their B cells not fully functional (9, 10). We then examined the sera of AbEp mice expressing Ii for the presence of ANAs (Fig. 1 C). We found that ANAs clearly were present in many (∼60%) mature AbEp,Ii+ mice. MHC–peptide repertoire in AbEp,Ii+ mice is significantly more diverse than in AbEp mice as a result of replacement of the covalently bound Eα peptide by a diverse set of peptides (11). AbEp,Ii+ mice still have ∼30% MHC class II molecules occupied by Eα peptide. This assumption is based on staining with AbEp-specific antibodies Y-Ae and with the 25–9-17 antibody that fails to recognize this complex (12). These data are in concordance with previous findings of B cell–mediated autoimmunity in AbEp,Ii+ mice (13). Thus, even partial limitation of MHC–peptide repertoire predisposes to autoimmunity.
MM14.4 TCR transgenic animals develop autoimmune dermatitis
Because the manifestations of autoimmunity in AbEp mice were relatively mild (which can be explained by the presence of suppressor T cells in these mice (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050198/DC1), we sought to find whether “autoimmunity-prone” TCRs generated in AbEp mice would cause an illness when expressed as monoclonal transgenes. For that, ConA-activated blasts from AbEp lymph nodes were fused to BW5147 thymoma cell line, and the resulting hybridomas were selected for the reactivity with the WT MHC class II Ab molecules. A TCR from one such CD4+ hybridoma (MM14.4) was cloned and microinjected into oocytes. Four independent MM14.4 TCR transgenic strains (500, 581, 675, and 690) expressed cointegrated MM14.4 TCRα (Vα2.5-Jα20 = TRAV14D-2-TRAJ26) and TCRβ (Vβ16.2-Dβ2.1-Jβ2.1 = TRBV3-TRBJ2-1). All of them developed lesions in the skin, which suggested that random transgene integration was not responsible for the disease. Infection did not cause the lesions because mice had no cutaneous pathogens, and nontransgenic littermates were always disease-free. Skin lesions (Fig. 2, d–k) were characterized by scaling and thickening of the epidermis, and mononuclear infiltrate (consisting primarily of lymphocytes) in the epidermis and dermis. Parakeratosis (thickened stratum corneum with retention of nuclei) was observed (Fig. 2, e, f, and h) as well as occasional pustules within the stratum corneum consisting of degenerating neutrophils and proteinaceous debris (Fig. 2 g). The epidermal hyperplasia was characterized by the loss of expression of keratins 1 and 10, and by gain of expression of keratin 6 (typical for hyperplasia; Fig. 2, i–k). These characteristics resemble human psoriasis. The hair growth also was affected in MM14.4 mice, which was different from human disease, although psoriatic lesions in the scalp accompanied by hair loss have been described in humans (14, 15). Skin infiltrates were found microscopically as early as 10–12 d post partum, and the onset of visible dermatitis was observed at about the time of weaning (3 wk). Dermatitis severity then peaked sharply, but subsided gradually in most mice.
Figure 2.

The features of psoriasiform dermatitis in MM14.4 transgenic mice. Nontransgenic littermates (a–c) had normal-appearing skin and hair. In contrast, transgenic mice had loss of hair and scaling skin (d–h). c and f are enlarged areas framed in b and e, respectively. Lesions were characterized by thickening of the epidermis and intense scaling, mononuclear infiltrates in the epidermis and dermis, occasional subcorneal pustules (g), and focal parakeratosis (f and h). Epidermal hyperplasia was marked by loss of keratin 1 and 10 (i and j) and acquisition of expression of keratin 6 (k), typical for hyperplasia. Bar, 100 μm.
CD8+ T cells are responsible for the development of the autoimmune dermatitis in MM14.4 mice
Because several independent MM14.4 transgenic strains were affected by psoriasiform dermatitis, it was apparent that the transgene-expressing T cells were responsible. We assumed, based on the CD4+ origin of the original MM14.4 cell, that CD4+ cells should be responsible for the dermatitis. However, to our surprise, several approaches pointed at CD8+ cells as the perpetrators. First, lymphocytes infiltrating the skin expressed the CD8 coreceptor (Fig. 3 A), were positive for TCRβ, and were negative for TCRδ and CD4 coreceptor (not depicted). Second, a genetic approach has been used to investigate the role of CD8+ cells. The analysis of several groups of mice derived from two different founders (581 and 690) showed that the disease developed in all mice independently of MHC class II expression (Table I, A). In these experiments, two types of KO mice were used: C2° mice lacking Aβb gene (8), and mice with complete deletion of MHC class II region (B6;129S-H2dlAb1-Ea/J) (C2complete KO; reference 16). This ensured that no cryptic MHC class II complexes (e.g., Aα/Eβ hybrid molecules that are not detectable under normal circumstances, but may exist at levels recognizable by T cells) were involved.
Figure 3.
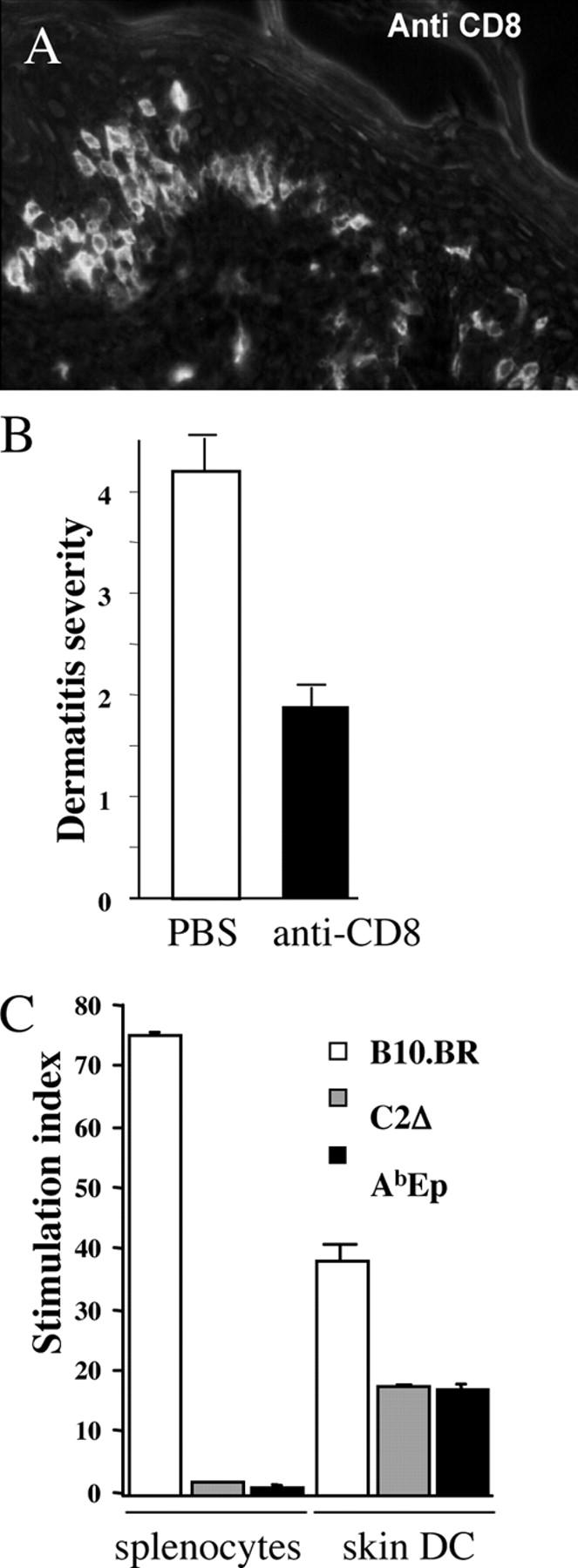
MM14.4 CD8+ T cells are responsible for the psoriasiform dermatitis. (A) CD8+ cells were found to infiltrate the epidermis (cryostat section, stained with anti-CD8 antibodies coupled to FITC; a 20× Fluotar objective). (B) Treatment of MM14.4 transgenic mice (strain 690) with anti-CD8 monoclonal antibodies decreased the relative dermatitis score (± SE). (C) Skin DCs isolated from AbEp or MHC class II–negative C2Δ mice, but not their splenocytes, triggered proliferation of purified CD8+ cells from 690 transgenic Rag-1–deficient mice. B10.BR (H-2k) cells were used as positive control because MM14.4 CD8+ cells have a strong alloreactivity to H2-Kk molecules. Combined data from two experiments.
