

Abstract
For CD8+ T cells, a relatively short antigen pulse seems sufficient for antigen-presenting cells to drive clonal expansion and differentiation. It is unknown whether the requirement for antigen is similarly ephemeral for CD4+ T cells. To study the dependence of a CD4+ T cell response on antigen persistence in a quantitatively and temporally controlled manner in vivo, we engineered a mouse line expressing a major histocompatibility complex class II–restricted epitope in dendritic cells under the control of a tetracycline-inducible promoter. Experiments tracking the proliferation of CD4+ T cells exposed to their cognate antigen in various amounts for different time periods revealed that the division of such cells was contingent on the presence of antigen throughout their expansion phase, even in the presence of an inflammatory stimulus. This previously unrecognized feature of a CD4+ T cell response contrasts with the proliferative behavior of CD8+ T cells that has been documented, and it implies that the two T cell subsets might require different strategies for efficient vaccination.
The exact way in which CD4+ T cell responses depend on the orchestrated activity of antigen presentation, costimulation, cytokines, and lymphocyte trafficking remains incompletely understood. In particular, the role that antigen plays in the expansion of antigen-specific T cells during an immune response in vivo has only recently been addressed. On the encounter of antigen-presenting DCs in the T lymphocyte zones of secondary lymphoid organs, T cells proliferate after a latency period of ∼24 h. They divide rapidly and, in parallel, express genes that enable them to become completely differentiated effector cells over the following days (1–5). Understanding how external signals guide the T cells' internal processes in vivo is critical both for better eliciting protective immune responses and for interfering with pathological ones.
In the case of CD8+ T cells, it seems that a relatively brief engagement of the TCR can be sufficient for initiating a program to drive cells through multiple rounds of division, as well as to acquire effector and memory functions. In a Listeria monocytogenes model, CD8+ T cell expansion continued despite the elimination of live bacteria with an antibiotic 24 h after infection (6). Similarly, naive CD8+ cells activated for 2–24 h in vitro and replated without the stimulus divided three to five times over the following days (7–10). Importantly, CD8+ T cells stimulated in vitro for 24 h divided at least seven times after a subsequent transfer into antigen-free hosts (11). Bevan and Fink (12) termed this phenomenon as being on “autopilot,” alluding to a transcriptional program that is induced on the initial antigen encounter and directs the cells toward their effector and memory functions without further antigenic stimulation. Although this idea has recently been challenged by experiments performed both in vitro (13) and in vivo (14), the current consensus is that the clonal selection of CD8+ T cells occurs within a short time span of TCR/MHC–peptide engagement.
For CD4+ T cells, the available data are less clear. Although an antigen pulse of several hours can be sufficient to support subsequent divisions (15), continuous antigen exposure considerably enhances ongoing proliferation (16–18). Thus, for a CD4+ T cell response, it is currently unknown whether the daughter cells from the first division need to reengage antigen-loaded DCs in the lymph node in order to continue dividing and whether clonal selection occurs during the initial antigen encounter or is a prolonged process.
To address antigen dependence during the CD4+ T cell expansion phase in vivo, we developed a mouse model in which an MHC class II–restricted epitope can be inducibly expressed in professional APCs. This model allowed us to induce and abrogate the expression of a T cell epitope in situ, avoiding potential changes in the APCs' maturation status because of antigen delivery or in vitro manipulation. We provide evidence that, rather surprisingly, CD4+ T cells are not programmed during the initial antigen encounter to divide a certain number of times, but rather proliferate according to the load and persistence of antigen they are exposed to throughout the expansion phase.
Results
Tetracycline (tet)-responsive antigen expression in professional APCs of transgenic mice
To study the influence of antigen dose and persistence on CD4+ T cell responses in vivo, we generated double transgenic mice expressing an antigenic peptide from moth cytochrome c (MCC) in a reversible manner. We used the tet-regulatable transgene expression system (Fig. 1 A) that we described previously (19). In brief, a first transgenic line named TIM (tet-inducible invariant chain with MCC) was designed to add the neoself T cell epitope MCC93-103 to the natural pool of self-peptides in cells that normally express MHC class II molecules. The Ek-restricted T cell epitope MCC93-103 replaced part of the class II–associated invariant chain (Ii) peptide region in order to deliver this antigen to the MHC class II loading pathway. The Ii–MCC construct was placed under the control of a tet-regulatable promoter.
Figure 1.

dox-controlled expression of a T cell epitope in vivo. (A) Schematic view of the double transgenic system. See the first two paragraphs of Results for details. (B) dox-dependent TIM reporter gene expression in lymphoid tissues. mRNA from the lymph nodes, spleen, and thymus of Ii-rTA+TIM+ animals supplied with standard (top) or 100 μg/ml dox-containing (bottom) drinking water was prepared, and TIM mRNA expression was assessed relative to HPRT by real-time RT-PCR. Data are from two and three animals, respectively (nd, not detectable). (C) In the spleen, the TIM reporter gene was mostly expressed in DCs. Splenocytes were sorted with mAbs against CD19, CD3, CD11b, and CD11c. cDNA was prepared, and TIM expression levels were determined by real-time RT-PCR and normalized to HPRT. Data from three animals are shown. The experiment was repeated twice with similar results. (D) TIM reporter mRNA quantitatively regulated by dox in the drinking water. Ii-rTA+TIM+ animals were exposed to the indicated doses of dox for 3 d, mRNA was prepared from subcutaneous lymph nodes, and TIM mRNA expression was assessed by real-time RT-PCR. Shown are values normalized to Ii-rTA mRNA. Each symbol represents a sample from one animal. The data were compiled from three independent experiments. (E) On transfer, AND CD4+ T cells proliferated in Ii-rTA+TIM+ recipients only in the presence of dox. Lymph node cells from AND+CD90.1+ animals were CFSE labeled and transferred into CD90.1− recipients of the indicated genotypes that were treated with 100 μg/ml dox as indicated, and subcutaneous lymph nodes and spleens were analyzed 60 h later. The histograms are gated on CD4+CD90.1+ cells, all other panels are gated on CD4+ cells. The experiment was repeated with similar results.
A second transgenic line carried the improved S2 mutant of the reverse tet transactivator (rtTAs-S2; reference 20) under the direction of a hybrid regulatory element consisting of the Eα enhancer and the Ii promoter. These control elements drive reporter gene expression at high levels in MHC class II–positive cells of transgenic mice (21). Transactivator expression was assessed in mice carrying this transgene (Ii-rTA) by Northern blot analysis. Like the endogenous invariant chain, the Ii-rTA mRNA was highly expressed in the lung, spleen, and thymus and was made at lower levels in the liver, heart, and kidney (unpublished data). Mice carrying Ii-rTA were crossed with the TIM reporter line, and double transgenic animals were administered the tet derivative doxycycline (dox) in their drinking water. This treatment led to the expression of the TIM gene, readily detectable by real-time RT-PCR in the lymph nodes, spleen and thymus, whereas no TIM mRNA was observed in the absence of dox (Fig. 1 B).
To establish which cell types express the inducible transgene, we sorted different cell populations from the spleens of dox-treated, double transgenic animals and quantitated TIM mRNA by real-time RT-PCR, relative to the housekeeping gene hypoxanthine phosphoribosyltransferase (HPRT). The highest level of TIM expression was found in DCs and was ∼20 times higher than in macrophages and 50 times higher than in B cells (Fig. 1 C). The low level of expression of a tet-inducible gene in B cells did not come as a surprise; several laboratories, including our own, have tried to achieve a tet-regulatable expression of diverse reporter genes in the mature B cells of transgenic mice, but have so far failed to do so for still obscure reasons (22, 23; unpublished data). However, because DCs play a pivotal role in CD4+ T cell priming to exogenous antigens (2, 5), this transgene expression pattern is very well suited for studying this process.
