

Abstract
Thymic stromal lymphopoietin (TSLP) is a cytokine that promotes CD4+ T cell homeostasis. We now demonstrate that TSLP is required to mount a normal CD4+ T cell–mediated inflammatory response. TSLP acts directly on naive, but not, memory CD4+ T cells, and promotes their proliferation in response to antigen. In addition, TSLP exerts an effect indirectly through DCs to promote Th2 differentiation of CD4+ T cells. Correspondingly, TSLP receptor (TSLPR) knockout (KO) mice exhibit strong Th1 responses, with high levels of interleukin (IL)-12, interferon-γ, and immunoglobulin (Ig) G2a, but low production of IL-4, -5, -10, -13, and IgE; moreover, CD4+ T cells from these animals proliferate less well in response to antigen. Furthermore, TSLPR KO mice fail to develop an inflammatory lung response to inhaled antigen unless supplemented with wild-type CD4+ T cells. This underscores an important role for this cytokine in the development of inflammatory and/or allergic responses in vivo.
Thymic stromal lymphopoietin (TSLP) is a cytokine that was identified as a growth factor in the supernatant of Z210R.1 thymic stromal cells that could support the development of immature NAG8/7 B cells to the B220+/IgM+ stage (1). The TSLP receptor complex is a heterodimer that is composed of the IL-7 receptor α chain (IL-7Rα) and a specific subunit, TSLPR (2, 3). TSLPR is most similar to the common cytokine receptor γ chain (γc), a protein that is a critical component of the receptors for IL-2, -4, -7, -9, -15, and -21 (4). TSLPR is expressed in many tissues, including liver, brain, testis, bone marrow, spleen, and thymus (2, 3, 5). Although TSLP shares some actions with IL-7 (6, 7) and mediates the activation of Stat5 proteins (8), there also is evidence for distinctive/nonredundant actions (9). TSLPR KO mice have normal lymphocyte numbers, but TSLPR/γc double KO mice display a greater defect than do γc KO mice, which indicates that TSLP contributes to lymphopoiesis independent of the contributions of IL-7 (10). Correspondingly, injection of TSLP into γc KO mice induces the expansion of T and B cells in vivo. TSLP enhances anti–CD3ɛ-induced proliferation and survival of CD4+, but not CD8+, single positive thymocytes and peripheral T cells in vitro, whereas IL-7 enhances the expansion of CD4+ and CD8+ T cells (10). Moreover, in adoptive transfer experiments, CD4+ T cells from TSLPR KO mice expand less efficiently than do WT CD4+ T cells in irradiated hosts, whereas CD8+ T cell expansion is not altered. Thus, murine TSLP has actions that are related to T and B cell lymphopoiesis (10).
Human TSLP can induce naive CD4+ T cells to generate the proallergic Th2 cytokines, IL-4, -5, and -13, while inhibiting the production of IL-10 and IFN-γ (11); however, this effect was an indirect effect of TSLP on DCs, rather than a direct effect on CD4+ T cells. Human DCs that are treated with TSLP also contribute to CD4+ T cell homeostasis (12). Furthermore, TSLP-activated DCs induce the differentiation of naive human CD8+ T cells into effectors that exhibit cytolytic activity and produce IL-5 and -13 (13). These reports suggested that human TSLP modulates immune function through activating DCs, but it was suggested that murine TSLP does not activate DCs (11).
We further investigate the role of murine TSLP in regulating the immune response. Our results indicate that TSLP enhances antigen-driven expansion of naive CD4+ T cells, but also can augment the activity of DCs. Moreover, we also found that TSLPR KO mice exhibit a markedly defective inflammatory lung response to inhaled antigen, which suggests an important role for TSLP in the development of this inflammatory response.
RESULTS
TSLP enhances proliferation of naive CD4+ T cells and generation of effector cells
Originally, murine TSLP was identified as a B cell growth factor (1, 6, 14), and subsequently was shown to act on CD4−CD8− double negative thymocytes in synergy with IL-1 (5). Subsequent work revealed that human TSLP can regulate CD4+ T cell function and homeostasis, in part by the activation of DCs (11, 12), and that murine TSLP plays an important role in promoting TCR-mediated CD4+ T cell homeostasis (10). To better understand the function of TSLP in immune responses and to clarify the CD4+ T cell populations on which it acts, we examined its effect on naive (CD62L+CD44low), central memory (CD62L+CD44hi), and effector memory (CD62L−CD44+) CD4+ T cell populations. TSLP significantly enhanced anti-CD3–induced proliferation of naive CD4+ T cells from BALB/c mice, but had little effect on the expansion of memory phenotype CD4+ T cells (Fig. 1 A).
Figure 1.

TSLP promotes the proliferation of naive CD4+ T cells. (A) Purified naive (CD4+CD62L+CD44−), central memory (CD4+CD62L+CD44+), and effector memory (CD4+CD62L−CD44+) T cells from WT BALB/c mice were activated for 48 h with 2 μg/ml anti-CD3 with or without 100 ng/ml TSLP and then pulsed with [3H]thymidine for 16 h. TSLP significantly increased the proliferation of naive CD4+ T cells (P = 0.01; top panel) but did not significantly affect memory CD4+ T cell proliferation (middle and bottom panels). (B) Defective CD69 induction in TSLPR KO mice. WT mice, and TSLPR KO mice expressing the DO11.10 transgene were injected i.p. with OVA and aluminum hydroxide (ALUM) and analyzed the next day. (C) Decreased antigen-induced CD4+ T cell proliferation in TSLPR KO mice. CD4+ T cells from DO11.10/WT and TSLPR KO mice were labeled with CFSE and transferred to WT hosts that were immunized with OVA the next day. On day 5, KJ1-26+ DO11.10/TSLPR KO cells had slower proliferation (mean fluorescence intensity (MFI) = 267 and percent gated 54%) than did DO11.10/WT cells (MFI = 12 and percent gated 98%) (cells from 7 out of 10 TSLPR KO mice showed a slower rate of CFSE loss than WT cells, whereas cells from 3 out of 10 did not). (D–F) WT and TSLPR KO mice (not expressing the DO11.10 transgene) were immunized with OVA (200 μg) and ALUM. Animals were killed on day 12 (D) or 60 (E) and splenocytes were cultured with OVA (0, 10, 50, and 200 μg/ml). Splenocytes from TSLPR KO mice displayed significantly lower proliferation in response to secondary encounter with antigen at all concentrations tested (P < 0.05 for all). (F) CD4+ T cells purified from mice 60 d after immunization were incubated with APCs (splenocytes that were depleted of T and NK cells) in the presence of different doses of OVA. CD4+ T cells from TSLPR KO mice displayed significantly lower proliferation in response to the OVA (200 μg/ml) in vitro (P < 0.01) than did CD4+ T cells from WT mice. Shown are means ± SEM for five experiments.
