

Abstract
Destruction of the host intestinal epithelium by donor effector T cell populations is a hallmark of graft-versus-host disease (GVHD), but the underlying mechanisms remain obscure. We demonstrate that CD8+ T cells expressing CD103, an integrin conferring specificity for the epithelial ligand E-cadherin, play a critical role in this process. A TCR transgenic GVHD model was used to demonstrate that CD103 is selectively expressed by host-specific CD8+ T cell effector populations (CD8 effectors) that accumulate in the host intestinal epithelium during GVHD. Although host-specific CD8 effectors infiltrated a wide range of host compartments, only those infiltrating the intestinal epithelium expressed CD103. Host-specific CD8 effectors expressing a TGF-β dominant negative type II receptor were defective in CD103 expression on entry into the intestinal epithelium, which indicates local TGF-β activity as a critical regulating factor. Host-specific CD8 effectors deficient in CD103 expression successfully migrated into the host intestinal epithelium but were retained at this site much less efficiently than wild-type host-specific CD8 effectors. The relevance of these events to GVHD pathogenesis is supported by the finding that CD103-deficient CD8+ T cells were strikingly defective in transferring intestinal GVHD pathology and mortality. Collectively, these data document a pivotal role for TGF-β–dependent CD103 expression in dictating the gut tropism, and hence the destructive potential, of CD8+ T cells during GVHD pathogenesis.
Graft-versus-host disease (GVHD) remains the primary complication of clinical BM transplantation (BMT) and a major impediment to the widespread application of this important therapeutic modality. The hallmark of GVHD is the infiltration of donor T lymphocytes into host epithelial compartments (1, 2) of the skin, intestine, and biliary tract (3). GVHD occurs when mature T cells contained in bone marrow inoculum are transplanted into immuno-incompetent hosts. Donor T cells directed to host histocompatibility antigens are activated in secondary lymphoid organs (4) by encountering host APCs (3, 5) or donor-derived APCs which cross-present host alloantigens (6). The newly generated T effector populations then migrate to peripheral host organs (7) and mediate target organ damage.
Intestinal injury is one of the earliest (8) and most common (9) features of GVHD. T cell infiltration into the intestinal epithelium is not only requisite for intestinal pathology during GVHD (4, 8), but also profoundly affects GVHD severity and overall mortality (10). Several lines of evidence point to a key role for CD8+ T cell effector populations (CD8 effectors) in this process. In experimental GVHD models, host-reactive CD8 effectors are associated with the most severe forms of intestinal pathology (11, 12) and predominate at the site of intestinal injury (2, 8). Analyses of clinical biopsies also reveal a predominance of CD8+ T cells within the epithelium at the site of intestinal GVHD lesions (13). The notion that host-reactive CD8 effectors play a central role in GVHD pathogenesis is strongly supported by clinical data demonstrating a reduced incidence of GVHD after BMT when CD8+ T cells are selectively depleted from the BM inoculum (14), but not when CD4+ T cells are eliminated (15).
It is often tacitly assumed that preferential targeting of the intestinal epithelium by CD8 effectors during GVHD reflects a recognition of tissue-specific MHC I–peptide complexes (16). However, recent studies indicate that the gut tropism of T effector populations is in large part determined by the pattern of adhesion molecules expressed on the cell surface (17–19). We previously demonstrated that the T cell integrin CD103 (also known as CD103/β7) plays a critical role in targeting epithelial allografts for destruction by CD8 effectors (20). CD103 confers specificity for the ligand E-cadherin (21, 22), a tissue-restricted molecule selectively expressed by cells composing epithelial layers (23). E-Cadherin is highly expressed by intestinal epithelial cells, which is relevant to GVHD pathogenesis (24). Moreover, this intestinal milieu is associated with high activity levels of TGF-β (25, 26), a cytokine known to promote CD103 expression by CD8+ T cells (27–29). Thus, these data raised the possibility that CD103 expression by CD8+ T cells may dictate the gut-specificity of GVHD pathology.
We used a TCR-transgenic (TCR-Tg) model of GVHD (30) to directly test the hypothesis that TGF-β–dependent CD103 expression promotes selective destruction of the intestinal epithelium by host-specific CD8 effectors (hsCD8eff). We report that CD103 is selectively expressed by hsCD8eff that infiltrate the intestinal epithelium during GVHD and, furthermore, that TGF-β plays a dominant role in the induction of such an expression. We further demonstrate that CD103 expression functions to promote the retention of CD8 effectors in the host intestinal epithelium and that such interactions are central to the development of GVHD-associated intestinal pathology and mortality. These findings document a key role for TGF-β–dependent CD103 expression in targeting the host intestinal epithelium for destruction by CD8+ T cells during GVHD pathogenesis. These data provide insight regarding mechanisms of intestinal GVHD pathology and point to novel targets for therapeutic intervention in this important clinical problem.
Results
Gut-specific expression of CD103 by hsCD8eff
The initial goal of this study was to determine whether CD103 is expressed by hsCD8eff that infiltrate host epithelial compartments during GVHD. To accomplish this, lethally irradiated BALB/c (Ld+) mice received 10 × 106 bone marrow cells (BMC) and 10 × 106 spleen cells (SC) from C57BL/6 (B6) (H-2b) donors plus trace numbers of SC from 2C TCR-Tg mice (30). Peripheral CD8 cells from 2C mice express a TCR with specificity for H-2Ld (31), which is readily detected using an mAb (1B2) directed to the clonotypic 2C TCR (32). Thus, the use of mAbs from CD8 and 1B2 in multicolor FACS analyses allowed the phenotypic attributes of donor CD8 effectors targeting host Ld (host-specific CD8+1B2+ cells) to be monitored in vivo during the course of GVHD.
Before transfer, the vast majority of host-specific CD8+1B2+ cells contained within the inoculum were CD44−CD11aloCD62Lhi, indicating a naive phenotype (unpublished data). After adoptive transfer into lethally irradiated recipients, CD8+1B2+ cells infiltrated host lymphoid (spleen and mesenteric LNs [MLN]) and nonlymphoid (kidney, liver, and intestine) compartments as early as day 5 after BMT and were readily detected by day 7 (Fig. 1). All such cells displayed an effector phenotype (CD44hiCD11ahiCD62Llo; Fig. 1 and not depicted). The migration of donor effector T cells into peripheral compartments correlated with the development of classic manifestations of GVHD such as diarrhea, weight loss, wasting, and mortality (mean survival time [MST] = 7.5 ± 0.3 d, range = 6–21 d).
Figure 1.

hsCD8eff infiltrate diverse host compartments during GVHD. Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with 106 SC from 2C TCR-Tg mice in combination with BMC and SC from B6 (H-2b) mice. Multicolor flow cytometry of cell suspensions with mAb 1B2 to the clonotypic 2C TCR (H-2Ld-specific) was used to identify and characterize host-specific CD8+1B2+ cells. Data shown are dot plots of 1B2 versus CD8 expression by lymphocyte populations (left) and histograms of CD44 expression by gated CD8+1B2+ lymphocytes (right; y axis, events) isolated from various host compartments at day 7 after BMT. Isotype control staining is represented by shaded histograms. Data are representative of six independent experiments.
As shown in Fig. 2, a major subset of hsCD8eff that infiltrated the host intestinal epithelium during GVHD expressed CD103, the percentage of which progressively increased over time. Thus, at day 7 after BMT, 8.4 ± 0.6% (n = 6) of the CD8+1B2+ T cells expressed CD103; this percentage increased to 31 ± 12.7% (n = 3) by day 10, to 62 ± 6.4% (n = 3) by day 15, and to 71.5 ± 5.5% (n = 2) by day 21. However, hsCD8eff that infiltrated other host compartments, including the spleen, kidney, liver, and MLN, expressed negligible levels of CD103 at all time points examined (Fig. 2). The mean percentage ± SE of CD8+1B2+ T cells in the spleen that expressed CD103 was 0.36 ± 0.2%, 1.2 ± 0.87%, 2.57 ± 1.1%, and 0.8 ± 0.2% at days 7, 10, 14, and 21, respectively (P < 0.05 for all values compared with the intestine). In experiments not shown, nontransgenic CD8 effectors (1B2−CD8+CD44hi cells) that infiltrated the host intestine also selectively expressed CD103, demonstrating that gut-specific expression of CD103 is not unique to TCR-Tg CD8 cells. Thus, these data revealed that CD103 is selectively expressed by hsCD8eff infiltrating the intestinal epithelium during the course of GVHD.
