

Abstract
Transcription factor, nuclear factor κB (NF-κB), is required for osteoclast formation in vivo and mice lacking both of the NF-κB p50 and p52 proteins are osteopetrotic. Here we address the relative roles of the two catalytic subunits of the IκB kinase (IKK) complex that mediate NF-κB activation, IKKα and IKKβ, in osteoclast formation and inflammation-induced bone loss. Our findings point out the importance of the IKKβ subunit as a transducer of signals from receptor activator of NF-κB (RANK) to NF-κB. Although IKKα is required for RANK ligand-induced osteoclast formation in vitro, it is not needed in vivo. However, IKKβ is required for osteoclastogenesis in vitro and in vivo. IKKβ also protects osteoclasts and their progenitors from tumor necrosis factor α–induced apoptosis, and its loss in hematopoietic cells prevents inflammation-induced bone loss.
Bone development and remodeling are highly regulated processes that involve synthesis of bone matrix by osteoblasts and coordinated bone resorption by osteoclasts (1). Osteoblasts originate from mesenchymal stem cells, whereas osteoclasts are derived from hematopoietic monocyte/macrophage precursors (1). Imbalanced osteoclast and osteoblast formation, activity, or survival can be caused by a variety of hormonal changes or perturbed production of inflammatory cytokines and growth factors, and result in skeletal abnormalities that are characterized by decreased (osteoporosis) or increased (osteopetrosis) bone mass (1). Increased osteoclast formation and activity is observed in many osteopenic disorders, including postmenopausal osteoporosis (2), lytic bone metastasis, or rheumatoid arthritis (3), and leads to accelerated bone resorption and crippling bone damage.
During osteoclast differentiation, osteoblastic/stromal cells provide a physical support for nascent osteoclasts and produce soluble and membrane-associated factors, such as macrophage-colony stimulating factor (M-CSF), and receptor activator of NF-κB ligand (RANKL) (4). RANKL (also called tumor necrosis factor–related activation-induced cytokine, osteoclast differentiation factor, osteoprotegerin ligand) is a member of the TNF cytokine family and an essential inducer of osteoclastogenesis and bone remodeling through its receptor RANK, a TNF-receptor (TNFR) family member (5, 6). Mice with a disrupted Rankl gene exhibit severe osteopetrosis (6, 7). Disruption of the Rank gene also results in lack of osteoclasts and ensuing osteopetrosis (8). Similar to RANKL, TNF-α is a potent osteoclastogenic factor that enhances proliferation and differentiation of osteoclast precursors through its type I receptor (TNFR1; reference 9). However, it remains controversial whether TNFα promotes osteoclastogenesis independently of RANKL (10, 11). RANK, like most other TNFR family members, including TNFR1, transduces its biochemical signals through recruitment of intracellular signal transducers, called TNF receptor-associated factors, which lead to activation of NF-κB and mitogen-activated protein kinase effector pathways (12–15). The relevance of these pathways to osteoclastogenesis is underscored by the osteopetrotic phenotypes of mice lacking TNF receptor–associated factor 6 (16); the NF-κB1/p50 and NF-κB2/p52 subunits of NF-κB (15, 17); or c-Fos (18), a component of the AP-1 transcription factor, whose expression is mitogen-activated protein kinase dependent (19).
NF-κB is a collection of dimeric transcription factors that recognize similar DNA sequences called κB sites. In mammals there are five NF-κB proteins: cRel, RelA and RelB, as well as NF-κB1/p50 and NF-κB2/p52. Although the Rel proteins contain transcriptional activation domains, such domains are absent in p50 and p52, whose activation function depends on heterodimerization with any of the three Rel proteins (20). As mentioned above, ablation of p50 and p52 results in a severe osteopetrotic phenotype, which most likely is due to the poor DNA binding activity of the remaining NF-κB subunits (15). NF-κB proteins reside in the cytoplasm of nonstimulated cells but rapidly enter the nucleus upon cell stimulation (21). This process, called NF-κB activation, depends on two pathways. The classic NF-κB signaling pathway involves activation of the IκB kinase (IKK) complex that phosphorylates the inhibitors of NF-κB (IκBs) and targets them to ubiquitin-dependent degradation (21). The IκBs retain most NF-κB dimers, with the exception of p52:RelB dimers, in the cytoplasm by masking their nuclear localization signals (21). The alternative NF-κB signaling pathway is responsible for activation of p52:RelB dimers, which are generated by processing of cytoplasmic p100:RelB dimers (21).
Currently, it is not entirely clear which of the two NF-κB activation pathways plays the dominant role in osteoclastogenesis. The IKK complex that is responsible for activation of the canonical NF-κB pathway consists of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ/NF-κB essential modulator (22). Gene disruption experiments demonstrated that IKKβ and IKKγ are important for IκB phosphorylation and degradation, whereas IKKα has different and nonoverlapping functions (21). Importantly, IKKα forms homodimers, not associated with IKKγ, that are required for phosphorylation-induced p100 processing and activation of the alternative pathway (23). Activation of the alternative pathway also depends on the IKKα-phosphorylating kinase, NF-κB–inducing kinase (NIK; refereneces 23, 24). It was observed that NIK-deficient osteoclast precursors do not respond to RANKL in an in vitro differentiation system that is devoid of osteoblasts (25). However, aly mice, which carry a point mutation in the Nik gene that prevents NIK activation, are not osteopetrotic (26); osteopetrosis also was not reported for Nik − / − mice (25). More recently, a peptide inhibitor of IKK, which prevents the association of IKKβ with IKKγ, and therefore, blocks activation of the classic pathway without affecting the alternative pathway, was shown to prevent inflammation-induced bone loss in vivo (27). These results suggest that the classic pathway is of greater importance for osteoclastogenesis. To determine the relative roles of the two pathways in osteoclastogenesis and inflammation-induced bone loss, we undertook a genetic approach based on the use of mouse strains that carry specific mutations in the Ikkα and Ikkβ genes. We found that IKKβ, but not IKKα, is essential for inflammation-induced bone loss and is required for osteoclastogenesis in vivo. However, the main function of IKKβ in osteoclastogenesis is to prevent TNFα-induced apoptosis of osteoclast precursors. Once TNFα-induced apoptosis is prevented through deletion of the Tnfr1 gene, IKKβ is no longer required for induction of inflammation-induced bone loss, but it is still needed for basal osteoclast function.