To analyze the role of TSLP in vivo, we crossed TSLPR KO mice to DO11.10 TCR transgenic (Tg) mice and analyzed TCR transgenic progeny that expressed (DO11.10/WT) or lacked (DO11.10/TSLPR KO) the TSLPR gene. Splenic CD4+ T cells from these mice expressed little, if any, CD69, an early T cell activation marker (Fig. 1 B, top). Injection of mice with OVA significantly induced CD69 on DO11.10/WT CD4+ T cells, but not on DO11.10/TSLPR KO CD4+ T cells (Fig. 1 B, bottom). This suggests that TSLP signaling is needed for the normal early activation of naive CD4+ T cells by antigen in vivo, although a developmental defect in the TSLPR KO mice is also possible. We next examined the ability of DO11.10/WT and TSLPR KO splenic T cells to proliferate in vivo by labeling them with (5,6-carboxyfluorescein diacetate-succinimidyl ester (CFSE) and transferring them to WT hosts that were immunized the next day. An examination of KJ1-26+ T cells at day 5 showed that TSLPR KO cells proliferated—as evidenced by the CFSE dilution—but at a slower rate than WT cells (Fig. 1 C), which indicated a role for TSLP in the expansion of CD4+ T cells.
We next examined the role of TSLP in TCR-driven generation of memory T cells from naive CD4+ T cells. WT and TSLPR KO mice on a BALB/c WT background (i.e., not expressing the DO11.10 transgene) were immunized with OVA. As evaluated by an in vitro antigen recall assay, splenocytes that were isolated from TSLPR KO mice 12 or 60 d after immunization had a lower proliferative response to secondary exposure to the antigen than did splenocytes from WT mice (Fig. 1, D and E). We next isolated CD4+ T cells 60 d after immunization and cultured these cells with APCs at a 1:1 ratio in the presence of OVA. CD4+ T cells from TSLPR KO mice showed much weaker proliferation in response to secondary antigen exposure than did cells from WT littermates (Fig. 1 F). These results suggest that TSLP is involved primarily in antigen-driven activation of naive CD4+ T cells and its absence substantially diminishes the generation of memory T cells.
TSLPR plays a role in the activation of DCs in mice
We next sought to clarify the effect of TSLP on the contribution of APCs to the CD4+ T cell expansion that occurs in response to secondary stimulation in vitro. WT and TSLPR KO mice were immunized with OVA, killed at day 60, and splenic CD4+ T cells were purified and incubated at a 1:1 ratio with APCs in the presence of OVA. WT CD4+ T cells cultured with WT APCs proliferated the most. Replacement of WT APCs with TSLPR KO APCs mildly, but significantly, reduced CD4+ T cell proliferation (Fig. 2 A). TSLPR KO CD4+ T cells showed the weakest proliferative response to antigen; this defect could not be corrected by introducing WT APCs (Fig. 2 A). Thus, TSLP is required for the proliferation of normal CD4+ T cells; the fact that proliferation of WT CD4+ T cells was decreased when TSLPR KO APCs were used suggests that TSLP signaling plays a role in promoting the optimal activity of APCs as well.
Figure 2.

TSLP activates murine DCs. (A) In vitro antigen recall response. CD4+ T cells and APCs from immunized WT and TSLPR KO mice were cultured with 200 μg/ml of OVA for 48 h and then pulsed with [3H]thymidine. Shown are means ± SEM for seven experiments. (B) Splenic CD11c+ cells were sorted from the spleens of WT BALB/c animals and incubated overnight with media, TSLP alone, 5 μg/ml of OVA323–339 peptide, or with OVA323–339 peptide plus TSLP. TSLP treatment increased the surface levels of CD80, MHC class II, and CD86. Numbers represent mean fluorescent intensity. (C) Splenic DCs were incubated with 5 μg/ml of OVA323–339 peptide alone or with TSLP before being washed, treated with mitomycin C, and cultured with purified CD4+ T cells from DO11.10 RAG2−/− mice at a 1:10 ratio. Antigen presentation of DCs was enhanced significantly by TSLP treatment (P = 0.005, shown are means ± SEM for five experiments). (D) CD4+ T cells and DCs were purified from DO11.01/WT and DO11.10/TSLPR KO unmanipulated mice, and these were cultured together in the indicated combinations at a ratio of 1:10 DC/T cell, with 5 μg/ml of OVA323–339 peptide, and [3H]thymidine incorporation determined at 48 h. Replacing WT APCs with TSLPR KO-derived DCs moderately, but significantly, reduced the antigen-driven proliferation of DO11.10 Tg/WT CD4+ T cells (P = 0.04). WT DCs provided no significant enhancement over the weak expansion of DO11.10/TSLPR KO CD4+ T cells. Shown are means ± SEM for seven experiments. (A and D) Asterisks indicate that the difference between WT CD4+/WT APCs or DCs and WT CD4+/KO APCs or DCs was statistically significant (P < 0.05).
Human TSLP was shown to activate DCs, but surprisingly it was suggested that murine TSLP did not show this ability (11). In view of our findings above, we investigated more formally the effect of murine TSLP on WT DCs. We first confirmed that WT DCs express TSLPR mRNA using RT-PCR (unpublished data). Moreover, the addition of TSLP moderately increased the cell surface expression of CD80, CD86, and MHC II (Fig. 2 B) on WT splenic CD11c+ DCs that were activated with OVA323–339 peptide for 24 h. As compared with untreated DCs, TSLP-treated DCs significantly enhanced OVA-mediated proliferation of DO11.10 TCR transgenic CD4+ T cells when the DCs and T cells were incubated in a 1:10 ratio (Fig. 2 C); this clearly demonstrated that mouse TSLP can activate DCs.
To examine further the effect of TSLP on the ability of DCs to activate CD4+ T cells, we isolated cells from naive DO11.10/WT and DO11.10/TSLPR KO spleens, and cultured DCs with CD4+ T cells at a 1:10 ratio in the presence of 5 μg/ml of OVA323–339 peptide. As expected, DO11.10/WT CD4+ T cells that were cultured with WT DCs showed substantial proliferation (Fig. 2 D). This was reduced slightly, but significantly, when DO11.10/TSLPR KO DCs were used (Fig. 2 D). The weak expansion seen when TSLPR KO CD4+ T cells were incubated with TSLPR KO DCs was not enhanced significantly when WT DCs were used. These results indicate that TSLP activates murine DCs, but that the presence of TSLPR on CD4+ T cells is more vital than its presence on DCs for optimal proliferation in response to the OVA323–339 peptide; this is consistent with the results shown in Fig. 2 A.
