

Abstract
Class switch recombination was the last of the lymphocyte-specific DNA modification reactions to appear in the evolution of the adaptive immune system. It is absent in cartilaginous and bony fish, and it is common to all tetrapods. Class switching is initiated by activation-induced cytidine deaminase (AID), an enzyme expressed in cartilaginous and bony fish that is also required for somatic hypermutation. Fish AID differs from orthologs found in tetrapods in several respects, including its catalytic domain and carboxy-terminal region, both of which are essential for the switching reaction. To determine whether evolution of class switch recombination required alterations in AID, we assayed AID from Japanese puffer and zebra fish for class-switching activity in mouse B cells. We find that fish AID catalyzes class switch recombination in mammalian B cells. Thus, AID had the potential to catalyze this reaction before the teleost and tetrapod lineages diverged, suggesting that the later appearance of a class-switching reaction was dependent on the evolution of switch regions and multiple constant regions in the IgH locus.
Antigen receptors found in amphibians, reptiles, birds, and mammals are products of lymphocyte-specific DNA modification reactions including V(D)J recombination, somatic hypermutation, and class switch recombination. Whereas V(D)J recombination is a site-specific recombination reaction that assembles Ig gene segments into a functional transcription unit, somatic hypermutation introduces point mutations in the assembled antibody gene. Both reactions alter the specificity of antibody combining sites and are found in all living jawed vertebrates (1). However, they are mechanistically distinct and catalyzed by different enzymes. V(D)J recombination is catalyzed by the recombination-activating genes 1 and 2, whereas somatic mutation is initiated by activation-induced cytidine deaminase (AID) (2–4).
Class switch recombination was the last of the lymphocyte-specific DNA modification reactions to emerge in evolution. It is a region-specific recombination reaction that does not alter the specificity of the antibody but replaces the constant region of the Ig heavy chain and, consequently, its effector function (5). Class switch recombination appears at the time of the divergence of the amphibians. It is absent in bony fish, including Japanese puffer and zebra fish, but is conserved in all tetrapods (6).
Despite the differences between somatic hypermutation and class switch recombination, the two reactions are catalyzed by AID (3, 4, 7, 8). AID initiates these reactions by deaminating cytosine residues in target DNA producing U:G mismatches that are recognized by uracil DNA glycosylase (UNG) or mismatch repair enzymes (9–13). The molecular mechanisms that mediate lesion formation, mismatch recognition, and initial processing appear to be common to both class switch and somatic hypermutation, but the requirements for completion of the two reactions are distinct. For example, efficient class switch recombination requires H2AX and ataxia teleangiectasia mutated protein, whereas somatic hypermutation does not (14–16). In addition, the carboxy-terminal domain of AID is required for class switch recombination but not for somatic hypermutation, whereas the amino terminus is required for efficient somatic hypermutation and dispensable for switch recombination (for review see reference 17).
The emergence of class switch recombination after somatic hypermutation might depend on alterations in AID, the structure of the Ig heavy chain locus, or both (1, 6). We report that AID from species that do not undergo class switching can complement AID deficiency in mammalian cells. Therefore, all of the features of AID required for Ig switch region targeting and the resolution of switch lesions were present before the emergence of the switch reaction and were conserved throughout evolution.
Results and discussion
Teleost AID is active in Escherichia coli and Saccharomyces cerevisiae
The presence of antibody affinity maturation, AID, and Ig loci enriched for mutation hotspots suggest that somatic hypermutation occurs in bony fish (for review see reference 6). However, there is no precise information on the catalytic activity or targeting of fish AID in vivo. To examine the mutator activity of AID orthologs from mouse (Mus musculus; m-AID), zebra fish (Danio rerio; z-AID), and Japanese puffer fish (Fugu rubripes; f-AID), we expressed the respective cDNAs in E. coli or S. cerevisiae. We assayed AID in both systems because AID from cold-blooded animals might be thermolabile (18). The E. coli assay is performed at 37°C and measures mutations in an inactive kanamycin allele (KanL94P) that becomes active by mutation (CCA to TCA or CTA) (19). m-AID induces a higher frequency of mutations than z-AID in this assay, whereas f-AID is indistinguishable from the negative control (Fig. 1 A). The S. cerevisiae assay is performed at 30°C and measures inactivation mutations in ura3, resulting in resistance to 5-fluoroorotic acid (5-FOA). In contrast to the E. coli assay, numerous mutations in ura3 result in loss of activity and resistance to 5-FOA (Tables S1 and S2, available at http://www.jem.org/cgi/content/full/jem.20051378/DC1). We found that z-AID was more active than m-AID in S. cerevisiae, whereas f-AID showed marginal levels of activity (Fig. 1 B). When the same experiment was performed in ung − S. cerevisiae, we observed an increase in the frequency of mutations, and f-AID activity was clearly above the background level. Increased mutation or altered patterns of mutation in the absence of UNG have been reported in E. coli, S. cerevisiae, chicken, and mammalian cells, implicating UNG in the recognition and repair of AID-dependent mutations (10, 13, 20, 21). In the absence of UNG, fewer AID-induced lesions are repaired, thereby increasing the sensitivity of the assay and underscoring the mutator activity of f-AID. We conclude that fish AID is catalytically active in E. coli and S. cerevisiae.
