

Abstract
T cell receptor (TCR) signaling plays an important role in early interleukin (IL)-4 production by naive CD4+ T cells. This “antigen-stimulated” early IL-4 is sufficient for in vitro Th2 differentiation. Here, we provide evidence that early IL-4 production by naive CD4+ T cells stimulated with cognate peptide requires TCR-induced early GATA-3 expression and IL-2 receptor signaling, both of which are controlled by the degree of activation of extracellular signal-regulated kinase (ERK). Stimulation of naive CD4+ T cells from TCR transgenic mice with low concentrations of peptide-induced IL-2–dependent STAT5 phosphorylation, IL-4-independent early GATA-3 expression, and IL-4 production. Neutralization of IL-2 abolished early IL-4 production without affecting early GATA-3 expression. In addition, naive CD4+ T cells from GATA-3 conditional KO mice failed to produce early IL-4 in response to TCR/CD28 stimulation. Stimulation with high concentrations of peptide abrogated early GATA-3 expression and IL-2–dependent STAT5 phosphorylation, and resulted in the failure to produce early IL-4. This high concentration–mediated suppression of early IL-4 production was reversed by blockade of the ERK pathway. A MEK inhibition rescued early GATA-3 expression and responsiveness to IL-2; these cells were now capable of producing early IL-4 and undergoing subsequent Th2 differentiation.
Naive CD4+ T cells can differentiate into at least two major distinct phenotypes, Th1 and Th2 cells, which are characterized by polarized patterns of cytokine production. Many factors have been reported to play a role in differentiation to the Th1 and Th2 phenotypes. Among these, the set of cytokines present during the priming period is particularly important. The presence of IL-4 is critical for in vitro Th2 differentiation (1, 2), whereas in vitro Th1 differentiation depends heavily on IFN-γ and/or IL-12 (3, 4). Recently, IL-2 has been established as playing a major role in Th2 differentiation (5). Mast cells (6, 7), eosinophils (8), basophils (9–11), and NK T cells (12) have been reported to produce IL-4; however, it is still debated whether these cell populations serve as exogenous sources of IL-4 during the priming period.
Naive CD4+ T cells are capable of producing IL-4 when they are stimulated with peptide/MHC class II complex on APCs; this endogenously produced IL-4 was shown to be sufficient for Th2 differentiation. When no polarizing cytokines are added to a culture, strength of signal through TCR may control Th cell differentiation (13). Thus, priming for IL-4–producing cells often is observed when a low concentration of antigen-derived peptide is used, whereas high concentrations of peptide were reported to induce IFN-γ–producing cells (14). Similarly, priming with ligands that interact with TCR less energetically than a WT peptide, so-called “altered peptide ligands,” often favors Th2 differentiation (15).
The basis of the “signal strength” effect on biasing differentiation to Th1 or Th2 subset has not been clarified, although evidence for the involvement of NFAT (16), extracellular signal-regulated kinase (ERK), and activated protein-1 (AP-1) (17) has been presented. We have reexamined the “dose effect” on priming for Th cell differentiation using rigorously purified naive CD4+ T cells from TCR transgenic mice. We show that low concentrations of peptide induce IL-4–independent induction of IL-4 mRNA beginning at 14–16 h after stimulation. This early IL-4 mRNA expression is associated with IL-4–independent early GATA-3 expression and is dependent on IL-2. However, IL-2 is not essential for early GATA-3 expression. Moreover, naive CD4+ T cells from GATA-3 conditional KO mice fail to express IL-4 mRNA in response to TCR stimulation indicating that early IL-4 transcription is GATA-3 dependent. High concentrations of peptide inhibit early IL-4 mRNA expression by abrogating early GATA-3 induction and by blocking IL-2R-mediated signaling, as shown by the failure of STAT5 phosphorylation, although IL-2 is produced abundantly. This high concentration–mediated inhibition is reversed by the blockade of ERK pathway by an inhibitor of mitogen-activated protein kinase kinase (MEK) allowing the cells to express GATA-3 and to respond to IL-2. This results in the restoration of early IL-4 mRNA expression and subsequent development into high-rate IL-4–producing cells. We conclude that TCR-mediated control of Th2 differentiation is dependent upon early IL-4 transcription determined by the independent actions of GATA-3 and IL-2, and that the dose effect reflects dose-dependent control of early transcription of GATA-3 and high dose desensitization of the IL-2R mediated by TCR-induced ERK activation.
RESULTS
Peptide concentration regulates priming of naive CD4+ T cells for IL-4–producing cells
We isolated highly pure naive (CD44lowCD62Lhigh) CD4+ T cells from 5C.C7 TCR transgenic RAG2−/− B10.A (line 94) mice (Fig. 1 A). Splenic DCs were purified by isolating CD11c+ cells followed by the removal of NK cells and NK T cells by cell sorting (Fig. 1 A), because NK cells and NK T cells were reported to produce large amounts of IFN-γ and IL-4, respectively (12, 18), which may bias T cell priming toward Th1 or Th2 differentiation. Naive line 94 CD4+ T cells and splenic DCs were co-cultured in the presence of 0.001–10 μM pigeon cytochrome c peptide (pPCC) without adding any exogenous cytokines. In some experiments, we used P13.9 cells as APCs, instead of splenic DCs. Intracellular staining for IL-4 and IFN-γ was performed following recall challenge with immobilized anti-CD3 plus anti-CD28 for 6 h. Stimulating naive line 94 cells with 0.001 μM pPCC with splenic DCs or P13.9 cells resulted in the development of cells that were capable of producing IL-4. When P13.9 cells were used, IL-4–producing cells also were induced with 0.01 μM pPCC. The frequency of IL-4 producers declined strikingly as the concentration of pPCC was increased (Fig. 1, B and C). This decline was not reversed by the inclusion of anti–IL-12 or anti–IFN-γ in the culture, which indicated that failure of T cells primed with high concentrations of pPCC to become IL-4 producers is not due to endogenous IFN-γ production (Fig. 1 B).
Figure 1.
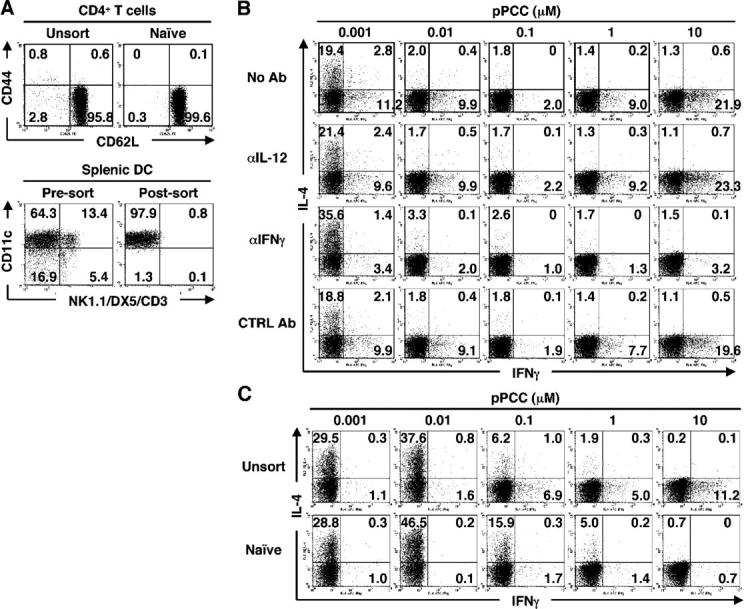
Peptide concentration regulates priming of naive CD4+T cells to develop into IL-4–producing cells. (A) Surface phenotypes of line 94 CD4+ T cells and of splenic DCs used for experiments. In this and in subsequent figures, the numbers in quadrants indicate the percent of cells in the given quadrant. (B) Naive line 94 CD4+ T cells were stimulated with splenic DCs and 0.001–10 μM pPCC in the presence or absence of anti–IL-12 (10 μg/ml), anti–IFN-γ (10 μg/ml), or rat IgG1 (10 μg/ml) plus rat IgG2a (10 μg/ml) as isotype controls. Intracellular staining for IL-4 and IFN-γ was performed. (C) Unsorted or naive line 94 CD4+ T cells were stimulated with P13.9 cells in the presence of 0.001–10 μM pPCC. Intracellular staining for IL-4 and IFN-γ was carried out after restimulation. All experiments were performed at least three times with consistent results.