Our intention was to create a system in which the expression of a designated T cell antigen could also be regulated in a quantitative manner. To evaluate this potential in the Ii-rTA–TIM system, we provided mice with varied amounts of dox in their drinking water and assessed TIM expression in lymph nodes by real-time RT-PCR. A close correlation between the dose of antibiotic and TIM expression, extending over three orders of magnitude, was found (Fig. 1 D). Again, we could not detect any TIM mRNA in the absence of dox.
We next exploited the ability of the TIM-encoded antigen to stimulate T cells as a sensitive readout for TIM expression to (a) establish whether the MCC peptide was expressed in a stimulatory manner; and (b) test for “leaky” expression in the absence of dox, which can hamper the proper interpretation of results. Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled naive T cells from AND TCR transgenic mice, which have a T cell repertoire skewed for an Ek–MCC specificity, were transferred into Ii-rTA+TIM+ double transgenic or nontransgenic control recipients. 60 h after transfer, a vigorous proliferation of such cells was observed in the lymph nodes and spleen of dox-treated recipients. Proliferating AND cells acquired an effector phenotype by up-regulating CD44 and synthesizing IFN-γ (Fig. 1 E, bottom). Such activation was dependent on the AND TCR, as T cells of irrelevant specificity were not triggered to proliferate (see Fig. 6; unpublished data). In the absence of dox, no activation could be detected beyond the background observed in the nontransgenic recipient (Fig. 1 E, top and middle). These data, obtained using the more relevant and sensitive assay of T cell activation, also confirm that TIM and Ek–MCC expression are absolutely dependent on dox.
Figure 6.

Primed CD4+ cells require antigen for continued proliferation in a double transfer model and under inflammatory conditions. (A) AND cells divide two to four times in a primary host. CFSE-labeled lymph node cells from CD90.1+ AND TCR transgenic mice were transferred into a double transgenic Ii-rTA+TIM+ recipient pretreated with 100 μg/ml dox. 24 h later, straight water was given. Another 24 h later, CD4+CD90.1+ lymph node cells were analyzed for CFSE dilution. (B) Primed AND T cells require antigen for proliferation in a secondary host. Unlabeled CD90.1+ AND cells were transferred into a double transgenic Ii-rTA+TIM+ primary recipient pretreated with 100 μg/ml dox. 24 h later, straight water was given. Another 24 h later, the lymph node cells were CFSE labeled and transferred into an untreated nontransgenic (top) or a dox-treated Ii-rTA+TIM+ (bottom) secondary host. 60 h later, subcutaneous lymph node cells were analyzed by flow cytometry. CFSE profiles of CD4+CD90.1− cells (left) and CD4+CD90.1+ AND cells (right) are depicted. One representative experiment out of three independent ones is shown. (C) Primed AND T cells require antigen for proliferation in a secondary host even under inflammatory conditions. The experiment was performed as described in B, but 10 nmol of the CpG-containing oligonucleotide 1668 were injected i.p. at the time of the first transfer. (D) Continued proliferation of CD4+ T cells depends on antigen persistence, even if they were primed under inflammatory conditions. CFSE-labeled lymph node cells from CD90.1+ AND TCR transgenic mice were transferred into mice that were treated with 100 μg/ml dox for 24 h according to the indicated schedules and injected i.p. with PBS (left) or 10 nmol of the CpG-containing oligonucleotide 1668 (right). Subcutaneous lymph nodes were removed 48, 72, and 120 h after transfer and were analyzed by flow cytometry. All panels are gated on CD4+CD90.1+ cells.
Finally, the dox requirement for TIM expression was verified by analyzing thymi of Ii-rTA+TIM+AND+ animals, given that differentiating T cells are exquisitely sensitive to a trace amount of antigen. Flow cytometric analysis showed that in the absence of dox, thymi from the triple transgenic Ii-rTA+TIM+AND+ and from a single transgenic AND animal were indistinguishable (unpublished data). However, when dox was present in the drinking water, AND thymocytes did not differentiate to the mature CD4+CD8− single positive stage, indicating a strong negative selection. These findings emphasize the absolute dependence of TIM transcription on dox in the thymus. Thus, we conclude that the expression of the neoself peptide in APCs of this system depends on the presence of dox, at least to the extent that RT-PCR, mature T cells, and thymocytes can detect it.
How antigen dose affects CD4+ T cell proliferation
The possibility of modifying the amount of antigen predominantly expressed by DCs allowed us to ask how antigen regulates the expansion of responding CD4+ T cells. Initially, the proliferative response of adoptively transferred AND T cells to different amounts of the MCC epitope was analyzed. Ii-rTA+TIM+ animals were exposed to increasing amounts of dox in the drinking water, CFSE labeled, AND transgenic T cells were transferred into them, and the proliferation of the transferred cells in lymph nodes was analyzed 60 h later. Although there was some mouse-to-mouse variability (comparing pairs of cage mates), AND T cell proliferation increased in a dose-dependent manner (Fig. 2 A). Plotting the relative TIM mRNA levels versus the fraction of divided cells from the same animal revealed that no proliferation was observed at a low dox dose though TIM mRNA was detectable. There was a sharp transition to maximum proliferation at intermediate doses of dox, suggesting that a threshold of TIM mRNA was required to achieve complete T cell activation in vivo (Fig. 2 B). The fact that the T cell response appeared to saturate at doses >30 μg/ml indicates that this system targets antigen presentation efficiently to professional APCs.
Figure 2.
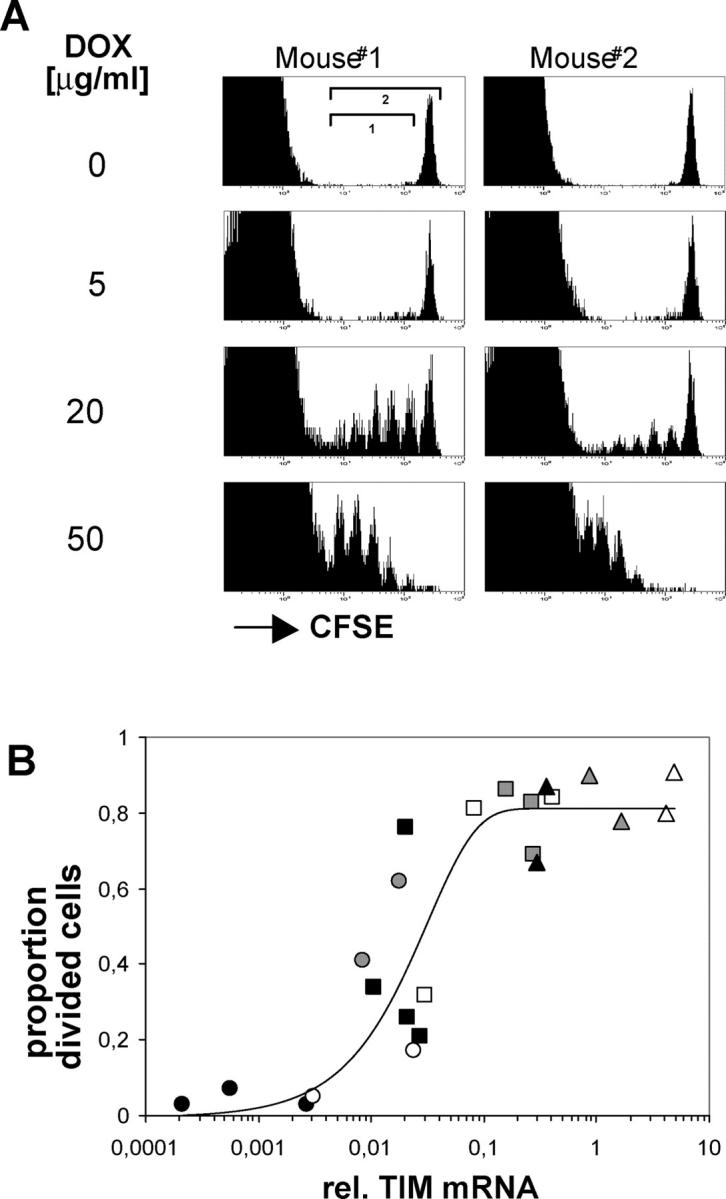
T cell responses vary with antigen dose. (A) Lymph node cells from AND TCR transgenic mice were CFSE labeled and transferred into two double transgenic Ii-rTA+TIM+ recipients that were exposed throughout the experiment to drinking water containing dox at the indicated concentrations. Lymph node cells were analyzed 60 h later. The plots are gated on CD4+ cells. (B) T cell activation threshold in vivo. Lymph nodes from the animals described in A were also analyzed for Ii-rtTA and TIM mRNAs by quantitative RT-PCR. The proportion of divided cells 60 h after transfer was determined by dividing the cell numbers in each peak by 2i, with i being the division number, and dividing this normalized number of divided cells (region 1) by the normalized total (region 2). Each data point stands for one animal, with each symbol representing one dose of dox in the drinking water (closed black circle, 5; open circle, 10; closed gray circle, 15; closed black square, 20; open square, 25; closed gray square, 50; closed black triangle,100; open triangle, 50; closed gray triangle, 200 [μg/ml each]). The data are compiled from three independent experiments.