Figure 1.

m- and z-AID are catalytically active in E. coli and S. cerevisiae. (A) Mutator assay in E. coli. Expression of m-, z-, and f-AID was induced for 3 h in exponentially growing cultures that were then assayed for kanamycin resistance. Points represent the mutation frequency of individual starting colonies. The mean values and standard deviations from three experiments are given. (B) Mutator assay in ung + and ung − S. cerevisiae. Expression of m-, f-, and z-AID was induced by galactose for 48 h at 30°C, and cultures were assayed for ura3 auxotrophy in 5-FOA–containing medium. Points represent the mutation frequency in cultures from individual starting colonies. The mean values and standard deviations are given. ung + background data were from four experiments. One experiment in the ung− background is shown.
Teleost AID catalyzes class switch recombination
Zebra fish and the Japanese puffer fish are teleosts, a group capable of mounting an adaptive immune response but lacking class switch recombination. A comparison of the sequences of bony fish AID orthologs with amphibians, birds, and mammals reveals that fish AID has a longer cytidine deaminase motif and extensive substitutions in the carboxy-terminal region that are required for class switch recombination (22). To determine whether AID expressed by species lacking class switch recombination can induce switching, we used retroviruses to express m-, z-, or f-AID in mouse AID−/− lymphocytes. To facilitate switching and retroviral infection, AID-deficient mouse B cells were stimulated with LPS and IL-4. Virally infected cells were identified by enhanced GFP expression, and Ig class switching was measured by cell surface IgG1 expression. Virally expressed z-AID induced class switching in AID−/− B cells with a similar frequency as m-AID (Fig. 2, A and B). f-AID–expressing cells showed lower levels of switching than m- or z-AID (Fig. 2, A and B), as would be expected from the E. coli and S. cerevisiae assay. To monitor protein expression levels, we repeated these experiments using amino-terminal FLAG-tagged m-, f-, and z-AID (tm-, tf-, and tz-AID). The percentage of switching to IgG1 triggered by tm-, tf-, and tz-AID was lower than with the native counterparts but clearly above background levels for m- and z-AID (Fig. 2 B). In agreement with the observation that teleost AID may be thermolabile, immunoblotting revealed that AID from fish, notably tf-AID, was less stable than tm-AID in mouse B cells grown at 37°C (Fig. 2 C) (18). We conclude that teleost AIDs can induce switching to IgG1 in mouse B cells and that differences in the intrinsic deoxycytidine deamination activity and/or the stability of different AID proteins correlate with switching activity.
Figure 2.

AID from bony fish induces class switch recombination in mouse AID−/− splenocytes. (A) Representative flow cytometry profiles of IL-4 and LPS-stimulated B cells from AID−/− mice infected with retroviruses encoding m-, f-, or z-AID or an empty vector control. The y axis shows GFP expression, and the x axis shows cell surface IgG1. The percentage of cells in each indicated quadrant is shown. (B) Percentage of switching to IgG1 by GFP+ cells. Points represent individual cultures. (C) Immunoblot analysis of protein extracts from AID−/− B cell cultures infected with retroviruses encoding FLAG-tagged AID. GFP expression was used as a loading control.