5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution revealed that IL-4 was produced mainly by cells that had undergone four or more divisions at 0.001 μM when splenic DCs were used as APCs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051304/DC1), and three or more divisions at 0.001–0.01 μM when P13.9 cells were used as APCs (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20051304/DC1). The peak number of cell divisions was five at all concentrations of peptide tested when splenic DCs were used as APCs (Fig. S1) and at ≥0.01 μM when P13.9 cells were used as APCs (Fig. S2). Thus, the failure of cells to divide a sufficient number of times cannot account for high concentrations of peptide not leading to the production of IL-4.
IFN-γ was produced by T cells cultured with splenic DCs at low and high concentrations of peptide; anti–IL-12 did not block the development of IFN-γ–producing cells, but anti–IFN-γ did (Fig. 1 B). When P13.9 cells were used, few, if any, IFN-γ–producing cells appeared, even without neutralization of IFN-γ or IL-12 (Fig. 1 C). Thus, splenic DCs and P13.9 cells differ in their capacity to induce naive CD4+ T cells to develop into IFN-γ producers, even in the absence of IL-12. Because splenic DCs were shown recently not to produce IFN-γ (18), this suggests that the APCs differ in their intrinsic capacity to stimulate naive CD4+ T cells to produce “early” IFN-γ. P13.9 cells did stimulate priming for IFN-γ production if small numbers of CD44high (memory) line 94 CD4+ T cells were present (Fig. 1 C). The latter cells presumably served as a source of “early” IFN-γ upon stimulation (unpublished data).
High peptide concentration-induced activation is inhibitory for TCR-induced early IL-4 mRNA expression
In subsequent experiments, P13.9 cells were used as APCs, because the antigen concentration effects held when splenic DCs or P13.9 cells were used to present pPCC. We examined the early expression of IL-4 mRNA in naive line 94 CD4+ T cells primed with various concentrations of pPCC. IL-4 mRNA was first detectable at 14 h in cells that were stimulated with 0.01 μM pPCC, and increased in level of expression by 20–24 h (Fig. 2 A). Increasing the concentration of pPCC to 0.1 μM diminished the amount of IL-4 mRNA by >10-fold, and IL-4 mRNA was barely detectable when 10 μM pPCC was used for priming. This early IL-4 mRNA expression was independent of IL-4 as shown by the failure of anti–IL-4 to diminish the level of IL-4 mRNA in cells that were stimulated for 24 h with 0.01 μM pPCC (Fig. 2 C).
Figure 2.

Peptide concentration-dependent inhibition of early IL-4 and GATA-3 mRNA expression. (A) Naive line 94 CD4+ T cells were stimulated with P13.9 cells in the presence of 0.001–10 μM of pPCC. Total RNA was harvested at indicated time points and relative mRNA levels for IL-4, GATA-3, IL-2, and IFN-γ were analyzed by real-time PCR. (B) Naive line 94 CD4+ T cells were stimulated with 0.01 or 10 μM pPCC on P13.9 cells. Cells were harvested 20 h after stimulation, and CD4 and GATA-3 were visualized by confocal microscopy. (C) Naive line 94 CD4+ T cells were stimulated with 0.01 μM pPCC on P13.9 cells in the presence or absence of anti–IL-4 (10 μg/ml), anti–IL-2 (10 μg/ml) plus anti-CD25 (10 μg/ml), or rat IgG1 (10 μg/ml) plus rat IgG2a (10 μg/ml) as isotype controls. Total RNA was harvested at indicated time points, and relative mRNA levels for IL-4 and GATA-3 were analyzed by a real-time PCR. (D) CFSE-labeled naive line 94 CD4+ T cells were stimulated as described in C. Intracellular IL-4 and IFN-γ were stained after restimulation. All experiments were carried out at least three times with consistent results.
The induction of GATA-3 mRNA followed a similar dose-dependent pattern to that of IL-4 mRNA (Fig. 2 A). GATA-3 mRNA first exceeded the level found in naive line 94 CD4+ T cells at 14 h of stimulation with 0.01 μM pPCC. GATA-3 mRNA levels increased at 24 h after stimulation. Cells stimulated with >1 μM pPCC failed to express GATA-3 mRNA. Analysis of GATA-3 protein by confocal microscopy indicated that it was expressed in the nucleus of line 94 CD4+ T cells stimulated for 20 h with 0.01 μM, but not 10 μM, pPCC (Fig. 2 B). The 24-h expression of GATA-3 mRNA and protein induction was not inhibited by anti–IL-4 (Fig. 2 C; not depicted).
IFN-γ mRNA levels were extremely low throughout the first 24 h of culture; there was no significant difference in IFN-γ mRNA expression among cell populations that were stimulated with different concentrations of pPCC (Fig. 2 A). IL-2 mRNA was induced by 6 h of culture and showed an entirely different concentration dependence from that of IL-4 mRNA (Fig. 2 A). IL-2 mRNA was maximal at 1 and 10 μM and >10-fold lower at 0.01 μM pPCC.
Expression of IL-4 and GATA-3 after 24 h is IL-4 dependent
Culturing naive line 94 CD4+ T cells with 0.01 μM peptide in the presence of anti–IL-4 did not affect the amount of IL-4 or GATA-3 mRNA at 24 h. However, the presence of anti–IL-4 prevented the increase in IL-4 and GATA-3 mRNA levels that were observed at 48 and 72 h when “early” IL-4 was not neutralized (Fig. 2 C). Furthermore, if IL-4 was added from the outset of the culture, the amount of IL-4 and GATA-3 mRNA that was expressed by the cells at all time points throughout 72 h was increased further (unpublished data). These results indicate that, in cells cultured under neutral conditions, the early “IL-4–independent” IL-4 that is produced is essential for the continued production of IL-4 and the continued expression of GATA-3 by these cells. Anti–IL-4 reduced the frequency of cells producing IL-4 in response to challenge with pPCC (0.01 μM) at 72 h from 35.6 to 4.8% (Fig. 2 D). This implied that the further induction of IL-4 by endogenous “early” IL-4 is essential for the differentiation of naive CD4+ T cells into cells that are capable of secreting large amounts of IL-4.
IL-2 is essential for early IL-4, but not GATA-3, induction
Adding anti–IL-2 and anti-CD25 to cells that were cultured under neutral conditions completely inhibited the early (24 h) appearance of IL-4 mRNA but had no inhibitory effect on early GATA-3 mRNA (Fig. 2 C) or protein expression (not depicted). At 48 h, IL-4 mRNA still was not observed and GATA-3 mRNA levels failed to increase (Fig. 2 C), presumably because of the lack of IL-4, which is essential for the further increase of GATA-3 mRNA (19). The effect of neutralizing IL-2 on early IL-4 mRNA expression could not be accounted for by its effects on cell growth, because there was no cell division during the first 24 h of culture—as measured by CFSE dilution—and cell yield at 24 h was not affected by the addition of anti–IL-2 and anti-CD25 (unpublished data). Together, these results imply that early IL-4 production depends on the induction of GATA-3 and on an IL-2–mediated event.
GATA-3–deficient naive CD4+ T cells fail to produce IL-4 in response to in vitro challenge
Although GATA-3 was demonstrated to play a critical role in Th2 differentiation (19–22), it has remained unclear whether GATA-3 is involved in IL-4–independent early IL-4 production. To address this question, we crossed mice that were homozygous for a “floxed” Gata3 gene (22) onto CD4-Cre transgenic mice (Gata3 f/fCD4-Cre mice) (23), so that the Gata3 gene could be deleted in peripheral T cells. These mice possess few CD4+ T cells (not depicted), and most of those that are present exhibit a memory or effector/memory phenotype (Fig. 3 A). We purified CD44lowCD62LhighCD4+ T cells from these mice and from mice that were homozygous for the floxed gene without CD4-Cre (Gata3 f/f) (Fig. 3 A). Such “naive” cells derived from Gata3 f/fCD4-Cre mice failed to express IL-4 mRNA 24 h after stimulation, whereas those derived from Gata3 f/f mice expressed considerable amounts of IL-4 mRNA in response to immobilized anti-CD3 and anti-CD28; this IL-4 mRNA expression was not inhibited by anti–IL-4 (Fig. 3 B). Thus, deletion of GATA-3 prevented early expression of IL-4 mRNA in response to TCR-mediated stimulation. Cells from Gata3 f/fCD4-Cre mice were equivalent to those from Gata3 f/f mice in their capacity to induce IL-2 mRNA in response to anti-CD3 and anti-CD28 (Fig. 3 B), which indicated that loss of GATA-3 does not influence the degree of T cell activation.
Figure 3.

