

Abstract
To investigate the role of Toll-like receptor (TLR)9 in the immune response to mycobacteria as well as its cooperation with TLR2, a receptor known to be triggered by several major mycobacterial ligands, we analyzed the resistance of TLR9−/− as well as TLR2/9 double knockout mice to aerosol infection with Mycobacterium tuberculosis. Infected TLR9−/− but not TLR2−/− mice displayed defective mycobacteria-induced interleukin (IL)-12p40 and interferon (IFN)-γ responses in vivo, but in common with TLR2−/− animals, the TLR9−/− mice exhibited only minor reductions in acute resistance to low dose pathogen challenge. When compared with either of the single TLR-deficient animals, TLR2/9−/− mice displayed markedly enhanced susceptibility to infection in association with combined defects in proinflammatory cytokine production in vitro, IFN-γ recall responses ex vivo, and altered pulmonary pathology. Cooperation between TLR9 and TLR2 was also evident at the level of the in vitro response to live M. tuberculosis, where dendritic cells and macrophages from TLR2/9−/− mice exhibited a greater defect in IL-12 response than the equivalent cell populations from single TLR9-deficient animals. These findings reveal a previously unappreciated role for TLR9 in the host response to M. tuberculosis and illustrate TLR collaboration in host resistance to a major human pathogen.
Toll-like receptors (TLRs) are thought to play a critical role in both innate resistance and the initiation of adaptive immunity to infectious agents (1, 2). TLRs are known to recognize distinct molecular structures on microbes, and in several cases (e.g., recognition of viruses by TLR3, TLR7, TLR8, and TLR9), different sets of TLRs have been associated with the response to different classes of microorganisms (1). Although the available evidence suggests that multiple rather than single TLRs are required for innate defense against most pathogens (for review see reference 2), it is not clear how signals from different TLRs are orchestrated in generating a protective response. In particular, there is controversy as to whether at the in vivo level multiple TLR interactions are required to trigger individual effector elements or whether TLR cooperation stems from the interaction of distinct effector mechanisms, each triggered by individual TLRs.
TLR signaling has been postulated to have a major involvement in the regulation of host resistance to Mycobacterium tuberculosis (3, 4), an important human pathogen that infects over one third of the world's population (5). Immunological control of M. tuberculosis infection has been shown to depend on Th1 CD4+ T cells as well as TNF, IFN-γ, and IL-12 (6–14). The latter cytokine, produced largely by APCs such as DCs and macrophages, is thought to function in mycobacterial immunity by both inducing and maintaining the Th1-mediated IFN-γ response (8, 9, 15). M. tuberculosis as well as other mycobacteria contain well-characterized TLR ligands that are potent in vitro stimuli of a number of proinflammatory cytokines, including TNF and IL-12 (16–19). A role for TLR signaling in host resistance to M. tuberculosis is further supported by the observation that mice deficient in MyD88, a major adaptor molecule required for signaling events by most TLR/IL-1R family members, show greatly enhanced susceptibility to aerosol infection with the pathogen, equivalent to that observed with IFN-γ–deficient mice (20, 11). Infected MyD88−/− animals, in addition to their loss of resistance, display impaired proinflammatory cytokine synthesis, which was found to correlate with decreased nitric oxide synthase 2 expression and diminished IFN-γ synthesis (20). In addition, MyD88-deficient APCs display a marked reduction in the synthesis of IL-12, TNF, and nitric oxide when exposed to M. tuberculosis in vitro (21, 22).
Although MyD88 appears to play a major role in resistance to M. tuberculosis, it has been difficult to attribute this requirement to the function of a single TLR. Thus, mice deficient in TLR2, TLR4, TLR6, IL-1, or IL-18, although in some cases displaying specific defects in antimycobacterial responses, exhibit only minor increases in susceptibility to low dose aerogenic challenge (23–28). For example, M. tuberculosis–infected TLR2−/− mice, although showing defective granuloma formation, display only a small elevation in pulmonary bacterial loads late in infection and survive for at least 150 d (28). Interestingly, however, when infected with unconventionally high doses of M. tuberculosis, TLR2−/− but not TLR4−/− mice exhibit greatly enhanced susceptibility compared with WT animals (23, 24). This loss in resistance is accompanied by alterations in proinflammatory cytokine and nitric oxide synthase 2 expression as well as the pulmonary granulomatous response (28). TLR2 also has been shown to play an important role in the regulation of mycobacterial-induced cytokine production by APCs in vitro (23, 28–30). Nevertheless, it is difficult to reconcile this evidence for TLR2 involvement with the minimal loss in resistance consistently observed in TLR2−/− mice challenged with low level physiological doses of M. tuberculosis.
One interpretation of the above findings is that host resistance to M. tuberculosis depends on a previously unevaluated TLR–ligand interaction. TLR9 is one such TLR/IL-1R family member whose involvement in control of M. tuberculosis infection has never been formally addressed. TLR9 was initially described as recognizing unmethylated CpG motifs in bacterial and viral DNA (31, 32) and was shown to be responsible for the immunostimulatory effects of these nucleic acids. In this regard, it is of interest that the original demonstration of the adjuvant properties of DNA emerged from studies on Mycobacterium bovis (bacillus of Calmette and Guerin [BCG]) (33) and that DNA from M. tuberculosis as well as other mycobacteria has subsequently been shown to contain highly immunostimulatory CpG motifs (34–37). TLR9 is known to be localized in endosomes as well as phagolysosomes, where it could be triggered by mycobacterial DNA after uptake of the pathogen (38–41). For these reasons we considered TLR9 to be an important candidate pattern-recognition receptor that might account for the MyD88 dependency of host resistance to M. tuberculosis.