TSLP-treated DCs reduce IFN-γ production by CD4+ T cells
Because TSLPR enhances naive CD4+ T cell proliferation, we examined if TSLP could influence TCR-induced cytokine production by these cells. Purified naive CD4+CD62L+ CD44low and effector memory CD4+CD62L−CD44hi splenic T cells from WT BALB/c mice were activated with anti-CD3 in the presence or absence of TSLP. On day 4, cells were stimulated with PMA plus ionomycin for 5 h, and intracellular levels of IFN-γ and IL-4 were determined. TSLP did not affect the level of IFN-γ or IL-4 in naive or memory CD4+ T cells under neutral conditions (Fig. 3 A) or when cells were activated using Th1 (IL-12 plus anti–IL-4) or Th2 (IL-4 plus anti–IFN-γ) polarizing conditions (Fig. 3 B).
Figure 3.

DCs activated with TSLP negatively regulate IFN-γ production by naive CD4+ T cells. In panels A, B, C, and E, cells were cultured as indicated and subsequently challenged with PMA+ ionomycin, and the intracellular levels of IFN-γ and IL-4 were measured by intracellular staining. Numbers indicate the percent of cells in the indicated quadrant. (A) Naive and memory CD4+ T cells (>99% pure) were isolated from WT animals and treated with anti-CD3 with or without TSLP for 4 d. The addition of TSLP had no effect on the levels of IFN-γ and IL-2 that were produced by CD4+ T cells. (B) Purified CD4+ T cells were activated with anti-CD3/anti-CD28 under Th1 (IL-12 and anti–IL-4) or Th2 (IL-4 and anti–IFN-γ) polarizing conditions with or without TSLP. IL-2 was added on day 2, and cells were allowed to grow for 1 wk. TSLP did not affect the IFN-γ or IL-4 production by these polarized cells. (C) Sorted splenic CD11c+ DCs were incubated with 5 μg/ml of OVA323–339 peptide alone or with TSLP before being washed, treated with mitomycin C, and cultured with purified CD4+ T cells from DO11.10 RAG2−/− mice at a 1:10 ratio from nonimmunized animals. TSLP-treated DCs reduced the levels of IFN-γ production by KJ1-26+CD4+ T cells, whereas the levels of IL-4 were not affected. (D) No significant difference was observed in the levels of IL-12 (p35) mRNA examined by RT-PCR in CD11c+ DCs that were incubated overnight with 5 μg/ml of OVA323–339 peptide alone or with TSLP. (E) Total splenocytes from DO11.10/WT and DO11.10/TSLPRKO mice were cultured for 4 d with 5 μg/ml of OVA323–339 peptide before being activated with PMA+ ionomycin for 5 h. KJ1-26+ TSLPR KO T cells produced more IFN-γ than did DO11.10/WT cells. (F) RNA was extracted from DO11.10/WT and DO11.10/TSLPRKO total splenocytes that were cultured for 4 d with 5 μg/ml of OVA323–339 peptide. RT-PCR revealed significantly lower levels of IL-4 mRNA in the spleens of TSLPR KO mice; asterisk indicates that the value was significantly lower (P < 0.05). The experiment was done four times with one or two mice in each experimental group in each experiment.
We next examined whether TSLP could influence the differentiation of CD4+ T cells indirectly by its action on DCs. Sorted WT splenic CD11c+ DCs were preincubated with OVA323–339 in the presence or absence of TSLP before being washed and incubated with congenic DO11.10 Tg CD4+ T cells on a RAG2−/− background. Treatment of DCs with TSLP plus OVA323–339 reduced the levels of IFN-γ production in KJ1-26+CD4+ DO11.10 Tg T cells (Fig. 3 C), and levels of IL-12 were below the level of detection sensitivity by ELISA (not depicted). Treatment of DCs with OVA323–339 plus TSLP versus OVA323–339 alone did not result in a statistically significant change in IL-12 mRNA levels (Fig. 3 D). We next examined the production of IFN-γ and IL-4 by TSLPR KO mice. Splenocytes from WT and TSLPR KO mice expressing the DO11.10 transgene were cultured in the presence of OVA323–339. After 4 d of culture, KJ1-26+CD4+ T cells from DO11.10/TSLPR KO mice consistently produced more IFN-γ than did the analogous T cells from DO11.10/WT mice (Fig. 3 E). Changes in IL-4 production were difficult to evaluate by intracellular staining because levels were very low in WT mice (Fig. 3 E), but RT-PCR indicated that significantly less IL-4 mRNA was expressed by the DO11.10/TSLPR KO splenocytes. These results suggest that TSLP can influence cytokine production by CD4+ T cells indirectly by modulating the activity of DCs.
TSLPR KO mice fail to develop a lung inflammatory response to OVA antigen
Having identified the effects of TSLP ON CD4+ T cell proliferation and differentiation, we next wanted to evaluate its importance in a pathophysiologic model system. Therefore, we examined the ability of TSLPR KO mice to control an inflammatory response, using an OVA-induced allergic asthma model (15). WT and TSLPR KO (F4 BALB/c) mice were immunized twice i.p. with OVA and challenged by intratracheal (i.t.) and intranasal (i.n.) administration of OVA. As expected (15, 16), WT mice receiving OVA had perivascular inflammation, marked peribronchiole cuffing with inflammatory cells, as well as goblet cell hyperplasia (Fig. 4, A versus B). These mice scored 3–3.5 out of 4 on the inflammation scale (see Materials and methods), with greater infiltration of eosinophils and neutrophils (Table I) than control animals. In sharp contrast, TSLPR KO mice treated with OVA had profoundly fewer cellular infiltrates and little, if any, goblet cell hyperplasia in the bronchi (Fig. 4, D versus C), with very few inflammatory cells (Table I). There were no obvious differences between control WT and TSLPR KO mice that were treated with PBS (Fig. 4, C versus A, top; Table I). Analogous to this increased inflammation, WT mice that were immunized with OVA had significantly greater airway hyperresponsiveness to the increasing doses of methacholine, whereas immunized TSLPR KO mice did not (Fig. 4 E). Moreover, cultured splenocytes from immunized WT mice produced significantly higher levels of IL-4 than did splenocytes from TSLPR KO mice, with no significant differences in the total amount of IFN-γ (Fig. 4 F).
Figure 4.