Naive CD4+ T cells lacking Gata3 gene fail to produce early IL-4. (A) CD44 and CD62L expression by unsorted and FACS-sorted CD4+ T cells from Gata3 f/f mice and Gata3 f/fCD4-Cre mice. (B) Naive CD4+ T cells from Gata3 f/f mice and Gata3 f/fCD4-Cre mice were stimulated with immobilized anti-CD3 (1 μg/ml) and anti-CD28 (3 μg/ml) in the presence or absence of 10 μg/ml anti–IL-4. Cells were harvested 24 h after stimulation and relative mRNA levels for IL-4 and IL-2 were measured by real-time PCR. *not detectable, N.D., not done. The experiment was performed twice with consistent results.
High concentrations of pPCC inhibit STAT5 phosphorylation in cells cultured for 24 h
High concentrations of peptide block early GATA-3 expression and inhibit the responsiveness of the cells to IL-2. Thus, when naive line 94 CD4+ T cells were cultured for 24 h with P13.9 cells that had been preloaded with 0.01 μM pPCC, the CD25+ cells that arose as a result of stimulation could be stained with an anti–phospho-STAT5 antibody (Fig. 4). This staining did not occur if the cells had been incubated in anti–IL-2 and anti-CD25 during the culture period, and was not observed in the CD25− cells; this indicated that staining for phospho-STAT5 is specific for IL-2–mediated event. Neutralizing IL-2 and blocking CD25 did not diminish the frequency of CD25+ cells at 24 h substantially. Although high concentrations of peptide caused the expression of >10-fold more IL-2 mRNA than did low concentrations (Fig. 2 A), naive CD4+ T cells stimulated with high concentrations of peptide failed to show phospho-STAT5 among their CD25+ cells; this implied that high concentrations of peptide “desensitized” the IL-2 receptor. Thus, the failure of naive CD4+ T cells that were stimulated with high concentrations of peptide to produce early IL-4 could be attributed to the failure to induce GATA-3 and to transduce IL-2R–mediated signaling.
Figure 4.

Stimulation with high concentrations of peptide renders T cells unresponsive to IL-2. Naive line 94 CD4+ T cells were stimulated with 0.01 or 10 μM pPCC on P13.9 cells in the presence or absence of anti–IL-2 (10 μg/ml) and anti-CD25 (10 μg/ml). Cells were harvested 24 h after stimulation; fixed; permeabilized; and then stained for CD4, CD25, and phospho-STAT5. The number in the upper right quadrant indicates the proportion of phospho-STAT5+ cells within CD25+ cells that arose as a result of activation. The experiment was performed three times with consistent results.
A MEK inhibitor allows naive CD4+ T cells that are stimulated with high concentrations of peptide to produce early IL-4
To examine whether mitogen-activated protein kinase (MAPK) pathways mediate high peptide concentration-dependent suppression of IL-4–independent early IL-4 production, naive line 94 CD4+ T cells were pretreated with specific inhibitors of p38 MAPK, c-Jun N-terminal kinase (JNK), or MEK, the upstream regulator of ERK, and stimulated with 10 μM pPCC. Inhibiting the p38 MAPK or JNK pathways did not allow such “high-dose” cells to express IL-4 mRNA at 24 h (Fig. 5 A), or to develop into IL-4-producers at 72 h (Fig. 5 B). However, a MEK inhibitor (U 0126) caused a dose-dependent increase in IL-4 mRNA expression at 24 h (Fig. 5 A), and resulted in priming for high-rate IL-4–producing cells (Fig. 5 B).
Figure 5.

Activation of the ERK pathway is responsible for high peptide concentration–mediated suppression of IL-4 production. (A) Naive line 94 CD4+ T cells that were pretreated with different concentrations of U 0126, SB 203580, or SP 600125 were stimulated with P13.9 cells that had been preloaded with 10 μM pPCC. Cells were harvested 24 h after stimulation, and the relative mRNA levels for IL-4 and GATA-3 were measured by real-time PCR. (B) Naive line 94 CD4+ T cells were stimulated as described in A. Intracellular staining for IL-4 and IFN-γ was carried out after restimulation. (C) Naive line 94 CD4+ T cells were pretreated with U 0126 (3 μM) or DMSO and stimulated with P13.9 cells that were preloaded with 10 μM pPCC. Cells were harvested 20 h after stimulation, and CD4 and GATA-3 were visualized by confocal microscopy. (D) Naive line 94 CD4+ T cells were stimulated as in (C). Cells were harvested 24 and 48 h after stimulation; fixed; permeabilized; and then stained for CD4, CD25, and phospho-STAT5. The number in the upper right quadrant indicates the proportion of phospho-STAT5+ cells within CD25+ cells that arose as a result of activation. Quadrant location was determined based on the dot plot analysis for the cells that were stimulated in the presence of anti–IL-2 (10 μg/ml) and anti-CD25 (10 μg/ml). All experiments were carried out at least three times with consistent results.
U 0126 pretreatment also restored early GATA-3 mRNA expression in a dose-dependent manner, whereas neither the p38 inhibitor nor the JNK inhibitor induced the expression of GATA-3 mRNA (Fig. 5 A). Restoration of GATA-3 protein expression by U 0126 was confirmed by confocal microscopic analysis (Fig. 5 C).
Moreover, blockade of the ERK pathway allowed cells that were stimulated with high concentrations of peptide to respond to IL-2. Thus, naive line 94 CD4+ T cells without U 0126 pretreatment failed to show IL-2–driven STAT5 phosphorylation at 24 h, although they did express phospho-STAT5 at 48 h (Fig. 5 D). By contrast, in cells that were pretreated with U 0126, a small, but significant, degree of IL-2–driven STAT5 phosphorylation was observed at 24 h; phosphorylation was enhanced further at 48 h (Fig. 5 D).
These results indicate that ERK activation, as a result of stimulation with high concentrations of peptide, suppresses early GATA-3 mRNA induction and IL-2R–mediated signaling, which jointly result in the failure of “high-dose” cells to produce early IL-4. Stimulation of line 94 CD4+ T cells with 10 μM pPCC induced striking phosphorylation of ERK, as shown by flow cytometric analysis with an anti–phospho-ERK antibody (Fig. 6; open line graphs). By contrast, little or no ERK phosphorylation was detected in cells that were stimulated with 0.01 μM pPCC at any time from 2 min to 2 h after stimulation (Fig. 6; open line graphs). Pretreatment of line 94 CD4+ T cells with U 0126 blocked TCR-induced ERK phosphorylation in response to low and high concentrations of pPCC (Fig. 6; shaded line graphs).
Figure 6.

Intense phosphorylation of ERK induced by high concentrations of peptide. Line 94 CD4+ T cells were pretreated with U 0126 (3 μM) or DMSO and stimulated with P13.9 cells that were preloaded with 0.01 or 10 μM pPCC. Cells were fixed at indicated time points, and intracellular staining for phospho-ERK was performed. The levels of phospho-ERK in U 0126- and DMSO-pretreated cells are shown in shaded and open line graphs, respectively. The experiment was carried out three times with consistent results.
Blockade of the ERK pathway inhibits priming for IL-4–producing cells at low concentrations of peptide
We examined how blockade of the ERK pathway influenced the priming of naive CD4+ T cells with different concentrations of peptide. Thus, CFSE-labeled naive line 94 CD4+ T cells that were pretreated with U 0126 were cultured for 72 h with P13.9 cells that had been loaded with 0.001–10 μM pPCC. U 0126 pretreatment strikingly enhanced the frequency of IL-4–producing cells at 0.1 μM or more pPCC, but dramatically suppressed the frequency of IL-4 producers at 0.01 μM or less pPCC, with only a relatively modest effect on cell division (Fig. 7 A).
Figure 7.