Figure 2.

Gut-specific expression of CD103 by hsCD8eff during GVHD. Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with 106 SC from 2C TCR-Tg mice in combination with BMC and SC from B6 (H-2b) mice. At the indicated time points, lymphocytes were isolated from the various host organs and subjected to four-color flow cytometry as described in the legend to Fig. 1. Data shown are histograms of CD103 expression by gated host-specific CD8+1B2+ lymphocytes (y axis, events). Isotype control staining is represented by shaded histograms. Data shown are representative of two to six independent experiments. Numbers (top) show the mean percentage (±SE) of intestinal CD8+ T cells that expressed CD103.
Gut-specific expression of CD103 by hsCD8eff is dependent on TGF-β activity
In vitro studies (28) indicate that the expression of CD103 by CD8 effectors is regulated by active TGF-β, and mice overexpressing a dominant negative type II receptor mutation targeted to T cells (33) are deficient in CD103 expression after in vitro activation in the presence of TGF-β (unpublished data). To determine whether CD103 expression by gut-infiltrating hsCD8eff is similarly regulated by TGF-β activity, we used 2C TCR-Tg mice expressing a dominant negative TGF-β type II receptor (DNR; reference 33). Thus, the substitution of 2C-DNR cells for wild-type 2C cells in our model allowed an assessment of the contribution of TGF-β signaling to the induction of CD103 expression by hsCD8eff infiltrating the intestinal epithelium. In these experiments, lethally irradiated BALB/c recipients were adoptively transferred with B6 BM and SC, as well as 2C-DNR cells. Similar to wild-type 2C, 2C-DNR cells migrated into the intestinal epithelium by day 7 after BMT and displayed an effector phenotype as indicated by up-regulation of CD44 (Fig. 3 A). However, in contrast to their wild-type counterparts (Fig. 2), 2C-DNR cells that migrated into the intestinal epithelium were completely devoid of CD103 expression (Fig. 3 A).
Figure 3.

Gut-specific expression of CD103 by hsCD8eff is dependent on TGF-β activity. (A) Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with 106 SC from either 2C TCR-Tg mice expressing 2C-DNR in combination with BMC and SC from B6 (H-2b) mice. Recipients were killed at day 7 after BMT and lymphocytes infiltrating the intestinal epithelium were subjected to a three-color FACS analysis. Data shown are histograms of CD44 (left) or CD103 (right) expression by gated host-specific CD8+1B2+ (y axis, events). (B) Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with an equal mixture (0.5 × 106) of SC from wild-type 2C (Thy1.1+) and 2C-DNR (Thy1.1−) mice in combination with BMC and SC from B6 (H-2b) mice. Lymphocytes infiltrating the intestinal epithelium were isolated at the indicated time points and subjected to a three-color FACS analysis. (left) dot plot shows Thy1.1 and 1B2 expression by gated CD8+ lymphocytes. (right) histograms show the percentage of CD103 expression by gated host-specific CD8+1B2+ cells of either a wild-type 2C (top, Thy1.1+1B2+ cells) or 2C-DNR (bottom, Thy1.1−1B2+ cells) origin (y axis, events). Isotype control staining is indicated by shaded histograms. Results are representative of three independent experiments.
To directly compare CD103 expression by wild type with 2C-DNR cells in the same mouse, equal numbers (0.5 × 106) of each cell population were transferred together into lethally irradiated BALB/c recipients. To discriminate between the two populations of TCR-Tg cells, wild-type 2C cells expressing the congenic marker Thy1.1 were generated, thus allowing the identification of wild-type 2C (CD8+1B2+Thy1.1+) and 2C-DNR (CD8+1B2+Thy1.1−) cells using four-color FACS analyses. A progressively increasing subset of wild-type 2C cells infiltrating the intestinal epithelium up-regulated CD103 (Fig. 3 B, top). In contrast, a notably lower percentage of 2C-DNR cells expressed CD103 at the same site at all time points examined (Fig. 3 B, bottom). At day 15 after BMT, the mean percentage (± SE) of CD103-expressing cells among wild-type 2C was 43.6 ± 12.4% (n = 3) as compared with 22.2 ± 8.7% (n = 3) for 2C-DNR cells (P = 0.01). Thus, these data support an important role for TGF-β activity in regulating CD103 expression by hsCD8eff that infiltrate the host intestinal epithelium.
CD103 expression promotes the retention of hsCD8eff in the intestinal epithelium
The known ligand of mouse CD103 is the epithelial cell-specific ligand, E-cadherin (21), and CD103 is known to promote adhesion to E-cadherin–expressing cells (34). Thus, these data suggested that CD103 expression promotes the retention of CD103+CD8+ effectors in the host intestinal epithelium during GVHD (Fig. 2). To test this hypothesis, 2C mice were crossed onto the CD103−/− background, and the resulting CD103−/− 2C effectors were compared with their wild-type counterparts for the capacity to migrate into the host intestinal epithelium. In these experiments, equal numbers (0.5 × 106) of wild-type 2C (Thy1.1+1B2+) cells and CD103−/− 2C (Thy1.1−1B2+) cells, combined with B6 BM and SC, were transferred into lethally irradiated BALB/c hosts (Thy1.2+/Thy1.2+). Host organs were harvested at days 6, 12, 21, and 28 after transfer, and the infiltrating lymphocytes were subjected to four-color FACS analyses. Before transfer, CD8+ T cells derived from both donor populations displayed a naive phenotype (unpublished data), but after transfer both populations rapidly acquired a CD62LloCD11ahi phenotype characteristic of T effector cells (see Fig. 5, A and B). Furthermore, wild-type and CD103−/− hsCD8eff secreted comparable levels of the effector cytokines TNF-α (see Fig. 5 C) and IFN-γ (see Fig. 5 D).
Figure 5.