TSLPR KO mice fail to mount an inflammatory response. (A–D) Periodic acid-Schiff–stained lung tissue sections of BALB/c WT and TSLPR KO mice that were sensitized (i.p.) and challenged (i.t. and i.n.) with OVA or PBS (i.p.). There were no obvious differences in the lung morphology between WT (A) and TSLPR KO (C) animals that were exposed to PBS. WT mice that received OVA displayed perivascular inflammation, peribronchiolar cuffing, and goblet cell hyperplasia (B), whereas TSLPR KO mice that were treated with OVA showed no obvious inflammation (D). (E) WT and TSLPR KO animals treated as shown above were tested for their airway hypersensitivity response using methacholine (0, 6, 12, 25, and 50 mg/ml). Unlike WT animals, TSLPR KO mice immunized and sensitized with OVA showed similar results to PBS control mice (six to seven animals per group, shown is one representative experiment out of three). *Statistical significance (P < 0.05) between the indicated dose and the 0 mg/ml. (F) Splenocytes from immunized WT and TSLPR KO mice were cultured in vitro with 200 μg/ml OVA for 3 d. Supernatants were examined for IFN-γ and IL-4 levels. The levels of IL-4 were significantly lower in the TSLPR KO animals versus the WT animals, whereas no statistical significance was observed for IFN-γ. The experiment was done four times with four mice in each experimental group in each experiment. *Statistical significance (P < 0.05).
Table I.
Absence of TSLPR blocks the development of lung inflammation and infiltration of inflammatory cells
| Analysis | Score | Monocytes | Lymphocytes | Neutrophils | Eosinophils | |
|---|---|---|---|---|---|---|
| Normal lungs Normal lungs High perivascular and peribronchial inflammation Slight inflammation |
% | % | % | % | ||
| WT + PBS | 0–1 | 87 ± 4 | 13 ± 4 | 0 | 0 | |
| KO + PBS | 0–1 | 95 ± 3 | 5 ± 3 | 0 | 0 | |
| WT + OVA | 3–3.5 | 42 ± 2 | 21 ± 3 | 16 ± 4 | 21 | |
| KO + OVA | 0.5–1.5 | 87 ± 2 | 11 ± 2 | 1 ± 1 | 1 ± 1 |
BALB/c WT and TSLPR KO mice were sensitized (i.p.) and challenged (i.t. and i.n.) with OVA or PBS (i.p.). Shown are levels of inflammation in the lungs observed in Fig. 4 by periodic acid-Schiff staining, as well as the distribution of infiltrating leukocytes in BAL. TSLPR KO mice that were challenged with OVA showed weak inflammation with much less infiltration of inflammatory cells than did WT animals. The increase in eosinophils and neutrophils was significant (P < 0.0001). The experiment was done four times with two to four mice in each experimental group in each experiment.
Corresponding to their having a defective “allergic” response, TSLPR KO mice also had significantly lower levels of IgE and higher IgG2a than did WT mice (Fig. 5 A). Levels of OVA-specific IgG1, IgG2b, and IgG3 were normal (Fig. 5 A). Furthermore, unlike WT mice, TSLPR KO mice that were sensitized to OVA and subsequently challenged with OVA did not show a statistically significant increase in IL-2, -4, -5, -10, -13, and IFN-γ mRNA as compared with PBS-treated mice (Fig. 5 B). OVA-induced protein levels of IL-5 and -13 were decreased in the bronchoalveolar lavage (BAL) fluid from TSLPR KO mice (Fig. 5 C). Levels of IL-4 in the BAL were below sensitivity levels (unpublished data). OVA induced an increase in IL-13 mRNA in the lungs of TSLPR KO mice, but only to the level seen in PBS-treated WT controls (Fig. 5 B). These mice also displayed a significant increase in IL-12 mRNA (Fig. 5 B). Furthermore, as compared with WT mice, the lungs of TSLPR KO mice that received OVA had significantly lower levels of CCL11 (eotaxin), CCL17 (TARC), and CXCL9 (Mig) (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050199/DC1). These results indicate that TSLP is required to mount an optimal pulmonary inflammatory response to OVA.
Figure 5.

TSLPR KO mice exhibit partially altered allergic/ inflammatory immunoglobulin and cytokine profiles in the lungs in OVA-treated mice. BALB/c WT and TSLPR KO mice were sensitized (i.p.) and challenged (i.t. and i.n.) with OVA or PBS (i.p.). (A) Sera were tested at the indicated dilutions and the levels of OVA-specific immunoglobulin in PBS- and OVA-treated mice. Antibody was captured with plate-bound ovalbumin (2 μg/ml) and detected with biotinylated isotype-specific secondary antibodies. OVA-TSLPR KO mice displayed significantly less IgE and more IgG2a than did WT treated littermates. (B) Cytokine levels in the lungs as determined by RT-PCR. (C) Cytokines protein levels in BAL as determined by ELISA. The results are expressed as means ± SEM for four experiments, two to four mice per group per experiment. (A and B) *P < 0.05 between PBS and OVA group of WT and PBS and OVA group of TSLPR KO mice.
The greatly diminished inflammatory response in the lungs of TSLPR KO mice may have resulted from decreased activation of CD4+ T cells and/or from a developmental defect in the TSLPR KO lungs. To study further the basis for the decreased inflammatory response in the lungs, WT mice were immunized twice with OVA, and CD4+ T cells were isolated from spleen and LNs and were transferred (i.v.) into similarly immunized TSLPR KO mice. These mice were challenged with OVA by an i.t. and i.n. route. CD4+ T cells from WT and TSLPR KO mice were transferred similarly into WT and TSLPR KO animals as positive and negative controls, respectively. An examination of the lungs revealed severe inflammation and infiltration of eosinophils and neutrophils in WT animals that received WT CD4+ T cells (scoring 3.5 out of 4) (Fig. 6 A, i and ii), whereas TSLPR KO supplemented with TSLPR KO CD4+ T cells showed little, if any, inflammation (0–0.5 out of 4) (Fig. 6 A, iii and iv). This was consistent with the results in Table I and Fig. 4. TSLPR KO mice that received WT CD4+ T cells developed goblet cell hyperplasia and peribronchiole cuffing, mostly in the large airways, but with some in the medium airways with infiltration of eosinophils and neutrophils (Fig. 6 A, v and vi); they scored 2.2 out of 4 (range, 1.5–2.75) on the inflammation scale. Thus, the TSLPR KO host could support, at least in part, an inflammatory response if WT CD4+ T cells were provided, with a concomitant significant increase in the levels of IFN-γ IL-4, -5, and -13 mRNA (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050199/DC1). Transferring primed CD4+ T cells from TSLPR KO to WT nonimmunized animals did not lead to the development of inflammation in the lungs of the host after i.t. and i.n. challenge (unpublished data). These results further confirm that TSLP is involved in the activation of CD4+ T cells and is consistent with a role for this cytokine in the development of the inflammatory response.
Figure 6.