A comparison of the CFSE profiles of T cells transferred into mice exposed to intermediate and high doses of dox (Fig. 2 A) suggested that the amount of antigen not only determined how many T cells entered the cell cycle, but also regulated how quickly they proceeded through the following divisions. To investigate this dose-dependent control of proliferation in more detail, we monitored responses over time to graded amounts of antigen. As before, CFSE-labeled AND T cells were transferred into double transgenic animals that were continuously exposed to various dox concentrations in the drinking water. During the following 6 d, subcutaneous lymph nodes were removed sequentially from a given animal, and the dilution of CFSE in AND T cells was analyzed. The data depicted in Fig. 3 support the idea that antigen dose regulates the magnitude of the T cell response by controlling both T cell recruitment into the response and the rate of subsequent divisions. First, whereas almost all of the AND T cells in animals exposed to 100 μg/ml dox had already divided two or three times by 36 h, only 50% of the antigen-specific cells had divided at all in mice exposed to a 10-fold lower dox concentration (Fig. 3, A [far left] and B [intermediate doses]). Second, many of the T cells in the animal exposed to 10 μg/ml dox divided fewer than six times and appear to continue cycling even after day 4 (Fig. 3 A). The presence of cells induced to divide, but fewer than seven times, suggests that the strength of the antigenic signal is able to continually have an impact on the course of the CD4+ T cell response. Such a notion contrasts with the “autopilot” idea (12) which argued that CD8+ T cells, once recruited into the cell cycle, were set to run through a programmed number of at least seven divisions independent of antigen persistence.
Figure 3.

The amount of antigen displayed by DCs determines both the initial induction of CD4+ T cells to proliferate and its rate. (A) At a low antigen dose, some CD4+ cells divided fewer than six times. Lymph node cells from CD90.1+ AND TCR transgenic mice were CFSE labeled and transferred into Ii-rTA+TIM+ animals that were supplied with 10, 30, or 100 μg/ml dox starting 2 d before transfer. The control animal (top) was an animal transgenic for TIM only and was fed with 100 μg/ml dox. Individual subcutaneous lymph nodes were removed 36, 60, 84, and 140 h after the transfer and analyzed by gating on CD4+CD90.1+ cells. Percentages indicate gated cells. (B) Induction of cell division 36 h after transfer correlated with antigen dose. The fraction of undivided cells at each time point after T cell transfer was calculated as described in Fig. 2 B and plotted against the dox dose. (C) Relative cell numbers of antigen-specific cells (CD90.1+) in the CD4+ T cell compartment increased corresponding to the dox dosage. The experiment was repeated with similar results.
Kinetics of antigen removal
The tet system offered the possibility of directly testing CD4+ T cells' continuous dependence on antigen because it permitted one to initiate a response and then punctually shut off the synthesis and presentation of the MCC epitope. It was initially necessary to establish the kinetics of TIM expression and MCC presentation in Ii-rTA+TIM+ double transgenic animals. Mice were exposed to 100 μg/ml dox for 24 h to allow for the highest TIM expression without variability, and the amount of TIM mRNA in excised lymph nodes was assessed at different times after the treatment. A short pulse of dox administration was chosen in order to avoid the potential problem of dox accumulation and persistence in tissues during long-term treatment. The maximal expression of TIM mRNA in the lymph nodes was achieved within 6 h of the exposure of the double transgenic mice to dox (Fig. 4 A). TIM transcripts disappeared with a half-life of 168 min (2.8 h), a value remarkably close to the previously reported 170-min half-life of dox in mouse serum (Fig. 4 A; reference 24). These data suggest, then, that TIM mRNA is quite unstable, turning over within minutes, and that its levels track serum dox concentrations very closely.
Figure 4.
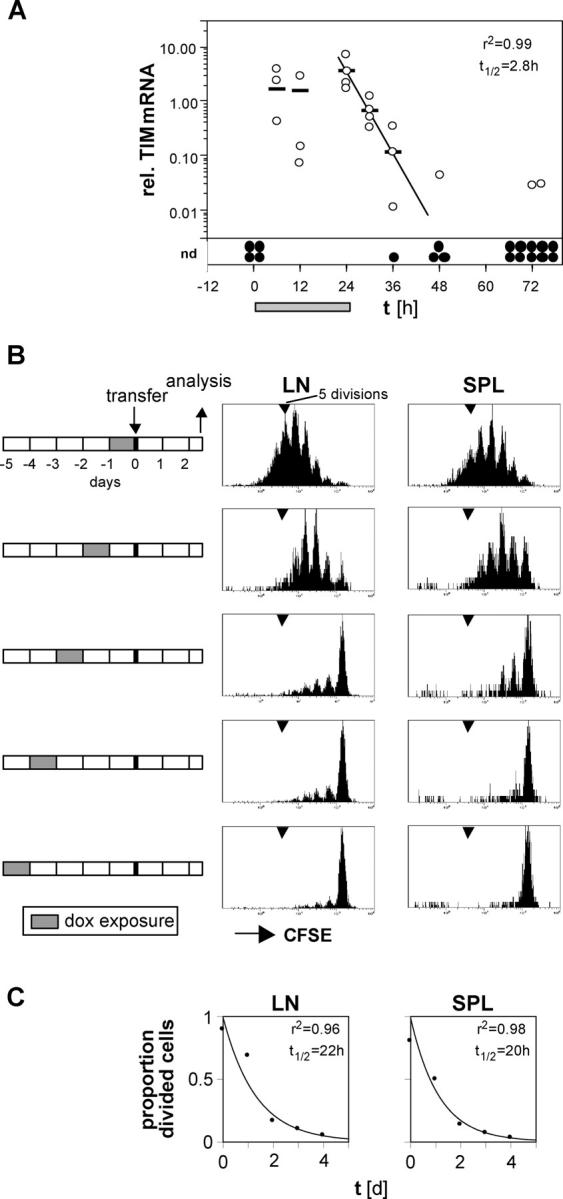
Half-life of TIM mRNA and Ek–MCC93-103 complexes in vivo. (A) Ii-rTA+TIM+ mice were exposed to 100 μg/ml dox in the drinking water for 24 h (gray bar). Subcutaneous lymph nodes were removed from four animals at each time point (except 12 that were taken at 72 h) and analyzed for TIM expression relative to Ii-rTA mRNA, as described for Figs. 1 and 2 (open circle, lymph node; bars, arithmetic means; closed circles, samples below the detection limit of the quantitative RT-PCR assay). The disappearance of the TIM mRNA within 24 h after dox withdrawal could be modeled by y = 3.0e−0.24x. The experiment was repeated twice with similar results (nd, not detectable). (B) Double transgenic animals were exposed to 100 μg/ml dox in the drinking water for 24 h from 0 to 5 d before the transfer of CFSE-labeled lymph node cells from CD90.1+ AND TCR transgenic mice. 60 h later, subcutaneous lymph nodes and spleen were analyzed. The histograms depict CD4+CD90.1+ cells. The experiment was repeated with similar results. (C) The proportion of divided cells was quantitated from the data shown in B as described for Fig. 2 B, modeled with y = 0.89e−0.75x and y = 0.87e−0.75x for the lymph nodes and spleen, respectively.