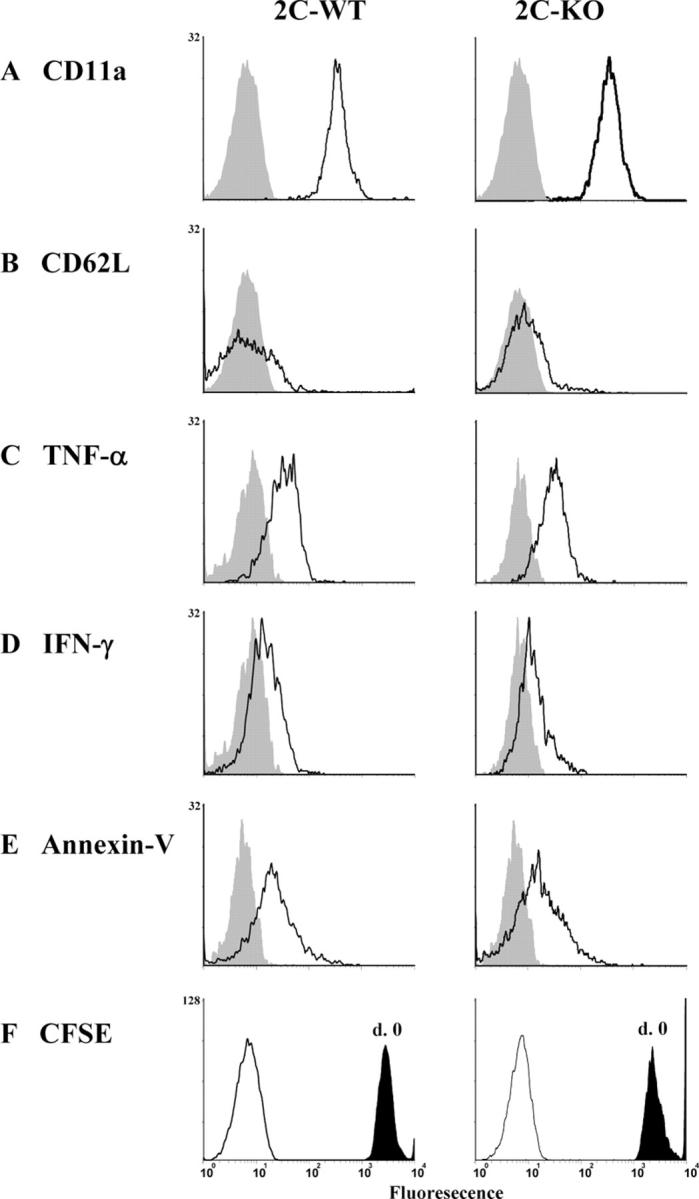
Properties of wild-type and CD103−/− host-specific CD8 cells that infiltrate the host intestinal epithelium during GVHD. (A–E) Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with an equal mixture (0.5 × 106) of SC from wild-type 2C (Thy1.1+) and 2C-CD103−/− (Thy1.1−) mice in combination with BMC and SC from B6 (H-2b) mice. Recipients were killed at the indicated time points and lymphocytes infiltrating various host organs were subjected to a three-color FACS analysis. Data shown are the expression of CD11a (A), CD62L (B), TNF-α (C), IFN-γ (D), and Annexin-V (E) by wild-type (left) or CD103−/− (right) host-specific CD8 cells (gated Thy1.1+1B2+ or Thy1.1−1B2+ cells, respectively) isolated from the intestinal epithelium at day 6. Shaded histograms represent isotype control staining. Results are representative of two to three independent experiments. (F) Equal numbers of CFSE-labeled splenic T cells from wild-type (Thy1.1+) and CD103−/− (Thy1.1−) 2C mice were transferred into lethally irradiated BALB/c mice in combination with BMC and SC from B6 (H-2b) mice. Lymphocytes were harvested from the intestine at day 6 after BMT and analyzed by four-color flow cytometry. Data shown are CSFE fluorescence by wild-type (left) or CD103−/− (right) host-specific CD8 cells (gated Thy1.1+1B2+ or Thy1.1−1B2+ cells, respectively) isolated from the intestinal epithelium. Shaded histograms show CSFE fluorescence at day 0. Y axis, events.
As shown in Fig. 4 A (top), CD103−/− hsCD8eff composed 22% of the total CD8 T cells infiltrating the host intestinal epithelium at day 6 after transfer. This percentage was comparable to that of the wild-type hsCD8eff (Fig. 4 A), indicating that CD103 expression is not required for the initial migration of hsCD8eff into the intestinal epithelium. However, the numbers of wild-type hsCD8eff increased dramatically over time compared with their CD103−/− counterparts at later time points (i.e., the ratio of wild type/CD103−/− hsCD8eff increased to 2:1 by day 21 and to 4:1 by day 28). The selective retention of wild-type versus CD103−/− hsCD8eff was also manifest in the spleen by day 28 after transfer (Fig. 4 A), but not in any of the nonlymphoid organs examined including the kidney, lung, and liver (Fig. 4 B). Experiments in which the wild-type 2C cells and CD103−/− 2C cells were transferred separately yielded similar results (unpublished data).
Figure 4.

CD103 expression promotes retention of hsCD8eff in the host intestinal epithelium. Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with an equal mixture (0.5 × 106) of SC from wild-type 2C (Thy1.11) and 2C-CD1032/2 (Thy1.12) mice in combination with BMC and SC from B6 (H-2b) mice. Lymphocytes infiltrating host organs were isolated at the indicated time points and subjected to a three-color FACS analysis. (A) Dot plots of Thy1.1 versus 1B2 expression by gated CD81 lymphocytes isolated from the intestinal epithelium (top) or spleen (bottom) at the indicated time points. (B) Dot plots of Thy1.1 versus 1B2 expression by gated CD8+ lymphocytes isolated from the indicated organs at day 28 after BMT. Numbers in quadrants denote the corresponding percentages among gated CD8+ T cells.
The data in Fig. 4 are consistent with a key role for CD103 in promoting the retention of hsCD8eff within the intestinal epithelium, but do not exclude the reasonable possibilities that CD103 expression may promote the proliferation of hsCD8eff at this site and/or protect such cells from apoptosis. To address the former possibility, equal numbers (0.5 × 106) of wild-type 2C (Thy1.1+1B2+) cells and CD103−/− 2C (Thy1.1−1B2+) cells were carboxy-fluorescein diacetate succinimidyl ester (CFSE) labeled and transferred into lethally irradiated BALB/c hosts (Thy1.2+/Thy1.2+) in combination with B6 BM and SC. Lymphocytes infiltrating the host intestine and spleen were harvested at days 3 and 6 after BMT and subjected to four-color FACS analyses. CD8+1B2+ were not detectable in the host intestine or spleen at day 3 after transplantation but were abundant in both compartments by day 6 (unpublished data). As shown in Fig. 5 F, wild-type and CD103−/− hsCD8eff that infiltrated the host intestinal epithelium demonstrated comparable levels of CFSE expression, indicating similar levels of cell division. It is important to note, however, that the CFSE dilution observed in this experiment potentially reflects the homeostatic- and/or alloantigen-driven expansion of CD8 cells occurring in the peripheral environment before the migration of CD8 cells into the host epithelium.
To determine whether CD103 expression protects hsCD8eff from apoptosis, lymphocytes infiltrating the intestinal epithelium were stained with Annexin-V, which detects cells in the early stages of apoptosis (35). As shown in Fig. 5 E, the frequency of CD103−/− hsCD8eff that bound Annexin-V was comparable with that of wild-type hsCD8eff, indicating equivalent apoptosis in the two cell populations. Thus, we find no evidence that CD103 expression promotes either the expansion or survival of hsCD8eff within the intestinal epithelium.
CD103 expression promotes destruction of the host intestinal epithelium by CD8+ T cells
Hosts transferred with wild-type 2C cells experienced more rapid mortality (MST = 7.5 ± 0.3 d, n = 4) than those receiving CD103−/− 2C cells (MST = 24.7 ± 5.9 d, n = 4) or 2C-DNR cells (MST = 16.3 ± 2.4 d, n = 4; unpublished data). These data suggested that CD103 expression by hsCD8eff is causally related to the development of GVHD pathology and mortality. However, the interpretation of survival data in this model was confounded by the presence of donor CD4+ T cells (derived from the initial B6 SC inoculum) which potentially contribute to GVHD pathogenesis (8). Furthermore, it was not clear that pathology mediated by a clonal population of CD8+ T cells directed to a single MHC–peptide complex would be representative of a normal (polyclonal) response.
To unambiguously assess the impact of CD103 expression on GVHD mediated by polyclonal CD8+ T cells, two groups of BALB/c mice were lethally irradiated and adoptively transferred with CD8-enriched SC (<0.5% CD4 contamination) obtained from either wild-type or CD103−/− donors (polyclonal B6 background) in combination with B6 BM. An additional control group received B6 BMC alone. Hosts from each group were monitored for survival, and intestinal specimens were analyzed histologically. As shown in Fig. 6 A, recipients of CD103−/− CD8-enriched T cells enjoyed dramatically prolonged survival times (MST > 48.6 ± 6.5 d, n = 10) compared with recipients of wild-type cells (MST = 8.2 ± 0.8 d, n = 11; P < 0.001). Indeed, recipients of CD103−/− CD8 cells exhibited mortality kinetics similar to recipients of BMC alone. Furthermore, as shown in Fig. 6 B, histopathologic evaluation of intestinal specimens from mice transferred with wild-type cells revealed more pronounced destruction than that seen in cohorts receiving CD103−/− cells (e.g., villus atrophy, architectural distortion, lymphocytic infiltration, and the appearance of apoptotic bodies). A quantitative analysis of the latter in the two groups revealed a fourfold increase in the number of apoptotic bodies in recipients of wild-type cells compared with recipients of CD103−/− cells. Analyses of liver pathology revealed a mild to moderate cellular infiltrate in both groups (Fig. 6 C).