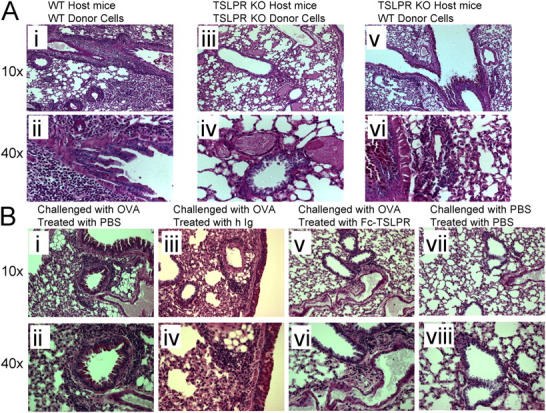
TSLP plays a role in the development of asthma. (A)TSLPR KO mice succeed in mounting an inflammatory response in the lung after receiving WT CD4+ T cells. Donor and recipient mice (F4 BALB/c) were sensitized with OVA (i.p.). Recipient mice were challenged with OVA (i.t.) 4 h before receiving 8 × 106 CD4+ T cells. CD4+ T cells from WT donor were transferred to WT (positive control, i and ii) and TSLPR KO (test subject, panels iii and iv) mice, whereas CD4+ T cells from TSLPR KO mice were given to TSLPR KO mice to act as negative control (v and vi). All mice were treated the next day with OVA (i.n.) and killed 24 h later. Shown are the periodic acid-Schiff staining of lung tissue sections. The WT positive control displayed perivascular inflammation, peribronchiolar cuffing, and goblet cell hyperplasia (i and ii), whereas TSLPR KO mice showed no signs of lung inflammation (iii and iv). However, TSLPR KO mice that were supplemented with CD4+ T cells (v and vi) exhibited inflammatory cells infiltration combined with peribronchiolar cuffing, and goblet cell hyperplasia in the large and medium airways. (B) Lung sections stained with periodic acid-Schiff show the effect of blocking TSLP using Fc-TSLPR fusion protein on the lung inflammation in WT animals that were immunized and sensitized with OVA. Introducing Fc-TSLPR fusion protein 4 h before each of the i.t. and i.n. OVA reduced the levels of inflammation in these animals (v and vi, score 1.6 with a range of 1–2.5) compared with animals that were treated with Ig (iii and iv, score 2.6 and a range of 2–3.5). WT mice that were treated with OVA (i and ii) or PBS (vii and viii) acted as positive and negative controls, respectively. The difference was statistically significant (P = 0.01). The experiment was done three times with two mice in each experimental group in each experiment.
Finally, we neutralized TSLP in the lungs of WT mice using 100 μg of Fc-TSLPR fusion protein or Ig (i.t. and i.n.), 4 h before OVA challenge. Although positive control mice (OVA only; Fig. 6 B, i and ii) or mice that received control Ig (Fig. 6 B, iii and iv) developed a moderate to severe level of inflammation and goblet cell hyperplasia, and scored 2.6 on the inflammation scale (range 2–3.5 out of 4) (Fig. 6 B, i), the introduction of Fc-TSLPR fusion consistently diminished the inflammatory response, with lower cellular infiltration and fewer goblet cells observed (score 1.6, range of 1–2.5) (Fig. 6 B, v and vi). Negative control mice that received PBS (Fig. 6 B, vii and viii) showed no inflammation.
DISCUSSION
The survival of effector T cells and their progression to a memory state has been an area of active investigation (17, 18). The critical role of the IL-7/IL-7Rα system has been underscored by the phenotypes of IL-7 and IL-7Rα KO mice (19, 20) and humans with IL-7Rα–deficient SCID (21). Thus, part of the phenotype in IL-7Rα–deficient mice and humans presumably results from the inactivation of TSLP signaling. Data have accumulated that TSLP-activated human DCs can induce the expansion and activation of allogenic naive CD4+ T cells to a central memory phenotype, whereas murine TSLP contributes to the generation of CD4+ T cells and can rescue, in part, lymphopoiesis in γc-deficient mice (10).
In this study, we discovered that TSLP preferentially enhances the proliferation and expansion of TCR-stimulated naive phenotype CD4+ T cells. Moreover, TSLP is important for the transition of naive CD4+ T cells into activated effector cells. Although we cannot exclude a developmental defect in TSLPR KO mice, our data suggest that the defect in these mice may be due, at least in part, to weaker antigen-driven activation. Specifically, TSLPR KO mice displayed no apparent abnormalities in lymphocyte numbers and subpopulation distribution (10), but there was weaker activation of TSLPR KO CD4+ T cells after immunization. Nevertheless, TSLPR KO mice produced OVA-specific IgG1 and IgG2b at similar levels to WT mice; this indicated that an active immune response is taking place in these mice and argues against a global developmental defect.
Memory phenotype CD4+ T cells express TSLPR mRNA (unpublished data), but showed no significant proliferative response to TSLP. The decreased responsiveness of splenocytes from immunized TSLPR KO mice—as compared with WT animals—to secondary exposure to antigen may be due to the weak initiation phase of the immunization process, with fewer cells differentiating into effector and memory cells. It seems that naive cells are poised to respond to TSLP activation, whereas IL-7 seems to play a greater role that is related to their differentiation into memory cells (18).
Murine DCs can promote the clonal-specific expansion of naive CD4+ T cells. We show that TSLP is involved in this process, but that TSLPR is more important to CD4+ T cells than to DCs. Although TSLP had no direct effect on the production of IFN-γ and IL-4 by CD4+ T cells under neutral or polarizing conditions, TSLP-treated DCs diminished the production of IFN-γ by CD4+ T cells, and correspondingly, more IFN-γ and less IL-4 were produced by DO11.10/TSLPR KO splenocytes than by DO11.10/WT cells. Similarly, splenocytes from immunized WT animals that were activated in vitro with OVA produced more IL-4 than did TSLPR KO cells. The lower number of effector cells in the TSLPR KO mice that produced higher levels of IFN-γ could explain the lack of difference in the amount of IFN-γ that was present in these cultures. Thus, the greatest effect of TSLP on DCs may be to augment the differentiation and cytokine production, rather than affecting the proliferation of naive CD4+ T cells directly.
The greatly diminished inflammatory response that was seen in the lungs of TSLPR KO mice that were challenged with OVA underscores the importance of the TSLP/TSLPR system in vivo. Although TSLP levels are increased in inflamed tonsils and in the skin of individuals with atopic dermatitis (11, 12), the importance of TSLP for these responses could not be assessed because no neutralization or knockout methodology was used. An immune response was confirmed in these mice by the presence of OVA-specific IgG1 and IgG2b; IgG2a was increased but IgE was not, which indicated a Th1-like phenotype in the absence of TSLP (22). This could result from higher levels of IFN-γ that are produced in the TSLPR KO CD4+ T cells. Because IL-13 is a mediator of asthma that affects eosinophilic infiltration, mucus secretion, and airway hyperresponsiveness in the lungs (23–25), the reduced IL-13 in the lungs of TSLPR KO mice might explain the absence of inflammation and IgE in these animals. Similarly, the absence of inflammatory CD4+ T cells in the lungs could explain the lack of IL-5, and subsequently, the absence of eosinophils in affected tissues. The absence of CCL11 and CCL17 in the lungs of TSLPR KO mice that were challenged with OVA helps to explain the absence of inflammation in these mice, because neutralizing these chemokines can abrogate lung eosinophilia and airway hypersensitivity response (26–28).