Table I.
Development of psoriasiform dermatitis in MM14.4 TCR transgenic mice
| Dermatitis incidence
|
|||||
|---|---|---|---|---|---|
| Strain 0.581
|
Strain 0.690
|
||||
| Genotype of MM14.4+ mice |
Females | Males | Females | Males | |
| (%) | (%) | (%) | (%) | ||
| A | C1+C2°AbEp+li° | 22/31 (71) | 9/18 (50) | 36/44 (82) | 19/34 (56) |
| C1+C2° (Aβ°) | 17/22 (77) | 12/24 (50) | 10/12 (83) | 7/11 (63) | |
| C1+C2complete KO | 9/9 (100) | 8/10 (80) | |||
| C1+C2°AbEp+li+ | 4/11 (36) | ||||
| C1°C2° | 0/15 (0) | ||||
| C1a°C2+li°a | 0/22 (0) | 0/18 (0) | |||
| C1+C2+li°a | 3/3 | ||||
| B | C1+C2° TCRα° | 3/3 | |||
| C1+C2° TCRα°β° | 3/3 | 1/2 | |||
| C1+C2° TCRδ° | 13/13 (100) | ||||
| C1+C2° TCRβ°TCRδ° | 24/26 (90) | 14/20 (70) | |||
| C1+C2° μMT | 8/9 (88) | 7/10 (70) | |||
Disease was determined macroscopically as described in Materials and methods. Two MM14.4 strains, 581 and 690, were very similar in the disease development. Females generally were more susceptible than males. (A) Dermatitis development requires MHC class I expression. The frequency of dermatitis was independent of AbEp expression, but was affected by coexpression of AbEp and Ii due to the negative selection in the thymus (see also Fig. 4 B). MHC class I seemed to be indispensable for disease development. (B) T cells expressing endogenous (nontransgenic) TCR chains and B cells are not required for dermatitis development.
These mice still have MHC class II, and thus, were bred to Ii KO mice to reduce negative selection of MM14.4 cells, and C1+C2+li° mice were used as controls. The dermatitis in these control mice was less severe than in C2°-KO mice, but was clearly detectable.
C1°, β2-microglobulin KO; C2°, KO of Aβ b; C1a°, KO of classic MHC class I molecules Kb and Db; C2complete KO, deletion of all class II genes within MHC.
By contrast, dermatitis was not observed in MM14.4 transgenic mice lacking MHC class I expression as a result of targeted mutation of the β2-microglobulin gene (β2m KO), or in mice lacking classic MHC class I molecules (Kb,Db KO mice; Table I, A). To confirm the importance of CD8+ T cells in disease development, we treated newborn MM14.4 transgenic mice with anti-CD8 monoclonal antibodies. Treated mice did not develop dermatitis, or its severity was attenuated compared with control mice (Fig. 3 B).
Finally, we tested directly the reactivity of CD8+ cells from MM14.4 transgenic mice against DCs isolated from skin of mice expressing AbEp or lacking MHC class II (C2° mice). As shown in Fig. 3 C, purified CD8+ T cells from lymph nodes of Rag1-deficient MM14.4 mice proliferated in response to DCs isolated from skin of AbEp and MHC class II-negative mice; this indicated that CD8+ cells recognize syngeneic MHC class I molecules loaded with a skin-specific peptide. Notably, CD8+ cells did not respond to splenocytes from the same donors. The CD8+ MM14.4+ cells were functional because they responded to splenocytes from B10.BR mice of the H-2k haplotype, to which MM14.4 has a strong alloreactivity (against Kk molecule, see Fig. 5). The overall conclusion from genetic and activation experiments was that CD8+ cells were the primary effectors causing dermatitis in MM14.4 mice. Given that original MM14.4 hybridoma was CD4-positive and reacted to Ab MHC class II molecules, we had to assume that MM14.4 TCR was capable of interacting with MHC class II and class I molecules.
Figure 5.

Dual MHC-reactivity and coreceptor-dependence of MM14.4 TCR. (A) CD4+, MM14.4 T cells recognize Ab MHC class II molecules, while CD8+, MM14.4 T cells recognize Kk MHC class I molecules. T cell lymphoma cells transfected with MM14.4 TCR chains alone (double-negative, DN) or cotransfected with CD4 or CD8 c-DNA were tested for IL-2 production in response to MHC class II and class I molecules (left). Reactivity of CD4+ cells was inhibited by anti-Ab and anti-CD4 mAb (middle), while reactivity of the CD8+ cells was inhibited by anti-Kk and anti-CD8 mAb (right). Color conversion of Alamar Blue by IL-2–dependent cell line was used to measure IL-2 in culture medium. CD8 was a CD8α,β dimer transcribed from a polycistronic matrix. (B) Recognition of MHC class II by MM14.4 required CD4 interaction with MHC class II molecules. CD4+ MM14.4-transfected T cells reacted to Ab-positive stimulators (B6 or WT Ab-transgenic, Ab WT) but not to stimulators that expressed Ab with inactivated CD4 binding site (Ab MUT; left). The presence or absence of CD4 did not matter for activation with plate-bound anti-CD3 antibodies (right).
MM14.4 is a dual–MHC-restricted TCR
Dual MHC class I and class II reactivity of MM14.4 could help to explain how a TCR that originates from CD4+ T cells may be responsible for an autoimmune disease that is caused by CD8+ T cells expressing the same TCR. However, one possible caveat to this line of reasoning was the fact that endogenous (nontransgenic) TCR chains are capable of modifying selection of T cells and the specificity of T cell responses (17, 18). Thus, an important question was whether expression of endogenous TCRs could explain MHC class I reactivity and cause the disease or contribute significantly to its progression. To address this issue we performed extensive breeding of MM14.4 mice to KO strains lacking genes encoding TCR chains, and observed KO mice for development of dermatitis. Mice lacking endogenous TCRα chains, TCRβ chains, or both developed dermatitis (Table I, B). Neither γδ T cells nor B cells was needed for development of the disease, because MM14+ mice carrying KO of TCRδ chain or of μ heavy chain developed skin lesions. Thus, we concluded that MM14.4 TCR-expressing cells are solely responsible for the disease. The disease was tolerant to mixing the genetic backgrounds of mice during breeding to multiple KO mouse strains (predominantly of the B6 genetic background). This alleviates the concern that autoimmunity in AbEp mice resulted from the combination of genetic backgrounds that contributed to the generation of these mice.