To find out how the biologically relevant gene product, the Ek–MCC complex detected by CD4+ T cells, disappeared from an animal, we supplied double transgenic animals with dox for 24 h and transferred CFSE-labeled AND T cells into them 0–4 d later (Fig. 4 B). T cell proliferation was vigorous when cells were transferred within 1 d of dox withdrawal. In contrast, few cells divided when transferred 2 d or more after dox removal, and division was essentially undetectable above background after 4 d. Based on these data, the half-life of this MHC–peptide complex, as detected by in vivo T cells, was ∼1 d in the subcutaneous lymph nodes and spleen (Fig. 4 C). This estimate reflects TIM mRNA and protein degradation, antigen processing, the survival of the stimulatory MHC–peptide complex at the cell surface, and DC longevity. Nevertheless, it was possible to effectively shut off most of the CD4+ T cell stimulatory capability in double transgenic mice within 48 h of dox removal.
Persistence of antigen is required for proliferation and effector function
These kinetics provided a system with which to test the influence of antigen persistence on the course of a CD4+ T cell response. If the presence of antigen does not matter to already recruited T cells, proliferation rates would be expected to be the same in animals from which antigen is removed at different times after the initiation of the response. However, this was not the case. AND T cells were labeled with CFSE and transferred into Ii-rTA+TIM+ animals exposed to dox continuously or for 24-h periods, timed as depicted in Fig. 5 A. The resulting responses were monitored by the serial removal of subcutaneous lymph nodes from a given mouse at regular intervals.
Figure 5.

Continued proliferation of CD4+ T cells depends on antigen persistence. (A) CFSE-labeled lymph node cells from CD45.1+ AND TCR transgenic mice were transferred into mice that were treated with 100 μg/ml dox for 24 h according to the indicated schedules. All recipients were double transgenic Ii-rTA+TIM+, except mouse 1, which carried the TIM gene only and was dox treated throughout. Subcutaneous lymph nodes were removed 36, 60, 84, and 108 h after transfer and were analyzed by flow cytometry. The 36-h and 60-h panels are gated on CD4+ cells, the others on CD4+CD45.1+ cells. One representative experiment out of five independent ones is shown. (B) Mean number of divisions of transferred cells from experiment A plotted against time is shown. (C) Proportion of transferred AND CD4+ cells among total CD4+ cells in subcutaneous lymph nodes from experiment A over time is shown.
In all Ii-rTA+TIM+ double transgenic recipients, most of the AND cells had divided once or twice by 36 h, attesting to a TCR engagement sufficient for initiated proliferation (Fig. 5 A, plotted in B as the number of divisions over time). These divisions persisted in the continually treated animal (Fig. 5, A and B, mouse 5), with five divisions occurring by 60 h, and two more by 84 h. However, in the animals in which dox had been removed at various times, proliferation suffered a halt: earliest in mice 2 and 3, later for mouse 4 (Fig. 5, A and B). The antigen dependence of T cell proliferation was also reflected in the relative number of antigen-specific cells in the lymph node; in addition, early antigen withdrawal could accelerate the contraction phase (Fig. 5 C). Thus, the number of divisions CD4+ T cells underwent in the first 4 d after the antigen encounter depended on antigen persistence at high levels during that time. It appears that CD4+ T cells stop dividing as soon as the antigen concentration falls below a certain threshold.
We independently confirmed the requirement for the continuous presence of antigen by applying a second experimental strategy. Congenically marked AND T cells were doubly transferred. In the primary recipient, a completely induced Ii-rTA+TIM+ animal, unlabeled cells were exposed to antigen for 48 h. This elicited two to four rounds of division, which were visualized in a parallel transfer of CFSE-labeled cells depicted in Fig. 6 A. The cells were then taken out, CFSE labeled, and transferred into secondary recipients that did or did not express the TIM transgene. To minimize the contribution of transferred APCs, the primary hosts were taken off dox 24 h before the second transfer, a feature of the system that made prolonged cell handling for sorting unnecessary. The secondary recipients were analyzed 60 h after transfer (Fig. 6 B). Here again, continued proliferation in the lymph nodes and spleens of the secondary hosts depended on antigen being expressed. The cells transferred into an antigen-free secondary recipient appeared to undergo perhaps one additional cycle (activated T cells do not label with CFSE well enough to allow an accurate estimate), but then stopped, whereas the continuous presence of antigen allowed the cells to progress a further four cycles or more (Fig. 6 B). Even exposures to antigen in the primary host for 60 or 84 h yielded similar results, indicating that, under these conditions, higher antigenic strength, in the form of longer term priming, does not propel CD4+ T cells into antigen-independent proliferation (unpublished data). These findings corroborate the conclusion from the dox-controlled antigen turn-off experiment.
We also examined whether an initial T cell encounter with activated DCs could alter the antigen dependence of CD4+ T cell proliferation. The experiment described in the previous paragraph was repeated except that the primary recipient was injected with DC-activating, unmethylated CpG-containing oligodeoxynucleotides at the time of T cell transfer. This treatment activated DCs, as indicated by the up-regulation of costimulatory molecules and the production of IL-12 (25, 26; unpublished data). Even under these conditions, the AND T cells proliferated in the dox-treated secondary recipient, but not in the untreated one (Fig. 6 C), indicating that CD4+ T cells primed by activated DCs still depend on the antigen for continued proliferation. This conclusion was supported by removing the antigen by dox withdrawal (Fig. 6 D). Ii-rTA+TIM+ animals were exposed to dox as in the experiment depicted in Fig. 5, but injected with either PBS- or CpG-containing oligonucleotides at the time of dox application so that the Ek–MCC complexes would be presented by activated DCs. The CFSE profiles of the two groups of animals were virtually indistinguishable (Fig. 6 D).
If antigen persistence is required for efficient clonal expansion, is this also true for the acquisition of effector characteristics? Or is the induction of, for example, IFN-γ synthesis, a reporter of productive priming (26–28), programmed early on antigen encounter and only realized later? As before, Ii-rTA+TIM+ animals were exposed to dox at different times relative to the transfer of CFSE-labeled AND T cells and analyzed 60 h later. As expected, the cells divided further in the animal where antigen persisted longer (Fig. 7). Whereas CD44 was expressed on all cells early on, IFN-γ was detected only with continuous antigen exposure (after more than five rounds of division). This finding suggests a requirement for antigen throughout the response not only for the expansion of CD4+ T cells, but also for the manifestation of their effector functions.