The transfer of WT CD4+ T cells to TSLPR KO mice had a profound effect on the ability of these mice to mount an inflammatory response. TSLPR KO lungs showed goblet cell hyperplasia and peribronchiole cuffing, accompanied by the infiltration of inflammatory cells; this was consistent with the increase in IFN-γ and IL-4, -5, and -13. Thus, the lungs of TSLPR KO mice are structurally normal, and the microenvironment within them can allow the development of an inflammatory response when functional preactivated CD4+ T cells are available. However, the lower inflammation in these mice compared with WT animals might be explained by the fact that WT mice possess more CD4+ T cells, because only a fraction of the WT CD4+ T cells that were transferred to TSLPR KO animals is expected to be OVA responsive. In addition, the high levels of IL-12 and the absence of CCL11 and CCL17 in the lungs of TSLPR KO animals activated with OVA might limit the inflammatory response in these animals.
In conclusion, our results reveal that TSLP plays a key role in promoting the proliferation of naive CD4+ T cells. Although DC activation by TSLP may contribute to this process, CD4+ T cells seem to be the more important target for TSLP. TSLP had little, if any, direct effect on cytokine production by CD4+ T cells, but influenced IFN-γ production by way of actions on DCs. In view of the importance of TSLP, we also investigated its role in an allergic inflammatory model in vivo, and unexpectedly found profoundly reduced inflammation in the lungs of TSLPR KO mice. These data indicate that TSLP signaling is crucial for generation of an inflammatory reaction in the lung and establish TSLP as a potential target for modulating inflammation, findings with potentially important clinical implications as well.
MATERIALS AND METHODS
Mice.
TSLPR KO mice have been described (10). DO11.10 transgenic Rag 2−/− mice on B10.D2 and BALB/c backgrounds (purchased from Taconic) were crossed to TSLPR KO mice on F1 129/BL/6 or BALB/c (F4) genetic backgrounds (29). WT BALB/c animals were from The Jackson Laboratory. All experiments were performed under protocols approved by the National Institute of Health Animal Use and Care Committee and followed the Using Animals in Intramural Research guidelines from the National Institutes of Health.
TSLP effect on naive versus memory CD4 T cells.
CD4+CD62L+ CD44low (naive), CD4+CD62L+CD44high (central memory), and CD4+ CD62L−CD44high (effector memory) phenotype T cells were isolated by cell sorting and were >99% pure. RNA was extracted from freshly sorted cells using TRIzol (Invitrogen). Purified cells also were suspended at 2 × 105 cells/well and activated with anti-CD3ɛ (2 μg/ml, BD Biosciences) with or without TSLP (100 ng/ml; R&D Systems) for 48 h before being pulsed with 1 μCi of [3H] thymidine (6.7 Ci/mmol, NEN Life Science Products) for the final 16 h of culture.
Immunization and antigen-recall response.
Mice were immunized by i.p. injection of 200 μg of OVA (Pierce Chemical Co.) in 100 μl of PBS that had been mixed with 100 μl of aluminum hydroxide (ALUM) as adjuvant and incubated at room temperature for 30 min before injection. Mice were killed 12 or 60 d after immunization. For assessing an antigen recall response, purified CD4+ T cells (105 cells/well) were cultured with an equal number of splenic APCs or total splenocytes (2 × 105 cells/well) that were treated with mitomycin C (50 μg/ml in PBS for 15 min at 37°C) and washed three times. These cells were incubated in 96-well flat-bottom plates for 48 h in RPMI 1640 medium containing 10% FBS, 2 mM L-glutamine, and antibiotics, and with 0, 10, 50, or 200 μg/ml of OVA. Wells were pulsed with 1 μCi of [3H]thymidine (6.7 Ci/mmol, NEN Life Science Products) for the final 16 h of culture. Where indicated, APCs included B cells, macrophages, and DCs, after depleting CD4+ T cells, CD8+ T cells, and NK cells by positive selection. WT and TSLPR KO mice that expressed the DO11.10 transgene were treated with 100 μg of OVA in 100 μl of PBS in 100 μl ALUM and analyzed the next day.
DC function.
TSLP was from R&D Systems and tested negative for endotoxin by the limulus amebocyte lysate test (BioWhittaker). To examine the effect of TSLP on murine DCs, total splenic CD11c+ DCs were isolated by sorting (>99% purity) from BALB/c WT mice, and cultured for 24 h at 106 cells/ml with OVA323–339 peptide (5 μg/ml; Bachem) with or without TSLP (100 ng/ml). The cells were lysed in TRIzol for RNA isolation or were washed, treated with mitomycin C (50 μg/ml, Sigma-Aldrich), washed three times, and incubated with DO11.10 RAG 2−/− CD4+ T cells at a 1:10 ratio. Cells were cultured for 48 h to allow measuring proliferation using [3H] thymidine incorporation, or for 4 d to examine intracellular levels of IFN-γ and IL-4 secretion, as described below.
“Mix and match” experiments were performed using CD4+ T cells that were purified by positive selection using specific magnetic beads (Miltenyi Biotec). CD4+ T cells and CD11c+ DCs were isolated by positive selection using labeled magnetic beads (Miltenyi Biotec) in the presence of 1% Fc block (BD Biosciences). The resulting CD4+ T cells were >90% pure and DCs were >80% pure. They were incubated together (1:10 ratio) at various combinations (e.g., WT CD4+ T cells with WT or TSLPR KO DCs and TSLPR KO CD4+ T cells with WT or TSLPR KO DCs) as indicated. All DCs were treated with mitomycin C before being incubated with CD4+ T cells. Wells were pulsed with 1 μCi of [3H]thymidine (6.7 Ci/mmol, NEN Life Science Products) for the final 16 h before harvesting.
Flow cytometric analyses.
Single-cell suspensions were prepared from thymus and spleen. Cells were washed with FACS buffer (phosphate buffered saline pH 7.4 containing 0.5% BSA and 0.02% sodium azide). 1 million cells were treated with Fc-block for 15 min before being incubated with the indicated fluorochrome-conjugated antibodies (all from BD Biosciences) for 20 min. Cells were washed twice with FACS buffer and analyzed. All antibodies used were from BD Biosciences, except KJ1-26 mAb, which was from Caltag Laboratories. All flow cytometric data presented are representative of at least three experiments with similar results.