RESULTS
RANKL-induced in vitro osteoclastogenesis requires IKKα and IKKβ
To determine the roles of the IKK catalytic subunits in osteoclastogenesis, we used mice that carry mutant forms of either subunit—IkkαAA and IkkβΔ mice. IkkαAA mice are homozygous for a knock-in mutant allele, in which the activation loop serines of IKKα—whose phosphorylation is required for its activation (28)—were replaced with alanines; this prevents IKKα activation by upstream stimuli, including RANK (29) and NIK (23). IkkαAA mice are viable, healthy, and fertile. These mice exhibit defective organization of secondary lymphoid organs and B cell maturation (30), a defect that is very similar to what was found in Nik − / − (31) or Nik aly/aly (26) mice. IkkβΔ mice were generated by crossing IkkβF/F mice, homozygotes for a “floxed” Ikkβ allele (32), with Mx1-Cre transgenic mice that express Cre recombinase from the IFN-inducible Mx-1 promoter (33). Injection of polyinosinic-polycytidylic acid (poly[IC]), which induces IFN production, into IkkβF/F:Mx1-Cre mice results in efficient deletion of the floxed third exon of the Ikkβ gene and generation of IKKβ-deficiency in IFN-responsive cells, including myeloid cells (34). To determine whether defective IKKα activation or complete loss of IKKβ affect osteoclastogenesis, we first used an in vitro differentiation system. BM hematopoietic progenitors that were isolated from WT, IkkαAA, and IkkβΔ mice were incubated with M-CSF and RANKL for 7 d, and stained for tartrate-resistant acid phosphatase (TRAP), a marker for osteoclasts. Wt cultures showed robust osteoclastogenesis and formed giant TRAP-positive cells, whereas osteoclast formation was absent in IkkαAA and IkkβΔ cultures (Fig. 1). Expression of several other osteoclast markers, including CatK and matrix metalloproteinase-9, also was defective in RANKL-treated IkkαAA and IkkβΔ BM cultures (unpublished data). These results suggest that IKKα and IKKβ are required for, and play nonredundant roles in, M-CSF and RANKL-induced osteoclastogenesis in vitro.
Figure 1.

IkkαAA and IkkβD BM cells exhibit defective RANKL- mediated osteoclastogenesis in vitro. Equal numbers of BM cells were isolated from WT, IkkαAA, and IkkβΔ mice and cultured in the presence of M-CSF (10 ng/ml) alone or M-CSF plus RANKL (50 ng/ml). Osteoclastogenesis was monitored after 7 d by TRAP staining (red color). Note the giant cells that appear after incubation of WT BM with M-CSF and RANKL.
RANKL-mediated NF-κB activation is impaired in IkkβΔ, but not IkkαAA, osteoclast progenitors
To gain insights into the possible mechanisms by which IKKα and IKKβ promote osteoclastogenesis in vitro, we examined RANKL-induced NF-κB activation in IkkαAA and IkkβΔ BM cultures. BM-derived precursors from WT, IkkαAA, and IkkβΔ mice were incubated with M-CSF and RANKL and analyzed for expression of IKKα and IKKβ by immunoblot analysis (Fig. 2 A). As expected, no IKKβ protein was detectable in IkkβΔ BM cells. Interestingly, the deletion of IKKβ also resulted in slightly decreased levels of IKKα (Fig. 2 A); this suggests that IKKα:IKKγ complexes or IKKα homodimers are not as stable as the heterotrimeric IKKα:IKKβ:IKKγ complexes (22). Electrophoretic mobility shift assays revealed marked induction of NF-κB DNA binding activity in response to RANKL treatment in WT and IkkαAA cultures, but only a weak response in IkkβΔ cultures (Fig. 2 B). Similarly, RANKL activated IKK in WT and IkkαAA cultures, but no IKK activity could be detected before or after RANKL treatment of IkkβΔ BM cells (Fig. 2 C). In addition, RANKL induced the nuclear translocation of all three Rel proteins in WT cells; this response was slightly reduced for RelB and cRel, but not for RelA, in IkkαAA cells (Fig. 2 D). By contrast, the basal level of all three Rel proteins in the nucleus and their RANKL-induced nuclear translocation were diminished severely in IkkβΔ cultures. Although IKKα plays a minor role in NF-κB activation, other experiments that were conducted with IkkαAA BM cells revealed its requirement for induction of NF-κB2/p100 processing to p52 in response to RANKL (Fig. 2 E), as previously shown for several other TNF family members (30).
Figure 2.

Biochemical analysis of RANKL signaling to NF-κB in IkkαAA and IkkβD osteoclast progenitors. (A) Western blot analysis of protein extracts from WT, IkkαAA, and IkkβΔ osteoclast precursors stimulated with RANKL for the indicated times. Immunoblot analysis was performed with anti-IKKβ, anti-IKKα, anti-IκBα, and anti-p38 (as loading control) antibodies. (B) NF-κB DNA binding activity was assayed at the indicated times in RANKL-stimulated WT, IkkαAA, and IkkβΔ osteoclast progenitors by electrophoretic mobility shift assay using a κB site oligonucleotide probe or a nuclear factor-y probe to control for loading and extract quality. (C) IKK activation by RANKL. Total protein lysates were prepared and IKK activity was measured by an immunocomplex kinase assay before and after RANKL stimulation of BM cells. IκBα(1–54) was used as a substrate. (D) Nuclear translocation of NF-κB subunits. Osteoclast progenitors from WT, IkkαAA, and IkkβΔ mice were incubated with RANKL for the indicated durations in the presence of M-CSF. Nuclear (nuc) and cytoplasmic (cyt) extracts were analyzed by immunoblotting using antibodies directed against NF-κB family members and an anti–poly(ADP-ribose)-polymerase antibody as a loading control. (E) p100 processing. Osteoclast progenitors from WT, IkkαAA, and IkkβΔ mice were incubated with RANKL for the indicated durations in the presence of M-CSF. Total protein extracts were analyzed for the presence of full-length p100 and its processed form, p52.
Defective osteoclastogenesis in IkkβΔ but not IkkαAA mice
To determine if IKKα and IKKβ play a role in osteoclastogenesis in vivo, bones from 4 mo-old IkkαAA and IkkβΔ mice were analyzed. To study the role of IKKβ in bone development we induced deletion of the Ikkβ gene in IkkβF/F:Mx1-Cre mice 9 d after birth. Histologic and histomorphometric analyses of long bone (tibias) sections from IkkαAA mice revealed no differences compared with WT bones, and no alterations were found in trabecular size and distribution or in the number of osteoclasts as determined by TRAP staining (Fig. 3 A–C). These results suggest that basal osteoclastogenesis is not affected by the loss of IKKα activation in vivo. By contrast, IkkβΔ mice showed an osteopetrotic phenotype. IkkβΔ mice are smaller and have a hunched back compared with their IkkβF/F littermates (unpublished data). Histologic analysis of tibias from these mice showed increased trabecular size and distribution which resulted in obliteration of the bone marrow cavity compared with IkkβF/F littermates or WT mice of the same age (Fig. 3 A). TRAP staining showed a greatly reduced number of osteoclasts in IkkβΔ bones (Fig. 3, B and C). Furthermore, quantitative histomorphometric analyses of IkkβΔ bones revealed a significant increase in bone volume due to increased trabecular number compared with bones of WT littermates (Fig. 3 C); this is diagnostic of reduced osteoclast-mediated bone resorption. The osteoclast parameters, number and surface, are reduced significantly in IkkβΔ mice (Fig. 3 C). It also seems that osteoblast number is reduced at 7 mo in the mutant mice, a defect that was observed in other osteopetrotic mouse models (35). The osteopetrotic phenotype of IkkβΔ mice becomes more dramatic with age (Fig. 3 C). These results suggest that IkkβΔ mice develop osteopetrosis that is due to defective osteoclast formation, and furthermore, indicate a critical role for Ikkβ in bone development.
Figure 3.