Teleost AID produces normal switch junctions in mammalian cells
Sμ–Sγ1 switch junctions frequently show regions of microhomology and the DNA region around the junction is enriched in somatic mutations (23). To determine whether switch junctions produced by teleost AID were similar to junctions produced by mammalian AID, we cloned Sμ–Sγ1 switch junctions from m- or z-AID expressing AID−/− B cells. We found no substantial differences in the amount of donor/acceptor homology or mutations at the switch junctions produced by mammalian and teleost AID. The mean length of overlap for m- and z-AID–generated junctions was 1.2 (n = 26) and 1.6 (n = 26; Fig. 3, A and B), and the mutation frequency in the vicinity of the junctions (±50 bp) was 0.4 and 0.7 × 10−2, respectively. Furthermore, the breakpoint distribution in Sμ and Sγ1 measured by scatter analysis was similar in m- and z-AID (Fig. 3 C). We conclude that z-AID generates normal Sμ–Sγ1 junctions, suggesting that the proteins required for switching, including nonhomologous end joining, mismatch repair, excision repair, and base excision repair enzymes, were recruited by z-AID in mouse B cells (for review see reference 24).
Figure 3.
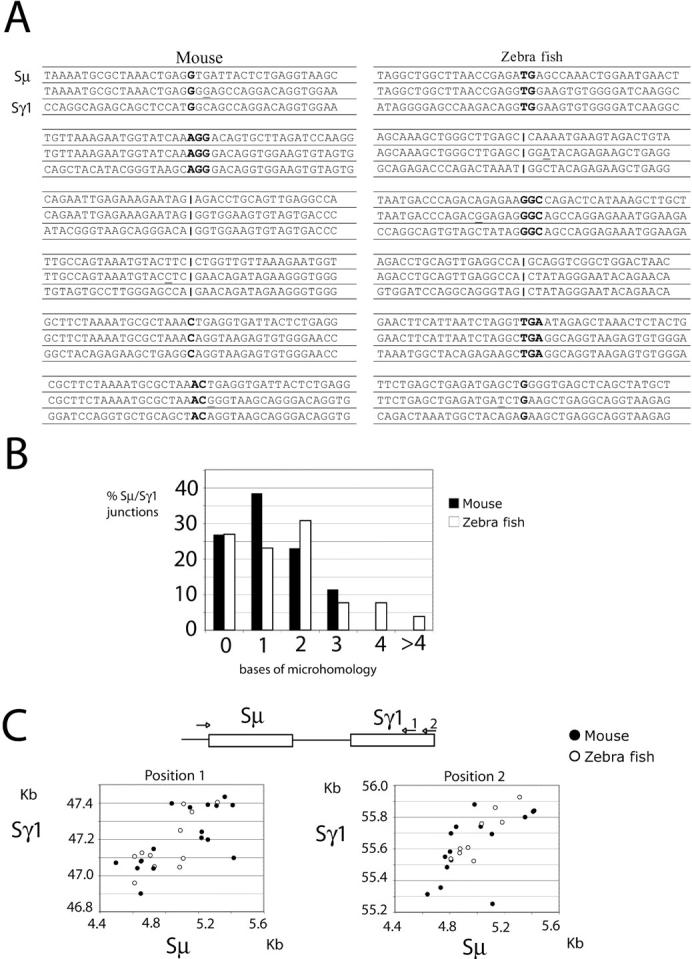
Normal Sμ–Sγ1 switch recombination junctions in z-AID–expressing mouse B cells. (A) Representative switch junctions cloned from mouse AID−/− B cells infected with m- or z-AID–encoding retroviruses. Overlap was determined by identifying the longest region at the switch junction of perfect uninterrupted donor/acceptor identity. Sμ and Sγ1 germ line sequences are shown above and below each junction sequence, respectively. Bold letters indicate regions of microhomology and underlined letters indicate mutations. (B) Length of microhomologies at Sμ–Sγ1 junctions (n = 26 for both m- and z-AID–derived junctions). (C) Scatter analysis of the Sμ–Sγ1 breakpoints derived from in vitro–stimulated B cells. The axes indicate the position relative to Sμ and Sγ1 sequences (available from GenBank/EMBL/DDBJ under accession nos. J00440 and AJ851869, respectively). Closed circles denote breakpoints induced by m-AID, and open circles denote breakpoints induced by z-AID. Data are pooled from two independent experiments.