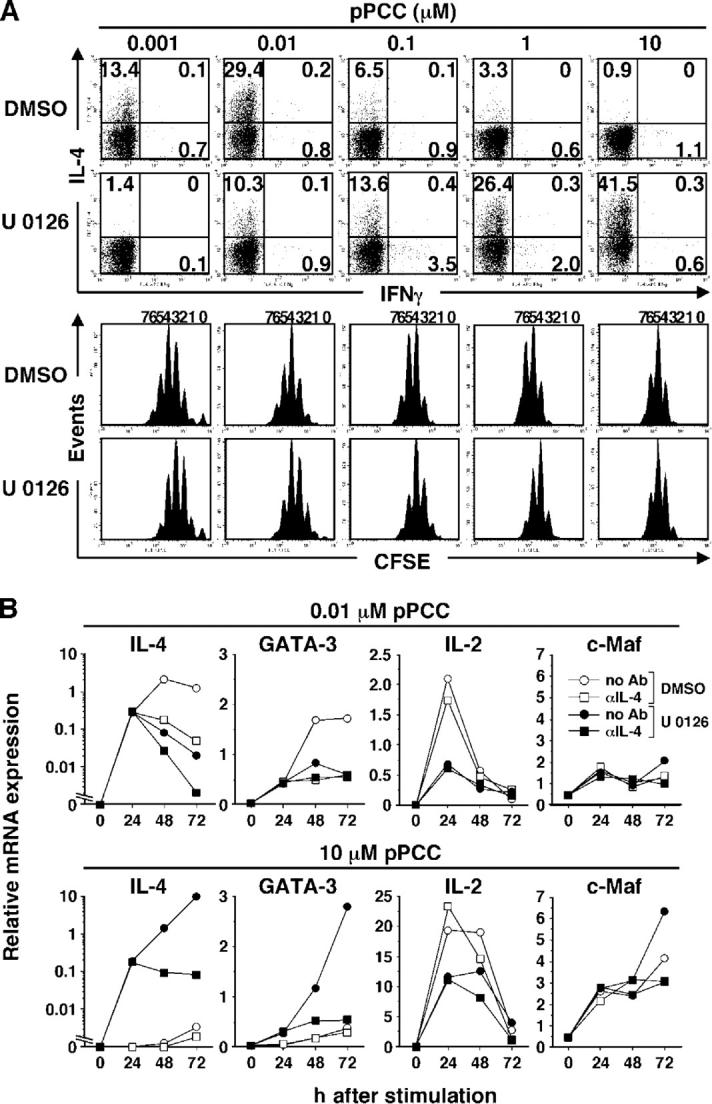
U 0126 allows IL-4 priming in response to high concentrations of pPCC but blocks such priming in response to low concentrations. (A) CFSE-labeled naive line 94 CD4+ T cells were pretreated with U 0126 (3 μM) or DMSO, and stimulated with P13.9 cells that were preloaded with 0.001–10 μM pPCC. Intracellular IL-4 and IFN-γ were stained after restimulation. (B) U 0126 (3 μM)- or DMSO-pretreated naive line 94 CD4+ T cells were stimulated with P13.9 cells that were preloaded with 0.01 or 10 μM pPCC in the presence or absence of 10 μg/ml anti–IL-4. Total RNA was isolated at indicated time points, and relative mRNA levels for IL-4, GATA-3, IL-2, and c-Maf were analyzed by real-time PCR. All experiments were carried out three times with consistent results.
To examine how blocking the ERK pathway exerted different effects on priming for IL-4–producing cells when low and high concentrations of peptide were used, we carried out a kinetic analysis of IL-4 and GATA-3 mRNA by real-time PCR in cells that were pretreated with U 0126 and stimulated with 0.01 or 10 μM pPCC. U 0126 pretreatment had no inhibitory effect on IL-4 or GATA-3 mRNA expression in cells that were stimulated with 0.01 μM pPCC at 24 h, but it strikingly diminished expression at 48 and 72 h (Fig. 7 B). In contrast, U 0126 pretreatment allowed IL-4–independent expression of IL-4 and GATA-3 mRNA in cells that were stimulated with 10 μM pPCC at 24 h; this resulted in a further enhancement of both mRNAs at 48 and 72 h that largely was IL-4 dependent (Fig. 7 B).
c-Maf was shown to play an important role in IL-4 production; its expression is induced by TCR stimulation (24, 25). Therefore, it was possible that blocking the ERK pathway might down-regulate TCR-induced c-Maf expression at low peptide concentration, causing the failure to sustain IL-4 mRNA expression in the late phase of activation. However, we did not observe any decrease in c-Maf mRNA expression in cells that were pretreated with U 0126 and stimulated with 0.01 μM pPCC (Fig. 7 B). Moreover, the level of c-Maf mRNA expression in cells that were stimulated with 10 μM pPCC was higher than that in cells with 0.01 μM pPCC (Fig. 7 B). Thus, it is unlikely that c-Maf is involved in TCR-induced early IL-4 production or that inhibition of c-Maf accounts for U 0126-mediated diminution in late IL-4 production at low peptide concentration.
Exogenous IL-2 rescues U 0126-mediated suppression of Th2 priming at low peptide concentration
The diminished priming for high-rate IL-4–producing cells at low peptide concentration seemed to be accounted for by the diminution in IL-2 production by cells that were treated with the MEK inhibitor. This occurred at high and low peptide concentrations; however, the “inhibited” amounts of IL-2 mRNA at high peptide concentration still exceeded the “uninhibited” amounts of IL-2 mRNA in cells that were stimulated with 0.01 μM pPCC (Fig. 7 A). To determine whether U 0126-mediated suppression of IL-4 production in “low-dose” stimulated cells was due to diminution in IL-2 production, we supplemented these cultures with different concentrations of IL-2 at 24 h of stimulation. Addition of 100 U/ml of IL-2 at 24 h restored IL-4 and GATA mRNAs at 48 and 72 h. This restoration was IL-4 dependent, which implied that IL-2 addition allowed endogenous IL-4 production, which in turn, was essential to up-regulate IL-4 and GATA-3 mRNA (Fig. 8 A). Addition of IL-2 at 24 h also restored priming for high-rate IL-4–producing cells that were pretreated with U 0126 and stimulated with 0.01 μM peptide. 30 units of IL-2 caused partial restoration, whereas 100 units fully restored priming for IL-4–producing capacity. IL-2–mediated restoration of priming for high-rate IL-4–producing cells was blocked completely by the inclusion of anti–IL-4 (Fig. 8 B).
Figure 8.
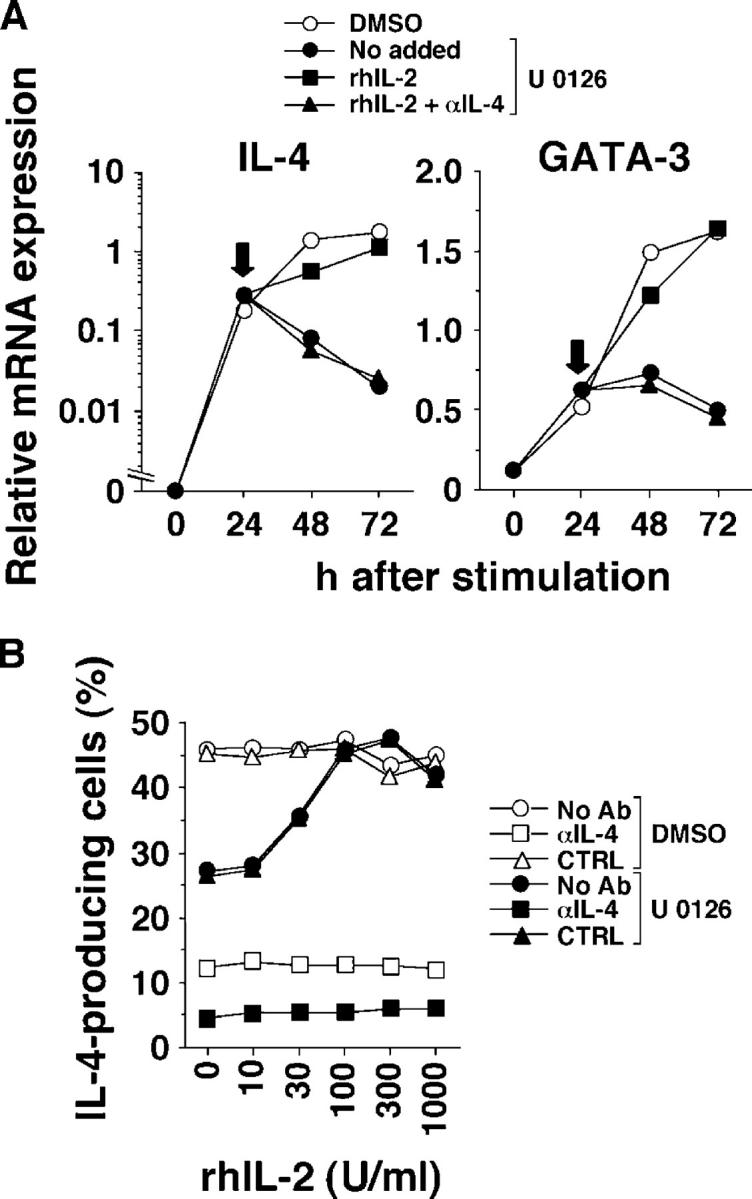
Exogenous IL-2 rescues U 0126-mediated suppression of Th2 priming at low peptide concentration. (A) U 0126 (3 μM)- or DMSO-pretreated naive line 94 CD4+ T cells were stimulated with P13.9 cells that were preloaded with 0.01 μM pPCC. At 24 h of stimulation (indicated by arrows), 100 U/ml rhIL-2 with or without 10 μg/ml anti–IL-4 were added to the cultures. Cells were harvested at indicated time points, and relative mRNA levels for IL-4 and GATA-3 were analyzed by real-time PCR. (B) Naive line 94 CD4+ T cells were stimulated as described in (A). Different concentrations of rhIL-2 with or without 10 μg/ml anti–IL-4 or rat IgG1 were added to the culture at 24 h. Intracellular IL-4 and IFN-γ were stained following restimulation. The frequency of IL-4–producing cells is shown. Both experiments were repeated three times with similar results.