Overall, we concluded that CD8+ T cells are critical for the autoimmune dermatitis in MM14.4 mice, and, moreover, that this receptor must have a dual MHC class I and class II restriction. To confirm that, we analyzed MM14.4 T cell selection in vivo and their reactivity in vitro.
Analysis of the selection of MM14.4 T cells in vivo revealed that these cells could be found in CD4+ and CD8+ compartments, and that MHC class I and class II contribute to selection. The mode of MM14.4 TCR selection was analyzed first using transgenic strains 581 and 690 crossed to mice lacking the Rag1 recombinase gene. The results obtained with strain 690 are representative of a common pattern of MM14.4 selection (Fig. 4 A). CD4+ and CD8+ cells were positively selected in MM14.4 TCR transgenic Rag-negative mice that expressed no endogenous class II molecules, but expressed AbEp and MHC class I. Although the selection of CD4+ cells was weak, in the absence of MHC class II the CD4+ population was almost completely absent, and confirmed that AbEp was used as a positively selecting ligand for this T cell subset. Positive selection into the CD8+ lineage was dramatically more efficient, and was independent of the presence of AbEp. At the same time, the thymi of mice in these two groups (C2°Ii°AbEp+ and C2°Ii°, Fig. 4 A, top two panels) were moderately sized and contained elevated numbers of double-negative cells. This suggested that some cells may be stimulated strongly enough to lose their coreceptors or to be eliminated by negative selection. Nevertheless, substantial numbers of MM14.4+ T cells were found in the periphery (Fig. 4 A, right) and were functional (Fig. 4 B). As expected, expression of the WT class II (Ab) molecules with Ii led to a very strong negative selection at the double-positive stage of thymocyte development; CD4 and CD8 lineages were reduced in relative and absolute numbers. Less severe depletion was detected in mice that did not have endogenous Ab molecules but were AbEp and Ii positive. The reactivity of MM14.4 cells selected by different MHC backgrounds correlated well with the antibody staining data (Fig. 4 B); reactivity of T cells to Ab (mediated by CD4+ cells, Fig. 5) was dependent on the presence of AbEp during selection and was obliterated by negatively selecting Ab molecules. At the same time, reactivity to allo MHC class I molecule Kk (used as a simple measure of CD8+ reactivity, Fig. 5) was independent of the presence of AbEp during selection, but was diminished severely by the WT Ab present during selection; these confirmed that negative selection had happened at the double-positive stage of thymic development.
Figure 4.

The pattern of MM14.4 T cell selection and lineage commitment reflects their dual MHC class I and MHC class II recognition. (A) CD4+ and CD8+ cells were positively selected in MM14.4.690 transgenics bred to Rag1-deficient background. The MHC and Ii genotypes are shown, as well as total cellularity [× 10−6] of the thymus and peripheral lymph nodes. Selection into CD4+ lineage was dependent on AbEp expression, while selection into CD8+ lineage was not. Expression of variable Ab/peptide repertoire in C2+Ii+ mice (Ab-positive, Ii-positive), AbEp+Ii+ mice, and C2+Ii° (Ab-positive, Ii-negative) mice led to negative selection of MM14.4+ T cells. (B) Reactivity of the lymph node cells from Rag1° MM14.4.690 transgenic mice with stimulators of variable MHC background confirms the pattern of positive and negative selection of the MM14.4 TCR. Mice used as donors of responding cells (FACS analysis is shown in A) were AbEp mice (Ab-negative, AbEp-positive, Ii-negative), C2° mice (mice with no MHC class II), or C2+Ii+ mice (Ab-positive, Ii-positive). Proliferation of 3 × 105 responders mixed with 5 × 105 of B6 (Ab-positive, left) or B10.BR (H-2Kk- positive, right) irradiated splenocytes. (C) Selection of CD8+ MM14.4+ T cells required expression of MHC class I molecules. MM14.4 mice lacking MHC class I because of genetic deletion of the β2-microglobulin had very few CD8 single-positive cells in the thymus and in the periphery independently of AbEp expression (top and middle), as compared with MHC class I–sufficient, MHC class II–negative mice (bottom). Numbers in brackets show absolute numbers of CD8+,Vα2+ cells (×10−6) in the thymi and lymph nodes. Coreceptor FACS dot-plots are shown for cells gated on Vα2-positive cells.
To show that the selection of CD8+MM14.4+ T cells was MHC class I dependent, we analyzed selection in MM14.4 transgenic mice lacking β2-microglobulin (C1° mice). It was clear that CD8+ single-positive cells in the thymus and CD8+ cells in the periphery were reduced very significantly in mice lacking MHC class I, and that the presence of AbEp did not allow positive selection of these cells (Fig. 4 B, note that the numbers in brackets show absolute number × 10−6 of CD8+Vα2+ single-positive cells and not the total cellularity of the organs). Mice used in this experiment were Rag-sufficient, but even so, the lack of positive selection of MM14.4 into the CD8 lineage was obvious. We also tested MM14.4 mice lacking only classic MHC class I molecules, Kb and Db. T cell selection in these mice is complicated by the inevitable presence of the WT MHC class II molecule, Ab. To diminish the negative effect of Ab, we crossed these mice to Ii-negative mice. Although lymph nodes from MHC class I–sufficient C2+Ii° MM14.4+ mice had ∼4 × 106 CD8+Vα2+ cells, these cells were practically absent from mice lacking classic MHC class I molecules.
The T cell selection patterns in MM14.4 transgenic mice correlated well with the incidence of dermatitis (Table I, A); the presence of the WT Ab molecules reduced the incidence of the disease, and the absence of MHC class I molecules alleviated the disease completely.
Transfection of cloned MM14.4 TCR chains into a T cell lymphoma allowed us to address directly the dual-MHC restriction of MM14.4 and the role of coreceptors in antigen recognition (Fig. 5). We found that recognition of MHC class II by MM14.4 was completely dependent on the CD4 expression by MM14.4 T cells (Fig. 5 A, left) and on the CD4–MHC class II interaction (Fig. 5 B). CD4+ MM14.4 cells recognized Ab molecules on B6 splenocytes independently of MHC class I because B6.β2m-negative splenocytes were as stimulatory to them as were WT splenocytes (Fig. 5 A, left). At the same time, the MM14.4 recognition of MHC class I molecules was CD8-dependent (Fig. 5 A, left); CD8+ MM14.4+ T cells did not react to MHC class II Ab molecules, but were activated by MHC class II–negative Ltk− cells of H-2k MHC haplotype. These reactivity patterns were confirmed by blocking with mAb against MHC molecules and against the coreceptors (Fig. 5 A, middle and right panels for CD4+ and CD8+ cells, respectively). In addition, MM14.4 cells expressing CD4 were unable to react to splenocytes from mice expressing mutant Ab molecules lacking the CD4 binding site (Ab MUT; reference 19; Fig. 5 B).
Experiments described in this section demonstrated that (a) MM14.4 clearly has dual restriction, and interacts with MHC class I and class II; (b) AbEp is a positively selecting ligand for CD4+MM14.4+ cells, but not for CD8+ MM14.4+ T cells; and (c) dual MHC restriction of MM14.4 is heavily dependent upon the coreceptors.