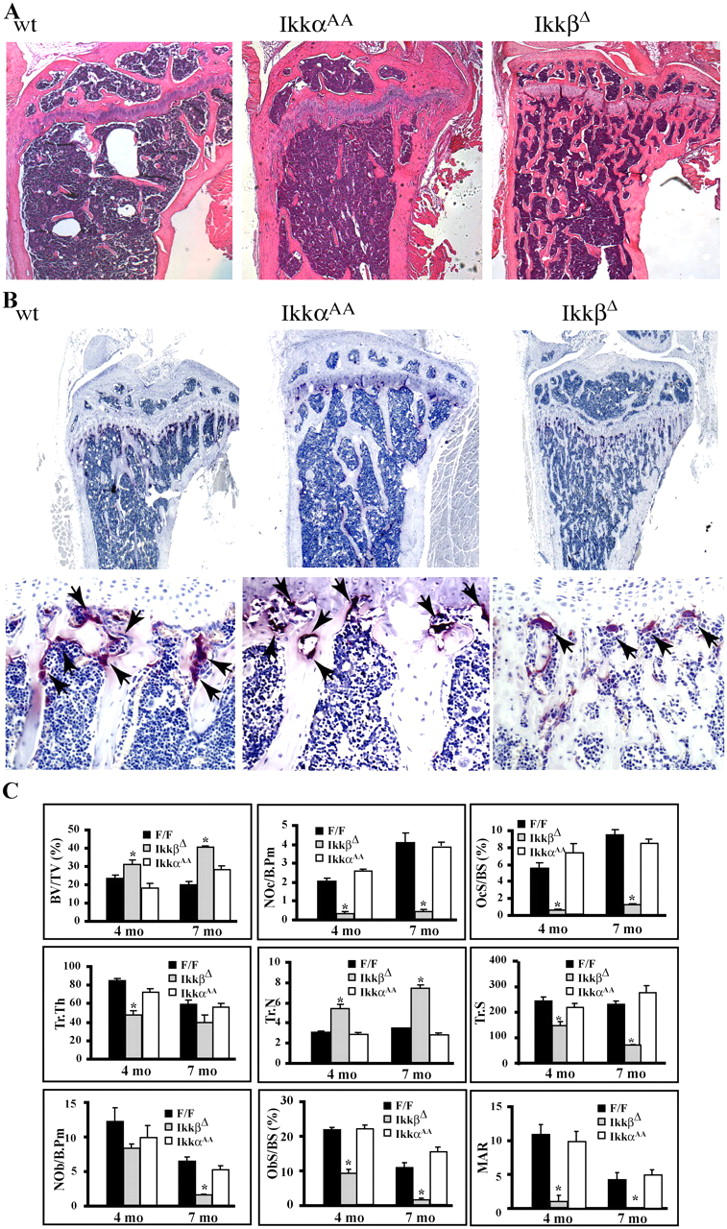
IkkβD, but not IkkαAA, mice are osteopetrotic. (A) Sections of the metaphyseal regions of the proximal tibia of 4-mo-old WT, IkkαAA, and IkkβΔ mice were subjected to H&E staining. (B) Decreased numbers of TRAP-positive (red-stained) multinucleated cells below the growth plate of IkkβΔ mice. TRAP-positive cells are marked by arrows. (C) Histomorphometric analysis of structural bone parameters in IkkβΔ (n = 6), IkkαAA(n = 6), and F/F (IkkβF/F, n = 6 ) mice at 4 and 7 mo of age. OcS/BS, osteoclast surface/bone surface; NOc/B.Pm, number of osteoclasts/bone perimeter/mm; Tr.Th, trabecular thickness (mm); Tr.N trabecular number/mm; Tr.S, trabecular separation/mm3; NOb/B.Pm, number of osteoblasts/bone perimeter/mm; ObS/BS, osteoblast surface/bone surface; MAR, mineral apposition rate. *P < 0.05.
Osteoblasts can rescue defective in vitro osteoclastogenesis of IkkαAA but not IkkβΔ BM cells
Osteoclasts also can be generated in vitro from BM hematopoietic precursors cultured in the presence of 1,25(OH)2-vitamin D3 and dexamethasone together with osteoblastic/stromal cells from mouse calvarias (36). In such a system, activated osteoblasts play the major role in osteoclast differentiation and provide M-CSF and RANKL directly. To examine whether the osteoclastogenic defects that are observed in IkkαAA and IkkβΔ BM cultures are caused by cell autonomous defects, we performed osteoclasts/osteoblast cocultures. Wt osteoblasts fully supported osteoclastogenesis of IkkαAA osteoclast precursors, but not IkkβΔ osteoclast precursors; this indicates a cell–autonomous osteoclast differentiation defect in IkkβΔ BM cells (Fig. 4 A, upper panels). Reciprocal cocultures of IkkαAA or WT BM cells with IkkαAA osteoblasts showed that IkkαAA osteoblasts had the same capacity as WT osteoblasts for supporting osteoclast differentiation (Fig. 4, A [lower panels] and B). Thus, IkkαAA osteoclasts can form normally as long as they receive osteoblast-derived signals; this suggests that these signals compensate for the defect in RANKL signaling.
Figure 4.

Osteoclastogenesis is impaired in IkkβD, but not in IkkαAA, BM cells that are cocultured with osteoblasts. (A) Equal numbers of BM cells from WT, IkkαAA, and IkkβΔ mice were plated in the presence of 5 × 105 cells/ml of primary calvarial osteoblasts (Ob) from WT or IkkαAA mice and cultured in the presence of 1,25(OH)2 vitamin D3 and dexamethasone for 6 d. Osteoclastogenesis was assayed by TRAP staining. (B) The extent of osteoclastogenesis (expressed as staining intensity ×10−5) was determined by TRAP staining, quantified, and expressed as staining intensity ×10−5.
IL-1 and TNFα rescue the osteoclastogenic defect of IkkαAA, but not IkkβΔ osteoclast progenitors
To identify potential signals that may compensate for the defect in RANKL signaling of IkkαAA osteoclast precursors, we examined the effect of the proinflammatory cytokines, IL-1 and TNFα. IL-1 and TNFα are potent osteoclastogenic factors and are likely to be involved with RANKL in inflammation-induced bone loss (10, 37). We cultured osteoclast progenitors from WT, IkkαAA, and IkkβΔ mice in the presence of IL-1 or TNFα, alone or together with RANKL. IL-1 and TNFα strongly augmented the osteoclastogenic response of WT BM to RANKL and led to the formation of numerous TRAP-positive giant cells (Fig. 5 A). Either IL-1 or TNFα, when combined with RANKL, completely rescued the osteoclastogenic defect of IkkαAA BM, although they did not induce differentiation on their own (Fig. 5 A and not depicted). However, the defect in osteoclast differentiation of IkkβΔ BM cultures could not be rescued by IL-1 or TNFα (Fig. 5, A and B). The bone-resorbing activity of these cells was assayed by an in vitro resorption pit assay and further quantified. IkkαAA, but not IkkβΔ, BM cells were able to resorb bone, once their differentiation defect was rescued by IL-1 (not depicted) or TNFα (Fig. 5 C). Furthermore, we found that when incubated with TNFα, either in the absence or presence of RANKL, most IkkβΔ osteoclast precursors were dead within 48 h (Fig. 5 A). This prompted us to examine whether IkkβΔ BM cells undergo apoptosis in response to TNFα. Terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) assay revealed the appearance of cells with distinct apoptotic morphology and fragmented DNA within 24 h of TNFα addition to IkkβΔ osteoclast precursors (Fig. 5, D and E). Very few such cells were detected in cultures that were not exposed to TNFα or in WT osteoclast precursors that were incubated with TNFα.