Class switch recombination requires switch region transcription, AID targeting, cytidine deamination, mismatch detection, and repair (17). In addition to deamination, AID is thought to be involved in targeting and switch lesion repair. Three lines of evidence support the existence of a specific targeting mechanism. First, only a small number of genes outside of antibody genes are somatically mutated (17). Second, B cells stimulated to undergo class switching in vitro mutate Ig switch regions but not Ig V regions (25). Third, deoxycytidine deamination activity and transcription of the switch regions is not sufficient for switching because AID-related deaminases, such as APOBEC-1, and catalytically active carboxy terminus AID mutants fail to induce switching to IgG1 in IL-4/LPS-stimulated B cells (26–28). Transcription exposes single-stranded DNA, which is the substrate of AID, and it is also essential for Ig gene targeting. The dual role of transcription in class switch recombination and hypermutation has made it difficult to identify the function of individual cis elements or transcription factors in the reaction. The only exception is the observation that an E47 binding site appears to enhance mutation of a transgenic substrate independently of its effects on the rate of transcription, and it has been proposed that AID may be targeted to Ig DNA by interaction with transcription factors like E47 (29). Finding that zebra fish AID is completely functional for class switching in mouse B cells suggests that the interaction domains required for targeting AID to Ig switch regions evolved before the appearance of class switch recombination. This apparent paradox could be explained if the protein interaction domains evolved in the context of other DNA modification reactions and were later adapted for class switch recombination.
We conclude that the molecular attributes required for AID-induced class switching did not coevolve with switch recombination and that the critical event for emergence of class switching in the last 400 million years was the evolution of switch regions and multiple constant region genes in the Ig heavy chain locus.
MATERIALS and METHODS
Fugu (F. rubripes) and zebra fish (D. rerio) full-length AID cDNA.
Fugu spleen total RNA (provided by M. Toshiaki, Fukui Prefectural University, Obama, Japan) was reverse transcribed into first-strand cDNA (GE Healthcare) and used as a template for amplification of the full-length AID transcript. The primers were Fugu-AIDs (5′-ATCCCGCCCGCCGAGTGTCAAA-3′) and Fugu-AIDas (5′-GACGAAGACGAGATGACGAAGATG-3′). All cDNA clones were derived from aberrantly spliced RNA, as a short intron was retained. To obtain a fugu AID cDNA with a correct translation frame, we used Fugu-AID-EcoRs (5′-TTTTGAATTCCACCATGATCACCAAGCTAGACAGTA-3′), Fugu-fusion1-as (5′-CCACAAATTTCTGCCAGCAATAGAAATAGTCTTTGTAGCTCATCACTGAG-3′), Fugu-fusion2-s (5′-GCGTGAGGATCTCAGTGATGAGCTACAAAGACTATTTCTATTGCTGGCAG-3′), and Fugu AID-Not Ias (5′-TTTTGCGGCCGCTCAGAATCCGAGGAGCTTAAGA-3′). The Zebra fish sequence used in this study was derived directly from a cDNA as previously described (22) and differs from the sequence deduced by genomic sequencing in which exon boundaries were incorrectly deduced (30).
Mutation in E. coli and S. cerevisiae.
We measured the deoxycytidine deaminase activity of AID in E. coli as described previously (19), with the exception that the codon usage in the AID sequences was not optimized for expression in prokaryotes. For experiments in S. cerevisiae, mouse, zebra fish, and fugu, AID cDNAs with a Kozak consensus sequence for the initiation of translation were cloned into a galactose-inducible S. cerevisiae expression vector (pESC-His; Stratagene) and transformed by lithium acetate into YPH500 (ura3−lys2−ade2−trp1−his1−leu2−) or ung− YPH500, engineered to contain a constitutively expressed ura3 gene in the HO locus (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051378/DC1). Targeted deletion of the ung gene was performed with a Trp-selectable cassette amplified from pUC18-TRP1 with the oligos ungLHA (5′-GTGGTGCATGAGAAGATTGCCAACAAATTCAGTTATGACGGTGTGAAATAAAGCATAGGCCACTAGTGGATCTG-3′) and ungRHA (5′-RHATCAAGGGTCCTTTGATTCTGACTCTAAGCGAGCATTTGCCCAGCTGAAGCTTCGTACGC-3′). Targeted integration was confirmed by PCR.
Colonies selected in his− ura− (his− ura− trp− for the ung - strain) medium were expanded in 2% glucose liquid medium (then washed in water), and 2 OD were then grown in 2% galactose/1% raffinose his− (his− trp− for the ung - strain) liquid medium for 48 h. During this period, the cultures were diluted periodically to keep the cells dividing. Cells were plated in ura− his− (his− ura− trp− for the ung - strain) 2% glucose medium with or without 750 μg/ml 5-FOA to count viable cells and cells with null mutations in ura3, respectively. All S. cerevisiae experiments were performed at 30°C.
Mouse B cell cultures and transduction.