Taken together, these data suggest that decreased amounts of IL-2, which results from the inhibition of the ERK pathway, is sufficient to allow the IL-4–independent “early” IL-4 production at low peptide concentration, but is not sufficient to sustain IL-4–dependent “late” IL-4 production.
MEK inhibition does not restore IL-4 production simply by lowering global T cell activation
Lck is activated in response to the ligation of TCR with antigen peptide–MHC class II complex, and plays an important role in the signal transduction through TCR. It was shown that agonistic peptide-induced activated ERK phosphorylates Ser59 of Lck and prevents it from inactivation by the SHP-1 phosphatase; this “protected” Lck is detectable as a 59-kD protein on SDS-PAGE (26). It is possible that blockade of the ERK pathway by a MEK inhibitor simply decreases the level of T cell activation by diminishing Lck activity, and allows naive CD4+ T cells that are stimulated with high concentrations of peptide to express early IL-4 mRNA. To determine whether the MEK inhibitor exerted its effect by simply diminishing the degree of T cell activation in response to 10 μM pPCC, we compared the relative amount of p59lck in cells that were stimulated with various concentrations of pPCC with that in cells that were pretreated with U 0126 and stimulated with 10 μM pPCC (Fig. 9 A). The relative amount of p59lck increased in a peptide concentration-dependent manner, and reached a plateau at 1 μM pPCC. U 0126 diminished the amount of p59lck in cells that were stimulated with 10 μM pPCC to an amount that was comparable to that induced by 0.1 μM pPCC. However, there were substantially greater amounts of p59lck in U 0126-pretreated cells that were stimulated with 10 μM pPCC compared with cells that were stimulated with 0.01 μM pPCC in the absence of inhibitor. Because the levels of IL-4 and GATA-3 mRNA in U 0126-pretreated cells that were stimulated with 10 μM pPCC were significantly higher than those in DMSO-pretreated cells that were stimulated with 0.1 μM pPCC and were comparable to those in cells that were stimulated with 0.01 μM pPCC (Fig. 9 B), it is unlikely that the effects of the MEK inhibitor can be ascribed to a global inhibition of T cell activation.
Figure 9.

Diminution in TCR-induced Lck activation by blocking the ERK pathway does not account for the restoration of early IL-4 mRNA expression at high peptide concentration. (A) Whole cell lysates from DMSO-treated naive line 94 CD4+ T cells that had been stimulated with various concentrations of pPCC on P13.9 cells, or those that were pretreated with 3 μM U 0126 and stimulated with 10 μM pPCC, were analyzed for Lck by Western blot. Comparable levels of ZAP70 indicate the equal loading of each sample. (B) U 0126 (3 μM)- or DMSO-pretreated naive line 94 CD4+ T cells were stimulated with 0.01–10 μM pPCC on P13.9 cells. Total RNA was isolated 24 h after stimulation, and relative mRNA levels for IL-4 and GATA-3 were analyzed by real-time PCR. Both experiments were carried out three times with consistent results.
DISCUSSION
In the present report, we provide evidence that peptide concentration regulates the level of IL-4–independent early GATA-3 expression and the degree of responsiveness to IL-2, both of which are indispensable for TCR-induced, IL-4–independent early IL-4 production by naive CD4+ T cells. Thus, low concentrations of peptide allow naive CD4+ T cells to express early GATA-3 and to respond to IL-2. Therefore, these cells are capable of producing early IL-4 and undergoing subsequent differentiation into high-rate IL-4–producing (Th2) cells. By contrast, stimulation with high concentrations of peptide abrogates early GATA-3 expression and the sensitivity to IL-2, and results in the failure to produce early IL-4. The preferential capacity of low peptide concentrations to induce Th2 responses also was observed by other investigators who used naive CD4+ T cells from different TCR transgenic mice (14, 27, 28). We also provide evidence that the degree of ERK activation accounts for peptide concentration–mediated early IL-4 transcription in naive CD4+ T cells.
The requirement of GATA-3 for early IL-4 transcription is based on the evidence that naive (CD44lowCD62Lhigh) CD4+ T cells purified from spleen and lymph nodes of Gata3 f/fCD4-Cre mice failed to induce early IL-4 mRNA when stimulated with immobilized anti-CD3 and anti-CD28, whereas such T cells from Gata3 f/f mice expressed considerable amounts of early “IL-4–independent” IL-4 mRNA. Moreover, there is much indirect evidence that early GATA-3 expression is required for low concentration–mediated induction of IL-4. Thus, the dose response curve of early GATA-3 and early IL-4 mRNA expression were the same, and GATA-3 mRNA appeared slightly earlier than did IL-4 mRNA (unpublished data). Although GATA-3 has been reported to play an important role in Th2 differentiation (19–22), our current work is the first to demonstrate clearly that GATA-3 is responsible for TCR-induced, IL-4–independent early IL-4 production by naive CD4+ T cells.
We have not clarified the mechanism by which TCR-mediated signaling induces early GATA-3 expression. Das et al. (29) reported that CD4+ T cells from NF-κB p50-deficient mice failed to express GATA-3 in the nucleus 96 h after stimulation with anti-CD3 and anti-CD28, and that these cells are impaired markedly in Th2 polarization. They also showed that CD4+ T cells that were pretreated with an inhibitor of NF-κB p50 did not express GATA-3 in the nucleus at 96 h. However, it remains unclear whether early GATA-3 induction is dependent on NF-κB p50. Previous reports have demonstrated that TCR-induced NF-κB activation is down-stream of protein kinase C θ activation (30), and that the degree of protein kinase C θ activation is directly dependent on the strength of TCR signaling (31). However, given the fact that early GATA-3 expression was suppressed in a peptide concentration–dependent manner, the degree of NF-κB activation may not account for TCR-induced early GATA-3 expression.
We showed in the present report that blockade of the ERK pathway allowed naive CD4+ T cells that were stimulated with high concentrations of peptide to induce GATA-3 mRNA and to express its protein in the nucleus. Komine et al. (32) reported that retroviral infection with Runx1 cDNA dramatically diminished the frequency of IL-4–producing cells by inhibiting GATA-3 mRNA expression under neutral and Th2-skewing conditions. This raised the possibility that the peptide concentration effect might be mediated by preferential induction of Runx1 at high peptide concentration. However, our real-time PCR analysis showed no significant difference in Runx1 mRNA levels between cells that were stimulated with low and high concentrations of peptide at 12–24 h of stimulation (unpublished data). Moreover, U 0126 pretreatment did not alter the levels of Runx1 mRNA expression at high peptide concentration. Thus, it is unlikely that Runx1 is responsible for high peptide concentration–mediated suppression of early GATA-3 expression.
Jorritsma et al. (17) showed that treatment of naive AND TCR transgenic CD4+ T cells with a MEK inhibitor (PD98059) during priming with relatively high concentrations (5 μg/ml) of moth cytochrome c peptide resulted in an increased ratio of nuclear JunB homodimers to JunB/c-Fos heterodimers that was capable of binding to the IL-4 promoter upon recall challenge with moth cytochrome c peptide. Therefore, they concluded that robust ERK activation inhibits IL-4 transcription by altering the pattern of distinct AP-1 complexes. However, we observed that 10 μM pPCC induced significantly higher nuclear JunB expression than did 0.01 μM pPCC at 12 to 24 h of priming, and that pretreatment with U 0126 did not alter the pattern of JunB expression (unpublished data). Moreover, 0.01 and 10 μM pPCC induced comparable levels of nuclear c-Fos expression, and pretreatment with U 0126 did not alter the pattern of c-Fos expression at low and high pPCC concentrations (unpublished data). Therefore, although it is possible that cells that have completed their differentiation to Th1 or Th2 phenotype have different AP-1 complexes that are capable of binding to the IL-4-promoter, it is unlikely that the formation of such different complexes is responsible for the peptide concentration–dependent early IL-4 production.