In this study, we show that DNA from M. tuberculosis is indeed a potent stimulus of TLR9-dependent proinflammatory cytokine production by both DCs and macrophages and that the in vitro responses of these cells to live mycobacteria are also partially dependent on TLR9. In addition, our data indicate that TLR9 plays an important role in the regulation of the mycobacteria-induced Th1 responses during M. tuberculosis infection in vivo. Finally, we demonstrate that mice doubly deficient in TLR9 and TLR2 show enhanced susceptibility to M. tuberculosis not observed in mice lacking either TLR2 or TLR9 alone. Taken together, these data reveal a role for TLR9 in the immune response to M. tuberculosis and provide an important example of TLR collaboration in host resistance to infection.
Results
DNA from M. tuberculosis induces proinflammatory cytokine responses through a TLR9-dependent pathway
To assess whether TLR9 plays a role in M. tuberculosis infection, we first asked if the stimulation of proinflammatory responses by mycobacterial DNA documented in previous studies (33–37) depends on this TLR as would be predicted. As shown in Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20051782/DC1, purified DNA from M. tuberculosis, BCG, or Escherichia coli stimulated IL-12p40 production by splenic CD11c+ DCs from WT mice. Importantly, this response was found to be TLR9-dependent as was M. tuberculosis DNA–induced IL-12p40, TNF, and IFN-α production by BM-derived DCs (BMDCs; Figs. S1, B and C, and S3) and TNF and IL-6 synthesis by BM-derived macrophages (BMM; Fig. S1, D and E). In keeping with previously published data demonstrating a requirement for endosomal acidification in immune stimulation by CpG oligonucleotides (42), the response of splenic DCs to mycobacterial DNA was found to be inhibited by chloroquine treatment (not depicted). Taken together, these results indicate that mycobacterial DNA is a potent stimulus for TLR9-dependent cytokine production by murine APCs.
The in vitro IL-12 response of APCs to M. tuberculosis bacilli is regulated by both TLR9 and TLR2, whereas TNF is controlled primarily by TLR2
Having demonstrated TLR9-dependent proinflammatory responses to mycobacterial DNA, we next asked whether TLR9 regulates cytokine production stimulated by live M. tuberculosis in freshly isolated splenic DCs. In addition, we simultaneously assessed the involvement of TLR2 because this receptor has previously been shown to influence proinflammatory cytokine production by BM-derived APC populations in response to M. tuberculosis infection (23, 28, 43).
As shown in Fig. 1 A, live M. tuberculosis induced IL-12p40 secretion by splenic DCs in a dose-dependent manner, and this response was markedly reduced in the absence of MyD88. Heat-killed mycobacteria also induced IL-12p40 production, indicating that this response is not dependent on DC infection. Nevertheless, in agreement with a previous study (44), M. tuberculosis–induced IL-12 synthesis was greatly impaired in the presence of cytochalasin D, suggesting at least a partial requirement for bacterial phagocytosis (Fig. 1 B). Interestingly, DCs from either TLR2- or TLR9-deficient mice displayed significantly reduced IL-12p40 production in response to live (Fig. 1 D) or heat-killed (not depicted) M. tuberculosis, with TLR9-deficient DCs showing the greater defect. However, DCs from mice lacking these single TLRs were clearly less impaired in their IL-12 responsiveness than DCs from MyD88−/− animals (Fig. 1 D). Chloroquine partially inhibited bacterial-induced IL-12 synthesis by WT DCs and completely blocked the response of TLR2−/− DCs to this stimulus (Fig. 1 C), supporting a major requirement for endosomal acidification in M. tuberculosis–stimulated IL-12 production.
Figure 1.

Role for both TLR9 and TLR2 in the MyD88-dependent IL-12p40 response of splenic DCs to M. tuberculosis. (A) Purified CD11c+ spleen cells from WT or MyD88−/− mice were exposed to live (MOI = 1:1 [M. tuberculosis 1] or 3:1 [M. tuberculosis 3]) or heat-killed (MOI = 1:1 H-K M. tuberculosis 1]) M. tuberculosis for 24 h. (B) WT DCs were treated with 5 μg/ml cytochalasin D or vehicle (DMSO) for 30 min and then incubated with live M. tuberculosis (MOI = 1:1) or 10 μg/ml LPS for 24 h. (C) DCs from WT or TLR2−/− mice were treated with 5 μg/ml chloroquine or vehicle (saline) for 30 min and then stimulated with live M. tuberculosis (MOI = 1:1) for 24 h. (D) DCs from WT, MyD88−/−, TLR2−/−, TLR9−/−, and TLR2/9−/− were exposed to live M. tuberculosis (MOI = 1:1) or TLR agonists as described in A. In all experiments, supernatants were harvested and IL-12p40 was determined by ELISA. Results are means ± SE of triplicate measurements. Experiments shown are representative of at least three performed. *, P < 0.05 between experimental and control groups in A, B, and C. **, P < 0.05 between TLR2−/− versus TLR9−/− values in D.
To test whether TLR9 and TLR2 have additive effects on M. tuberculosis–induced cytokine production, we generated TLR2/9-deficient mice and tested the response of DCs from these double KO (DKO) animals. Importantly, M. tuberculosis–exposed splenic DCs from TLR2/9−/− mice displayed a profound reduction in IL-12p40 synthesis, greater than that seen with either of the single KO animals and comparable to that seen with DCs from MyD88-deficient mice (Fig. 1 D). TLR2/TLR9 cooperation was also observed in the IL-12p40 as well as p70 response of BMDCs to M. tuberculosis (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20051782/DC1), although in agreement with previously published data in a similar in vitro system (43), levels of the IL-12 heterodimer were much lower than the single p40 chain. Also consistent with previous findings (45), no influence of TLR2, TLR9, or MyD88 on type I IFN production (IFN-α) by DCs stimulated with live M. tuberculosis was detected in these experiments (Fig. S2 D).