Figure 7.

Antigen persistence required for CD4+ effector cell differentiation. Lymph node cells from CD45.1+ AND TCR transgenic mice were CFSE labeled and transferred into Ii-rTA+TIM+ recipients exposed to 100 μg/ml dox in the drinking water for 24 h as indicated and analyzed 60 h later by flow cytometry. The histograms were gated on CD4+CD45.1+ cells, all other panels on CD4+ cells. The experiment was repeated with similar results.
Discussion
The experiments described here address the issue of whether antigen-specific responses by CD4+ T cells depend on the continued engagement of the TCR by cognate ligand, or on the initial conditions in which the antigen is encountered. We developed a transgenic mouse system in which a T cell antigen presented by MHC class II molecules presented on DCs can be finely regulated in vivo, as a function of the dox dose. Expression levels can be set over a 3-log dynamic range, and the fast decay of the transgene-encoded mRNA permits dynamic control of these levels. This system allowed us to evaluate the role of antigen in tuning the response in vivo. Because the supply of MHC class II–binding peptide is the sole variable in the system, CD4+ T cell responses could be studied in the absence of contributions from CD8+ T or B cells. We found that CD4+ T cell proliferation depended on, and continuously adjusted to, antigen persistence throughout the expansion phase.
When antigen was encountered at a low dose, three parameters varied relative to stimulation with a maximally effective dose: the induction of cell division was retarded, it involved fewer cells, and the progression through successive division cycles was slower (Fig. 3). Delayed, less frequent, and slower entries into the cell cycle was somewhat expected. The dampening of these parameters can be explained by the low frequency with which T cells encounter DCs that present sufficient numbers of peptides to activate them and a lower probability of reaching an activating threshold leading to the observed delay. This interpretation implies that antigenic signals can be read sequentially and somehow integrated over time, perhaps by building up the amount of an intracellular mediator (29, 30). One caveat to this interpretation might be that we do not currently have precise information on how lower dox concentrations regulate antigen presentation. It is possible that the number of TIM-expressing APCs is diminishing and/or the number of relevant Ek–MCC complexes is being homogenously reduced on all DCs. Based on our previous studies using the tet system, we anticipate the latter to be the case (19, 31).
On the other hand, the observation that a low dose of peptide caused a slower rate of proliferation of those cells that had entered the cell cycle was less expected. Kaech and Ahmed (11) suggested that the proportion of cells induced to divide is the only limiting step controlling the magnitude of a CD8+ T cell response, with each cell running on “autopilot” once cycling has been initiated (12). Here, we showed that a lower antigenic stimulus made for a slower pace of CD4+ T cell division. Whereas cells that encountered high amounts of antigen ran as tight populations through highly synchronized divisions, the CFSE profiles at low doses were, even after 4 d of stimulation, spread out over at least seven divisions (Fig. 3). Thus, it appears that the dividing cells still relied on antigen for proliferation and did not merely execute a program set in place soon after antigen encounter.
The proposition that the vigor with which activated CD4+ T cells cycle is controlled by the degree of continuous TCR triggering was tested most directly by antigen withdrawal. Removing the antigenic stimulus by punctual gene turn-off or by the transfer of antigen-stimulated T cells into antigen-free hosts led to quite an abrupt cessation of proliferation, with no sign of TCR-independent proliferation of previously activated CD4+ lymphocytes. In the transfer experiments, for example, proliferating cells appeared to merely finish the one cycle in which they were engaged (Fig. 6). This strict antigen dependence of the CD4+ response came as some surprise, in light of several in vitro experiments that showed antigen-independent cell division after an initial antigen pulse and emphasized the importance of cytokines in supporting continued division (15–17, 32). These results were slightly unclear, however, as an influence of continued TCR engagement was also detected in some of these and other studies (15–18). Although cytokine requirements, which our experiments did not address, might also play an important role in regulating the expansion phase in vivo, the present results demonstrate that the continued engagement of the TCR is a key element controlling the in vivo response of CD4+ T cells. These results are consistent with recent findings using the L. monocytogenes model, which propose that engendering a maximal CD4+ T cell response requires a longer window of infection unmitigated by antibiotic intervention than does a CD8+ T cell response (33, 34).
It is interesting to consider our conclusion in light of recent observations using time-lapse two-photon microscopy on T cell–DC interactions in lymph nodes. In the absence of antigen, T cells make transient contacts with multiple DCs in the T cell zones of secondary lymphoid organs (35–38). When antigen is present, these interactions are prolonged (37, 39) and are probably accompanied by increased Ca2+ levels in the T cells (36). During the following 3–24-h time interval, long-lasting T cell–DC clusters (>60 min) can be observed; this was followed by the resumption of shorter contacts and cell division (37, 39). Our data would suggest that the T cell–DC interactions that occur from 30 h onwards, although less intense than those provoked by the initial encounter with cognate antigen, are no less important for the continuation of an effective response.
The original autopilot idea, proposed in the context of CD8+ T cell responses, portrayed antigen recognition exclusively as the initiator of the immune response. A brief exposure (2–24 h, depending on the system) activated a genetic program that empowered the antigen-specific T cells and their progeny to proliferate and acquire effector functions quite autonomously over the following days. Only extrinsic factors like cytokines and other mediators would contribute to the second phase of expansion (6–9, 11, 12). Why is there a difference with the present data? Trivial explanations might rest on the different experimental systems used, and it would be important to test the responses of CD8+ T cells in an analogous system. Although we acknowledge this caveat, we favor the conclusion that CD4+ and CD8+ T cells differ in a fundamental aspect of their activation by antigen: CD8+ T cells slide into autopilot, whereas CD4+ T cells “stay on manual” by continuously adjusting the extent of their response to the parameters of antigen presentation.
There are a number of precedents for CD4+ and CD8+ T cells differing in their responses to antigenic challenge (40). In responses to pathogens that access both the classical MHC class I and II antigen presentation pathways, the number of specifically activated CD8+ cells exceeds those of CD4+ cells (influenza [41], LCMV [42], vaccinia [43], Sendai [44], L. monocytogenes [33, 45], and EBV [46, 47]). The proliferation of naive CD8+ T cells in lymphopenic hosts is generally more vigorous than that of CD4+ cells (48). CD8+ T cells also develop effector functions more readily. For example, 8 d after an L. monocytogenes infection, 85% of specific CD8+ but only 7% of CD4+ T cells produced IFN-γ (45). Finally, costimulatory requirements, via members of the CD28 or TNF receptor family appear different; CD4+ T cell responses are more hampered by compromised signals via CD28, CD40, or Ox40L than CD8+ T cells and vice versa for 4-1BB (49, 50). It is tempting to speculate that these divergent behaviors are caused by a common molecular mechanism that early on uncouples the cell cycle from TCR and costimulatory input in CD8+ but not CD4+ T cells, and programs the responding cells to divide a preset number of cycles and to differentiate quickly into effectors.
Why would the proliferation of CD4+ T cells be dependent on the continued presence of antigen, whereas that of CD8+ T cells appears not to be? Teleologically, it makes sense that cells with a helper and gatekeeper role in the immune response be more tightly controlled by the presence of antigen than the more effector-focused CD8+ T cells. One of the functions of CD4+ T cells is to activate DCs via the CD40-CD154 interactions for efficient CD8+ T cell priming (51, 52). Implicit in the high degree of antigenic control over the CD4+ response is that tresponse is indirectly maintained adequate to the antigenic challenge. It has been reported using several systems that DCs are swiftly eliminated or inhibited by CD8+ T cells as soon as the latter acquire their effector functions (53–56). CD4+ Th1 cells, on the other hand, have been shown to increase the DCs' longevity via CD40–CD40L interactions (57) and to render DCs resistant to CD8+ T cell–mediated killing by up-regulating the granzyme B inhibitor SPI-6 (58). Thus, keeping the activation and numbers of CD4+ T cells tied to the antigenic load ensures that the ensemble of CD4+ T cells, CD8+ T cells, and DCs responds adequately.