LeGros et al. (1) originally demonstrated that IL-4 and IL-2 are required for priming of CD4+ T cells to develop into IL-4–producing cells; however, the precise role of IL-2 in Th2 priming was not clarified. Recently, Cote-Sierra et al. (5) showed that IL-2 mediates its effect by stabilizing the accessibility of the Il4 gene after 48 h of priming. In the present report, we found that when naive line 94 CD4+ T cells were primed with 0.01 μM pPCC, induction of early IL-4 mRNA expression required IL-2, whereas early GATA-3 mRNA expression was independent of IL-2. We also observed that naive CD4+ T cells from IL-2−/− 5C.C7 TCR transgenic RAG2−/− mice failed to express “24-h” IL-4 mRNA in response to 0.01 μM pPCC, and that addition of exogenous IL-2 restored IL-4 mRNA expression. By contrast, IL-2−/− cells expressed levels of GATA-3 mRNA that were comparable to WT cells; addition of exogenous IL-2 did not enhance its expression (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20051304/DC1). Although the importance of STAT5, especially STAT5a, in Th2 differentiation has been reported (33, 34), it remains to be elucidated whether IL-2–driven STAT5 activation is involved in early IL-4 production.
In conclusion, peptide concentration–mediated strength of TCR signaling regulates early IL-4 production by naive CD4+ T cells by controlling the levels of ERK activation which, in turn, influence early GATA-3 expression and responsiveness to IL-2. Weak and transient ERK activation that is induced by low concentrations of peptide allows naive CD4+ T cells to express early GATA-3 and to respond to endogenous IL-2, both of which are required for early IL-4 production. This endogenously produced early IL-4 is required for priming of CD4+ T cells to develop into high-rate IL-4–producing (Th2) cells. By contrast, intense and sustained ERK activation that is induced by high concentrations of peptide inhibits early GATA-3 expression and transiently desensitizes the IL-2R; this results in the failure of naive CD4+ T cells to produce early IL-4 and to undergo subsequent Th2 differentiation.
Biologically, these results imply that low concentration challenge of individuals without overt activation of Toll-like receptors would lead to preferential priming of naive CD4+ T cells to develop into a Th2 phenotype, and thus, predispose to allergic inflammatory response. Bottomly and colleagues argued that low concentrations of LPS may be required to allow mice to develop an antigen-stimulated airway hypersensitivity response in vivo (35, 36). Addition of LPS (10 ng/ml) did not alter the overall antigen concentration effects described here (unpublished data). This suggests that peptide concentration/strength of TCR signaling may be a central element in determining in vivo priming for Th2 responses, and thus, bias toward allergic inflammation.
MATERIALS AND METHODS
Mice.
RAG2−/− B10.A mice transgenic for a 5C.C7 TCR specific for pPCC on I-Ek (line 94 mice), IL-2−/− 5C.C7 TCR transgenic RAG2−/− B10.A mice (line 110 mice), and B10.A mice were obtained from Taconic. Gata3 f/f mice generated in our lab (22) were crossed onto CD4-Cre transgenic mice on a C57BL/6 background (23). All mice were bred and maintained under specific pathogen-free conditions in the National Institute of Allergy and Infectious Diseases Animal Facility and used at 8–12 wk of age.
Culture media.
RPMI 1640 was purchased from Biosource and supplemented with 10% FCS, 2-ME, glutamine, penicillin, streptomycin, and sodium pyruvate (referred to as cRPMI).
Reagents.
FITC-anti-CD3, -CD8, -CD11b, -CD16/32, -CD24, -CD25, -B220, -NK1.1, -DX5, -Ly-6C/6G, -I-Ak, -I-Ab; AlexaFluor 488–anti-CD4; PE–anti-CD62L, –IL-4, –goat-anti–mouse Ig, -streptavidin; CyChrome-anti-CD44, -CD4; APC–anti-CD4, –IFN-γ; biotinylated anti-CD11c; purified anti–phospho-STAT5 (clone 47), -ZAP70 (clone 29); neutralizing anti–IL-2; blocking anti-CD25; rat IgG1; and rat IgG2a for isotype controls were purchased from BD Biosciences. Anti–IL-4, anti–IL-12, anti–IFN-γ, anti-CD3, and anti-CD28 were purified from ascites by Harlen. Anti–phospho-ERK mAb (E10) was purchased from Cell Signaling Technology. Mitomycin C, SB 203580, and SP 600125 were purchased from Calbiochem. Anti–GATA-3 mAb (HG3-31) was purchased from Santa Cruz Biotechnology, Inc. CFSE and AlexaFluor 647 donkey-anti–mouse IgG were purchased from Invitrogen. U 0126 and DAPI were purchased from Promega and Sigma-Aldrich, respectively. pPCC (residues 88-104; KAERADLIAYLKQATAK) was synthesized by American Peptide Company.
Isolation of naive CD4+ T cells and splenic DCs.
CD4+ T cells were isolated from lymph nodes of line 94 mice or line 110 mice by negative selection using FITC-conjugated mAbs to CD8, B220, I-Ak, CD16/32, NK1.1, and CD24, followed by incubating with anti-FITC Microbeads (Miltenyi Biotec) and passing through a MACS LS column (Miltenyi Biotec). Isolated cells were stained with PE–anti-CD62L and CyChrome anti-CD44, and then CD44lowCD62Lhigh CD4+ T cells were sorted by FACS Vantage III SE (Becton Dickinson) and used as naive CD4+ T cells. In some experiments, naive CD4+ T cells were labeled with 1.25 μM CFSE before use.
When we used Gata3 f/fCD4-Cre mice and Gata3 f/f mice, CD4+ T cells were isolated from spleen and lymph nodes by passing through a Mouse T Cell Enrichment Column (R&D Systems), followed by negative selection as described above, except FITC-anti-I-Ab was used instead of FITC-anti-I-Ak, and FITC–anti-DX5, -CD11b and -Ly-6C/6G were added. Isolated cells were stained with PE–anti-CD62L, Cy-Chrome–anti-CD44, and APC–anti-CD4. CD4+CD44lowCD62Lhigh cells were sorted by FACS Vantage III SE (Becton Dickinson) and used as naive CD4+ T cells.
DCs were isolated as follows. Spleens of B10.A mice were treated with Liberase Brendzyme 2 and DNase I (Roche Applied Science), and CD11c+ cells were isolated by positive selection using anti-CD11c (N418) Microbeads (Miltenyi Biotec) and a MACS LS column. Isolated cells were stained with biotinylated anti-CD11c (HL3) followed by PE-streptavidin and FITC–anti-DX5, –anti-NK1.1, and –anti-CD3. CD11c+, DX5−, NK1.1−, and CD3− cells were sorted by FACS Vantage III SE (Becton Dickinson), and used as splenic DCs.
Priming of naive CD4+ T cells.
Naive CD4+ T cells (5 × 105) from line 94 mice or line 110 mice were cultured in 1 ml of cRPMI in a 48-well plate with P13.9 fibroblast cells (1.25 × 105) stably expressing I-Ek, CD80, and CD54 (37) that had been treated with 25 μg/ml mitomycin C and loaded with 0.001–10 μM pPCC, or with splenic DCs (5 × 104) in the presence of 0.001–10 μM pPCC. In some experiments, naive CD4+ T cells were pretreated with U 0126 (MEK inhibitor), SB 203580 (p38 MAPK inhibitor), SP 600125 (JNK inhibitor), or DMSO (Sigma-Aldrich) at 37°C for 1 h. The final concentration of DMSO was 0.03%; this amount of DMSO did not affect T cell growth or viability (unpublished data). When indicated in figure legends, the following were added to the priming culture: neutralizing mAbs to IL-2, IL-4, IL-12, or IFN-γ; blocking mAb to CD25 (10 μg/ml for all mAbs); or isotype-matched control mAbs.
Naive CD4+ T cells (1 × 105) from Gata3 f/fCD4-Cre mice or Gata3 f/f mice were stimulated with immobilized anti-CD3 (1 μg/ml) and anti-CD28 (3 μg/ml) in the presence of recombinant human IL-2 (100 U/ml) with or without anti–IL-4 (10 μg/ml).