Figure 6.
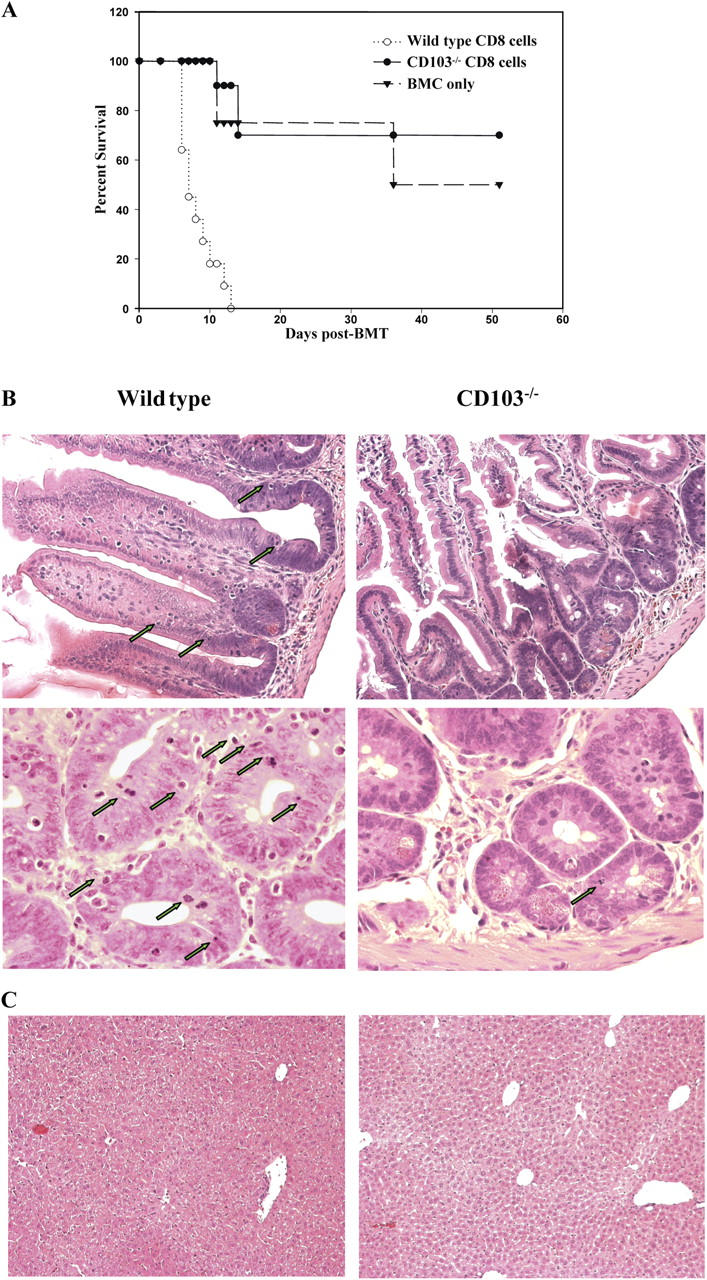
CD103 expression promotes destruction of the host intestinal epithelium by CD8+ T cells during GVHD. Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with 10 × 106 B6 BMC alone or in combination with 10 × 106 CD8-enriched SC from either wild-type or CD103−/− mice on the B6 (H-2b) background. (A) Survival of recipients of B6 BMC alone (dashed line, n = 4), or in combination with CD8+ T cells from wild-type (dotted line; MST = 8.2 ± 0.8 d, n = 11) or CD103−/− (continuous line; MST > 48.6 ± 6.5 d, n = 10) donors (P < 0.001). (B) Representative H&E sections of intestinal specimens at day 14 after BMT from recipients of wild-type (left) or CD103−/− (right) CD8+ T cells, at low (top, 20×) and high (bottom, 40×) magnification. Arrows denote apoptotic bodies. (C) Representative H&E sections of liver specimens at day 14 after BMT in recipients of wild-type (left) or CD103−/− (right) CD8+ T cells (magnification, 20×).
As shown in Fig. 7, FACS analyses of donor-origin lymphocytes infiltrating the intestinal epithelium at day 14 after BMT in recipients of wild-type lymphocytes revealed a vast predominance of CD8 effectors (80–90% of infiltrating lymphocytes), ∼40% of which expressed CD103. Note that CD103 expression by gut-infiltrating lymphocytes was almost entirely confined to the CD8 subset (Fig. 7). There was a corresponding twofold decrease in the abundance of CD103−/− cells in the intestinal epithelium as compared with wild-type counterparts (unpublished data). Thus, these data are consistent with a key role for CD103 in promoting both the retention and pathogenic potential of polyclonal host-reactive CD8 effectors that infiltrate the host intestinal epithelium during GVHD.
Figure 7.

Expression of CD103 by polyclonal CD8 effectors infiltrating the host intestinal epithelium during GVHD. Lethally irradiated BALB/c (H-2d) hosts were adoptively transferred with 10 × 106 B6 BMC and 10 × 106 CD8-enriched SC from either wild-type (right) or CD103−/− (left) B6 (H-2b) mice. Lymphocytes were isolated from the host intestinal epithelium at the indicated times and subjected to four-color FACS analyses using FITC-conjugated mAb to H-2Kb in combination with anti–CD8-PerCP, anti–CD103-PE, and anti–CD44-APCs. Data shown are dot plots of CD103 versus CD8 expression by gated donor (H-2Kb+) lymphocytes infiltrating the host intestinal epithelium. Donor CD8+ T cells were uniformly CD44hi, consistent with a CD8 effector phenotype (not depicted). Results are representative of two independent experiments. Numbers in quadrants denote the percentage of total cells within that area.
Discussion
The infiltration and destruction of the host intestinal epithelium by donor T cells is a hallmark of GVHD. Although the afferent pathways underlying the development of GVHD are increasingly well defined (3, 36), the critical efferent pathways by which T effector populations attack and destroy host tissues remain obscure. The present study documents a key role for CD103 in promoting intestinal GVHD pathology mediated by CD8+ T cells. These data provide insight into efferent pathways of GVHD and identify novel targets for therapeutic intervention in this important clinical problem.
In our studies, a TCR-Tg GVHD model was used to monitor the acquisition of CD103 expression by hsCD8eff during the course of GVHD. These studies revealed that CD103 is selectively expressed by hsCD8eff that infiltrate the host intestinal epithelium. Although hsCD8eff populated diverse host compartments including the spleen, kidney, liver, and lymph nodes, only those that infiltrated the intestinal epithelium expressed notable levels of CD103 (Fig. 2). The overall frequency of CD103+ hsCD8eff in the intestine progressively increased with time concomitant with the development of classic GVHD pathology at this site.
That a causal relationship exists between CD103+ hsCD8eff and the destruction of the intestinal epithelium was demonstrated by the results of transfer experiments using CD8+ T cells from CD103 knockout mice. These data revealed that CD103 expression is required for the efficient transfer of GVHD mortality and intestinal pathology by CD8+ T cells (Fig. 6). This defect did not reflect an inherent incompetence of CD8 cells from CD103 knockout mice because CD103−/− hsCD8eff are activated in response to host alloantigens and traffic to the intestinal epithelium as efficiently as their wild-type counterparts (Fig. 4 A; and Fig. 5, A–C). Moreover, recipients of CD103−/− CD8+ T cells developed mild liver pathology similar to cohorts transferred with wild-type CD8+ T cells (Fig. 6 C). These data are consistent with our prior observation that CD8+ T cells in CD103−/− mice mount normal effector responses to alloantigen, traffic normally to peripheral sites of inflammation, and reject skin allografts with kinetics identical to wild-type controls (20).