Culture under polarizing conditions.
Purified CD4+ T cells were activated with anti-CD3 (2 μg/ml) and anti-CD28 (1 μg/ml) and cultured in conditions favoring Th1 (1 ng/ml IL-12 and 10 μg/ml anti–IL-4) or Th2 (1 ng/ml IL-4 and 20 μg/ml anti–IFN-γ) in the presence or absence of TSLP (100 ng/ml). IL-2 (100 U/ml) was added on day 2 and cells were allowed to expand for 1 wk.
Intracellular staining for IFN-γ and IL-4 levels.
Cultured cells were activated with PMA (10 ng/ml) and ionomycin (1 μg/ml) (both from Sigma-Aldrich) for 5 h in the presence of Golgi Block (BD Biosciences). Cells were washed and stained for anti-CD4 (BD Biosciences) and/or for the DO11.10-specific transgene with KJ1-26. Intracellular staining was performed using Cytofix/Cytoperm kit, and anti–IFN-γ and IL-4 (all from BD Biosciences) according to the manufacturer's instructions.
Sensitization and airway challenge to mice.
OVA (100 μg in 100 μl of PBS) was mixed with 100 μl of Imject ALUM and 200 μl was injected i.p. on days 0 and 7 to sensitize mice. On day 13, mice were challenged i.t. with 50 μg of OVA in 30 μl PBS. On day 14, 50 μg of OVA in 25 μl PBS was administered i.n. Control mice were treated similarly with PBS instead of OVA. On day 16, mice were killed, bled, and BAL was performed using 0.5 ml of PBS. Cells from BAL fluid were isolated by cytospin, and leukocytes were identified by staining using Wright/Giemsa. Lungs were used for extracting RNA or for generating tissue sections for microscopic analysis. Lung tissue sections were stained with periodic acid-Schiff or Wright/Giemsa (Volu-Sol, UT). Serum levels of OVA-specific immunoglobulin were measured by ELISA (BD Biosciences). Inflammation was scored on a scale of 0 to 4 as follows: (0) normal lungs, no goblet cell hyperplasia; (1) minor perivascular inflammation around large blood vessels; (2) moderate perivascular and peribronchial inflammation, minimal evidence of goblet cell hyperplasia; (3) increased perivascular and peribronchial inflammation with increased goblet cell hyperplasia beginning in smaller airways; (4) severe formation of perivascular, peribronchial, and interstitial inflammation as well as goblet cell hyperplasia in small and large airways.
Airway hyperresponsiveness responses of WT and TSLPR KO mice that were immunized and challenged with PBS and OVA were examined by challenging the mice with methacholine at 0, 6, 12, 25, and 50 mg/ml of PBS and measuring “enhanced pause” using whole body plethysmography (Buxco Electronics) following the manufacturer's instructions.
To test the effect of blocking TSLP on the development of inflammation in the lungs, presensitized WT BALB/c mice were treated i.t. and i.n. with 100 μg (25 μl of PBS) of human Ig or Fc-TSLPR (R&D Systems) (the extracellular domain of murine TRSLPR fused to human Fc fragment) 4 h before OVA challenge through the same route. Mice were analyzed as above to determine degree of lung inflammation.
Adoptive transfer of CD4+ T cells.
WT and TSLPR KO mice that were backcrossed to BALB/c background for four generations were designated as donors and recipients, and immunized by intraperitoneal injections of OVA (100 μg) in 100 μl of PBS, which were mixed with equal volume of ALUM, and 200 μl was injected on days 0 and 7). On day 13, recipient mice were sensitized i.t. with 50 μg of OVA in 30 μl PBS, followed 4 h later by i.v. transfer of 8 × 106 CD4+ T cells extracted from spleen and lymph nodes of donor mice. On day 14, 50 μg of OVA in 25 μl PBS was administered i.n. Mice were killed and analyzed the next day for signs of lung inflammation, using the criteria presented described before.
To determine the ability of DO11.10/WT and TSLPR KO splenic CD4+ T cells to proliferate in vivo, these cells were isolated from spleens, labeled with CFSE (5 μM for 10 min at 37°C), and transferred (8 × 106 cells) to WT hosts that were immunized the next day with OVA (100 μg) in ALUM or PBS. On day 5, lymph nodes were extracted and cells were stained with anti–KJ1-26 mAb.
Real-time PCR.
RNA was extracted from single-cell suspensions or from lung tissue using TRIzol (Invitrogen). RNA was reverse transcribed using the RNA PCR gold kit (Applied Biosystems). Levels of IL-2, -4, -5, -10, and -13; IFN-γ; TSLP; CCL2; CCL7; CCL11; CCL17; CXCL9; CCL22; and TSLPR mRNA relative to 18S rRNA were measured by RT-PCR using “gene expression assays” ready-made primers from Applied Biosystems.
Online supplemental materials.
Fig. S1 shows the amount of mRNA, as determined by RT-PCR, of selected chemokines in the lungs of mice tested in an asthma model. Fig. S2 shows, in an asthma model, the increase in lung inflammatory cytokine levels in TSLPR KO mice into which WT CD4 T cells were transferred adoptively. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050199/DC1.
Acknowledgments
We thank R. Schwartz, P.L. Schwartzberg, and C.L. Mackall for their critical comments, discussions, and valuable suggestions. We also would like to thank H. Norris and A. Boesen for their help in examining the asthma model.
This research was supported by the Intramural Research Programs of the National Heart, Lung, and Blood Institute and the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
The authors have no conflicting financial interests.
Abbreviations used: γc, receptor γ chain; ALUM, aluminum hydroxide; BAL, bronchoalveolar lavage; CFSE, 5,6-carboxyfluorescein diacetate-succinimidyl ester; IL-7Rα, IL-7 receptor α chain; i.n., intranasal; i.t., intratracheal; Tg, transgenic; TSLP, thymic stromal lymphopoietin.