Mouse, zebra fish, and fugu AID with a Kozak consensus sequence for the initiation of translation were cloned into the pMX-PIE. Amino-terminal FLAG and hemagglutinin-tagged AIDs were derived by PCR by adding the following sequence 5′ of the second codon of AID by PCR: 5′-ATGGACTACAAGGACCGATGACAAGGGAGGATATCCGTATGATGTTCCTGATTATGCTAGCCTCGGAGGA-3′. Splenocytes from 2–5-mo-old AID−/− BALB/cByJ mice were isolated, stimulated with LPS and IL-4, and transduced as described previously (27). Infection efficiency was ∼30%, as determined by GFP expression by flow cytometry.
Protein analysis.
For Western blot assays, cells were lysed in 50 mM Tris, pH 8.0, 400 mM NaCl, 0.5% NP-40, 10% glycerol and ∼300 μg of protein lysate run separated on SDS polyacrylamide gels. Anti-FLAG M2 peroxidase–conjugated (Sigma-Aldrich) and anti-GFP rabbit IgG (Molecular Probes), combined with a peroxidase-conjugated anti–rabbit secondary antibody, were used to visualize FLAG-tagged AID and enhanced GFP, respectively.
Amplification and cloning of Sμ–Sγ1 junctions.
For the cloning of Sμ–Sγ1 from AID−/− splenocytes transduced with retroviruses, genomic DNA was prepared according to standard procedures from samples that had been transduced 48 h earlier. Switch junctions were amplified from 200–500 ng of DNA per 50-μl reaction with oligos Sμ3 (5′-AATGGATACCTCAGTGGTTTTTAATGGTGGGTTTA-3′) and γ1-R (5′-CAATTAGCTCCTGCTCTTCTGTGG-3′) and cloned, as described previously (14). Colonies were then screened by PCR with oligos Sμ-nested (5′- CTAATTTAGAATCAGTAAGGAGGGAC-3′) and γ1-R and sequenced with an M13rev oligo.
Flow cytometry.
Mouse B cells were stained with biotin-labeled anti–mouse IgG1 antibodies and visualized with streptavidin APC (all from Becton Dickinson). Dead cells were excluded on the basis of forward side scatter. Data were acquired on a FACSCalibur and analyzed with CellQuest software (both from Becton Dickinson).
Online supplemental material.
Fig. S1 describes the AID-expressing plasmids and the ura3 reporter for AID activity in S. cerevisiae. Table S1 shows oligos used to generate the ura3 mutation reporter construct. Table S2 shows the mutations found in the ura3 gene from 25 independent 5-FOA resistant S. cerevisiae clones recovered after induction of m-AID. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051378/DC1.
Acknowledgments
We thank Dr. Miyadai Toshiaki for providing fugu RNA and Drs. Thiago Carvalho, Revati Masilamani, Kevin McBride, Nina Papavasiliou, and Almudena Ramiro for critical readings of the manuscript.
This work was supported by the Swedish Research Council and grants from the National Institutes of Health (1000978-01-CCL3000671-67552-113620) and the Leukemia Society (both to M.C. Nussenzweig). M.C. Nussenzweig is an investigator with the Howard Hughes Medical Institute.
The authors have no conflicting financial interests.
References
- 1.Flajnik, M.F. 2002. Comparative analyses of immunoglobulin genes: surprises and portents. Nat. Rev. Immunol. 2:688–698. [DOI] [PubMed] [Google Scholar]
- 2.Schatz, D.G. 2004. V(D)J recombination. Immunol. Rev. 200:5–11. [DOI] [PubMed] [Google Scholar]
- 3.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 5.Kataoka, T., T. Kawakami, N. Takahashi, and T. Honjo. 1980. Rearrangement of immunoglobulin gamma 1-chain gene and mechanism for heavy-chain class switch. Proc. Natl. Acad. Sci. USA. 77:919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stavnezer, J., and C.T. Amemiya. 2004. Evolution of isotype switching. Semin. Immunol. 16:257–275. [DOI] [PubMed] [Google Scholar]
- 7.Yoshikawa, K., I.M. Okazaki, T. Eto, K. Kinoshita, M. Muramatsu, H. Nagaoka, and T. Honjo. 2002. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 296:2033–2036. [DOI] [PubMed] [Google Scholar]
- 8.Okazaki, I.M., K. Kinoshita, M. Muramatsu, K. Yoshikawa, and T. Honjo. 2002. The AID enzyme induces class switch recombination in fibroblasts. Nature. 416:340–345. [DOI] [PubMed] [Google Scholar]