The present data document a dominant role for the cytokine TGF-β in regulating CD103 expression by CD8+ T cells during the course of GVHD. Thus, hsCD8eff expressing DNR differentiated to effector status and efficiently migrated into the host epithelium, but were strikingly defective as compared with wild-type hsCD8eff in the capacity to up-regulate CD103 (Fig. 3 C). It is unclear whether the residual CD103 expression observed for CD8 cells expressing the DNR mutation (Fig. 3 C) reflects an inherent leakiness of this mutation or, alternatively, that additional factors operate to control CD103 expression by CD8 effectors during GVHD. We favor the former possibility because in vitro studies indicate that residual CD103 expression by DNR CD8+ T cells is completely abrogated by neutralizing anti–TGF-β mAb (unpublished data). It is conceivable, however, that the factors operating to control CD103 expression in vitro are distinct from in vivo factors. Unfortunately, our efforts to use neutralizing anti–TGF-β mAb in vivo resulted in the accelerated mortality of the mice because of generalized lymphocytic infiltration probably resulting from the loss of the systemic immunosuppressive activity of TGF-β required for appropriate T cell homeostasis (unpublished data).
The source of TGF-β activity that drives CD103 expression by hsCD8eff during GVHD is unclear. Given the gut-specific localization of CD103+ hsCD8eff (Fig. 2), it is likely that CD103 expression is controlled by TGF-β activity present locally in the intestinal milieu. Consistent with this possibility, intestinal epithelial cells are known to secrete TGF-β (26, 37); furthermore, local inflammation in the intestine is associated with increased levels of intestinal TGF-β mRNA (38) and protein (25). An important implication of these data is that blocking local (intestinal) TGF-β may provide an effective means for therapeutic intervention in the CD103 pathway. However, the present data do not exclude the possibility that CD8 effectors encounter TGF-β activity and are induced to express CD103 before entry into the intestinal epithelium, but are present in extra-intestinal compartments at levels below the limits of detection for FACS-based assay systems.
Our finding that CD103 expression is required for the efficient accumulation of hsCD8eff in the host intestinal epithelium provides a plausible mechanistic basis for the striking CD103 dependence of GVHD gut pathology. Thus, hsCD8eff that were unable to express CD103 (CD103−/− background) successfully migrated into the host intestinal epithelium but were retained there less efficiently than wild-type counterparts (Fig. 4 A). These data are consistent with the findings of Teshima et al. (39), who recently demonstrated that intestinal GVHD pathology mediated by CD8+ T cells requires, at least in part, recognition of MHC–peptide complex expressed within the target epithelium. Given that CD103 recognizes E-cadherin, which is selectively expressed E-cadherin within the intestinal epithelium (24), the simplest explanation for these data is that CD103 functions to promote the adhesion of hsCD8eff in the gut epithelium. Consistent with this possibility, CD103 is known to promote the adhesion of CD103-expressing intraepithelial lymphocytes to mucosal epithelial cells (34).
Theoretically, the deficient accumulation of CD103−/− hsCD8eff in the gut epithelium could also reflect a role for CD103 in promoting the intraepithelial expansion of CD8 effectors or, conversely, in conferring the resistance of such cells to apoptotic events. However, recent studies indicate that the engagement of CD103 does not notably promote T cell proliferation in vitro (unpublished data) and the data depicted here (Fig. 5 F) demonstrate that the expansion of CD103−/− hsCD8eff infiltrating the intestine during GVHD is comparable with that of wild-type hsCD8eff. In addition, there were no detectable differences in apoptosis between wild-type and CD103−/− hsCD8eff at this site (Fig. 5 E). The simplest explanation for these data is that CD103 critically functions to promote the retention of hsCD8eff within the intestinal epithelium during GVHD. Precisely how such retention promotes GVHD pathology and mortality remains to be determined.
CD8 effectors are associated with a variety of tissue-specific immune phenomena including not only intestinal GVHD pathology, but also organ-specific autoimmune disease (40), differential immunogenicity of organ allografts (41), and virus-associated immune pathology (42–44). It is often tacitly assumed that such processes reflect T cell recognition of MHC I–peptide complexes uniquely expressed within the target tissues. The present data challenge this concept by demonstrating that TGF-β–dependent CD103 expression can be the salient factor limiting intestinal GVHD pathology. Thus, even though the 2C TCR confers specificity for H-2Ld associated with the p2Ca peptide (45) expressed in the host intestinal epithelium, CD103−/− CD8+ T cells expressing the 2C TCR did not inflict considerable intestinal epithelial damage. Furthermore, tissue destruction mediated by wild-type CD8 effectors expressing the 2C TCR was confined to the intestinal epithelium (Fig. 6, B and C) even though H-2Ld/p2Ca is ubiquitously expressed by all tissue compartments (45, 46).
Based on the present findings, we propose an alternative model to explain the gut specificity of GVHD pathology. After migration into the host intestinal epithelium, hsCD8eff encounter local TGF-β activity and are induced to express CD103. CD103 expression, in turn, endows CD8 effectors with the capacity to engage E-cadherin, which is abundantly expressed by cells composing epithelial compartments (47). As a consequence, hsCD8eff expressing CD103 are efficiently retained within the intestinal epithelium and accumulate over time, eventually leading to the destruction of the underlying epithelium. This process apparently is not required for the destruction of other epithelial compartments because recipients of CD103−/− cells eventually develop severe alopecia and dermatologic injury (unpublished data), as well as liver pathology comparable to that of wild-type counterparts (Fig. 6 C). As discussed above, the gut-specific induction of CD103 expression can be explained by the unique capacity of the intestinal epithelium for the local production and/or processing of TGF-β (25).
Intraepithelial accumulation of CD8 effectors has the potential to promote selective destruction of the underlying epithelium either directly by the cytolysis of adjacent epithelial cells or by the recruitment of additional effector populations via the production of chemokines and/or cytokines (48–50). However, a recent study indicated that intestinal GVHD pathology is mediated by noncytolytic mechanisms; e.g., the targeted disruption of perforin or FasL pathways in mouse GVHD models does not abrogate intestinal injury (51). Based on these data, we postulate that host-specific CD103+CD8+ effectors in the gut epithelium act as sentinels that recruit other effector populations with pathogenic potential to the site, i.e., TNF-α–producing CD4+ T cells (52) and/or monocytes (53). Consistent with this possibility, the total number of non-CD8 cells infiltrating the intestine in mice transferred with wild-type hsCD8eff was increased compared with that of mice transferred with CD103−/− hsCD8eff (mean [× 106] ± SE = 0.44 ± 0.14 for recipients of wild-type cells vs. 0.25 ± 0.06 for recipients of CD103−/− cells at 2 wk after BMT; n = 4).
In summary, our results indicate that TGF-β−dependent CD103 expression plays a pivotal role in promoting destruction of the host intestinal epithelium by CD8+ T cells during GVHD pathogenesis. With regard to clinical applications, it will be important to determine whether anti-CD103 antibody has the same beneficial effect as the disruption of TGF-β signaling or CD103 gene expression, and whether such intervention can prevent GVHD pathology without compromising the beneficial graft-versus-leukemia effect (54). It will also be important to determine whether similar pathways are operative in other CD8-dependent immune phenomena characterized by tissue-specific immune pathology, especially organ-specific autoimmune disorders and the predisposition of the gut to immune inflammation.
Materials and Methods
Mice.