Figure 5.

IL-1 or TNFα rescue RANKL-induced osteoclast differentiation of IkkαAA, but not IkkβD, osteoclast progenitors. (A) Equal numbers of BM cells from WT, IkkαAA, IkkβΔ, and IkkβΔ :Tnfr1 − / − mice were cultured in the presence of M-CSF (20 ng/ml) and RANKL (50 ng/ml) for 7 d. TNFα (20 ng/ml) or IL-1 (10 ng/ml) alone or in combination with RANKL were added. Osteoclastogenesis was assayed by TRAP staining. Incubation of IkkβΔ BM cells with TNFα resulted in death of all cells within 48 h. (B) Numbers of osteoclasts per field (%). Results shown are averages ± SD (n = 4). (C) The bone-resorbing activity of WT, IkkαAA, IkkβΔ, Tnfr1 − / −, and IkkβΔ :Tnfr1 − / − osteoclasts was analyzed in vitro by quantification of resorption pit areas on calcium phosphate films. (D) TUNEL assay of BM cells from WT and IkkβΔ mice that were incubated for the indicated times with TNFα (1 ng/ml). (E) Quantification of the TUNEL assay results. The percentage of TUNEL positive cells in four representative fields was determined by cell counting. n.d., not depicted.
Deletion of Tnfr1 rescues IkkβΔ osteoclast progenitors from TNFα-induced apoptosis, but does not prevent osteopetrosis
TNFR1 contains a death domain and is capable of engaging the apoptotic machinery (38). To further examine the mechanism that underlies TNFα-induced death of Ikkβ-deficient preosteoclasts, we crossed IkkβΔ mice with Tnfr1 − / − mice to generate IkkβF/F:Mx1-Cre:Tnfr1−/− double mutants. After poly(IC) injection, BM cultures that were isolated from these mice were stimulated with RANKL alone or with RANKL plus TNFα; osteoclast differentiation was analyzed by TRAP staining. Loss of TNFR1 prevented TNFα-induced death of IkkβΔ osteoclast progenitors (Fig. 5 A, bottom). Furthermore, the absence of TNFR1 rescued the inability of IkkβΔ cells to become TRAP-positive, but did not allow them to differentiate fully into multinucleated giant osteoclasts (Fig. 5 A) with the ability to resorb bone (Fig. 5 C). Thus, IKKβ is required for the prevention of TNFα-induced death and is needed for formation of fully functional bone-resorbing osteoclasts.
Next we analyzed femurs of 4-mo-old WT, IkkβΔ, Tnfr1 − / −, and IkkβΔ :Tnfr1 − / −mice. Ikkβ deletion in both strains was induced as early as 9 d after birth. Histochemical staining revealed osteopetrosis in IkkβΔ :Tnfr1 − / − bones (Fig. 6 A). However, TRAP-positive cells could be detected easily in IkkβΔ :Tnfr1 − / − mice, whereas they were scarce in IkkβΔ mice (Fig. 6 B). Analysis of deoxypiridinoline cross-links in urine samples—which reflects osteoclast activity in vivo—revealed that in IkkβΔ :Tnfr1 − / − mice, osteoclast resorption activity remained severely attenuated, as in IkkβΔ mice. Normal levels of deoxypiridinoline cross-links were found in urine samples from IkkαAA or Tnfr1 − / − mice.
Figure 6.

Osteoclast precursors that lack IKKβ are sensitive to TNFα-mediated apoptosis. (A) Sections of the metaphyseal regions of femurs of 4-mo-old Tnfr1 − / − and IkkβΔ :Tnfr1 − / − mice were subjected to H&E staining. (B) TRAP staining of the metaphyseal regions of 4-mo-old WT, IkkβΔ, Tnfr1 − / −, and IkkβΔ :Tnfr1 − / − mice. The numbers refer to osteoclasts per field. (C) Deoxypiridinoline (DPD) cross-links in the urine of WT, IkkβΔ, Tnfr1 − / −, and IkkβΔ :Tnfr1 − / −mice were measured as an indicator of in vivo osteoclast activity. The values were normalized for muscle creatinine that also is excreted in the urine to account for urine concentration (n = 6). (D) H&E, F4/80 (red), and DAPI (blue) staining of sections from long bones of 4-mo-old WT, IkkβΔ, Tnfr1 − / −, and IkkβΔ :Tnfr1 − / − mice. (E) F4/80 (red) and TUNEL (green) double staining and DAPI (blue) staining of sections from long bones of WT and IkkβΔ mice. (F) F4/80 staining of liver sections of WT and IkkβΔ mice.
Immunohistochemical analyses by hematoxylin-eosin (H&E) staining revealed a small number of osteoclast precursors in the vicinity of the growth plate that were positive for F4/80 staining in IkkβΔ mice, whereas many more such cells were seen in WT mice (Fig. 6 D). Remarkably, the loss of TNFR1 in IkkβΔ :Tnfr1 − / − double mutant mice restored the presence of F4/80 positive cells next to the growth plate (Fig. 6 D). Double staining experiments revealed that in IkkβΔ mice, most of the few F4/80-positive cells that were present were apoptotic, based on TUNEL staining (Fig. 6 E). This further supports the hypothesis that the decreased osteoclast number in IkkβΔ mice is due to increased apoptosis of their precursors. Control experiments showed that in other tissues, such as the liver, the number of F4/80-positive cells did not change in the absence of Ikkβ (Fig. 6 F). Despite the decreased apoptosis of osteoclast precursors and restoration of TRAP-positive cells, the IkkβΔ :Tnfr1 − / − double mutant mice remain osteopetrotic. This suggests that osteoclasts that lack IKKβ and TNFR1 are not fully functional under basal conditions; this interpretation is consistent with the severely reduced bone-resorbing activity in these mice (Fig. 6 C).
Absence of IKKβ protects mice from inflammation-induced bone loss, in a manner dependent on TNFR1
IKKβ plays a crucial role in osteoclast differentiation under physiologic conditions. We next analyzed whether IKKβ or IKKα is involved in inflammation-induced bone loss. We used an established model of endotoxin-induced bone resorption (39, 40). IkkαAA, IkkβΔ, Tnfr1 − / −, IkkβΔ :Tnfr1 − / −, and WT mice were injected with 500 μg LPS in saline into the synovial space of the hind limb knee joint, whereas the contralateral knee was injected with saline alone; mice were killed 5 d after injection. Ikkβ deletion was induced 10 d before knee injection. Before sacrificing the mice, their ability to move and flex their hind limb was assessed by video cinematography (Videos 1 and 2, available at http://www.jem.org/cgi/content/full/jem.20042081/DC1). The staining of the joint bones (femurs and tibia) with H&E, TRAP, and F4/80 showed a considerably lower number of osteoclasts and osteoclast precursors in IkkβΔ mutant mice compared with WT controls or IkkαAA mice (Fig. 7, A–D; not depicted for IkkαAA). Although LPS-injected WT, IkkαAA, and Tnfr1 − / − mice completely lost the ability to flex the hind limb, no such aberrations were evident in IkkβΔ mice, which retained normal flexibility and movement of the LPS-treated hind limb (Videos 1 and 2). The loss of TNFR1 restored inflammation-induced bone loss in mice that lack IkkβΔ (Fig. 7), although the bone damage is not as extensive as in WT mice (compare resorption sites indicated by arrows in Fig. 7 A). The degree of inflammation-induced bone loss was quite similar in IkkβΔ :Tnfr1 − / − and Tnfr1 − / − mice. These results suggest that under strong inflammatory conditions, which are likely to result in massive cytokine production, IKKβ is no longer needed for osteoclastogenesis once its survival function has been rendered unnecessary by ablation of TNFR1.