- 9.Phung, Q.H., D.B. Winter, A. Cranston, R.E. Tarone, V.A. Bohr, R. Fishel, and P.J. Gearhart. 1998. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient in the MSH2 mismatch repair protein. J. Exp. Med. 187:1745–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rada, C., J.M. Di Noia, and M.S. Neuberger. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 16:163–171. [DOI] [PubMed] [Google Scholar]
- 11.Schrader, C.E., W. Edelmann, R. Kucherlapati, and J. Stavnezer. 1999. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiesendanger, M., B. Kneitz, W. Edelmann, and M.D. Scharff. 2000. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2–MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 191:579–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103. [DOI] [PubMed] [Google Scholar]
- 14.Reina-San-Martin, B., S. Difilippantonio, L. Hanitsch, R.F. Masilamani, A. Nussenzweig, and M.C. Nussenzweig. 2003. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 197:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reina-San-Martin, B., H.T. Chen, A. Nussenzweig, and M.C. Nussenzweig. 2004. ATM is required for efficient recombination between immunoglobulin switch regions. J. Exp. Med. 200:1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan-Hammarstrom, Q., S. Dai, Y. Zhao, I.F. van Dijk-Hard, R.A. Gatti, A.L. Borresen-Dale, and L. Hammarstrom. 2003. ATM is not required in somatic hypermutation of VH, but is involved in the introduction of mutations in the switch mu region. J. Immunol. 170:3707–3716. [DOI] [PubMed] [Google Scholar]
- 17.Barreto, V.M., A.R. Ramiro, and M.C. Nussenzweig. 2005. Activation-induced deaminase: controversies and open questions. Trends Immunol. 26:90–96. [DOI] [PubMed] [Google Scholar]
- 18.Conticello, S.G., C.J. Thomas, S.K. Petersen-Mahrt, and M.S. Neuberger. 2005. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 22:367–377. [DOI] [PubMed] [Google Scholar]
- 19.Ramiro, A.R., P. Stavropoulos, M. Jankovic, and M.C. Nussenzweig. 2003. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4:452–456. [DOI] [PubMed] [Google Scholar]
- 20.Poltoratsky, V.P., S.H. Wilson, T.A. Kunkel, and Y.I. Pavlov. 2004. Recombinogenic phenotype of human activation-induced cytosine deaminase. J. Immunol. 172:4308–4313. [DOI] [PubMed] [Google Scholar]
- 21.Di Noia, J., and M.S. Neuberger. 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 419:43–48. [DOI] [PubMed] [Google Scholar]
- 22.Zhao, Y., Q. Pan-Hammarstrom, Z. Zhao, and L. Hammarstrom. 2005. Identification of the activation-induced cytidine deaminase gene from zebrafish: an evolutionary analysis. Dev. Comp. Immunol. 29:61–71. [DOI] [PubMed] [Google Scholar]
- 23.Dunnick, W., G.Z. Hertz, L. Scappino, and C. Gritzmacher. 1993. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 21:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhuri, J., and F.W. Alt. 2004. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 4:541–552. [DOI] [PubMed] [Google Scholar]
- 25.Manser, T. 1987. Mitogen-driven B cell proliferation and differentiation are not accompanied by hypermutation of immunoglobulin variable region genes. J. Immunol. 139:234–238. [PubMed] [Google Scholar]
- 26.Ta, V.T., H. Nagaoka, N. Catalan, A. Durandy, A. Fischer, K. Imai, S. Nonoyama, J. Tashiro, M. Ikegawa, S. Ito, et al. 2003. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat. Immunol. 4:843–848. [DOI] [PubMed] [Google Scholar]
- 27.Barreto, V., B. Reina-San-Martin, A.R. Ramiro, K.M. McBride, and M.C. Nussenzweig. 2003. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol. Cell. 12:501–508. [DOI] [PubMed] [Google Scholar]
- 28.Fugmann, S.D., J.S. Rush, and D.G. Schatz. 2004. Non-redundancy of cytidine deaminases in class switch recombination. Eur. J. Immunol. 34:844–849. [DOI] [PubMed] [Google Scholar]
- 29.Michael, N., H.M. Shen, S. Longerich, N. Kim, A. Longacre, and U. Storb. 2003. The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity. 19:235–242. [DOI] [PubMed] [Google Scholar]
- 30.Saunders, H.L., and B.G. Magor. 2004. Cloning and expression of the AID gene in the channel catfish. Dev. Comp. Immunol. 28:657–663. [DOI] [PubMed] [Google Scholar]