Because macrophages play a major role in the response to mycobacteria, we also investigated TLR2/TLR9 involvement in proinflammatory cytokine production by these cells. BMM from TLR2- or TLR9-deficient mice showed significant reductions in bacteria-stimulated IL-12 synthesis (Fig. 2 A). Moreover, DKO BMM showed reduced cytokine responses comparable to those seen in MyD88−/− APC populations (Fig. 2), suggesting that TLR2 and TLR9 act in concert in signaling IL-12 responses by these cells. In contrast, TNF and IL-6 production by macrophages appeared to be controlled primarily by TLR2 and not TLR9 (Fig. 2, B and C). TLR2 also appeared to preferentially regulate the low level TNF response observed in BMDCs (Fig. S2 C) and splenic DCs (not depicted).
Figure 2.
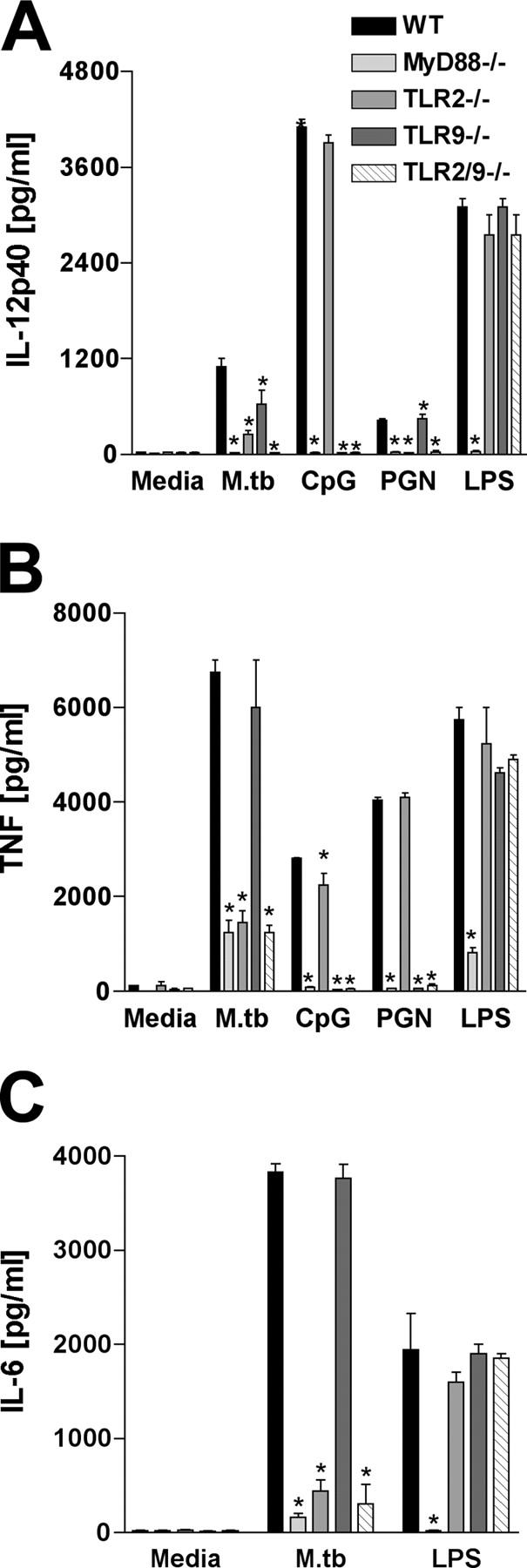
Role of TLR9 and TLR2 in proinflammatory cytokine production by M. tuberculosis–stimulated macrophages. BMM from WT, MyD88−/−, TLR2−/−, TLR9−/−, and TLR2/9−/− were stimulated with M. tuberculosis (MOI = 1:1), 15 μg/ml CpG, 5 μg/ml PGN, or 100 ng/ml LPS for 24 h. (A) IL-12p40, (B) TNF, and (C) IL-6 production was measured in the culture supernatants by ELISA. Results are means ± SE of triplicate measurements. Experiments shown are representative of two performed. *, P < 0.05 between WT versus KO values.
TLR2 and TLR9 cooperate in host resistance to M. tuberculosis infection
To assess and compare the respective roles of TLR2 and TLR9 in resistance to infection in vivo, we measured survival, bacterial loads, and histopathology in TLR2−/−, TLR9−/−, TLR2/9−/−, MyD88−/−, and WT control mice infected by the aerosol route with a low dose (50–100 CFUs) of virulent M. tuberculosis. At this challenge level, all WT animals survived for at least 200 d, whereas all MyD88−/− animals succumbed within 75 d as reported previously (20, 21). Most single TLR2−/− and TLR9−/− mice survived for the full 200-d period of the experiment, with some attrition occurring after day 100 (Fig. 3 A). In contrast, infected TLR2/9−/− mice began to die much earlier, and all succumbed within 120 d (Fig. 3 A).
Figure 3.