Figure 7.
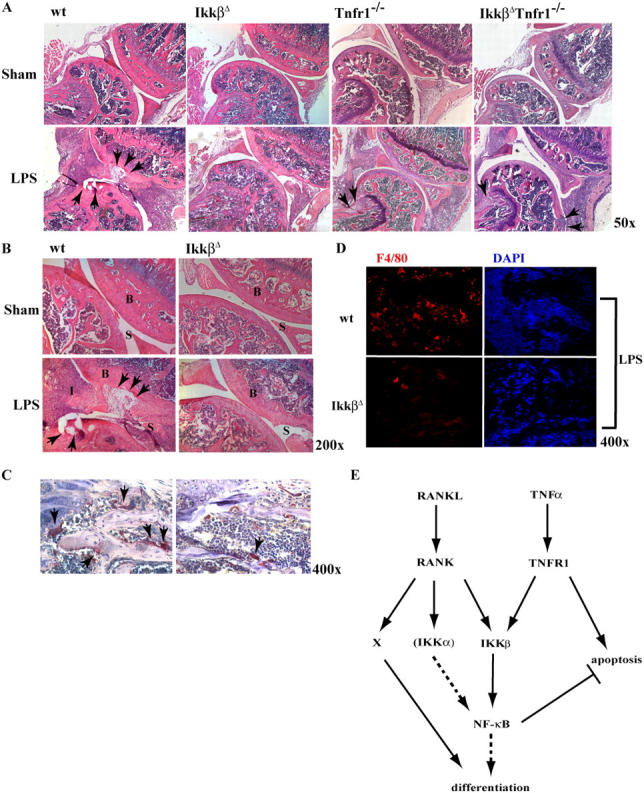
Lack of IKKβ prevents inflammation-induced bone loss. 2-mo-old WT (n = 11), IkkβΔ (n = 7), Tnfr1 − / − (n = 4), and IkkβΔ :Tnfr1 − / − (n = 4) mice were injected once with LPS (500 μg/ml) or saline control into the synovial space of their hind leg joints. (A) After 5 d the mice were killed and the limbs were fixed, decalcified, sectioned, and analyzed by H&E staining. The arrows show regions of bone loss. (B) Higher magnification of the H&E staining of the joints of WT and IkkβΔ mice. B, bone; I, inflammation; S, synovial space. (C) The presence of osteoclasts (arrows) in WT and IkkβΔ mice that were treated with LPS was analyzed by TRAP staining. (D) F4/80 (red) and DAPI (blue) staining of sections from joints of WT and IkkβΔ mice that were treated with LPS. (E) Schematic model of RANKL and TNFα signaling during osteoclastogenesis and inflammation-induced bone loss. X represents a pathway other than IKK/NF-κB that is activated by RANKL binding to RANK and is essential for production of functional osteoclasts. This pathway is not activated by TNFα binding to TNFR1.
DISCUSSION
Previous studies that were based on ablation of the p50 and p52 NF-κB members outlined an important role for these transcription factors in osteoclast differentiation (15). NF-κB is activated by RANKL, a cytokine whose expression and binding to the receptor, RANK, are essential for osteoclastogenesis. NF-κB also is activated by the proinflammatory cytokines, TNFα and IL-1 (21), which enhance inflammation-induced bone loss, although they are not essential for developmental osteoclastogenesis (41, 42). The activation of NF-κB by all of these cytokines depends on integrity of the IKK complex (21); recent results show that a small peptide that can prevent binding of the IKKα and IKKβ catalytic subunits to the IKKγ regulatory subunit can inhibit inflammation-induced bone loss (27). Although these results provide further support for the role of NF-κB in osteoclastogenesis, it has not been established which of the two IKK catalytic subunits plays a more critical role in basal osteoclastogenesis and inflammation-induced bone loss. For instance, it recently was described that the NIK- and IKKα-dependent alternative pathway is required for RANKL-induced osteoclast differentiation in vitro (25). Our results demonstrate a critical role for IKKβ, but not IKKα, in basic osteoclastogenesis in vivo and in inflammation-induced bone loss.
Although IKKα and IKKβ are important transducers of osteoclastogenic signals which emanate from RANKL in vitro, the mutation that prevents IKKα activation by its upstream kinase, NIK (23), had no effect on osteoclast formation in vivo and did not increase bone density. Furthermore, IkkαAA mice were fully sensitive to inflammation-induced bone loss (unpublished data). These results are consistent with the small effect of the IkkαAA mutation on RANKL-induced translocation of NF-κB proteins to the nucleus, as well as the absence of an osteopetrotic phenotype in mice defective in NIK, the upstream activator of IKKα (25). Like BM progenitors from Nik − / − mice, IkkαAA BM progenitors do not differentiate in response to RANKL when cultured in the absence of osteoblasts. However, either osteoblasts or the proinflammatory cytokines, IL-1β and TNFα, together with RANKL induce osteoclastogenesis of IkkαAA BM progenitors. Thus, although IKKα contributes to RANKL-induced differentiation in vitro, its function in vivo is dispensable because of the action of other factors that activate the IKKβ-driven classic NF-κB pathway. During normal bone development, these factors are likely to be derived from osteoblasts, whereas during inflammation these factors could be TNFα and IL-1β.
In contrast with IKKα, our findings illustrate a critical function for IKKβ. IKKβ-deficient BM progenitors do not form osteoclasts in vitro in response to RANKL or when cocultured with osteoblasts. Furthermore, their inability to respond to RANKL cannot be complemented by IL-1 or TNFα, and IkkβΔ mice are osteopetrotic. IKKβ-deficient BM progenitors are extremely sensitive to TNFα and undergo extensive apoptosis, despite the presence of the myeloid survival factor, M-CSF. Because TNFα-induced apoptosis of IKKβ-deficient preosteoclasts is prevented by the loss of TNFR1, we propose that one of the mechanisms by which IKKβ-dependent NF-κB activation contributes to osteoclastogenesis in vivo, especially during inflammation, is through prevention of TNFα-induced apoptosis of osteoclast progenitors. In the absence of IKKβ, such cells become very sensitive to TNFα and are eliminated when TNFα is produced in sufficiently large amounts. Nonetheless, although the death of Ikkβ-deficient osteoclast progenitors is prevented by loss of TNFR1 and IkkβΔ :Tnfr1 − / − double mutants display close to normal numbers of TRAP-positive cells in their bones (Fig. 6 B), these mice become osteopetrotic (Fig. 6 A) if Ikkβ deletion is induced early during bone development. The explanation for these results is that IKKβ-deficient osteoclasts remain defective in bone resorption, even when their TNFα-induced elimination does not take place. Our in vitro results (Fig. 5 A) suggest that IkkβΔ :Tnfr1 − / − progenitors cannot give rise to fully differentiated osteoclasts, although they do become TRAP-positive in response to RANKL. Morphologically, such cells look like monocytes, rather than multinucleated giant cells; this suggests that their ability to undergo cell fusion is eliminated. Similar defects in basal osteoclast functions were observed in Traf6 − / − and Src − / − mice (16, 43). These mice are osteopetrotic as a result of the presence of osteoclasts that are unable to form ruffled borders, and therefore, are defective in bone resorption.