Quantitative RT-PCR.
Stimulated cells were lysed in TRIzol (Invitrogen) and total RNA was isolated using RNeasy Midi Kit (QIAGEN) following the manufacturer's instructions. One microgram of total RNA was reverse-transcribed to cDNA using SuperScript II First-Strand Synthesis System for RT-PCR (Invitrogen). Quantitative PCR was performed on a 7900HT sequence detection system (Applied Biosystems). The primer and probe sets for detecting murine IL-2, IL-4, and IFN-γ (FAM-MGB probe), and TaqMan Ribosomal RNA Control Reagents for detecting the 18S ribosomal RNA (VIC-MGB probe) were purchased from Applied Biosystems. The sequences of primer and MGB probe for GATA-3 (34) and c-Maf (38) are described elsewhere. The levels of mRNA for cytokines and transcription factors were normalized to that of 18S ribosomal RNA.
Intracellular staining for IL-4/IFN-γ, phospho-STAT5, and phospho-ERK.
Primed T cells were harvested 72 h after stimulation, washed extensively with HBSS, and cultured overnight in 2 ml of cRPMI containing 100 U/ml rhIL-2 in a 24-well plate. Cells were harvested, washed with HBSS, and restimulated with immobilized anti-CD3 and anti-CD28 (3 μg/ml of each mAb) for 6 h. For the last 2 h, 2 μM monensin (Calbiochem) was added. Cells were harvested; fixed with 4% paraformaldehyde (PFA); permeabilized with 0.5% Triton X-100 and 0.1% BSA in PBS; and stained with PE–anti-IL-4, CyChrome–anti-CD4, and APC–anti-IFN-γ.
For phospho-STAT5 staining, 1 × 106 line 94 CD4+ T cells that were pretreated with U 0126 (3 μM) or DMSO were stimulated with 2.5 × 105 P13.9 cells that were preloaded with 0.01 or 10 μM pPCC in the presence or absence of anti–IL-2 plus anti-CD25. Cells were harvested 24 and 48 h after stimulation; fixed and permeabilized as described above; and then stained with anti–phospho-STAT5 mAb followed by FITC-anti-CD25 (7D4), PE-anti–mouse Ig, and CyChrome–anti-CD4. CD25 expression was detected on cell surface, not in cytoplasm, by a confocal microscope (unpublished data).
For phospho-ERK staining, 1 × 106 line 94 CD4+ T cells that were pretreated with U 0126 (3 μM) or DMSO were added to 2.5 × 105 P13.9 cells that were preloaded with 0.01 or 10 μM pPCC in a 1.5-ml microcentrifuge tube, spun at 12,000 rpm for 10 s, and incubated at 37°C. Cells were fixed with 4% PFA at various time points, permeabilized as described above, and stained with anti–phospho-ERK mAb followed by PE-anti–mouse Ig and CyChrome–anti-CD4.
Stained cells were acquired by a FACSCalibur (Becton Dickinson); the data were analyzed using a CELLQuest software (Becton Dickinson) by gating on CD4+ T cells.
Confocal microscopic analysis for GATA-3 protein expression.
U 0126 (3 μM)– or DMSO-pretreated line 94 CD4+ T cells (1 × 106) were stimulated with P13.9 cells that were preloaded with 0.01 or 10 μM pPCC. Cells were harvested 20 h after stimulation, mounted on a coverslip, and fixed with 4% PFA. Cells were permeabilized as described above, and stained with anti–GATA-3 mAb followed by AlexaFluor 488–anti-CD4, AlexaFluor 647 donkey–anti–mouse IgG, and DAPI. Stained cells were visualized with a Leica TCS-NT/SP confocal microscope with a 63× oil immersion objective. 20 to 30 z sections separated by 0.2 μm were acquired. Reconstitutions of images were performed by using an Imaris software system (BitPlane) and Photoshop software (Adobe Systems).
Western blot analysis for p56/p59lck.
Line 94 CD4+ T cells were stimulated as described in “phospho-ERK staining.” After 20 and 60 min of incubation, culture media was removed, and cells were treated with 40 μl of lysis buffer (1% NP-40, 10 mM Tris-HCl, 2 mM EDTA, 140 mM sodium chloride, 1 mM sodium orthovanadate) freshly supplemented with protease inhibitors (Complete Mini; Roche Applied Science) for 30 min on ice. After removal of debris by centrifugation at 12,000 rpm for 15 min, 20 μl of 3 × SDS sample buffer (Cell Signaling Technology) was added to whole cell lysates, and 20 μl of aliquots were used for electrophoresis on an 8% polyacrylamide gel (Invitrogen). Fractionated proteins were transferred onto a nitrocellulose membrane (Bio-Rad Laboratories). Lck was detected with rabbit anti-Lck polyclonal serum (BD Biosciences) followed by horseradish peroxidase–labeled goat anti–rabbit IgG (Bio-Rad), and visualized with SuperSignal West Dura Extended Duration Substrate (Pierce Chemical Co.). To confirm an equal loading, the probed membrane was stripped with Restore Western Blot Stripping Buffer (Pierce Chemical Co.) and reprobed with anti-ZAP70 mAb (BD Biosciences) followed by horseradish peroxidase–labeled goat anti–mouse IgG (Bio-Rad), and visualized with SuperSignal West Pico Extended Duration Substrate (Pierce Chemical Co.).
Online supplemental material.
Figs. S1 and S2 show the dot plot analysis for CFSE versus IL-4 and the histograms of CFSE dilution corresponding to Fig. 1, B and C, respectively. Fig. S3 shows the failure of IL-2−/− naive line 94 CD4+ T cells to produce early IL-4 in response to low concentrations of pPCC. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051304/DC1.
Acknowledgments
We thank Dr. R. Germain for providing the P13.9 cells and for many helpful discussions, Dr. C. Wilson for providing CD4-Cre transgenic mice, S. Tanksley for her excellent cell sorting, Drs. O. Schwartz and J. Kabat for assistance with confocal microscopic analysis, and Dr. I. Stefanova for technical advice for the detection of Lck by Western blot and critical reading of our manuscript. We thank all of the members of the Paul laboratory for their helpful discussion.
H. Yamane was a recipient of the Post-Doctoral Fellowship from Uehara Memorial Foundation.
The authors have no conflicting financial interests.
Abbreviations used: AP-1, activated protein-1; CFSE, 5,6-carboxyfluorescein diacetate succinimidyl ester; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase kinase ; MEK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase; PFA, paraformaldehyde; pPCC, pigeon cytochrome c peptide.