Biased MHC class II/peptide repertoire favors selection of TCRs with highly degenerate specificity
Thus, we have established a possible link between expression of extremely limited MHC class II/peptide repertoire and generation of autoimmune CD8+ T cells. We reasoned that biased MHC–peptide repertoire is likely to allow positive selection of a larger proportion of TCRs that is less dependent on recognition of a specific selecting peptide. Therefore, it is likely that the degeneracy of T cell selection by a biased peptide repertoire will result in increased numbers of TCRs with multiple cross-reactivities, including those with recognition of both MHC classes. To test this hypothesis, we performed a comparative analysis of the occurrence of dr-TCRs in B6 and AbEp mice using dual MHC restriction as a measure of TCR promiscuity (Table II). To ensure that only one TCRα chain per T cell was expressed, we crossed AbEp mice to B6.TCRα-negative mice, and then intercrossed them to produce AbEp mice with a single copy of the TCRα locus. Before the fusion, T cells were activated in vitro with irradiated splenocytes expressing disparate MHC class II molecules. Because the coreceptors may be important for activation of low-affinity TCRs, to provide dual-reactive TCRs with the coreceptor needed for MHC class I recognition, we made the fusion of activated T cells and BWZ CD8+ lymphoma cell line (20). The first round of hybridoma screening revealed MHC class II–reactive individual hybridoma cultures (total of 5,664 cultures tested, 4,437 were MHC class II reactive), which were tested individually for activation by a panel of 11 fibroblastoid cell lines (21) which originated from different mouse strains and expressed MHC class I but not class II molecules. Positive cultures (54 cultures) were subcloned several times and karyotyped to exclude a possibility of a triple-cell fusion. 14 of 15 TCRs from dual-restricted cells were cloned, sequenced, expressed in a T cell lymphoma cell line, and their unique specificities were confirmed by an in vitro activation of the transfectants. The results of these experiments are shown in Table II. Clearly, AbEp mice selected more (at least by an order of magnitude) dr-TCRs than did their control counterparts. We found no apparent bias in TCR Vα and Vβ region usage in dual-restricted T cells (Table III).
Table II.
Mice with restricted MHC class II–peptide repertoire produce increased numbers of TCRs with dual MHC (class II/class I) restriction
| Dual restricted hybridoma clones
|
|||||||
|---|---|---|---|---|---|---|---|
| Group | Normal fusion partner |
Activated with |
Total no. of hybridomas tested |
Hybridomas reactive to MHC class II |
Before cloning |
After cloning |
Frequency of dual-restricted clonesa |
| I | B6 TCRα+/− | Abm12 | 2,278 | 2,185 | 12 | 2 | |
| II | B6 TCRα+/− | H-2q | 480 | 364 | 5 | 0 | |
| Total | 2,758 | 2,549 | 2 | 1:1,275 | |||
| III | AbEp TCRα+/− | B6 | 1,920 | 1,886 | 36 | 12 | |
| IV | AbEp | ConA | 986 | 2 | 1 | 1 | |
| Total | 2,906 | 1,888 | 13 | 1:145 | |||
Estimation of relative frequencies of dual MHC class I/ MHC class II–restricted T cells in control (B6) mice and in AbEp mice by direct testing of MHC class II–reactive hybridomas against a panel of MHC class I–positive, class II–negative fibroblastoid cell lines. Mice used in groups I–III had one copy of the TCRα locus, and BW5147 TCR-negative, CD8-positive cell line was used as a fusion partner in these groups. Mice in group IV had two copies of TCRα locus, but no evidence of an additional productive rearrangement of the TCRα locus was found in MM14.4 hybridoma belonging to this group (not depicted). BW5147 TCR-negative, coreceptor-negative cell line was used as fusion partner in this group.
Frequency calculated per total number of MHC class II–reactive hybridomas.
Table III.
TCRα and β chain usage by dual-restricted hybridomas
| Origin | Hybridoma clone | Vα | Jα | Vβ | Jβ | MHC I |
|---|---|---|---|---|---|---|
| B6, TCRα+/−
group I |
4F5 | TRAV10(D) | TRAJ17 | TRBV14 | TRBJ2-1 | Kbm1, H-2u,s |
| IG2A | TRAV6D-4 | TRAJ31 | TRBV2 | TRBJ1-1 | H-2q | |
| AbEp, TCRα+/−
group III |
17a3 | TRAV5D-4 | TRAJ45 | TRBV12-2 | TRBJ2-2 | Dd |
| 19d4 | TRAV7-3 | TRAJ43 | TRBV19 | TRBJ2-3 | H-2q | |
| 21d1 | TRAV14D-3 | TRAJ33 | TRBV13-2 | TRBJ2-7 | Dq | |
| 24d5 | TRAV14D-1 | TRAJ49 | TRBV1 | TRBJ2-4 | H-2k | |
| 27b3 | TRAV8D-1 | TRAJ47 | TRBV3 | TRBJ2-7 | Kbm3 | |
| 37b1 | TRAV3-1 | TRAJ50 | TRBV13-1 | TRBJ2-1 | Kbm1, H-2s | |
| 40a1 | TRADV16D | TRAJ32 | TRBV14 | TRBJ2-1 | Kbm1, H-2s | |
| 42a2 | TRAV13-1 | TRAJ48 | TRBV12-2 | TRBJ2-4 | Kbm3 | |
| 44a4 | TRAV3(D)-3 | TRAJ6 | TRBV12-1 | TRBJ2-7 | H-2k | |
| 49c6 | TRAV5D-4 | TRAJ22 | TRBV19 | TRBJ1-1 | H-2k | |
| 8-1-6 | TRAV4D-3 | TRAJ30 | TRBV15 | TRBJ2-7 | H-2u | |
| AbEp, ConA group IV |
MM14.4 | TRAV14D-2 | TRAJ26 | TRBV3 | TRBJ2-1 | Kk |
All cells reacted to Ab. Their MHC class I cross-reactivity is shown in the right column. Where the exact molecule has not been identified, the whole MHC haplotype is shown. Groups are as per Table II.
Thus, limited MHC class II/peptide repertoire supports selection of T cells carrying TCRs with highly degenerate MHC recognition.
DISCUSSION
Autoimmunity linkage with MHC alleles is well-established. However, the exact mechanisms of such linkage are not clear. It also is unclear how the well-documented involvement of MHC class I–reactive CD8+ T cells in the pathogenesis of autoimmune diseases fits with autoimmunity linkage to MHC class II genes. Our study suggests answers to these problems.
Limitation of MHC–peptide repertoire leads to autoimmunity
MHC alleles associated with autoimmunity are believed to be unable to provide adequate negative selection because of a low affinity of MHC interactions with potentially hazardous peptides (22, 23) or a low avidity of TCR interactions with MHC–peptide complexes (24, 25). Because autoimmunity-associated MHC class II molecules have structural features which limit the pool of peptides that they can bind (3–5), we hypothesized that the key to such association is in the biased peptide repertoire presented by autoimmunity-prone MHC molecules. This biased (or limited) peptide repertoire leads to incomplete negative selection, which normally contributes to the shaping of the TCR repertoire by removing highly specific autoreactive receptors (26) and promiscuous receptors (27). It also may support positive selection of T cells carrying promiscuous receptors. TCRs with promiscuous receptors must be produced constantly in normal mice by a random association of germline-encoded segments of the TCR and other diversification mechanisms accompanying recombination. However, because the size of the T cell compartment is limited, the TCRs that are best fit for interactions with MHC–peptide complexes have a selective advantage and fill it to capacity. The limitation of peptide diversity places T cells with regular TCRs at a disadvantage and increases the relative input in the TCR pool of the otherwise outnumbered nonconventional TCRs.