Thus, in addition to the prevention of TNFα-induced apoptosis, IKKβ is required for terminal osteoclast differentiation. Although IKKβ-dependent NF-κB activation is essential for this process, it is not sufficient; potent NF-κB activating cytokines, such as TNFα, cannot substitute for RANKL (Fig. 7 E). Most likely, another pathway or factor, designated X in Fig. 7 E, needs to be switched on—along with IKKβ and NF-κB—for terminal osteoclast differentiation to take place. Nonetheless, our results illustrate the potential ability of IKKβ inhibition to prevent inflammation-driven bone destruction. Again, the mechanism through which IKKβ inhibition prevents inflammation-induced bone loss involves sensitization of osteoclast progenitors to TNFα-induced apoptosis, because IkkβΔ :Tnfr1 − / − mice are fully susceptible to inflammation-induced bone loss. The bone-resorbing ability of osteoclasts in IkkβΔ :Tnfr1 − / − mice seems to be restored after LPS injection. Because these cells do not respond to TNFα (as a result of the loss of TNFR1), the inflammation-induced factor that may stimulate their bone-resorbing activity could be IL-1β, which is known to be induced by LPS administration (44). However, in vitro IL-1β is unable to induce the formation of multinucleated bone-resorbing cells when given together with RANKL once IKKβ is absent (Fig. 5 A). Alternatively, IkkβΔ :Tnfr1 − / − osteoclast progenitors may be only partially defective in their ability to respond to RANKL; this defect may be eliminated when high levels of RANKL are present along with other proinflammatory cytokines. LPS-induced inflammation results in induction of RANKL along with other cytokines (45).
Our results support a role for IKKβ as an important regulator of bone homeostasis and a mediator of inflammation-induced bone loss. Our results also suggest that the major mechanism through which deletion or inhibition of IKKβ exerts its therapeutic effect in inflammation-induced bone loss is by predisposing osteoclast precursors to TNFα-induced apoptosis. A schematic model that summarizes our findings is presented in Fig. 7 E. Binding of RANKL to its receptor, RANK, induces a cascade of events that leads to activation of IKKβ and at least one more factor—that together with IKKβ—is required for induction of terminal osteoclast differentiation. During inflammation, proinflammatory cytokines, such as TNFα and IL-1β, are induced and strongly potentiate RANKL-induced osteoclastogenesis, although such factors cannot induce osteoclast differentiation on their own. TNFα signaling through TNFR1 has the potential to induce apoptosis through caspase 8, a process that is prevented by IKKβ-dependent NF-κB activation (46). Once IKKβ is inhibited, TNFα-induced apoptosis results in elimination of Ikkβ-deficient osteoclast progenitors, and thereby, prevents inflammation-induced bone destruction. Thus, IKKβ inhibition presents a logical strategy for prevention of numerous bone-resorbing disorders that are triggered by inflammation, such as rheumatoid arthritis.
MATERIALS AND METHODS
Mice.
IkkαAA and IkkβF/F mice were generated as described (29, 32). To delete IKKβ in hematopoietic cells, IkkβF/F mice were crossed with Mx1-Cre transgenic mice (33), and IkkβF/F:Mx1-Cre progeny were injected three times with poly(IC) every 2 d. Injections started at day 9 after birth for in vivo analysis, whereas injections were performed 10 d before killing mice or collecting BM cells, respectively. Deletion of Ikkβ was confirmed by PCR, whereas the absence of IKKβ protein was examined by immunoblotting. These mice are referred to as IkkβΔ mice. All experimental procedures were approved by the Animal Subjects Committee at the University of California San Diego, according to U.S. National Institutes of Health guidelines.
Histologic and histomorphometric analyses.
Tissues were fixed in PBS-buffered 4% formaldehyde, embedded in paraffin, sectioned at 5 μm, and stained as indicated using standard techniques. Calcified tissues were decalcified in EDTA (0.5 M, pH 8) for 12 d before embedding. TRAP staining was performed using a leukocyte acid phosphatase kit (Sigma-Aldrich). For histomorphometry, tibiae were embedded in methacrylate (Echnovit; Heraeus Kulzer) without previous decalcification and 3–4-μm sections were stained with Goldner trichrome. Histomorphometry of metaphyses was performed using an Axioskop 2 microscope (Carl Zeiss MicroImaging, Inc.) and OsteoMeasure Analysis System (OsteoMetrics) according to international standards (47). TUNEL assay and immunohistochemistry were as described previously (48, 49).
Osteoclast culture and activity assay.
BM cells from 6-wk-old mice were plated in the presence of 5 ng/ml recombinant M-CSF for 24 h Nonadherent cells were replated in the presence of recombinant M-CSF (10 ng/ml) and recombinant-RANKL (50 ng/ml) for 7 d and then fixed and stained for TRAP activity. For biochemical analysis, BM cells were plated in the presence of M-CSF (10 ng/ml) for 6 d, collected, and counted. Osteoclast activity was assayed in vitro after differentiation on a calcium phosphate film (BioCoat Bone Cell Culture System, Osteologic) and the resorpted area was quantified using AxioVision 4.3 software. Osteoclast activity in vitro was measured in urine samples using RatLaps ELISA (Nordic Bioscience Diagnostics).
Osteoblast–osteoclast cocultures.
Primary osteoblasts were isolated from calvarias of neonatal (2–4-d-old) WT and IkkαAA mice and were digested for 10 min in modified Eagle's medium (α-MEM) which contained 0.1% collagenase and 0.2% dispase. Cells from two to five mice were combined as an osteoblastic cell population and plated at a density of 5 × 105 cells/ml in α-MEM with 10% FCS for 24 h. Cocultures were performed as described (36, 50). Briefly, BM cells (106 per well) were added to primary osteoblasts (5 × 105 cells per well) and cultured in α-MEM which contained 10% FCS, 10−8 M 1,25(OH)2-vitamin D3, and 10−7 M dexamethasone in 24-well plates. Intensity of TRAP staining was measured by Image Pro Plus 5.1.
Subcellular fractionation and immunoblot analysis.