Materials and Methods
Transgenes.
The TIM transgene consists of seven tetO sequences, a CMV minimal promoter, rabbit β-globin intron 2, the mouse invariant chain cDNA in which amino acids 88–89 have been replaced by MCC93–103 (DLIAYLKQATK), and the rabbit β-globin 3′-UTR with a poly(A)+ RNA site. Its generation has been previously described (19). For the Ii-rTA transgene, the rTAS-S2 open reading frame from the plasmid pUHD52-1 (a gift from H. Bujard [University of Heidelberg, Heidelberg, Germany] and W. Hillen [University of Erlangen, Erlangen, Germany]) was cloned into pBS and from there via ClaI sites into pDOI-6 (21). The resulting pDOI6-rtTAS-S2 plasmid contains the Eαk enhancer, the Ii promoter, its first exon (with three ATGs removed) and intron, the rTAS-S2 open reading frame, and a rabbit β-globin exon 3 with a poly(A)+ RNA site.
Mice.
Part of the pBR322 backbone was removed from the pDOI6-rtTAS-S2 plasmid, and the resulting fragment was injected into FVB/N oocytes. Four independent lines were generated and transactivator expression in different tissues was assessed by Northern blot. Its expression in the liver was found to vary among the lines and the one with the lowest liver expression was backcrossed with C57BL/6 (The Jackson Laboratory) five times and crossed with the TIM line. A dox-dependent TIM expression in the thymus and spleen was found by RT-PCR for all lines. Double transgenic animals were backcrossed at least five generations to B10.BR (The Jackson Laboratory) for experiments. AND TCR transgenic mice (gift from S. Hedrick, University of California, San Diego, San Diego, CA; reference 59) and maintained on the B10.BR background. AND TCR transgenic animals congenic for the CD45.1 and CD90.1 markers were generated by introducing the loci from B6.SJL-Ptprc a Pep3b/BoyJ and B6.PL-Thy1 a/CyJ animals (The Jackson Laboratory), respectively. Animals were treated with dox (Sigma-Aldrich) by adding the indicated concentrations to the drinking water, including 40 mg aspartame (Equal; reference 60). Bottles were exchanged every 3 d. All animals were housed and bred under specific pathogen-free conditions at the Harvard Medical School Center for Animal Research and Comparative Medicine, and experiments were conducted in compliance with federal and institutional guidelines and with the approval of the Institutional Animal Care and Use Committee at Harvard University.
CFSE labeling and cell transfer.
Lymph node cells from AND animals were resuspended at 20 × 106 cells/ml in 37°C Ca2+-Mg2+–free PBS with 0.1% BSA, and 2 μl/ml 5 mM CFSE (V-12883; Molecular Probes) in DMSO was added while vortexing. Cells were incubated for 10 min at 37°C, resuspended in cold RPMI 1640 with 10% FCS, and washed five times in DMEM before i.v. transfer of the equivalent of 2–4 × 106 Vβ3+CD4+ cells into recipients. In one experiment, 10 nmol of the unmethylated CpG-containing oligodeoxynucleotide 1668 was injected i.p. (25).
Surgical removal of lymph nodes.
s.c. inguinal and brachial lymph nodes were surgically removed from anesthetized mice (50 μg/g ketamine [Bedford Laboratories]; 10 μg/g xylazine [Lloyd Laboratories]) via a 5-mm incision in the skin. The wound was closed with a 9-mm Reflex wound clip (Roboz Surgical Instruments), and the animal was treated with an analgesic (2.5 μg/g Flunixamine; Fort Dodge Animal Health) after the procedure and again 12 h later. Lymph nodes were transferred to cold medium for flow cytometry analysis or were snap frozen in liquid N2 for RNA analysis.
Flow cytometry.
Lymph node and RBC-lysed spleen suspensions were preincubated with 2.4G2 culture supernatant to block Fc-γ receptor binding, stained in DMEM, 10 mM Hepes, and 2% FCS, and analyzed on a cytometer (model Coulter XL) using Expo32 software (both from Beckman Coulter). ModFit software (Verity) was used to quantify CFSE peaks. Cells were sorted on a MoFlo cell sorter with Summit software (DakoCytomation). mAb's (BD Biosciences unless indicated otherwise) used were as follows: CD3 (17A2), CD4 (RM4-5), CD11b (M1/70.15; Caltag), CD11c (HL3), CD19 (ID3), CD44 (IM7), CD45.1 (A20), CD62L (MEL-14), CD90.1 (HIS51; eBioscience), and CD90.2, and FITC-labeled anti-mVβ3 (KJ25; purified and labeled in the laboratory). Intracellular staining was performed with the CytoFix/CytoPerm kit (BD Biosciences) using anti–mIFN-γ (XMG1.2) and an isotype control (R3-34; BD Biosciences).
RNA preparation and real-time PCR.
RNA from sorted cell populations was isolated using TRIzol (Invitrogen) following the manufacturer's instructions. From frozen organs, RNA was isolated by a modified LiCl method (61). Organs were homogenized first for 30 s in 2 ml 3 M LiCl/6 M urea/10 mM NaOAc, pH 5.2, plus 50 μl with 20% SDS using a high speed Polytron homogenizer (Kinematica). After incubating for >12 h at 4°C, the solution was centrifuged for 10 min at 12,000 g, and the pellet was resuspended in 3 M LiCl/6 M urea/10 mM NaOAc, pH 5.2, incubated for >30 min at 4°C, and centrifuged as before. The pellet was resuspended in 500 μl 10 mM Tris, pH 8/2 mM EDTA/200 mM NaCl/0.5% SDS/200 μg/ml proteinase K (freshly added from 20 mg/ml stock; Roche) and incubated 30 min at 37°C. RNA was extracted with phenol/chloroform, ethanol precipitated, DNA removed with the DNA-free kit (Ambion), and randomly primed cDNA was prepared with M-MLV-RT using standard procedures. Real-time PCR was performed using TaqMan universal PCR master mix (Applied Biosystems) and the following oligos and probes: HPRT-sense (GAC CGG TCC CGT CAT GC), HPRT-antisense (CAG TCC ATG AGG AAT AAA CAC TTT TTC), HPRTP-probe (VIC-CCG CAG TCC CAG CGT CGT GAT T-TAMRA), rTAS-S2-sense (TAG AGG GAT CCG CCC TAG A), rTAS-S2-antisense (CAT GGT GAA TTC GAT ATC AAG CTT), rTAS-S2-probe (FAM-CAC GGC TGC ACC TTT CTG GC-TAMRA), TIM-sense (GGA TCC TGA GAA CTT CAG GCT C), TIM-antisense (TTG GTC ATC CAT GGC TCT AGC), and TIM-probe (FAM-AAC GTG CTG GTT GTT GTG CTG TCT CAT C-TAMRA). Samples were analyzed on Prism 7700 (Applied Biosystems) or Mx3000p (Stratagene) instruments with 55°C as an annealing temperature, except for TIM with 60°C.
Acknowledgments
We thank Drs. H. Bujard and W. Hillen for the pUHD52-1 plasmid; P. Demougin, Q.M. Pham, and V. Tran for help with cloning and maintaining the mouse colony; Dr. C. Laplace for help with the figures; Dr. A.W. Goldrath for advice; and Drs. L.T. Nguyen and C.J. Luckey for critically reading the manuscript.