In several studies, autoimmunity was obliterated by transgenic expression of “good” MHC molecules along with “bad” (autoimmunity-prone) host MHC (28–30). One explanation for these results is that transgenic MHC molecules caused negative selection of TCRs with promiscuous recognition of multiple MHC molecules (28). It also is possible that expression of an additional and different MHC protein stimulates robust positive selection, and resulting T cells outnumber the potentially autoreactive T cells selected by a disease-prone MHC. This is a likely scenario in the case of the HLAQ062 molecule being protective for type 1 diabetes (31, 32). Its recently determined crystal structure suggests an expanded peptide repertoire presented by this molecule compared with the repertoire presented by homologous DQ molecules associated with type 1 diabetes (32).
Our experimental approach involved studies of mice with an extremely limited MHC class II/peptide repertoire (8). These mice developed multiple autoimmune reactions (Fig. 1), and when a TCR from such mice was overexpressed as a transgene (MM14.4), they developed an organ-specific autoimmunity (Fig. 2). A CD4+ T cell–mediated autoimmune reaction in independently generated AbEp mice also has been reported (33).
Autoimmunity was found only in older AbEp mice and was rather mild and suggested that the activity of self-reactive T cells could be controlled by suppressor T cells. Closer examination showed that AbEp mice have a robust population of suppressor T cells (Fig. S1, A and B, Table S1, and supplemental Materials and methods, available at http://www.jem.org/cgi/content/full/jem.20050198/DC1) which suppressed dermatitis development in MM14.4 transgenic mice upon adoptive transfer (Fig. S1 C). These data imply that autoimmunity observed in AbEp mice is quenched by active suppression.
AbEp mice are an extreme case of the biased MHC class II/peptide repertoire. Autoimmunity-prone MHC alleles in humans and rodents have a much broader peptide repertoire. In our own experiments and experiments of Ignatowicz's group (13), AbEp mice expressing Ii—and having ∼70% of the covalently bound Eα peptide replaced by other peptides—developed autoimmune responses (Fig. 1). Thus, even partial, incomplete restriction of the MHC–peptide repertoire may promote autoimmunity; this suggests that this may be a significant part of the mechanism by which autoimmunity-prone MHC molecules function.
Dual MHC class I and II restriction of TCRs selected by biased peptide repertoire may explain MHC class II linkage of MHC class I–driven autoimmunity
To our surprise, MM14.4 TCR originally cloned from a CD4+ hybridoma was found in transgenic mice, predominantly expressed by the CD8 lineage T cells, which also were responsible for the dermatitis. Further examination showed that MM14.4 is a truly dual-MHC class I/MHC class II–restricted TCR (Figs. 4 and 5). Spontaneous infiltrates around the pancreatic islets in non-TCR transgenic AbEp mice (Fig. 1 B) consisted predominantly of CD8+ T cells; these indicated that other autoimmune TCRs behave similarly to M14.4 and that AbEp mice select more of such receptors.
Receptors with dual-MHC restriction can be selected by normal MHC–peptide repertoire (reference 34, Table II), but that is more of an exception. Direct comparison of the relative frequencies of the dr-TCRs in B6 and AbEp mice showed that the latter select ∼10 times more dr-TCRs than do normal mice (Table II). It is likely that dr-receptors have a bias toward recognition of broad framework MHC determinants, which allows them to interact with both MHC classes. The TCR structural features that allow these interactions have not been determined. The V region usage in α and β chains seems to be broad (Table III). Similarly, sequences of the CDR3α regions do not contain an obvious motif (Table S2, available at http://www.jem.org/cgi/content/full/jem.20050198/DC1). The only unusual feature is the presence of a bulky tryptophan residue in the CDR3 regions of the β chains of many of these receptors (Table S3, available at http://www.jem.org/cgi/content/full/jem.20050198/DC1). The significance of this observation remains to be determined.
Under normal circumstances, dr-receptors are likely to be removed by unrestricted MHC–peptide repertoire due to a high overall avidity of interactions with both MHC classes and coreceptors. The CD8+ compartment in mice lacking CD4 molecules contained increased numbers of MHC class II–reactive cells compared with normal CD4-sufficient mice (35). Conversely, a biased peptide repertoire allows dual-restricted TCRs to slip through negative selection by MHC class II, and promotes their positive selection into the CD4 lineage. In addition, under conditions of a biased peptide repertoire, other factors, such as MHC-peptide complex density and the presence of functional coreceptors, may compensate for the lack of TCR–peptide interactions. Dependence of the T cell activation on the coreceptors was associated with a low affinity of TCR–MHC interaction (36). Coreceptors may contribute to positive selection by enforcing signaling through the TCR (37) or by increasing the overall avidity of the TCR-coreceptor-MHC–peptide complex (38, 39).
It is well-established that CD8+ T cells participate in the development of organ-specific autoimmune diseases, such as type 1 diabetes. However, diabetes-prone animals do not have unique MHC class I molecules, whereas MHC class II is linked clearly to diabetes development. It is possible that selection of promiscuous diabetogenic receptors (40) follows the same rules that we have observed in mice with biased MHC class II/peptide repertoire; however, this issue awaits further examination. Dual-restricted receptors are likely to be an extreme example of TCR promiscuity, and it would be naive to claim that they are the exclusive source of autoreactive TCRs. Nevertheless, because biased peptide repertoire selects more of these receptors and leads to various forms of autoimmune reactions (including dermatitis caused by MM14.4), they may make an important contribution to the autoimmune pool of TCRs. It is intriguing that a substantial subset of the naturally occurring dual MHC-restricted TCRs described so far (Table S4, available at http://www.jem.org/cgi/content/full/jem.20050198/DC1; the list contains TCRs with proven or potential dual MHC class restriction) is involved in autoimmune reactions.
Thus, a biased MHC class II–peptide repertoire facilitates selection of aberrant, highly promiscuous TCRs. This is an important prerequisite for the development of autoimmunity; however, the clinical outcome undoubtedly is influenced by additional factors, such as suppressor T cell activity, that reflect the complicated genetics of autoimmunity.
MATERIALS AND METHODS
Mice and genotyping.
Mice expressing no endogenous MHC class II and Ii (C2°) and C2° mice expressing AbEp complexes (AbEp mice) were a gift from P. Marrack (National Jewish Medical and Research Center, Denver, CO). BALB/cJ, C57BL/6J (B6), B10.BR-H2k2 H2-T18a/SgSnJ (B10.BR), C3H/HeJ, B6.C-H2bm12/KhEg (bm12), B6.129P2-β2mtm1Unc (β2m KO), C57BL/6J-Rag1tm1Mom (Rag1 KO), B6.129S-Tcratm1Mom (TCRα KO), B6.129S-Tcrbtm1Mom/J (TCRβ KO), B6.129S-Tcrbtm1MomTcrdtm1Mom/J (TCRβ,δ KO), B6.129-H2dlAb1-Ea/J (C2complete KO), B6.129.S2-Igh-6tm1Cqn/J (B cell− mice), B6.129S6-Iitm1Liz/J (Ii KO), and B6.129-H2-DMatm1Luc (DM KO) mice were obtained from The Jackson Laboratory. Mice lacking classic MHC class I molecules, Kb and Db (41), were provided by D. Roopenian (The Jackson Laboratory). TCRα KO mice were genotyped by Southern blotting as described (42). Mice lacking Rag1 and β2m were typed by staining with anti-Ig (to detect the loss of B cells) and anti–MHC class I antibodies, respectively. Mice expressing transgenic MHC class II Ab molecules (AbWT) and Ab with disrupted CD4-binding site (AbMUT; reference 19) were maintained at the Jackson Laboratory animal research facility. Animal research protocols were approved by The Jackson Laboratory Animal Care and Use Committee.