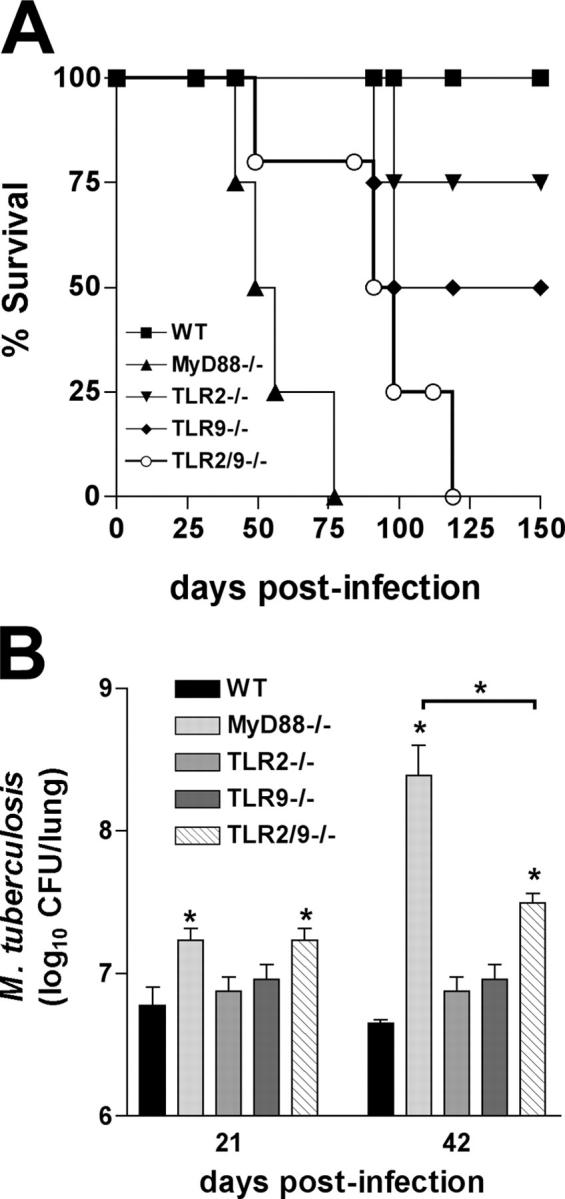
Interaction of TLR9 and TLR2 in host resistance to aerosol M. tuberculosis infection. (A) WT, TLR2-, TLR9-, TLR2/9-, and MyD88-deficient mice were aerogenically infected with 50–100 CFUs/mouse (n = 6 animals per group), and survival was monitored. The results shown are representative of two independent experiments. Statistical analysis revealed that the MyD88-, TLR9-, and TLR2/9-deficient mice were significantly more susceptible (P < 0.01) than WT animals and that the survival curve of the TLR2/9−/− mice is significantly different from that of the TLR9 (P = 0.027), TLR2 (P = 0.0053), or MyD88 (P = 0.002) animal groups. (B) Lungs from infected animals were harvested at 21 and 42 d after infection, and mycobacterial loads were determined. Results are mean ± SE of measurements from four animals. The experiment shown is representative of two performed. *, differences in CFUs between the KO versus WT groups that are statistically significant (P < 0.05).
In several previous studies, a more profound effect of TLR2 deficiency on host resistance to M. tuberculosis was revealed at higher challenge doses (23, 28). In analogous fashion, TLR9-deficient mice were shown to be highly susceptible to challenge with 500 CFUs/mouse, with all of the animals now succumbing by day 70 (Fig. 4 A).
Figure 4.

Increased susceptibility of TLR9−/− mice to high dose M. tuberculosis infection. (A) WT, MyD88-, and TLR9-deficient mice were aerogenically infected with 500 CFUs/mouse (n = 5 animals per group) instead of the usual 50–100 CFU challenge, and survival was monitored. The results shown are representative of two independent experiments. Statistical analysis revealed that the MyD88- and TLR9-deficient mice were significantly more susceptible (P < 0.001) than WT animals and that the survival curve of the TLR9 mice is significantly different (P = 0.0042) from that of the MyD88 animal group. (B) Lungs from infected animals were harvested at 14 and 21 d after infection, and mycobacterial loads were determined. Results are mean ± SE of measurements from four animals. *, a statistically significant difference (P < 0.05) in CFUs between TLR9−/− versus WT mice.
The observed effects of TLR deficiency on host resistance correlated with changes in pulmonary bacterial load measured at days 21 and 42 after infection with a low dose inocula (Fig. 3 B). Thus, as reported previously (20, 21), MyD88−/− mice showed greatly increased bacterial burdens in comparison to WT animals, a difference that approached 2 logs by day 42 after infection. In contrast, the single TLR2−/− and TLR9−/− mice showed only minor increases in mycobacterial loads at both time points (Fig. 3 B), and it was only at day 100 after infection that these differences in CFUs reached statistical significance (not depicted). Importantly, at day 21 after infection, TLR2/9−/− animals displayed increases in pulmonary bacterial counts equivalent to those observed in the MyD88−/− mice, and at day 42, the TLR2/9−/− animals still maintained significantly higher CFUs when compared with WT or each of the single TLR-deficient mice, albeit lower than the bacterial counts in the MyD88−/− animals (Fig. 3 B). Similar alterations in CFUs were observed in the spleens from the same animals (not depicted). Although single TLR9−/− mice challenged with a low dose of M. tuberculosis failed to display significantly higher pathogen loads than WT animals at days 21 and 42, they exhibited statistically significant increases in CFUs when challenged with the higher dose (500 CFUs) inocula (Fig. 4 B), and this loss in resistance correlated with diminished ex vivo IL-12p40 production detected in lung homogenates (not depicted).
Cooperative effects of TLR2 and TLR9 on the pulmonary histopathologic response to M. tuberculosis
To determine whether the increased susceptibility of TLR9−/− and TLR2/9−/− mice is reflected in the tissue response to M. tuberculosis, we examined the lungs of the animals at day 42 after infection. As reported previously (20), lungs from MyD88−/− animals exhibited widespread necrosis with few distinct granulomas (Fig. 5, B and G) and showed marked increases in acid-fast–stained bacilli (Fig. 5 L) when compared with WT mice (Fig. 5, A, F, and K). Also in agreement with previous studies (28), lungs from infected TLR2−/− mice showed increased inflammation with exuberant polymorphonuclear infiltrates, interstitial pneumonitis, and general disruption of granuloma morphology (Fig. 5, C, H, and M).