This work was funded by a grant from the National Institutes of Health (RO1 AI51530; to C. Benoist and D. Mathis, the William T. Young Chairs in Diabetes Research), and Joslin's National Institute of Diabetes and Digestive and Kidney Diseases–funded Diabetes Endocrine Reseach Center cores (P30 DK36836). R. Obst was supported by fellowships from the German Research Council (OB 150/2-1) and the Cancer Research Institute. H.-M. van Santen was supported by fellowships from the Cancer Research Institute and the Leukemia and Lymphoma Society.
The authors have no conflicting financial interests.
Abbreviations used: CFSE, carboxyfluorescein diacetate succinimidyl ester; dox, doxycycline; HPRT, hypoxanthine phosphoribosyltransferase; Ii, invariant chain; MCC, moth cytochrome c; tet, tetracycline; TIM, tetracycline-inducible invariant chain with MCC.
R. Obst and H.-M. van Santen contributed equally to this work.
H.-M. van Santen's present address is Centro Biología Molecular Severo Ochoa, Consejo Superior de Investigaciones Científicas, Universidad Autónoma de Madrid, Madrid 28049, Spain.
References
- 1.Guermonprez, P., J. Valladeau, L. Zitvogel, C. Thery, and S. Amigorena. 2002. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 20:621–667. [DOI] [PubMed] [Google Scholar]
- 2.Itano, A.A., and M.K. Jenkins. 2003. Antigen presentation to naive CD4 T cells in the lymph node. Nat. Immunol. 4:733–739. [DOI] [PubMed] [Google Scholar]
- 3.Catron, D.M., A.A. Itano, K.A. Pape, D.L. Mueller, and M.K. Jenkins. 2004. Visualizing the first 50 hr of the primary immune response to a soluble antigen. Immunity. 21:341–347. [DOI] [PubMed] [Google Scholar]
- 4.Huang, A.Y., H. Qi, and R.N. Germain. 2004. Illuminating the landscape of in vivo immunity: insights from dynamic in situ imaging of secondary lymphoid tissues. Immunity. 21:331–339. [DOI] [PubMed] [Google Scholar]
- 5.Trombetta, S.E., and I. Mellman. 2005. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 23:975–1028. [DOI] [PubMed] [Google Scholar]
- 6.Mercado, R., S. Vijh, S.E. Allen, K. Kerksiek, I.M. Pilip, and E.G. Pamer. 2000. Early programming of T cell populations responding to bacterial infection. J. Immunol. 165:6833–6839. [DOI] [PubMed] [Google Scholar]
- 7.van Stipdonk, M.J., G. Hardenberg, M.S. Bijker, E.E. Lemmens, N.M. Droin, D.R. Green, and S.P. Schoenberger. 2003. Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4:361–365. [DOI] [PubMed] [Google Scholar]
- 8.van Stipdonk, M.J., E.E. Lemmens, and S.P. Schoenberger. 2001. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat. Immunol. 2:423–429. [DOI] [PubMed] [Google Scholar]
- 9.Wong, P., and E.G. Pamer. 2001. Antigen-independent CD8 T cell proliferation. J. Immunol. 166:5864–5868. [DOI] [PubMed] [Google Scholar]
- 10.Wong, P., and E.G. Pamer. 2004. Disparate in vitro and in vivo requirements for IL-2 during antigen-independent CD8 T cell expansion. J. Immunol. 172:2171–2176. [DOI] [PubMed] [Google Scholar]
- 11.Kaech, S.M., and R. Ahmed. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bevan, M.J., and P.J. Fink. 2001. The CD8 response on autopilot. Nat. Immunol. 2:381–382. [DOI] [PubMed] [Google Scholar]
- 13.Curtsinger, J.M., C.M. Johnson, and M.F. Mescher. 2003. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J. Immunol. 171:5165–5171. [DOI] [PubMed] [Google Scholar]
- 14.Storni, T., C. Ruedl, W.A. Renner, and M.F. Bachmann. 2003. Innate immunity together with duration of antigen persistence regulate effector T cell induction. J. Immunol. 171:795–801. [DOI] [PubMed] [Google Scholar]
- 15.Lee, W.T., G. Pasos, L. Cecchini, and J.N. Mittler. 2002. Continued antigen stimulation is not required during CD4+ T cell clonal expansion. J. Immunol. 168:1682–1689. [DOI] [PubMed] [Google Scholar]
- 16.Bajénoff, M., O. Wurtz, and S. Guerder. 2002. Repeated antigen exposure is necessary for the differentiation, but not the initial proliferation, of naive CD4+ T cells. J. Immunol. 168:1723–1729. [DOI] [PubMed] [Google Scholar]
- 17.Gett, A.V., F. Sallusto, A. Lanzavecchia, and J. Geginat. 2003. T cell fitness determined by signal strength. Nat. Immunol. 4:355–360. [DOI] [PubMed] [Google Scholar]
- 18.Schrum, A.G., and L.A. Turka. 2002. The proliferative capacity of individual naive CD4+ T cells is amplified by prolonged T cell antigen receptor triggering. J. Exp. Med. 196:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Santen, H.M., C. Benoist, and D. Mathis. 2004. Number of T reg cells that differentiate does not increase upon encounter of agonist ligand on thymic epithelial cells. J. Exp. Med. 200:1221–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urlinger, S., U. Baron, M. Thellmann, M.T. Hasan, H. Bujard, and W. Hillen. 2000. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. USA. 97:7963–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Santen, H.M., C. Benoist, and D. Mathis. 2000. A cassette vector for high-level reporter expression driven by a hybrid invariant chain promoter in transgenic mice. J. Immunol. Methods. 245:133–137. [DOI] [PubMed] [Google Scholar]
- 22.Witherden, D., N. van Oers, C. Waltzinger, A. Weiss, C. Benoist, and D. Mathis. 2000. Tetracycline-controllable selection of CD4+ T cells: half-life and survival signals in the absence of major histocompatibility complex class II molecules. J. Exp. Med. 191:355–364. [DOI] [PubMed] [Google Scholar]
- 23.Hess, J., A. Werner, T. Wirth, F. Melchers, H.M. Jack, and T.H. Winkler. 2001. Induction of pre-B cell proliferation after de novo synthesis of the pre-B cell receptor. Proc. Natl. Acad. Sci. USA. 98:1745–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Böcker, R., and C.J. Estler. 1981. Comparison of distribution of doxycycline in mice after oral and intravenous application measured by a high-performance liquid chromatographic method. Arzneimittelforschung (Drug Res.). 31:2116-2117. [PubMed] [Google Scholar]
- 25.Sparwasser, T., E.S. Koch, R.M. Vabulas, K. Heeg, G.B. Lipford, J.W. Ellwart, and H. Wagner. 1998. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur. J. Immunol. 28:2045–2054. [DOI] [PubMed] [Google Scholar]
- 26.Spörri, R., and C. Reis e Sousa. 2005. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat. Immunol. 6:163–170. [DOI] [PubMed] [Google Scholar]
- 27.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujii, S., K. Liu, C. Smith, A.J. Bonito, and R.M. Steinman. 2004. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J. Exp. Med. 199:1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedl, P., and M. Gunzer. 2001. Interaction of T cells with APCs: the serial encounter model. Trends Immunol. 22:187–191. [DOI] [PubMed] [Google Scholar]
- 30.Faroudi, M., R. Zaru, P. Paulet, S. Muller, and S. Valitutti. 2003. T lymphocyte activation by repeated immunological synapse formation and intermittent signaling. J. Immunol. 171:1128–1132. [DOI] [PubMed] [Google Scholar]