TCR cloning and transgenic mice.
Lymph node T cells from AbEp mice were stimulated with ConA and fused with the TCR-negative BW5147 thymoma. Hybridomas were tested for IL-2 production after stimulation with B6 splenocytes. A CD4+ subclone MM14.4 was chosen for future experiments. MM14.4 expressed Vα2 as determined by staining with a panel of mAb obtained from BD Biosciences. The Vβ region (Vβ16.2) was determined by PCR with a set of β-specific primers (43). cDNA for α and β chains were prepared by RT PCR, cloned into a TA vector (Invitrogen), and sequenced. Primers were designed to amplify the Vα2.5-Jα20 and Vβ16.2-Dβ2.1-Jβ2.1 regions from MM14.4 genomic DNA. The resulting fragments were subcloned into pTα and pTβ genomic TCR-cassette vectors (a gift from D. Mathis, Harvard Medical School, Boston, MA) as described previously (44). The new constructs were tested for expression by transfection (along with CD4 cDNA) into the 4G4 cell line, which expresses CD3 but no endogenous α or β TCR chains. The reactivity of the transfectants was not different from that of the initial MM14.4 hybridoma and was blocked by anti-Ab mAb. These results confirmed that this was the correct and functional construct. DNA fragments encoding α and β chains were coinjected into (B6xBALB/cBy)F1 oocytes fertilized by C2°,AbEp+ transgenic males. Of seven independent founders, integration of both TCR chains was detected in five of them.
Dual-restricted TCRs from hybridomas.
Lymph node cells from mice carrying one copy of TCRα-locus with normal MHC class II (Ab) or AbEp were activated in vitro with allo MHC class II (bm12 or Aq) and WT Ab MHC class II, respectively, and were fused to CD8+ BWZ lymphoma cell line (a gift from N. Shastri, University of California, Berkeley, CA; reference 20). All hybridomas were tested first in IL-2 production assay (described in the T cell activation section) for reactivity with the immunizing MHC class II molecules in the presence or absence of blocking anti–MHC class II antibodies. Positive MHC class II–reactive cultures were tested for activation by a panel of 11 SV40-transformed MHC class II–negative, MHC class I–positive fibroblasts from different mouse strains (21). Positive cultures were subcloned multiple times, and karyotyped to ensure that they did not result from fusion of three cells. TCR chains were cloned exactly as described for MM14.4.
Dermatitis severity was determined as follows: 1, small lesions (≤1 cm2) on the ventral trunk; 2, larger lesions, but still localized only to the ventral trunk; 3, scattered lesions on the ventral and dorsal sides; 4, lesions on the entire trunk and head; 5, similar to 4 but with visible exudate.
Monoclonal antibodies and FACS analysis.
All fluorochrome-labeled mAbs were purchased from BD Biosciences except for anti–CD8-Quantum red and anti–mouse Ig Fc fragment polyclonal antibodies labeled with FITC (Sigma-Aldrich) and anti–CD4-Red613 (GIBCO BRL). Purified anti–MHC class II antibodies and anti-CD3ɛ mAb 145-2C11 were obtained from the Yale University Hybridoma Facility. Data collection on the FACSCalibur flow cytometer (Beckton Dickinson) and subsequent analysis was performed using CELLQuest software.
In vivo elimination of CD8+ T cells was done by i.p. injection of 50 μg/mouse of anti-CD8 mAb YTS169.4 (45) every other day for 2 wk starting at 7–10 d post partum.
Histology and immunohistochemistry.
Tissues were fixed and stained with hematoxylin-eosin by standard methods. Cryosections of pancreas, thyroids, salivary glands, and skin were stained with FITC-conjugated anti-CD4 (Caltag), anti-CD8, and anti-CD3 (BD Biosciences). Antikeratin staining was done on paraffin sections. Endogenous peroxidase was quenched with 3% H2O2, and sections were stained overnight with antisera to keratins 1, 6, and 10 (Babco); washed in PBS; and incubated with biotinylated secondary antibody and streptavidin–horseradish peroxidase. Positive reaction was detected with diaminobenzidine (Vector Laboratories), and sections were counterstained with hematoxylin. ANA staining was performed by indirect immunofluorescence using HEP-2–coated slides (RHI Gene Inc.).
T cell and skin DC purification.
CD4+ and CD8+ T cells were isolated from lymph node cells by treatment with mAbs against MHC class II (Y3JP; reference 46) and anti-CD8 (53–6.72; reference 47) or anti-CD4 (GK1.5; reference 48), respectively, for 45 min at 4°C (107 cells per ml of culture supernatant) followed by negative selection with the mixture of magnetic beads (anti–mouse IgG, anti–mouse IgM, anti–rat IgG) from PerSeptive Biosystems according to the manufacturer's protocol. For T cell depletion, a mixture of anti-CD8 and anti-CD4 (GK1.5) mAbs was used, followed by treatment with anti–rat Ig magnetic beads. DCs were purified from ventral and dorsal skin of AbEp and C2Δ mice as described previously (49).
T cell activation.
T cell proliferation was measured by 3H-thymidine incorporation after 3 d of cocultivation of 2 × 105 spleen or lymph node cells with 2 × 105 irradiated (2,000 rad) stimulator splenocytes in 96-well plates (Becton Dickinson) in 150 μl of culture medium [Click's EHAA Medium (Irvine Scientific) supplemented with 5% FCS (Sigma-Aldrich), 20 mM L-glutamine (GIBCO BRL), 5 × 10−5 M β-mercaptoethanol (Bio-Rad Laboratories) and 100 U/ml penicillin/streptomycin mixture (GIBCO BRL)]. 2 × 105 of purified CD8+ T cells from Rag-negative MM14.4 mice were activated with 5 × 104 skin DCs. The range of proliferation was 1–1.8 × 102 cpm in DCs or T cells alone, or T cell + splenocyte controls, and 2–4 × 103 in cocultures of responder T cells and skin DCs. 2.5 × 104 T cell hybridomas or TCR transfectants were activated by cultivation with 105 stimulator cells or by plate-bound anti-CD3 mAb; IL2 production into the medium was measured by proliferative response of the IL-2–dependent cell line CTLL using Alamar Blue (Biosource International) color conversion assay.
Western blotting was performed on lysates of CD4-enriched lymph node cells. Lysates were prepared using 1% NP-40 (Sigma-Aldrich) in 0.1 M Tris buffer (pH 7.4) supplemented with “complete mini” protease inhibitor cocktail (Roche Diagnostics Corp.). Lysates were loaded at different volumes to match the cell equivalents of CD4+ cells as determined by FACS staining after negative selection of B cells and CD8+ cells with magnetic beads. Donkey anti–rabbit horseradish peroxidase–conjugated antibodies (Jackson ImmunoResearch Laboratories) were used as secondary antibodies. Blots were developed with the enhanced chemiluminescence reagent (Amersham Biosciences).
Online supplemental material.