Figure 5.
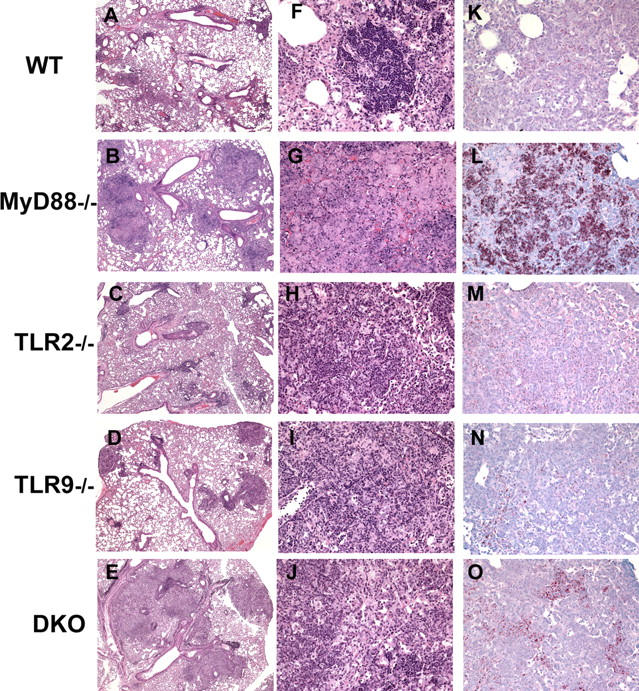
Lungs from TLR2/9−/− mice display exacerbated pulmonary pathology and increased acid-fast bacilli. Formalin-fixed, paraffin-embedded pulmonary tissue sections from day 42 infected mice were stained with hematoxylin and eosin (A–J). Note the increased inflammation in TLR2−/− and TLR2/9−/− lungs (A and E), with the more extreme pathology in the latter group. Acid-fast bacilli in lung tissue were stained with the Ziehl-Neelsen method (K–O). Representative sections from infected WT (A, F, and K), MyD88−/− (B, G, and L), TLR2−/− (C, H, and M), TLR9−/− (D, I, and N), and TLR2/9−/− (E, J, and O) mice are shown. Original magnification is 5 (A–E), 20 (F–J), and 10 (K–O).
In contrast, lungs from TLR9−/− mice showed no major difference in overall histopathology (Fig. 5, D and I) or granuloma numbers (not depicted) when compared with lungs from similar infected WT animals. And like lungs from TLR2−/− mice, lungs from TLR9−/− mice showed only marginal increases in acid-fast–stained bacteria (Fig. 5, K–N). However, a striking difference was seen in the lungs of the day 42 infected TLR2/9−/− animals in comparison with either of the single TLR-deficient or WT mice. Sections from the former animals displayed widespread inflammation (Fig. 5, E and J), even more extreme than that seen in the TLR2−/− mice (Fig. 5, C and H) and closely resembling that observed in the MyD88−/− animals (Fig. 5, B and G). Moreover, lungs from the TLR2/9−/− animals exhibited focal necrosis, a process rarely seen in WT or single TLR-deficient mice but common in MyD88−/− mice. In addition, numerous acid-fast bacilli were clearly evident in lung tissue from the double TLR-deficient hosts (Fig. 5 O), consistent with the increased pulmonary CFUs observed in these animals (Fig. 3 B).
In contrast to TLR2, TLR9 controls IFN-γ production by CD4+ T cells in M. tuberculosis–infected animals
Because IL-12–dependent IFN-γ is a major mediator of resistance to M. tuberculosis and our data indicated a major influence of TLR9 on bacteria-induced IL-12 production in vitro (Fig. 1 D), we asked whether defects in in vivo IFN-γ responses are also evident in TLR9 as well as in TLR2/9−/− mice. When measured at 30 d after infection by intracellular cytokine staining after ex vivo anti-CD3 stimulation, ∼40% of pulmonary CD4+ cells from WT animals were found to secrete IFN-γ, and this response was diminished by 60–70% in the equivalent population from MyD88−/− mice (Fig. 6, A and B). Importantly, TLR9−/− and TLR2/9−/− but not TLR2-deficient mice showed significant reductions in IFN-γ+ CD4+ cells as well as in lung IFN-γ and IL-12p40 mRNA expression, although the observed decreases were less than those seen in MyD88−/− mice (Fig. 6, B–D). In contrast, none of the KO mice showed deficiencies in IFN-γ+ CD8+cells (Fig. 6 B), and it is possible the retention of this cell population in the TLR9−/− and TLR2/9− contributes to their partial resistance. Measurement of changes in IL-12p70 production was not possible because the heterodimer is not present in sufficient quantity in sera or lung homogenates to allow detection by ELISA.
Figure 6.