- 31.Labrecque, N., L.S. Whitfield, R. Obst, C. Waltzinger, C. Benoist, and D. Mathis. 2001. How much TCR does a T cell need? Immunity. 15:71–82. [DOI] [PubMed] [Google Scholar]
- 32.Jelley-Gibbs, D.M., N.M. Lepak, M. Yen, and S.L. Swain. 2000. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen-dependent and late cytokine-driven expansion and differentiation. J. Immunol. 165:5017–5026. [DOI] [PubMed] [Google Scholar]
- 33.Corbin, G.A., and J.T. Harty. 2004. Duration of infection and antigen display have minimal influence on the kinetics of the CD4+ T cell response to Listeria monocytogenes infection. J. Immunol. 173:5679–5687. [DOI] [PubMed] [Google Scholar]
- 34.Williams, M.A., and M.J. Bevan. 2004. Shortening the infectious period does not alter expansion of CD8 T cells but diminishes their capacity to differentiate into memory cells. J. Immunol. 173:6694–6702. [DOI] [PubMed] [Google Scholar]
- 35.Underhill, D.M., M. Bassetti, A. Rudensky, and A. Aderem. 1999. Dynamic interactions of macrophages with T cells during antigen presentation. J. Exp. Med. 190:1909–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gunzer, M., A. Schäfer, S. Borgmann, S. Grabbe, K.S. Zänker, E.B. Bröcker, E. Kämpgen, and P. Friedl. 2000. Antigen presentation in extracellular matrix: interactions of T cells with dendritic cells are dynamic, short lived, and sequential. Immunity. 13:323–332. [DOI] [PubMed] [Google Scholar]
- 37.Mempel, T.R., S.E. Henrickson, and U.H. von Andrian. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 427:154–159. [DOI] [PubMed] [Google Scholar]
- 38.Miller, M.J., A.S. Hejazi, S.H. Wei, M.D. Cahalan, and I. Parker. 2004. T cell repertoire scanning is promoted by dynamic dendritic cell behavior and random T cell motility in the lymph node. Proc. Natl. Acad. Sci. USA. 101:998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller, M.J., O. Safrina, I. Parker, and M.D. Cahalan. 2004. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J. Exp. Med. 200:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seder, R.A., and R. Ahmed. 2003. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat. Immunol. 4:835–842. [DOI] [PubMed] [Google Scholar]
- 41.Powell, T.J., D.M. Brown, J.A. Hollenbaugh, T. Charbonneau, R.A. Kemp, S.L. Swain, and R.W. Dutton. 2004. CD8+ T cells responding to influenza infection reach and persist at higher numbers than CD4+ T cells independently of precursor frequency. Clin. Immunol. 113:89–100. [DOI] [PubMed] [Google Scholar]
- 42.Homann, D., L. Teyton, and M.B.A. Oldstone. 2001. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 7:913–919. [DOI] [PubMed] [Google Scholar]
- 43.Harrington, L.E., R.R. Most, J.L. Whitton, and R. Ahmed. 2002. Recombinant vaccinia virus-induced T-cell immunity: quantitation of the response to the virus vector and the foreign epitope. J. Virol. 76:3329–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cauley, L.S., T. Cookenham, T.B. Miller, P.S. Adams, K.M. Vignali, D.A. Vignali, and D.L. Woodland. 2002. Virus-specific CD4+ memory T cells in nonlymphoid tissues express a highly activated phenotype. J. Immunol. 169:6655–6658. [DOI] [PubMed] [Google Scholar]
- 45.Foulds, K.E., L.A. Zenewicz, D.J. Shedlock, J. Jiang, A.E. Troy, and H. Shen. 2002. CD4 and CD8 T cells are intrinsically different in their proliferative responses. J. Immunol. 168:1528–1532. [DOI] [PubMed] [Google Scholar]
- 46.Maini, M.K., N. Gudgeon, L.R. Wedderburn, A.B. Rickinson, and P.C. Beverley. 2000. Clonal expansions in acute EBV infection are detectable in the CD8 and not the CD4 subset and persist with a variable CD45 phenotype. J. Immunol. 165:5729–5737. [DOI] [PubMed] [Google Scholar]
- 47.Rickinson, A.B., and E. Kieff. 2001. Epstein-Barr virus. In Fields Virology. D.M. Knipe and P.M. Howley, editors. Lippincott Williams & Wilkins, Philadelphia. 2575-2627.
- 48.Tan, J.T., E. Dudl, E. LeRoy, R. Murray, J. Sprent, K.I. Weinberg, and C.D. Surh. 2001. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc. Natl. Acad. Sci. USA. 98:8732–8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whitmire, J.K., and R. Ahmed. 2000. Costimulation in antiviral immunity: differential requirements for CD4+ and CD8+ T cell responses. Curr. Opin. Immunol. 12:448–455. [DOI] [PubMed] [Google Scholar]
- 50.Croft, M. 2003. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat. Rev. Immunol. 3:609–620. [DOI] [PubMed] [Google Scholar]
- 51.Lanzavecchia, A. 1998. Licence to kill. Nature. 393:413–414. [DOI] [PubMed] [Google Scholar]
- 52.Quezada, S.A., L.Z. Jarvinen, E.F. Lind, and R.J. Noelle. 2004. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 22:307–328. [DOI] [PubMed] [Google Scholar]
- 53.Loyer, V., P. Fontaine, S. Pion, F. Hetu, D.C. Roy, and C. Perreault. 1999. The in vivo fate of APCs displaying minor H antigen and/or MHC differences is regulated by CTLs specific for immunodominant class I-associated epitopes. J. Immunol. 163:6462–6467. [PubMed] [Google Scholar]
- 54.Hermans, I.F., D.S. Ritchie, J. Yang, J.M. Roberts, and F. Ronchese. 2000. CD8+ T cell-dependent elimination of dendritic cells in vivo limits the induction of antitumor immunity. J. Immunol. 164:3095–3101. [DOI] [PubMed] [Google Scholar]
- 55.Ronchese, F., and I.F. Hermans. 2001. Killing of dendritic cells: a life cut short or a purposeful death? J. Exp. Med. 194:F23–F26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong, P., and E.G. Pamer. 2003. Feedback regulation of pathogen-specific T cell priming. Immunity. 18:499–511. [DOI] [PubMed] [Google Scholar]
- 57.Miga, A.J., S.R. Masters, B.G. Durell, M. Gonzalez, M.K. Jenkins, C. Maliszewski, H. Kikutani, W.F. Wade, and R.J. Noelle. 2001. Dendritic cell longevity and T cell persistence is controlled by CD154-CD40 interactions. Eur. J. Immunol. 31:959–965. [DOI] [PubMed] [Google Scholar]
- 58.Medema, J.P., D.H. Schuurhuis, D. Rea, J. van Tongeren, J. de Jong, S.A. Bres, S. Laban, R.E.M. Toes, M. Toebes, T.N.M. Schumacher, et al. 2001. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte–induced apoptosis: differential modulation by T helper type 1 and type 2 cells. J. Exp. Med. 194:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaye, J., M.L. Hsu, M.E. Sauron, S.C. Jameson, N.R. Gascoigne, and S.M. Hedrick. 1989. Selective developmsent of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 341:746–749. [DOI] [PubMed] [Google Scholar]
- 60.Green, E.A., and R.A. Flavell. 2000. The temporal importance of TNFα expression in the development of diabetes. Immunity. 12:459–469. [DOI] [PubMed] [Google Scholar]
- 61.Auffray, C., and F. Rougeon. 1980. Purification of mouse immunoglobulin heavy-chain messenger RNAs from total myeloma tumor RNA. Eur. J. Biochem. 107:303–314. [DOI] [PubMed] [Google Scholar]