Supplemental Materials and methods and Fig. S1 show evidence that AbEp mice have elevated levels of T suppressor cells that express FoxP3 and are capable of blocking dermatitis in MM14.4 TCR transgenic mice upon transfer into newborn mice. Table S1 shows age-dependent accumulation of activated T cells in AbEp mice. Tables S2 and S3 show V and CDR3 regions of α and β chains, respectively, of cloned TCRs with dual-MHC restriction. Table S4 lists examples of TCRs with known or strongly implied dual MHC restriction and their relevance to autoimmunity. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050198/DC1.
Acknowledgments
We are thankful to the late C.A. Janeway Jr. for discussion of our initial results and to C. Doyle for the gift of mice lacking CD4 binding by Ab.
The work was supported by National Institutes of Health grant nos. AI46782 (to A.V. Chervonsky) and AR 43801 (to J.P. Sundberg), and by core grant CA 34196.
The authors have no conflicting financial interests.
Abbreviations used: ANA, antinuclear antibody; dr-TCR, dual–MHC-restricted TCR; Ii, invariant chain.
N.N. Logunova, C. Viret, and L.A. Pobezinsky contributed equally to this work.
C. Viret's present address is Institut National de la Santé et de la Recherche Medicale Unit 548 and CEA-DRDC, 38054 Grenoble Cedex 9, France.
References
- 1.Nepon, G.T. 1998. Major histocompatibility complex-directed susceptibility to rheumatoid arthritis. Adv. Immunol. 68:315–332. [DOI] [PubMed] [Google Scholar]
- 2.Nepon, G.T., and W.W. Kwok. 1998. Molecular basis for HLA-DQ associations with IDDM. Diabetes. 47:1177–1184. [DOI] [PubMed] [Google Scholar]
- 3.Dessen, A., C.M. Lawrence, S. Cupo, D.M. Zaller, and D.C. Wiley. 1997. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 7:473–481. [DOI] [PubMed] [Google Scholar]
- 4.Corper, A.L., T. Stratmann, V. Apostolopoulos, C.A. Scott, K.C. Garcia, A.S. Kang, I.A. Wilson, and L. Teyton. 2000. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science. 288:505–511. [DOI] [PubMed] [Google Scholar]
- 5.Latek, R.R., A. Suri, S.J. Petzold, C.A. Nelson, O. Kanagawa, E.R. Unanue, and D.H. Fremont. 2000. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. 12:711–720. [DOI] [PubMed] [Google Scholar]
- 6.Carrasco-Marin, E., J. Shimizu, O. Kanagawa, and E.R. Unanue. 1996. The class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J. Immunol. 156:450–458. [PubMed] [Google Scholar]
- 7.Woulfe, S.L., C.P. Bono, D.A. Zacheis, T.A. Kirschmann, C.S. Baudino, R.W. Karr, and B.D. Schwartz. 1995. Negatively charged residues interacting with the p4 pocket confer binding specificity to DRB1*0401. Arthritis Rheum. 38:1744–1753. [DOI] [PubMed] [Google Scholar]
- 8.Ignatowicz, L., J. Kappler, and P. Marrack. 1996. The repertoire of T cells shaped by a single MHC/peptide ligand. Cell. 84:521–529. [DOI] [PubMed] [Google Scholar]
- 9.Shachar, I., and R.A. Flavell. 1996. Requirement for invariant chain in B cell maturation and function. Science. 274:106–108. [DOI] [PubMed] [Google Scholar]
- 10.Benlagha, K., S.H. Park, R. Guinamard, C. Forestier, L. Karlsson, C.H. Chang, and A. Bendelac. 2004. Mechanisms governing B cell developmental defects in invariant chain-deficient mice. J. Immunol. 172:2076–2083. [DOI] [PubMed] [Google Scholar]
- 11.Ignatowicz, L., G. Winslow, J. Bill, J. Kappler, and P. Marrack. 1995. Cell surface expression of class II MHC proteins bound by a single peptide. J. Immunol. 154:3852–3862. [PubMed] [Google Scholar]
- 12.Chervonsky, A.V., R.M. Medzhitov, L.K. Denzin, A.K. Barlow, A.Y. Rudensky, and C.A. Janeway Jr. 1998. Subtle conformational changes induced in major histocompatibility complex class II molecules by binding peptides. Proc. Natl. Acad. Sci. USA. 95:10094–10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaszewska-Mastalarz, A., P. Muranski, B. Chmielowski, P. Kraj, and L. Ignatowicz. 2000. Altered selection of CD4+ T cells by class II MHC bound with dominant and low abundance self-peptides. J. Immunol. 165:6099–6106. [DOI] [PubMed] [Google Scholar]
- 14.Wright, A.L., and A.G. Messenger. 1990. Scarring alopecia in psoriasis. Acta Derm. Venereol. 70:156–159. [PubMed] [Google Scholar]
- 15.Stanimirovic, A., M. Skerlev, T. Stipic, T. Beck, A. Basta-Juzbasic, and D. Ivankovic. 1998. Has psoriasis its own characteristic trichogram? J. Dermatol. Sci. 17:156–159. [DOI] [PubMed] [Google Scholar]
- 16.Madsen, L., N. Labrecque, J. Engberg, A. Dierich, A. Svejgaard, C. Benoist, D. Mathis, and L. Fugger. 1999. Mice lacking all conventional MHC class II genes. Proc. Natl. Acad. Sci. USA. 96:10338–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He, X., C.A. Janeway Jr., M. Levine, E. Robinson, P. Preston-Hurlburt, C. Viret, and K. Bottomly. 2002. Dual receptor T cells extend the immune repertoire for foreign antigens. Nat. Immunol. 3:127–134. [DOI] [PubMed] [Google Scholar]
- 18.Padovan, E., G. Casorati, P. Dellabona, C. Giachino, and A. Lanzavecchia. 1995. Dual receptor T-cells. Implications for alloreactivity and autoimmunity. Ann. N. Y. Acad. Sci. 756:66–70. [DOI] [PubMed] [Google Scholar]
- 19.Riberdy, J.M., E. Mostaghel, and C. Doyle. 1998. Disruption of the CD4-major histocompatibility complex class II interaction blocks the development of CD4(+) T cells in vivo. Proc. Natl. Acad. Sci. USA. 95:4493–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanderson, S., and N. Shastri. 1994. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol. 6:369–376. [DOI] [PubMed] [Google Scholar]
- 21.Pfizenmaier, K., G. Trinchieri, D. Solter, and B.B. Knowles. 1978. Mapping of H-2 genes associated with T cell-mediated cytotoxic responses to SV40-tumour-associated specific antigens. Nature. 274:691–693. [DOI] [PubMed] [Google Scholar]
- 22.Liu, G.Y., P.J. Fairchild, R.M. Smith, J.R. Prowle, D. Kioussis, and D.C. Wraith. 1995. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 3:407–415. [DOI] [PubMed] [Google Scholar]
- 23.Fairchild, P.J., R. Wildgoose, E. Atherton, S. Webb, and D.C. Wraith. 1993. An autoantigenic T cell epitope forms unstable complexes with class II MHC: a novel route for escape from tolerance induction. Int. Immunol. 5:1151–1158. [DOI] [PubMed] [Google Scholar]
- 24.Ridgway, W.M., M. Fasso, C.G. Fathman. 1999. A new look at MHC and autoimmune disease. Science. 284:749–751. [DOI] [PubMed] [Google Scholar]