Influence of TLR9 on the generation of IFN-γ–producing CD4+ T cells and Th1-associated cytokines in M. tuberculosis–infected mice. (A) Lung cells isolated from day 30 infected mice were stimulated with anti-CD3 mAb, and intracellular IFN-γ production was determined by flow cytometry after gating lymphocyte populations by forward and side scatter parameters. The FACS profiles of anti-CD4– and anti-CD8–stained lymphocytes in A are from pooled cells from two mice and are representative of results from four animals per group. The majority (85–95%) of the IFN-γ plus CD4− cells shown in the dot plots in the top panel were determined to be CD8+ T cells (unpublished data). Based on its nonspecific staining with multiple antibodies, the CD4 dim IFN-γ+ population in the lung preparations from infected MyD88 KO mice is likely to represent dead cells, consistent with their abundance in sections of the same tissue (Fig. 5, B and G). Percentage of CD4+ and CD8+ T cells that stain positively for IFN-γ calculated from the experiments shown in A. Relative expression of mRNAs for IFN-γ (C) and IL-12p40 (D) determined in lungs at 30 d after infection. Results are mean ± SE of measurements from three animals. (E) Purified splenic CD4+ T cells from the same mice described in A were cocultured with BMDCs infected with different MOIs for 72 h. IFN-γ was assayed by ELISA in culture supernatants. The means ± SE of measurements from triplicate wells are presented. The experiment shown was performed twice with similar results. *, significantly different values (P < 0.05) between WT and KO cells.
To confirm that these differences reflect alterations in M. tuberculosis–specific Th1 priming, we tested the recall responses of splenic CD4+ T cells from day 30 infected mice using BMDCs from WT mice that had been infected in vitro with different doses of live mycobacteria as APCs. As shown in Fig. 6 E, major defects in M. tuberculosis–specific IFN-γ responses were observed in the TLR9−/−, MyD88−/−, and TLR2/9−/− mice but not in the TLR2−/− animals.
Discussion
The greatly enhanced susceptibility of MyD88−/− mice to M. tuberculosis is perhaps the strongest evidence for a role of TLR/IL-1R signaling in host resistance to this pathogen. Nevertheless, in previous studies it has been difficult to assign this defect to the role of any individual TLR/IL-1R family member. Because mice deficient in each of the known TLRs have not yet been systematically screened, the possibility remains that one yet-to-be-investigated TLR accounts for MyD88-dependent resistance. An alternative explanation proposed by others (2, 46) as a general concept for TLR function is that multiple TLRs act in concert in determining pathogen control. In this study, we have identified a previously unrecognized TLR–ligand interaction that contributes to the innate and adaptive immune response to M. tuberculosis and provided clear-cut evidence for its cooperation with a second TLR signal in host resistance to this bacterium.
Although mycobacterial DNA has long been linked to the adjuvant properties of BCG as well as mycobacterial extracts (34–36), it is only with the recent discovery of immunostimulatory CpG motifs (31) and their recognition by TLR9 (47) that the basis of this effect of mycobacteria on the immune system has been properly appreciated. Nevertheless, the role of TLR9/DNA interaction on host resistance to M. tuberculosis or to other medically important mycobacteria has never been systematically examined. The findings presented here confirm that mycobacterial DNA does indeed stimulate proinflammatory cytokine synthesis through TLR9 and further establish a role for this TLR in the IL-12 and Th1 responses to live M. tuberculosis in vitro as well as in vivo. Although these defects were associated with only minor increases in pulmonary bacterial loads and decreases in host survival at low dose challenge, TLR9−/− mice displayed markedly enhanced susceptibility when exposed to higher dose bacterial inocula (Fig. 4) as described previously for TLR2−/− animals (23, 28). However, in direct contrast to TLR2−/− mice, infected TLR9−/− animals did not show major alterations in granulomatous pathology or in vitro TNF production by APCs. Thus, although TLR9−/− and TLR2−/− mice show comparable changes in resistance to M. tuberculosis infection at both low and high dose infection, they exhibit distinct immune response defects to this pathogen in vivo.
It is likely that TLR9 triggering by mycobacteria requires bacterial uptake and phagolysosomal fusion, as drugs that inhibit these two processes dampened the TLR9-dependent IL-12 response of APCs. Therefore, we hypothesize that M. tuberculosis triggers TLR9 by releasing DNA from either bacteria dying within phagolysosomes or through the uptake of dead bacilli. An alternative possibility that cannot be ruled out at present is that mycobacteria possess TLR9 ligands distinct from genomic bacterial DNA. A recently described precedent is the stimulation of TLR9 by hemozoin pigment from malaria (48).
Although TLR9 alone had distinct but partial effects on host resistance to M. tuberculosis, these were clearly enhanced in mice doubly deficient in TLR9 and TLR2 as reflected in both increased bacterial load and reduced mean survival. In addition, DCs from TLR2/9 DKO mice showed greater impairment in their mycobacteria-induced IL-12 responses than did the equivalent populations from each of the single TLR−/− animals. In each of the above parameters, the observed effects of the absence of TLR9 and TLR2 appeared to be additive in the TLR2/9 DKO mice, although the pulmonary pathology and bacterial burden in these animals as measured by both CFU and acid-fast bacilli in situ staining showed evidence of synergy (Figs. 3 and 5, M–O). Nevertheless, in terms of the other parameters analyzed, the phenotype of the TLR2/9 DKO animals is more complex. For example, redundancy was observed in the regulation of IL-12p40 production by TLR2 and TLR9 in BMDCs (Fig. S2 A). Moreover, as discussed above, in several cases individual immune deficiencies appeared to be linked to single TLRs. For example, M. tuberculosis–induced TNF production appears to be controlled by TLR2 and recall IFN-γ responses by TLR9.
It has recently been proposed in several studies (46, 49) that TLR signals synergize in the triggering of IL-12 and other Th1-promoting mediators by DCs. This hypothesis, based largely on experiments examining the interactions of multiple TLR ligands on DC responses, argues that the induction and maintenance of an effective immune response against microbes depends on the recognition of a “pathogen code” by a combination of different TLRs. Our results partially support this concept because some but not all of the M. tuberculosis–induced TLR effects were synergistic in vivo. Nevertheless, it is likely that specific combinations of TLRs act in concert by instead stimulating distinct responses that together are required for effective microbial control, a mechanism also requiring a pathogen code. This mechanism also would depend on induction thresholds related to both the dose and kinetics of infection.