2C TCR-Tg (H-2b) mice (31) that express a TCR with specificity for an 8-mer peptide (p2Ca) in association with H-2Ld (46) were originally obtained from Ted Hansen (Washington University School of Medicine, St. Louis, MO). 2C mice were routinely maintained by backcrossing heterozygous 2C males with B6 females and screening the offspring for expression of the clonotypic 2C TCR by a FACS of peripheral blood using the 1B2 mAb. Mice expressing a DNR (33) and 2C mice bred onto the DNR background (2C-DNR) were used in some experiments. B6 (H-2b) and BALB/c (H-2d) mice were purchased from the Jackson Laboratory. 2C CD103−/− mice were generated by breeding 2C (H-2b) and CD103−/− B6 (H-2b) mice (55), followed by backcrossing the 2C CD103+/− F1 generation with the CD103−/− parental strain to generate 2C CD103−/− mice, as identified by surface staining for the TCR transgene and PCR for the homozygous CD103−/− mutation. Wild-type congenic 2C Thy1.1+ mice were obtained by interbreeding 2C Thy1.2+/Thy1.2+ and B6 Thy1.1+/Thy1.1+ animals to generate a 2C Thy1.1+/Thy1.2+ F1 generation. All mice were maintained under specific pathogen-free conditions in the animal facility at the University of Maryland, Baltimore. All animal studies were approved by the Institutional Review Board.
Antibodies.
1B2 mAb (anti-clonotypic 2C TCR, mouse IgG1; reference 32) was obtained from hybridoma culture supernatant. Biotinylated rat anti–mouse IgG1 was purchased from Caltag. Directly conjugated anti–mouse antibodies used include CD8-FITC, CD8-PerCP, Thy1.1-PerCP, Thy1.2-PE, CD44-FITC, CD44-PE, CD62L-PE, M290 (anti-CD103)-PE, CD11a-PE, CD25-PE, IFN-γ–PE, TNF-α–PE, and the species- and isotype-matched controls (all purchased from BD Biosciences).
Adoptive transfer and BMC.
Lethally irradiated (8 Gy, 137Cs source) BALB/c (H-2d) mice were reconstituted within 4–6 h by a single 0.5-ml intravenous inoculum containing 10 × 106 B6 BMC and 10 × 106 B6 SC with either 106 2C SC or 106 2C-DNR cells. In some experiments, equal numbers (0.5 × 106) of wild-type Thy1.1+ 2C and Thy1.1− 2C-DNR SC were mixed together and adoptively transferred in combination with B6 BMC and SC into lethally irradiated BALB/c hosts. For adoptive transfer experiments to determine the role of CD103 in lymphocyte migration to the intestine, equal numbers (0.5 × 106) of wild-type Thy1.1+ 2C SC and CD103−/− Thy1.1− 2C SC were mixed and adoptively transferred into a group of lethally irradiated BALB/c hosts. In some experiments, 106 wild-type or CD103−/− 2C cells were separately transferred into lethally irradiated BALB/c mice in combination with B6 BM and SC.
To examine the survival and organ pathology in recipients of CD103-deficient CD8 cells, two groups of lethally irradiated BALB/c hosts received 10 × 106 B6 BMC and either 10 × 106 CD8-enriched B6 SC or 10 × 106 CD8-enriched B6 CD103−/− SC. In these experiments, donor SC were enriched for naive CD8+ T cells by treatment with an mAb to CD4 (RL172.4), followed by incubation in 1/10 Low-Tox M rabbit complement (Accurate Chemical and Scientific). The resulting cell suspensions contained <0.5% CD4+ T cells. To ensure that equivalent numbers of CD8 cells were transferred in different experiments, FACS analysis using anti-CD8 and TCR-α/β was performed on each preparation to assess the degree of enrichment for CD8+ T cells. Additional experiments were performed by transferring 10 × 106 B6 BMC alone. Mice were randomly grouped before and after irradiation and were given acidified drinking water to prevent infection. Control irradiated untreated mice were also included in each experiment.
Lymphocyte isolation.
Lymphocytes infiltrating the intestinal epithelium during GVHD were isolated as previously described (56) with modifications. The small intestine was flushed with PBS. After removal of the Peyer's patches and fat, the intestine was divided longitudinally and cut into 2-mm sections. The mucosa was scraped and dissociated by mechanical disruption on a stirring platform for 15 min in RPMI 1640 with 10% FCS and 1 mM dithioerythritol. Tissue debris and cell aggregates were removed by passage over a glass wool column in RPMI 1640 with 10% FCS. The lymphocytes were obtained by centrifugation on Lympholyte-M (Cedarlane Laboratories Limited), and the cells were suspended in complete medium.
Lymphocytes were isolated from the kidney as previously described (29). In brief, the kidney was minced, and the resulting fragments were incubated for 30 min in medium containing collagenase (Worthington Biochemical), soybean trypsin inhibitor (Sigma-Aldrich), and DNase (Roche). Lymphocytes were isolated by centrifugation on Lympholyte-M. Cells were isolated from the MLN and spleens of recipients by mincing with forceps and passing the resulting cell suspension through nylon mesh with a 100-μm pore size. Lung- and liver-infiltrating lymphocytes were harvested as previously described (57) with modifications. After perfusion with PBS, liver and lung tissue was minced and incubated with stirring at 37°C for 30 min in medium containing collagenase, soybean trypsin inhibitor, and DNase. Lymphocytes were isolated by centrifugation on Lympholyte-M. Lymphocyte populations were washed twice in FACS buffer before antibody staining and FACS analyses.
Histology.
Host organs were harvested at the designated times, fixed in 10% buffered formalin, and embedded in paraffin. 6-μm sections were stained with hematoxylin and eosin (H&E) and blindly analyzed.
FACS analyses.
For three-color FACS analyses, harvested cells were stained with PerCP-conjugated mAb to CD8b in combination with FITC- and PE-conjugated mAbs to markers of interest. For four-color FACS analyses, cells were stained with anti–CD8a-FITC and anti–Thy1.1-PerCP in combination with PE- and APC-conjugated antibodies. Species and isotype-matched mAbs of irrelevant specificity were used as controls for nonspecific fluorescence. After staining, cells were fixed with 0.5% paraformaldehyde and analyzed using a FACSCalibur (Becton Dickinson).
For intracellular cytokine staining, harvested cells were activated with PMA and ionomycin, followed by the addition of 4 μl/ml monensin (Golgi Stop; BD Biosciences) for an additional 6 h at 37°C. Cells were washed and resuspended in staining buffer followed by surface staining. Cells were then washed and resuspended in Cytoperm/Cytofix solution (BD Biosciences) for 20 min on ice and subsequently incubated on ice for 30–60 min with appropriate dilutions of anti-cytokine or isotype control antibodies. For Annexin-V staining, lymphocytes were stained in FACS buffer at 4°C with FITC-conjugated mAb to CD8b and APC-conjugated mAb to Thy1.1. Cells were then washed and incubated in Annexin-V binding buffer with PE/Annexin-V at 1:20 dilution and 7-amino-actinomycin D (to exclude nonviable cells) for 15 min at room temperature. The cells were washed, resuspended in Annexin-V binding buffer, and analyzed within 1 h.
Lymphocyte populations were gated by forward scatter/side scatter analysis to exclude monocytes and debris. WinMDI 2.8 software (http://facs.scripps.edu/software.html) was used for the analysis and graphic display of flow cytometry data. The percentage of positive cells for a given marker was based on cutoff points chosen to exclude >99% of the negative control population.
Statistical analyses
Data are expressed as mean ± SEM. To assess statistical significance, Student's t tests were performed using SigmaPlot 2000 software (SPSS Inc.). P < 0.05 was considered statistically significant.
Acknowledgments
We are indebted to Drs. Stephen T. Bartlett, Jan Cerny, Donna L. Farber, and Robert G. Fenton for their critical reading of this manuscript and helpful comments.