- 25.Ridgway, W.M., and C.G. Fathman. 1999. MHC structure and autoimmune T cell repertoire development. Curr. Opin. Immunol. 11:638–642. [DOI] [PubMed] [Google Scholar]
- 26.Kisielow, P., H. Bluthmann, U.D. Staerz, M. Steinmetz, and H. von Boehmer. 1988. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 333:742–746. [DOI] [PubMed] [Google Scholar]
- 27.Huseby, E.S., F. Crawford, J. White, J. Kappler, and P. Marrack. 2003. Negative selection imparts peptide specificity to the mature T cell repertoire. Proc. Natl. Acad. Sci. USA. 100:11565–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lund, T., L. O'Reilly, P. Hutchings, O. Kanagawa, E. Simpson, R. Gravely, P. Chandler, J. Dyson, J.K. Picard, A. Edwards, et al. 1990. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-A beta-chain or normal I-E alpha-chain. Nature. 345:727–729. [DOI] [PubMed] [Google Scholar]
- 29.Slattery, R.M., L. Kjer-Nielsen, J. Allison, B. Charlton, T.E. Mandel, and J.F. Miller. 1990. Prevention of diabetes in non-obese diabetic I-Ak transgenic mice. Nature. 345:724–726. [DOI] [PubMed] [Google Scholar]
- 30.Bohme, J., B. Schuhbaur, O. Kanagawa, C. Benoist, and D. Mathis. 1990. MHC-linked protection from diabetes dissociated from clonal deletion of T cells. Science. 249:293–295. [DOI] [PubMed] [Google Scholar]
- 31.Todd, J.A., and L.S. Wicker. 2001. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity. 15:387–395. [DOI] [PubMed] [Google Scholar]
- 32.Siebold, C., B.E. Hansen, J.R. Wyer, K. Harlos, R.E. Esnouf, A. Svejgaard, J.I. Bell, J.L. Strominger, E.Y. Jones, and L. Fugger. 2004. Crystal structure of HLA-DQ0602 that protects against type 1 diabetes and confers strong susceptibility to narcolepsy. Proc. Natl. Acad. Sci. USA. 101:1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oono, T., Y. Fukui, S. Masuko, O. Hashimoto, T. Ueno, T. Sanui, A. Inayoshi, M. Noda, M. Sata, and T. Sasazuki. 2001. Organ-specific autoimmunity in mice whose T cell repertoire is shaped by a single antigenic peptide. J. Clin. Invest. 108:1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arsov, I., and S. Vukmanovic. 1999. Dual MHC class I and class II restriction of a single T cell receptor: distinct modes of tolerance induction by two classes of autoantigens. J. Immunol. 162:2008–2015. [PubMed] [Google Scholar]
- 35.Tyznik, A.J., J.C. Sun, and M.J. Bevan. 2004. The CD8 population in CD4-deficient mice is heavily contaminated with MHC class II-restricted T cells. J. Exp. Med. 199:559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee, D.S., C. Ahn, B. Ernst, J. Sprent, and C.D. Surh. 1999. Thymic selection by a single MHC/peptide ligand: autoreactive T cells are low-affinity cells. Immunity. 10:83–92. [DOI] [PubMed] [Google Scholar]
- 37.Hernandez-Hoyos, G., S.J. Sohn, E.V. Rothenberg, and J. Alberola-Ila. 2000. Lck activity controls CD4/CD8 T cell lineage commitment. Immunity. 12:313–322. [DOI] [PubMed] [Google Scholar]
- 38.Doyle, C., and J.L. Strominger. 1987. Interaction between CD4 and class II MHC molecules mediates cell adhesion. Nature. 330:256–259. [DOI] [PubMed] [Google Scholar]
- 39.Salter, R.D., R.J. Benjamin, P.K. Wesley, S.E. Buxton, T.P. Garrett, C. Clayberger, A.M. Krensky, A.M. Norment, D.R. Littman, and P. Parham. 1990. A binding site for the T-cell co-receptor CD8 on the alpha 3 domain of HLA-A2. Nature. 345:41–46. [DOI] [PubMed] [Google Scholar]
- 40.Serreze, D.V., T.M. Holl, M.P. Marron, R.T. Graser, E.A. Johnson, C. Choisy-Rossi, R.M. Slattery, S.M. Lieberman, and T.P. DiLorenzo. 2004. MHC class II molecules play a role in the selection of autoreactive class I-restricted CD8 T cells that are essential contributors to type 1 diabetes development in nonobese diabetic mice. J. Immunol. 172:871–879. [DOI] [PubMed] [Google Scholar]
- 41.Vugmeyster, Y., R. Glas, B. Perarnau, F.A. Lemonnier, H. Eisen, and H. Ploegh. 1998. Major histocompatibility complex (MHC) class I KbDb −/− deficient mice possess functional CD8+ T cells and natural killer cells. Proc. Natl. Acad. Sci. USA. 95:12492–12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mombaerts, P., A.R. Clarke, M.A. Rudnicki, J. Iacomini, S. Itohara, J.J. Lafaille, L. Wang, Y. Ichikawa, R. Jaenisch, M.L. Hooper, et al. 1992. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature. 360:225–231. [DOI] [PubMed] [Google Scholar]
- 43.Casanova, J.L., P. Romero, C. Widmann, P. Kourilsky, and J.L. Maryanski. 1991. T cell receptor genes in a series of class I major histocompatibility complex-restricted cytotoxic T lymphocyte clones specific for a Plasmodium berghei nonapeptide: implications for T cell allelic exclusion and antigen-specific repertoire. J. Exp. Med. 174:1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kouskoff, V., K. Signorelli, C. Benoist, and D. Mathis. 1995. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods. 180:273–280. [DOI] [PubMed] [Google Scholar]
- 45.Cobbold, S.P., A. Jayasuriya, A. Nash, T.D. Prospero, and H. Waldmann. 1984. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature. 312:548–551. [DOI] [PubMed] [Google Scholar]
- 46.Janeway, C.A., Jr., P.J. Conrad, E.A. Lerner, J. Babich, P. Wettstein, and D.B. Murphy. 1984. Monoclonal antibodies specific for Ia glycoproteins raised by immunization with activated T cells: possible role of T cellbound Ia antigens as targets of immunoregulatory T cells. J. Immunol. 132:662–667. [PubMed] [Google Scholar]
- 47.Ledbetter, J.A., and L.A. Herzenberg. 1979. Xenogeneic monoclonal antibodies to mouse lymphoid differentiation antigens. Immunol. Rev. 47:63–90. [DOI] [PubMed] [Google Scholar]
- 48.Dialynas, D.P., D.B. Wilde, P. Marrack, A. Pierres, K.A. Wall, W. Havran, G. Otten, M.R. Loken, M. Pierres, J. Kappler, and F.W. Fitch. 1983. Characterization of the murine antigenic determinant, designated L3T4a, recognized by monoclonal antibody GK1.5: expression of L3T4a by functional T cell clones appears to correlate primarily with class II MHC antigen-reactivity. Immunol. Rev. 74:29–56. [DOI] [PubMed] [Google Scholar]
- 49.Tokura, Y., J. Yagi, M. O'Malley, J.M. Lewis, M. Takigawa, R.L. Edelson, and R.E. Tigelaar. 1994. Superantigenic staphylococcal exotoxins induce T-cell proliferation in the presence of Langerhans cells or class II-bearing keratinocytes and stimulate keratinocytes to produce T-cell-activating cytokines. J. Invest. Dermatol. 102:31–38. [DOI] [PubMed] [Google Scholar]