Although our in vivo data reveal a major cooperative interaction between TLR9 and TLR2 in host resistance to mycobacteria, this is clearly not the case for other TLR combinations. Thus, TLR2/4−/− mice have been found to display unimpaired resistance to M. tuberculosis as well as BCG infection (45, 50). Moreover, in a recent experiment, we failed to observe increased susceptibility to M. tuberculosis in animals deficient in TIRAP, an adaptor molecule required for MyD88-dependent signaling by both TLR2 and TLR4, relative to mice singly deficient in TLR2 (unpublished data). It is of interest that both TLR2−/− and TLR9−/− mice display clearly enhanced susceptibility to high dose M. tuberculosis infection, a property that does not appear to be shared by TLR4−/− animals (23). This observation suggests that screening of different TLR/IL-1R–deficient mice by high dose challenge could be used as a strategy for detecting additional signaling receptors that cooperate with TLR2 and TLR9 in explaining the MyD88-dependent control of M. tuberculosis infection. Having shown cooperation between two TLR/IL-1R family members in host resistance to this pathogen, the next obvious step is to determine whether even greater effects on control of M. tuberculosis will be evident in mice with the appropriate triple receptor deficiency.
The findings of this study in the murine tuberculosis model have several implications for the investigation of the role of TLR/IL-1R in susceptibility of humans to mycobacterial infection and disease. The involvement of these receptors has been suggested from genetic studies correlating single nucleotide polymorphisms within the TLR2 gene, with disease severity in patients infected with M. leprae (51) or susceptibility to infection in populations exposed to M. tuberculosis (52, 53). Our observations that TLR9 also contributes to host resistance to mycobacterial infection suggests that polymorphisms in this gene (e.g., TLR9 single nucleotide polymorphisms at −1,237 and −2,428; reference 54) should also be examined in the same type of genetic study and predicts that more extreme disease susceptibility will be seen in patients with simultaneous mutations in both TLR2 and TLR9. At a more general level, the evidence that different TLRs cooperate in determining host resistance to infection in murine experimental models supports the notion that complex polygenic analyses involving the interaction of multiple rather than single TLR gene family alleles might be required to reveal major functions for the TLR/IL-1R system in innate immunity to human infectious diseases.
MATERIALS AND METHODS
Experimental animals.
WT control C57BL/6 mice were purchased from Taconic Farms. Breeding pairs of MyD88−/−, TLR2−/−, and TLR9−/− mice were obtained from S. Akira (Osaka University, Osaka, Japan) via D. Golenbock (University of Massachusetts Medical School, Worcester, MA) and R. Seder (National Institutes of Health [NIH], Bethesda, MD). The MyD88−/− and TLR9−/− mice had been backcrossed to C57BL/6 for 10 generations, and the TLR2−/− animals had been backcrossed for five generations. TLR2/9−/−animals were generated by mating TLR2−/− with TLR9-deficient animals. These F1 animals were then intercrossed to derive homozygous TLR2/9−/− mice identified by PCR of tail snips (unpublished data). Because of the known influence of 129/SvJ genes in host resistance to intravenous M. tuberculosis infection (55) and the finite although remote possibility that the relevant genes may have been retained in the TLR2−/− parents of this cross, we compared the resistance of C57BL/6 and B6/129F2 mice under the conditions of low dose aerosol infection used. In agreement with studies by other investigators (56–59), we observed no significant differences in survival and bacterial loads between the two mouse groups (unpublished data). Animals were bred and maintained at an American Association of Laboratory Animal Care–accredited facility at the National Institute of Allergy and Infectious Diseases [NIAID], NIH. Mice of both sexes between 8- and 14-wk old were used in all experiments.
TLR agonists and other reagents.
The synthetic lipoprotein Pam3Cys (S-[2,3-bis(palmitoyloxy)-(2-RS)-propyl]-N-palmitoyl-(R)-Cys-(S)-Ser-Lys4-OH, trihydrochloride) was obtained from EMC Microcollections. Peptideoglycan (Staphylococcus aureus), ultra-pure LPS (E. coli 0111:B4), endotoxin-free E. coli DNA (K12), and CpG oligo DNA (1826) were purchased from Invivogen. Purified genomic DNA from M. tuberculosis H37Rv was provided by J. Belisle (Colorado State University, Fort Collins, CO) under the NIH, NIAID contract NO1 AI-75320 “Tuberculosis Research Materials and Vaccine Testing.” Purified genomic DNA from Mycobacterium sp (BCG) was purchased from American Type Culture Collection (no. 19015D). Chloroquine and cytochalasin D were obtained from Sigma-Aldrich.
APCs and cell cultures.
Splenic CD11c+ cells were obtained by incubation of splenic cell suspensions with anti-CD11c MicroBeads (Miltenyi Biotec) for 15 min at 4°C followed by a washing step in PBS/bovine serum albumin and then sorted in an AutoMACS (isolation mode POSSEL_S; Miltenyi Biotec). Analysis of the sorted cells showed purity >95%.
BMDCs were generated as originally described elsewhere (60 ). In brief, BM cells were removed from the femurs and tibias of mice and cultured in RPMI 1640 (GIBCO BRL) supplemented with 2 mM l-glutamine, heat-inactivated 10% FCS, 100 μg/ml penicillin, 100 μg/ml streptomycin, 5 × 10−5 M 2-mercaptoethanol (complete media; all from Sigma-Aldrich), plus 20 ng/ml GM-CSF (GIBCO BRL). On days 3 and 6, complete media was added containing 10 ng/ml GM-CSF. BMDCs were used at days 6–7 of culture.