G.A. Hadley was supported by grant AI36532 from the National Institutes of Health.
The authors have no conflicting financial interests.
Abbreviations used: B6, C57BL/6; BMC, BM cells; BMT, BM transplantation; CD8 effectors, CD8+ T cell effector populations; CFSE, carboxy- fluorescein diacetate succinimidyl ester; DNR, dominant negative TGF-β type II receptor; GVHD, graft-versus-host disease; H&E, hematoxylin and eosin; hsCD8eff, host-specific CD8 effectors; MLN, mesenteric LNs; MST, mean survival time; SC, spleen cells; TCR-Tg, TCR transgenic.
References
- 1.Nakhleh, R.E., D.C. Snover, S. Weisdorf, and J.L. Platt. 1989. Immunopathology of graft-versus-host disease in the upper gastrointestinal tract. Transplantation. 48:61–65. [DOI] [PubMed] [Google Scholar]
- 2.Schattenfroh, N.C., R.A. Hoffman, S.A. McCarthy, and R.L. Simmons. 1995. Phenotypic analysis of donor cells infiltrating the small intestinal epithelium and spleen during graft-versus-host disease. Transplantation. 59:268–273. [PubMed] [Google Scholar]
- 3.Ferrara, J.L., and H.J. Deeg. 1991. Graft-versus-host disease. N. Engl. J. Med. 324:667–674. [DOI] [PubMed] [Google Scholar]
- 4.Murai, M., H. Yoneyama, T. Ezaki, M. Suematsu, Y. Terashima, A. Harada, H. Hamada, H. Asakura, H. Ishikawa, and K. Matsushima. 2003. Peyer's patch is the essential site in initiating murine acute and lethal graft-versus-host reaction. Nat. Immunol. 4:154–160. [DOI] [PubMed] [Google Scholar]
- 5.Shlomchik, W.D., M.S. Couzens, C.B. Tang, J. McNiff, M.E. Robert, J. Liu, M.J. Shlomchik, and S.G. Emerson. 1999. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 285:412–415. [DOI] [PubMed] [Google Scholar]
- 6.Teshima, T., and J.L. Ferrara. 2002. Understanding the alloresponse: new approaches to graft-versus-host disease prevention. Semin. Hematol. 39:15–22. [DOI] [PubMed] [Google Scholar]
- 7.von Andrian, U.H., and C.R. Mackay. 2000. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 343:1020–1034. [DOI] [PubMed] [Google Scholar]
- 8.Guy-Grand, D., and P. Vassalli. 1986. Gut injury in mouse graft-versus-host reaction. Study of its occurrence and mechanisms. J. Clin. Invest. 77:1584–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takatsuka, H., T. Iwasaki, T. Okamoto, and E. Kakishita. 2003. Intestinal graft-versus-host disease: mechanisms and management. Drugs. 63:1–15. [DOI] [PubMed] [Google Scholar]
- 10.Duffner, U., B. Lu, G.C. Hildebrandt, T. Teshima, D.L. Williams, P. Reddy, R. Ordemann, S.G. Clouthier, K. Lowler, C. Liu, et al. 2003. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp. Hematol. 31:897–902. [DOI] [PubMed] [Google Scholar]
- 11.Mowat, A.M., and M.V. Felstein. 1990. Experimental studies of immunologically mediated enteropathy. V. Destructive enteropathy during an acute graft-versus-host reaction in adult BDF1 mice. Clin. Exp. Immunol. 79:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mowat, A.M., M.V. Felstein, A. Borland, and D.M. Parrott. 1988. Experimental studies of immunologically mediated enteropathy. Development of cell mediated immunity and intestinal pathology during a graft-versus-host reaction in irradiated mice. Gut. 29:949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy, J., J.L. Platt, and D.J. Weisdorf. 1993. The immunopathology of upper gastrointestinal acute graft-versus-host disease. Lymphoid cells and endothelial adhesion molecules. Transplantation. 55:572–578. [DOI] [PubMed] [Google Scholar]
- 14.Champlin, R., W. Ho, J. Gajewski, S. Feig, M. Burnison, G. Holley, P. Greenberg, K. Lee, I. Schmid, J. Giorgi, et al. 1990. Selective depletion of CD8+ T lymphocytes for prevention of graft-versus-host disease after allogeneic bone marrow transplantation. Blood. 76:418–423. [PubMed] [Google Scholar]
- 15.Nagler, A., R. Condiotti, C. Nabet, E. Naparstek, R. Or, S. Samuel, and S. Slavin. 1998. Selective CD4+ T-cell depletion does not prevent graft-versus-host disease. Transplantation. 66:138–141. [DOI] [PubMed] [Google Scholar]
- 16.Steinmuller, D. 1984. Tissue-specific and tissue-restricted histocompatibility antigens. Immunol. Today. 5:234–240. [DOI] [PubMed] [Google Scholar]
- 17.Butcher, E.C., and L.J. Picker. 1996. Lymphocyte homing and homeostasis. Science. 272:60–66. [DOI] [PubMed] [Google Scholar]
- 18.Campbell, D.J., and E.C. Butcher. 2002. Rapid acquisition of tissue-specific homing phenotypes by CD4+ T cells activated in cutaneous or mucosal lymphoid tissues. J. Exp. Med. 195:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mora, J.R., M.R. Bono, N. Manjunath, W. Weninger, L.L. Cavanagh, M. Rosemblatt, and U.H. Von Andrian. 2003. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 424:88–93. [DOI] [PubMed] [Google Scholar]
- 20.Feng, Y., D. Wang, R. Yuan, C.M. Parker, D.L. Farber, and G.A. Hadley. 2002. CD103 expression is required for destruction of pancreatic islet allografts by CD8+ T cells. J. Exp. Med. 196:877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karecla, P.I., S.J. Bowden, S.J. Green, and P.J. Kilshaw. 1995. Recognition of E-cadherin on epithelial cells by the mucosal T cell integrin alpha M290 beta 7 (alpha E beta 7). Eur. J. Immunol. 25:852–856. [DOI] [PubMed] [Google Scholar]
- 22.Lefrancois, L., C.M. Parker, S. Olson, W. Muller, N. Wagner, M.P. Schon, and L. Puddington. 1999. The role of β-7 integrins in CD8 T cell trafficking during an antiviral immune response. J. Exp. Med. 189:1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aberle, H., H. Schwartz, and R. Kemler. 1996. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J. Cell. Biochem. 61:514–523. [DOI] [PubMed] [Google Scholar]
- 24.Dogan, A., Z.D. Wang, and J. Spencer. 1995. E-cadherin expression in intestinal epithelium. J. Clin. Pathol. 48:143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dignass, A.U., J.L. Stow, and M.W. Babyatsky. 1996. Acute epithelial injury in the rat small intestine in vivo is associated with expanded expression of transforming growth factor alpha and beta. Gut. 38:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barnard, J.A., G.J. Warwick, and L.I. Gold. 1993. Localization of transforming growth factor beta isoforms in the normal murine small intestine and colon. Gastroenterology. 105:67–73. [DOI] [PubMed] [Google Scholar]
- 27.Robertson, H., W.K. Wong, D. Talbot, A.D. Burt, and J.A. Kirby. 2001. Tubulitis after renal transplantation: demonstration of an association between CD103+ T cells, transforming growth factor beta1 expression and rejection grade. Transplantation. 71:306–313. [DOI] [PubMed] [Google Scholar]
- 28.Hadley, G.A., E.A. Rostapshova, D.M. Gomolka, B.M. Taylor, S.T. Bartlett, C.I. Drachenberg, and M.R. Weir. 1999. Regulation of the epithelial cell-specific integrin, CD103, by human CD8+ cytolytic T lymphocytes. Transplantation. 67:1418–1425. [DOI] [PubMed] [Google Scholar]