References
- 1.Friend, S.L., S. Hosier, A. Nelson, D. Foxworthe, D.E. Williams, and A. Farr. 1994. A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Exp. Hematol. 22:321–328. [PubMed] [Google Scholar]
- 2.Park, L.S., U. Martin, K. Garka, B. Gliniak, J.P. Di Santo, W. Muller, D.A. Largaespada, N.G. Copeland, N.A. Jenkins, A.G. Farr, et al. 2000. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: formation of a functional heteromeric complex requires interleukin 7 receptor. J. Exp. Med. 192:659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandey, A., K. Ozaki, H. Baumann, S.D. Levin, A. Puel, A.G. Farr, S.F. Ziegler, W.J. Leonard, and H.F. Lodish. 2000. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat. Immunol. 1:59–64. [DOI] [PubMed] [Google Scholar]
- 4.Leonard, W.J. 2001. Cytokines and immunodeficiency diseases. Nat. Rev. Immunol. 1:200–208. [DOI] [PubMed] [Google Scholar]
- 5.Sims, J.E., D.E. Williams, P.J. Morrissey, K. Garka, D. Foxworthe, V. Price, S.L. Friend, A. Farr, M.A. Bedell, N.A. Jenkins, et al. 2000. Molecular cloning and biological characterization of a novel murine lymphoid growth factor. J. Exp. Med. 192:671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ray, R.J., C. Furlonger, D.E. Williams, and C.J. Paige. 1996. Characterization of thymic stromal-derived lymphopoietin (TSLP) in murine B cell development in vitro. Eur. J. Immunol. 26:10–16. [DOI] [PubMed] [Google Scholar]
- 7.Leonard, W.J., E.W. Shores, and P.E. Love. 1995. Role of the common cytokine receptor gamma chain in cytokine signaling and lymphoid development. Immunol. Rev. 148:97–114. [DOI] [PubMed] [Google Scholar]
- 8.Isaksen, D.E., H. Baumann, B. Zhou, S. Nivollet, A.G. Farr, S.D. Levin, and S.F. Ziegler. 2002. Uncoupling of proliferation and Stat5 activation in thymic stromal lymphopoietin-mediated signal transduction. J. Immunol. 168:3288–3294. [DOI] [PubMed] [Google Scholar]
- 9.Leonard, W.J. 2002. TSLP: finally in the limelight. Nat. Immunol. 3:605–607. [DOI] [PubMed] [Google Scholar]
- 10.Al-Shami, A., R. Spolski, J. Kelly, T. Fry, P.L. Schwartzberg, A. Pandey, C.L. Mackall, and W.J. Leonard. 2004. A role for thymic stromal lymphopoietin in CD4(+) T cell development. J. Exp. Med. 200:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soumelis, V., P.A. Reche, H. Kanzler, W. Yuan, G. Edward, B. Homey, M. Gilliet, S. Ho, S. Antonenko, A. Lauerma, et al. 2002. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 3:673–680. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe, N., S. Hanabuchi, V. Soumelis, W. Yuan, S. Ho, R. de Waal Malefyt, and Y.J. Liu. 2004. Human thymic stromal lymphopoietin promotes dendritic cell-mediated CD4+ T cell homeostatic expansion. Nat. Immunol. 5:426–434. [DOI] [PubMed] [Google Scholar]
- 13.Gilliet, M., V. Soumelis, N. Watanabe, S. Hanabuchi, S. Antonenko, R. de Waal-Malefyt, and Y.J. Liu. 2003. Human dendritic cells activated by TSLP and CD40L induce proallergic cytotoxic T cells. J. Exp. Med. 197:1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin, S.D., R.M. Koelling, S.L. Friend, D.E. Isaksen, S.F. Ziegler, R.M. Perlmutter, and A.G. Farr. 1999. Thymic stromal lymphopoietin: a cytokine that promotes the development of IgM+ B cells in vitro and signals via a novel mechanism. J. Immunol. 162:677–683. [PubMed] [Google Scholar]
- 15.Keane-Myers, A., M. Wysocka, G. Trinchieri, and M. Wills-Karp. 1998. Resistance to antigen-induced airway hyperresponsiveness requires endogenous production of IL-12. J. Immunol. 161:919–926. [PubMed] [Google Scholar]
- 16.Keane-Myers, A.M., W.C. Gause, F.D. Finkelman, X.D. Xhou, and M. Wills-Karp. 1998. Development of murine allergic asthma is dependent upon B7-2 costimulation. J. Immunol. 160:1036–1043. [PubMed] [Google Scholar]
- 17.Seddon, B., P. Tomlinson, and R. Zamoyska. 2003. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 4:680–686. [DOI] [PubMed] [Google Scholar]
- 18.Kaech, S.M., J.T. Tan, E.J. Wherry, B.T. Konieczny, C.D. Surh, and R. Ahmed. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198. [DOI] [PubMed] [Google Scholar]
- 19.von Freeden-Jeffry, U., P. Vieira, L.A. Lucian, T. McNeil, S.E. Burdach, and R. Murray. 1995. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J. Exp. Med. 181:1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peschon, J.J., P.J. Morrissey, K.H. Grabstein, F.J. Ramsdell, E. Maraskovsky, B.C. Gliniak, L.S. Park, S.F. Ziegler, D.E. Williams, C.B. Ware, et al. 1994. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 180:1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puel, A., S.F. Ziegler, R.H. Buckley, and W.J. Leonard. 1998. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat. Genet. 20:394–397. [DOI] [PubMed] [Google Scholar]
- 22.Pulendran, B. 2004. Modulating TH1/TH2 responses with microbes, dendritic cells, and pathogen recognition receptors. Immunol. Res. 29:187–196. [DOI] [PubMed] [Google Scholar]
- 23.Wills-Karp, M., J. Luyimbazi, X. Xu, B. Schofield, T.Y. Neben, C.L. Karp, and D.D. Donaldson. 1998. Interleukin-13: central mediator of allergic asthma. Science. 282:2258–2261. [DOI] [PubMed] [Google Scholar]
- 24.Fallon, P.G., C.L. Emson, P. Smith, and A.N. McKenzie. 2001. IL-13 overexpression predisposes to anaphylaxis following antigen sensitization. J. Immunol. 166:2712–2716. [DOI] [PubMed] [Google Scholar]
- 25.Grunig, G., M. Warnock, A.E. Wakil, R. Venkayya, F. Brombacher, D.M. Rennick, D. Sheppard, M. Mohrs, D.D. Donaldson, R.M. Locksley, and D.B. Corry. 1998. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 282:2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalo, J.A., C.M. Lloyd, L. Kremer, E. Finger, A.C. Martinez, M.H. Siegelman, M. Cybulsky, and J.C. Gutierrez-Ramos. 1996. Eosinophil recruitment to the lung in a murine model of allergic inflammation. The role of T cells, chemokines, and adhesion receptors. J. Clin. Invest. 98:2332–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalo, J.A., C.M. Lloyd, D. Wen, J.P. Albar, T.N. Wells, A. Proudfoot, A.C. Martinez, M. Dorf, T. Bjerke, A.J. Coyle, and J.C. Gutierrez-Ramos. 1998. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J. Exp. Med. 188:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawasaki, S., H. Takizawa, H. Yoneyama, T. Nakayama, R. Fujisawa, M. Izumizaki, T. Imai, O. Yoshie, I. Homma, K. Yamamoto, and K. Matsushima. 2001. Intervention of thymus and activation-regulated chemokine attenuates the development of allergic airway inflammation and hyperresponsiveness in mice. J. Immunol. 166:2055–2062. [DOI] [PubMed] [Google Scholar]
- 29.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]