Cells were resuspended in buffer L1 (50 mM Tris-Cl, pH 8.0, 2 mM EDTA, 0.1% NP-40, 10% glycerol) that contained protease inhibitors, incubated for 5 min at 4°C, and centrifuged for 5 min at 4,000 revolutions/min in a microcentrifuge. Cytoplasmic supernatants were stored and nuclear pellets were extracted further in buffer L2 (20 mM Hepes-KOH, pH 7.6, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 25 mM β-glycerophosphate) which contained protease inhibitors. After lysis, nuclei were centrifuged at 15,000 revolutions/min and the supernatant was collected for further analysis. Nuclear (5 μg) and cytoplasmic (20 μg) extracts were electrophoresed, transferred to nitrocellulose membranes, and immunoblotted with anti-IKKα (Imgenex), anti-IKKβ (UBI), anti-RelA, anti-RelB, and anti-c-Rel (Santa Cruz Biotechnology, Inc.) antibodies. p100 processing in total extracts was assayed using anti–NF-κB/p52 K-27 antibody (Santa Cruz Biotechnology, Inc.).
IKK and gel shift assays.
IKK immunocomplex kinase assay was as described (51), except that an IKKγ antibody (BD Biosciences) was used for immunoprecipitation. Electrophoretic mobility shift assay for NF-κB was described previously (23).
Inflammation-induced bone loss.
2- to 3-mo-old mice were given an intrajoint injection of Escherichia coli LPS (Sigma-Aldrich), 500 μg in saline, and a vehicle control into the contralateral joint, 5 d after the last poly(IC) treatment. 5 d later, mice were killed and joint histology was examined in the Histologic and histomorphometric analyses section.
Statistical analysis.
Data are expressed as mean ± SEM. Differences were analyzed by Student's t test.
Online supplemental material
IkkβF/F (Video 1) and IkkβΔ (Video 2) mice were given an intrajoint injection of E. coli LPS (Sigma-Aldrich), 500 μg in saline, (left hind limb) and a vehicle control (right hind limb). The two videos show the ability of the mice to move and stretch their hind limbs. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20042081/DC1.
Acknowledgments
The authors would like to thank Drs. A. Hoebertz and L. Kenner for helpful discussion and A. Chang for LPS knee joint injections.
S. Maeda, J.M. Park, L.-C. Hsu, and Y. Cao were supported by postdoctoral fellowships from the Japan Society for the Promotion of Science, Bristol-Myers Squibb Foundation Research Fellowship at the Irvington Institute, the Cancer Research Institute, and an American Association for Cancer Research–Genentech BioOncology Career Development Award for Cancer Research. Support was provided by National Institutes of Health grants nos. ES06376 and AI43477 (to M. Karin). In addition, M. Karin is an American Cancer Society Research Professor.
The authors have no conflicting financial interests.
Abbreviations used: H&E, hematoxylin-eosin; IκB, inhibitor of NF-κB; IKK, IκB kinase; M-CSF, macrophage-colony stimulating factor; NIK, NF-κB–inducing kinase; poly(IC), polyinosinic-polycytidylic acid; RANKL, receptor activator of NF-κB ligand; TNFR, TNF receptor; TRAP, tartrate-resistant acid phosphatase; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling.
References
- 1.Karsenty, G., and E.F. Wagner. 2002. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell. 2:389–406. [DOI] [PubMed] [Google Scholar]
- 2.Manolagas, S.C., and R.L. Jilka. 1995. Bone marrow, cytokines, and bone remodeling. Emerging insights into the pathophysiology of osteoporosis. N. Engl. J. Med. 332:305–311. [DOI] [PubMed] [Google Scholar]
- 3.Gravallese, E.M. 2002. Bone destruction in arthritis. Ann. Rheum. Dis. 61(Suppl 2):ii84–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suda, T., N. Takahashi, N. Udagawa, E. Jimi, M.T. Gillespie, and T.J. Martin. 1999. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 20:345–357. [DOI] [PubMed] [Google Scholar]
- 5.Kong, Y.Y., H. Yoshida, I. Sarosi, H.L. Tan, E. Timms, C. Capparelli, S. Morony, A.J. Oliveira-dos-Santos, G. Van, A. Itie, et al. 1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 397:315–323. [DOI] [PubMed] [Google Scholar]
- 6.Lacey, D.L., E. Timms, H.L. Tan, M.J. Kelley, C.R. Dunstan, T. Burgess, R. Elliott, A. Colombero, G. Elliott, S. Scully, et al. 1998. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 93:165–176. [DOI] [PubMed] [Google Scholar]
- 7.Anderson, D.M., E. Maraskovsky, W.L. Billingsley, W.C. Dougall, M.E. Tometsko, E.R. Roux, M.C. Teepe, R.F. DuBose, D. Cosman, and L. Galibert. 1997. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 390:175–179. [DOI] [PubMed] [Google Scholar]
- 8.Dougall, W.C., M. Glaccum, K. Charrier, K. Rohrbach, K. Brasel, T. De Smedt, E. Daro, J. Smith, M.E. Tometsko, C.R. Maliszewski, et al. 1999. RANK is essential for osteoclast and lymph node development. Genes Dev. 13:2412–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang, Y.H., A. Heulsmann, M.M. Tondravi, A. Mukherjee, and Y. Abu-Am. 2001. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J. Biol. Chem. 276:563–568. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi, K., N. Takahashi, E. Jimi, N. Udagawa, M. Takami, S. Kotake, N. Nakagawa, M. Kinosaki, K. Yamaguchi, N. Shima, et al. 2000. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 191:275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam, J., S. Takeshita, J.E. Barker, O. Kanagawa, F.P. Ross, and S.L. Teitelbaum. 2000. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest. 106:1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsao, D.H., T. McDonagh, J.B. Telliez, S. Hsu, K. Malakian, G.Y. Xu, and L.L. Lin. 2000. Solution structure of N-TRADD and characterization of the interaction of N-TRADD and C-TRAF2, a key step in the TNFR1 signaling pathway. Mol. Cell. 5:1051–1057. [DOI] [PubMed] [Google Scholar]
- 13.Inoue, J., T. Ishida, N. Tsukamoto, N. Kobayashi, A. Naito, S. Azuma, and T. Yamamoto. 2000. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp. Cell Res. 254:14–24. [DOI] [PubMed] [Google Scholar]
- 14.Baud, V., Z.G. Liu, B. Bennett, N. Suzuki, Y. Xia, and M. Karin. 1999. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 13:1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iotsova, V., J. Caamano, J. Loy, Y. Yang, A. Lewin, and R. Bravo. 1997. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat. Med. 3:1285–1289. [DOI] [PubMed] [Google Scholar]
- 16.Lomaga, M.A., W.C. Yeh, I. Sarosi, G.S. Duncan, C. Furlonger, A. Ho, S. Morony, C. Capparelli, G. Van, S. Kaufman, et al. 1999. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13:1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franzoso, G., L. Carlson, L. Xing, L. Poljak, E.W. Shores, K.D. Brown, A. Leonardi, T. Tran, B.F. Boyce, and U. Siebenlist. 1997. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 11:3482–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grigoriadis, A.E., Z.Q. Wang, M.G. Cecchini, W. Hofstetter, R. Felix, H.A. Fleisch, and E.F. Wagner. 1994. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 266:443–448. [DOI] [PubMed] [Google Scholar]