- 29.Wang, D., R. Yuan, Y. Feng, R. El-Asady, D.L. Farber, R.E. Gress, P.J. Lucas, and G.A. Hadley. 2004. Regulation of CD103 expression by CD8+ T cells responding to renal allografts. J. Immunol. 172:214–221. [DOI] [PubMed] [Google Scholar]
- 30.Dey, B., Y.G. Yang, F. Preffer, A. Shimizu, K. Swenson, D. Dombkowski, and M. Sykes. 1999. The fate of donor T-cell receptor transgenic T cells with known host antigen specificity in a graft-versus-host disease model. Transplantation. 68:141–149. [DOI] [PubMed] [Google Scholar]
- 31.Sha, W.C., C.A. Nelson, R.D. Newberry, D.M. Kranz, J.H. Russell, and D.Y. Loh. 1988. Selective expression of an antigen receptor on CD8-bearing T lymphocytes in transgenic mice. Nature. 335:271–274. [DOI] [PubMed] [Google Scholar]
- 32.Kranz, D.M., D.H. Sherman, M.V. Sitkovsky, M.S. Pasternack, and H.N. Eisen. 1984. Immunoprecipitation of cell surface structures of cloned cytotoxic T lymphocytes by clone-specific antisera. Proc. Natl. Acad. Sci. USA. 81:573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lucas, P.J., S.J. Kim, S.J. Melby, and R.E. Gress. 2000. Disruption of T cell homeostasis in mice expressing a T cell–specific dominant negative transforming growth factor β II receptor. J. Exp. Med. 191:1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cepek, K.L., S.K. Shaw, C.M. Parker, G.J. Russell, J.S. Morrow, D.L. Rimm, and M.B. Brenner. 1994. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. 372:190–193. [DOI] [PubMed] [Google Scholar]
- 35.van Engeland, M., L.J. Nieland, F.C. Ramaekers, B. Schutte, and C.P. Reutelingsperger. 1998. Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 31:1–9. [DOI] [PubMed] [Google Scholar]
- 36.Shlomchik, W.D. 2003. Antigen presentation in graft-vs-host disease. Exp. Hematol. 31:1187–1197. [DOI] [PubMed] [Google Scholar]
- 37.Barnard, J.A., R.D. Beauchamp, R.J. Coffey, and H.L. Moses. 1989. Regulation of intestinal epithelial cell growth by transforming growth factor type beta. Proc. Natl. Acad. Sci. USA. 86:1578–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCabe, R.P., H. Secrist, M. Botney, M. Egan, and M.G. Peters. 1993. Cytokine mRNA expression in intestine from normal and inflammatory bowel disease patients. Clin. Immunol. Immunopathol. 66:52–58. [DOI] [PubMed] [Google Scholar]
- 39.Teshima, T., R. Ordemann, P. Reddy, S. Gagin, C. Liu, K.R. Cooke, and J.L. Ferrara. 2002. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat. Med. 8:575–581. [DOI] [PubMed] [Google Scholar]
- 40.Liblau, R.S., F.S. Wong, L.T. Mars, and P. Santamaria. 2002. Autoreactive CD8 T cells in organ-specific autoimmunity: emerging targets for therapeutic intervention. Immunity. 17:1–6. [DOI] [PubMed] [Google Scholar]
- 41.Poindexter, N.J., N.S. Steward, S. Shenoy, M.D. Jendrisak, M.W. Flye, T.K. Howard, and T. Mohanakumar. 1995. Cytolytic T lymphocytes from human renal allograft biopsies are tissue specific. Hum. Immunol. 44:43–49. [DOI] [PubMed] [Google Scholar]
- 42.Zhou, S., R. Ou, L. Huang, G.E. Price, and D. Moskophidis. 2004. Differential tissue-specific regulation of antiviral CD8+ T-cell immune responses during chronic viral infection. J. Virol. 78:3578–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murali-Krishna, K., J.D. Altman, M. Suresh, D.J. Sourdive, A.J. Zajac, J.D. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 8:177–187. [DOI] [PubMed] [Google Scholar]
- 44.Butz, E.A., and M.J. Bevan. 1998. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 8:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Udaka, K., T.J. Tsomides, and H.N. Eisen. 1992. A naturally occurring peptide recognized by alloreactive CD8+ cytotoxic T lymphocytes in association with a class I MHC protein. Cell. 69:989–998. [DOI] [PubMed] [Google Scholar]
- 46.Udaka, K., T.J. Tsomides, P. Walden, N. Fukusen, and H.N. Eisen. 1993. A ubiquitous protein is the source of naturally occurring peptides that are recognized by a CD8+ T-cell clone. Proc. Natl. Acad. Sci. USA. 90:11272–11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeichi, M. 1988. The cadherins: cell-cell adhesion molecules controlling animal morphogenesis. Development. 102:639–655. [DOI] [PubMed] [Google Scholar]
- 48.Conlon, K., A. Lloyd, U. Chattopadhyay, N. Lukacs, S. Kunkel, T. Schall, D. Taub, C. Morimoto, J. Osborne, J. Oppenheim, et al. 1995. CD8+ and CD45RA+ human peripheral blood lymphocytes are potent sources of macrophage inflammatory protein 1 alpha, interleukin-8 and RANTES. Eur. J. Immunol. 25:751–756. [DOI] [PubMed] [Google Scholar]
- 49.Mosmann, T.R., L. Li, and S. Sad. 1997. Functions of CD8 T-cell subsets secreting different cytokine patterns. Semin. Immunol. 9:87–92. [DOI] [PubMed] [Google Scholar]
- 50.Price, D.A., P. Klenerman, B.L. Booth, R.E. Phillips, and A.K. Sewell. 1999. Cytotoxic T lymphocytes, chemokines and antiviral immunity. Immunol. Today. 20:212–216. [DOI] [PubMed] [Google Scholar]
- 51.Marks, L., N.H. Altman, E.R. Podack, and R.B. Levy. 2004. Donor T cells lacking Fas ligand and perforin retain the capacity to induce severe GvHD in minor histocompatibility antigen mismatched bone-marrow transplantation recipients. Transplantation. 77:804–812. [DOI] [PubMed] [Google Scholar]
- 52.Brown, G.R., E. Lee, and D.L. Thiele. 2002. TNF-TNFR2 interactions are critical for the development of intestinal graft-versus-host disease in MHC class II-disparate (C57BL/6J-->C57BL/6J x bm12)F1 mice. J. Immunol. 168:3065–3071. [DOI] [PubMed] [Google Scholar]
- 53.Fowler, D.H., J. Foley, J. Whit-Shan Hou, J. Odom, K. Castro, S.M. Steinberg, J. Gea-Banacloche, C. Kasten-Sportes, R.E. Gress, and M.R. Bishop. 2004. Clinical “cytokine storm” as revealed by monocyte intracellular flow cytometry: correlation of tumor necrosis factor alpha with severe gut graft-versus-host disease. Clin. Gastroenterol Hepatol. 2:237–245. [DOI] [PubMed] [Google Scholar]
- 54.Appelbaum, F.R. 2001. Haematopoietic cell transplantation as immunotherapy. Nature. 411:385–389. [DOI] [PubMed] [Google Scholar]
- 55.Schon, M.P., A. Arya, E.A. Murphy, C.M. Adams, U.G. Strauch, W.W. Agace, J. Marsal, J.P. Donohue, H. Her, D.R. Beier, et al. 1999. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J. Immunol. 162:6641–6649. [PubMed] [Google Scholar]
- 56.Guy-Grand, D., C. Griscelli, and P. Vassalli. 1978. The mouse gut T lymphocyte, a novel type of T cell. Nature, origin, and traffic in mice in normal and graft-versus-host conditions. J. Exp. Med. 148:1661–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Masopust, D., V. Vezys, A.L. Marzo, and L. Lefrancois. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 291:2413–2417. [DOI] [PubMed] [Google Scholar]