References
- 1.LeGros, G., S.Z. Ben-Sasson, R. Seder, F.D. Finkelman, and W.E. Paul. 1990. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-4 and IL-2 are required for in vitro generation of IL-4-producing cells. J. Exp. Med. 172:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swain, S.L., A.D. Weinberg, M. English, and G. Huston. 1990. IL-4 directs the development of Th2-like helper effectors. J. Immunol. 145:3796–3806. [PubMed] [Google Scholar]
- 3.Hsieh, C.-S., S.E. Macatonia, C.S. Tripp, S.F. Wolf, A. O'Garra, and K.M. Murphy. 1993. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 260:547–549. [DOI] [PubMed] [Google Scholar]
- 4.Seder, R.A., R. Gazzinelli, A. Sher, and W.E. Paul. 1993. IL-12 acts directly on CD4+ T cells to enhance priming for IFNγ production and diminish IL-4 inhibition of such priming. Proc. Natl. Acad. Sci. USA. 90:10188–10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cote-Sierra, J., G. Foucras, L. Guo, L. Chiodetti, H.A. Young, J. Hu-Li, J. Zhu, and W.E. Paul. 2004. Interleukin 2 plays a central role in Th2 differentiation. Proc. Natl. Acad. Sci. USA. 101:3880–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown, M.A., J.H. Pierce, C.J. Watson, J. Falco, J.N Ihle, and W.E. Paul. 1987. B cell stimulatory factor-1/interleukin-4 mRNA is expressed by normal and transformed mast cells. Cell. 50:809–818. [DOI] [PubMed] [Google Scholar]
- 7.Plaut, M., J.H. Pierce, C.J. Watson, H.J. Hanley, R.P. Nordan, and W.E. Paul. 1989. Mast cell lines produce lymphokines in response to cross-linkage of FcɛRI or to calcium ionophores. Nature. 339:64–67. [DOI] [PubMed] [Google Scholar]
- 8.Moqbel, R., S. Ying, J. Barkans, T.M. Newman, P. Kimmitt, M. Wakelin, L. Taborda-Barata, Q. Meng, C.J. Corrigan, S.R. Durham, and A.B. Kay. 1995. Identification of messenger RNA for IL-4 in human eosinophils with granule localization and release of the translated product. J. Immunol. 155:4939–4947. [PubMed] [Google Scholar]
- 9.Seder, R.A., W.E. Paul, A.M. Dvorak, S.J. Sharkis, A. Kagey-Sobotka, Y. Niv, F.D. Finkelman, S.A. Barbieri, S.J. Galli, and M. Plaut. 1991. Mouse splenic and bone marrow cell populations that express high-affinity Fcɛ receptors and produce interleulin 4 are highly enriched in basophils. Proc. Natl. Acad. Sci. USA. 88:2835–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voehringer, D., K. Shinkai, and R.M. Locksley. 2004. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 20:267–277. [DOI] [PubMed] [Google Scholar]
- 11.Min, B., M. Prout, J. Hu-Li, J. Zhu, D. Jankovic, E.S. Morgan, J.F. Urban Jr., A.M. Dvorak, F.D. Finkelman, G. LeGros, and W.E. Paul. 2004. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med. 200:507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshimoto, T., and W.E. Paul. 1994. CD4pos, NK1.1pos T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J. Med. Exp. 179:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Constant, S.L., and K. Bottomly. 1997. Induction of Th1 and Th2 CD4+ T cell response: the alternative approaches. Annu. Rev. Immunol. 15:297–322. [DOI] [PubMed] [Google Scholar]
- 14.Constant, S., C. Pfeiffer, T. Pasqualini, and K. Bottomly. 1995. Extent of T cell receptor ligation can determine the functional differentiation of naïve CD4+ T cells. J. Exp. Med. 182:1591–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao, X., C. Grant, S. Constant, and K. Bottomly. 1997. Induction of IL-4-producing CD4+ T cells by antigenic peptides altered for TCR binding. J. Immunol. 158:4237–4244. [PubMed] [Google Scholar]
- 16.Brogdon, J.L., D. Leitenberg, and K. Bottomly. 2002. The potency of TCR signaling differentially regulates NFATc/p activity and early IL-4 transcription in naïve CD4+ T cells. J. Immunol. 168:3825–3832. [DOI] [PubMed] [Google Scholar]
- 17.Jorritsma, P.J., J.L. Brogdon, and K. Bottomly. 2003. The role of TCR-induced extracellular signal-regulated kinase activation in the regulation of early IL-4 expression in naïve CD4+ T cells. J. Immunol. 170:2427–2434. [DOI] [PubMed] [Google Scholar]
- 18.Laouar, Y., F.S. Sutterwala, L. Gorelik, and R.A. Flavell. 2005. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat. Immunol. 6:600–607. [DOI] [PubMed] [Google Scholar]
- 19.Ouyang, W., S.H. Ranganath, K. Weindel, D. Bhattacharya, T.L. Murphy, W.C. Sha, and K.M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 9:745–755. [DOI] [PubMed] [Google Scholar]
- 20.Zheng, W.-P. and R.A. Flavell. 1997. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 89:587–596. [DOI] [PubMed] [Google Scholar]
- 21.Zhang, D.-H., L. Yang, L. Cohn, L. Parkyn, R. Homer, P. Ray, and A. Ray. 1999. Inhibition of allergic inflammation in a murine model of asthma by expression of a dominant-negative mutant of GATA-3. Immunity. 11:473–482. [DOI] [PubMed] [Google Scholar]
- 22.Zhu, J., B. Min, J. Hu-Li, C.J. Watson, A. Grinberg, Q. Wang, N. Killeen, J.F. Urban Jr., L. Guo, and W.E. Paul. 2004. Conditional deletion of GATA-3 reveals its critical role in Th1/Th2 responses. Nat. Immunol. 5:1157–1165 [DOI] [PubMed] [Google Scholar]
- 23.Lee, P.P., D.R. Fitzpatrick, C. Beard, H.K. Jessup, S. Lehar, K.W. Makar, M. Perez-Melgosa, M.T. Sweetser, M.S. Schlissel, S. Nguyen, et al. 2001. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 15:763–774. [DOI] [PubMed] [Google Scholar]
- 24.Ho, I.-C., M.R. Hodge, J.W. Rooney, and L.H. Glimcher. 1996. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 85:973–983. [DOI] [PubMed] [Google Scholar]
- 25.Kim, J.I., I.-C. Ho, M.J. Grusby, and L.H. Glimcher. 1999. The transcription factor c-Maf controls the production of interleukin-4 but not other Th2 cytokines. Immunity. 10:745–751. [DOI] [PubMed] [Google Scholar]
- 26.Stefanova, I., B. Hemmer, M. Vergelli, R. Martin, W.E. Biddison, and R.N. Germain. 2003. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 4:248–254. [DOI] [PubMed] [Google Scholar]
- 27.Noben-Trauth, N., J. Hu-Li, and W.E. Paul. 2002. IL-4 secreted from individual naïve CD4+ T cells acts in an autocrine manner to induce Th2 differentiation. Eur. J. Immunol. 32:1428–1433. [DOI] [PubMed] [Google Scholar]
- 28.Ise, W., M. Totsuka, Y. Sogawa, A. Ametani, S. Hachimura, T. Sato, Y. Kumagai, S. Habu, and S. Kaminogawa. 2002. Naïve CD4+ T cells exhibit distinct expression patterns of cytokines and cell surface molecules on their primary responses to varying doses of antigen. J. Immunol. 168:3242–3250. [DOI] [PubMed] [Google Scholar]
- 29.Das, J., C.-H. Chen, L. Yang, L. Cohn, P. Ray, and A. Ray. 2001. A critical role for NF-κB in Gata3 expression and TH2 differentiation in allergic airway inflammation. Nat. Immunol. 2:45–50. [DOI] [PubMed] [Google Scholar]
- 30.Sun, Z., C.W. Arendt, W. Ellmeier, E.M. Schaeffer, M.J. Sunshine, L. Gandhi, J. Annes, D. Petrzilka, A. Kupfer, P.L. Schwartzberg, and D.R. Littman. 2000. PKC-θ is required for TCR-induced NF-κB activation in mature but not immature T lymphocyte. Nature. 404:402–407. [DOI] [PubMed] [Google Scholar]
- 31.Isakov, N., and A. Altman. 2002. Protein kinase Cθ in T cell activation. Annu. Rev. Immunol. 20:761–794. [DOI] [PubMed] [Google Scholar]
- 32.Komine, O., K. Hayashi, W. Natsume, T. Watanabe, Y. Seki, N. Seki, R. Yagi, W. Suzuki, H. Tamauchi, K. Hozumi, S. Habu, M. Kubo, and M. Satake. 2003. The Runx1 transcription factor inhibits the differentiation of naïve CD4+ T cells into the Th2 lineage by repressing GATA3 expression. J. Exp. Med. 198:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagami, S., H. Nakajima, A. Suto, K. Hirose, K. Suzuki, S. Morita, I. Kato, Y. Saito, T. Kitamura, and I. Iwamoto. 2001. Stat5A regulates T helper cell differentiation by several distinct mechanisms. Blood. 97:2358–2365. [DOI] [PubMed] [Google Scholar]
- 34.Zhu, J., J. Cote-Sierra, L. Guo, and W.E. Paul. 2003. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 19:739–748. [DOI] [PubMed] [Google Scholar]
- 35.Eisenbarth, S.C., D.A. Piggott, J.W. Huleatt, I. Visintin, C.A. Herrick, and K. Bottomly. 2002. Lipopolysaccharide-enhanced, Toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 196:1645–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piggott, D.A., S.C. Eisenbarth, L. Xu, S.L. Constant, J.W. Huleatt, C.A. Herrick, and K. Bottomly. 2005. MyD88-dependent induction of allergic Th2 responses to intranasal antigen. J. Clin. Invest. 115:459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding, L., P.S. Linsley, L.Y. Huang, R.N. Germain, and E.M. Shevach. 1993. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J. Immunol. 151:1224–1234. [PubMed] [Google Scholar]
- 38.Guo, L., J. Hu-Li, and W.E. Paul. 2004. Probabilistic regulation of IL-4 production in Th2 cells: accessibility at the Il4 locus. Immunity. 20:193–203. [DOI] [PubMed] [Google Scholar]