- 19.Karin, M. 1995. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270:16483–16486. [DOI] [PubMed] [Google Scholar]
- 20.May, M.J., and S. Ghosh. 1997. Rel/NF-kappa B and I kappa B proteins: an overview. Semin. Cancer Biol. 8:63–73. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-kappaB puzzle. Cell. 109(Suppl):S81–96. [DOI] [PubMed] [Google Scholar]
- 22.Rothwarf, D.M., and M. Karin. 1999. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE:RE1. [DOI] [PubMed]
- 23.Senftleben, U., Y. Cao, G. Xiao, F.R. Greten, G. Krahn, G. Bonizzi, Y. Chen, Y. Hu, A. Fong, S.C. Sun, and M. Karin. 2001. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 293:1495–1499. [DOI] [PubMed] [Google Scholar]
- 24.Xiao, G., E.W. Harhaj, and S.C. Sun. 2001. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell. 7:401–409. [DOI] [PubMed] [Google Scholar]
- 25.Novack, D.V., L. Yin, A. Hagen-Stapleton, R.D. Schreiber, D.V. Goeddel, F.P. Ross, and S.L. Teitelbaum. 2003. The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J. Exp. Med. 198:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinkura, R., K. Kitada, F. Matsuda, K. Tashiro, K. Ikuta, M. Suzuki, K. Kogishi, T. Serikawa, and T. Honjo. 1999. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat. Genet. 22:74–77. [DOI] [PubMed] [Google Scholar]
- 27.Jimi, E., K. Aoki, H. Saito, F. D'Acquisto, M.J. May, I. Nakamura, T. Sudo, T. Kojima, F. Okamoto, H. Fukushima, et al. 2004. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat. Med. 10:617–624. [DOI] [PubMed] [Google Scholar]
- 28.Delhase, M., M. Hayakawa, Y. Chen, and M. Karin. 1999. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 284:309–313. [DOI] [PubMed] [Google Scholar]
- 29.Cao, Y., G. Bonizzi, T.N. Seagroves, F.R. Greten, R. Johnson, E.V. Schmidt, and M. Karin. 2001. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 107:763–775. [DOI] [PubMed] [Google Scholar]
- 30.Bonizzi, G., M. Bebien, D.C. Otero, K.E. Johnson-Vroom, Y. Cao, D. Vu, A.G. Jegga, B.J. Aronow, G. Ghosh, R.C. Rickert, and M. Karin. 2004. Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J. 23:4202–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin, L., L. Wu, H. Wesche, C.D. Arthur, J.M. White, D.V. Goeddel, and R.D. Schreiber. 2001. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 291:2162–2165. [DOI] [PubMed] [Google Scholar]
- 32.Li, Z.W., S.A. Omori, T. Labuda, M. Karin, and R.C. Rickert. 2003. IKK beta is required for peripheral B cell survival and proliferation. J. Immunol. 170:4630–4637. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn, R., F. Schwenk, M. Aguet, and K. Rajewsky. 1995. Inducible gene targeting in mice. Science. 269:1427–1429. [DOI] [PubMed] [Google Scholar]
- 34.Hsu, L.C., J.M. Park, K. Zhang, J.L. Luo, S. Maeda, R.J. Kaufman, L. Eckmann, D.G. Guiney, and M. Karin. 2004. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 428:341–345. [DOI] [PubMed] [Google Scholar]
- 35.Amling, M., L. Neff, M. Priemel, A.F. Schilling, J.M. Rueger, and R. Baron. 2000. Progressive increase in bone mass and development of odontomas in aging osteopetrotic c-src-deficient mice. Bone. 27:603–610. [DOI] [PubMed] [Google Scholar]
- 36.Jimi, E., I. Nakamura, H. Amano, Y. Taguchi, T. Tsurukai, M. Tamura, N. Takahashi, and T. Suda. 1996. Osteoclast function is activated by osteoblastic cells through a mechanism involving cell-to-cell contact. Endocrinology. 137:2187–2190. [DOI] [PubMed] [Google Scholar]
- 37.Abu-Amer, Y., J. Erdmann, L. Alexopoulou, G. Kollias, F.P. Ross, and S.L. Teitelbaum. 2000. Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J. Biol. Chem. 275:27307–27310. [DOI] [PubMed] [Google Scholar]
- 38.Baud, V., and M. Karin. 2001. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 11:372–377. [DOI] [PubMed] [Google Scholar]
- 39.Chiang, C.Y., G. Kyritsis, D.T. Graves, and S. Amar. 1999. Interleukin-1 and tumor necrosis factor activities partially account for calvarial bone resorption induced by local injection of lipopolysaccharide. Infect. Immun. 67:4231–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takayanagi, H., K. Ogasawara, S. Hida, T. Chiba, S. Murata, K. Sato, A. Takaoka, T. Yokochi, H. Oda, K. Tanaka, et al. 2000. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 408:600–605. [DOI] [PubMed] [Google Scholar]
- 41.Li, P., H. Allen, S. Banerjee, S. Franklin, L. Herzog, C. Johnston, J. McDowell, M. Paskind, L. Rodman, J. Salfeld, et al. 1995. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 80:401–411. [DOI] [PubMed] [Google Scholar]
- 42.Rothe, J., W. Lesslauer, H. Lotscher, Y. Lang, P. Koebel, F. Kontgen, A. Althage, R. Zinkernagel, M. Steinmetz, and H. Bluethmann. 1993. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 364:798–802. [DOI] [PubMed] [Google Scholar]
- 43.Soriano, P., C. Montgomery, R. Geske, and A. Bradley. 1991. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 64:693–702. [DOI] [PubMed] [Google Scholar]
- 44.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 45.Wada, N., H. Maeda, Y. Yoshimine, and A. Akamine. 2004. Lipopolysaccharide stimulates expression of osteoprotegerin and receptor activator of NF-kappa B ligand in periodontal ligament fibroblasts through the induction of interleukin-1 beta and tumor necrosis factor-alpha. Bone. 35:629–635. [DOI] [PubMed] [Google Scholar]
- 46.Karin, M., and A. Lin. 2002. NF-kappaB at the crossroads of life and death. Nat. Immunol. 3:221–227. [DOI] [PubMed] [Google Scholar]
- 47.Parfitt, A.M., M.K. Drezner, F.H. Glorieux, J.A. Kanis, H. Malluche, P.J. Meunier, S.M. Ott, and R.R. Recker. 1987. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2:595–610. [DOI] [PubMed] [Google Scholar]
- 48.Park, J.M., F.R. Greten, Z.W. Li, and M. Karin. 2002. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 297:2048–2051. [DOI] [PubMed] [Google Scholar]
- 49.Chen, L.W., L. Egan, Z.W. Li, F.R. Greten, M.F. Kagnoff, and M. Karin. 2003. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 9:575–581. [DOI] [PubMed] [Google Scholar]
- 50.Udagawa, N., N. Takahashi, T. Akatsu, H. Tanaka, T. Sasaki, T. Nishihara, T. Koga, T.J. Martin, and T. Suda. 1990. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA. 87:7260–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DiDonato, J.A., M. Hayakawa, D.M. Rothwarf, E. Zandi, and M. Karin. 1997. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 388:548–554. [DOI] [PubMed] [Google Scholar]