BMM were generated as described previously (61). In brief, BM cells were washed and resuspended in DMEM containing glucose, supplemented with 2 mM l-glutamine, 10% FCS, 10 mM Hepes, 100 μg/ml streptomycin, 100 U/ml penicillin (all from Sigma-Aldrich), and 20–30% L929 cell-conditioned medium (as a source of M-CSF), and incubated for 7 d at 37°C, 5% CO2.
M. tuberculosis infections.
For in vitro exposure of APCs to M. tuberculosis, the virulent strain H37Rv was prepared from frozen stocks as described previously (62). In some experiments, mycobacteria were killed by heating at 60°C for 1 h. DCs and BMM were exposed to different multiplicities of infection (MOIs) of mycobacteria or TLR agonists in complete media for 18–24 h. In one set of experiments, splenic DCs were first preincubated with 5 μg/ml chloroquine or cytochalasin D for 30 min at 37°C. Cells were then exposed to mycobacteria (MOI = 1) or 10 μg/ml LPS as a control. Supernatants were then harvested, and ELISAs for IL-12p40, TNF, and IL-6 (R&D Systems) as well as for IFN-α (PBL Biomedical Laboratories) were performed.
For in vivo mycobacterial infection, mice were infected as described previously (58). In brief, animals were placed in a closed, nose-only aerosolization system (CH Technologies) and exposed for 15 min to nebulized M. tuberculosis to deliver 50–100 bacteria/mouse (low dose inocula). In a different set of experiments, mice were infected with ∼500 bacteria/mouse (high dose inocula). To assess mycobacterial load, lungs and spleens were harvested at several times after infection, and tissue homogenates were diluted in PBS/Tween-20 and cultured on 7H11 agar plates as described previously (63). Colony counts were determined 21 d later.
Flow cytometry.
Single cell suspensions from individual mice were immunostained as described previously (15). In brief, for intracellular detection of IFN-γ, total lung cells were stimulated with 10 μg/ml of plate-bound anti-CD3 at 37°C for 6 h, and brefeldin A was added during the last 2 h. Cells were then surface stained with mAb to CD4 (clone RM4-5) or CD8 (clone 53-6.7), fixed, and permeabilized. Intracellular IFN-γ was detected with anti–IFN-γ mAb (clone XMG1.2). Data were collected using a FACSCalibur (BD Immunocytometry Systems) with CELLQuest (BD Biosciences) and analyzed with FlowJo software (Tree Star). All mAbs were obtained from BD Biosciences.
Measurement of cytokine gene expression in lung tissue.
Total RNA was isolated from lungs, and real-time RT-PCR was performed on an ABI Prism 7900 sequence detection system (Applied Biosystems) using SYBR Green PCR Master Mix (Applied Biosystems) after RT of 1 μg RNA using Superscript II reverse transcriptase (Invitrogen). The relative level of gene expression was determined by the comparative threshold cycle method as described by the manufacturer, whereby each sample was normalized to hprt and expressed as a fold change compared with untreated controls. The following primer pairs were used: for hprt: GTTGGTTACAGGCCAGACTTTGTTG (forward) and GAGGGTAGGCTGGCCTATAGGCT (reverse); il-12p40: CTCACATCTGCTGCTCCACAAG (forward) and AATTTGGTGCTTCACACTTCAGG (reverse); ifn-γ: AGAGCCAGATTATCTCTTTCTACCTCAG (forward) and CTTTTTTCGCCTTGCTGCTG (reverse).
CD4+ T cell recall response assay.
Spleen cells from M. tuberculosis–infected WT, MyD88−/−, TLR2−/−, TLR9−/−, and TLR2/9−/− mice were incubated with anti-CD4 MicroBeads (Miltenyi Biotec) for 15 min at 4°C, washed, and then sorted in the AutoMACS (Miltenyi Biotec). The purified CD4+ T cells (106 cell/ml) were then cocultured with M. tuberculosis–infected BMDCs (5 × 105 cells/ml) for 72 h. In parallel, 10 μg/ml of plate-bound anti-CD3– (clone 145-2C11; BD Biosciences) stimulated CD4+ T cells were cultured for 72 h as a positive control. IFN-γ (R&D Systems) levels in culture supernatants were then determined by ELISA.
Histopathology.
Lungs were fixed by inflating the tissues with neutral buffered formalin, sectioned, and then stained with hematoxylin and eosin or by the Ziehl-Neelsen method to detect acid-fast mycobacteria.
Statistics.
Student's t test was used to determine the significance of differences between groups. Survival curves were generated using the Kaplan-Meier method, and the significance of differences was calculated by the log-rank test. Values of P < 0.05 were considered statistically significant.
Online supplemental material.
Fig. S1 demonstrates the TLR9 dependence of proinflammatory cytokine responses by APCs stimulated with mycobacterial DNA. Fig. S2 demonstrates a marked decrease in M. tuberculosis–stimulated IL-12 but not TNF production by TLR2/9−/− BMDCs when compared with the equivalent cell populations from single TLR−/− or WT animals. Fig. S3 shows that M. tuberculosis–stimulated type I IFN (IFN-α) production by DCs does not require TLR2, TLR9, or MYD88 in contrast with the TLR9/MyD88 dependence of the same cytokine response stimulated by mycobacterial DNA. Figs. S1–S3 are available at http://www.jem.org/cgi/content/full/jem.20051782/DC1.
Acknowledgments
We are grateful to Sara Hieny, Patricia Casper, and Sandy White for their invaluable technical assistance and Drs. Ulrike Wille-Reece and Robert Seder for generously providing the TLR9−/− mice breeders used in this study. We also thank Drs. Giorgio Trinchieri, Ricardo T. Gazzinelli, David Segal, and Karen L. Elkins for their critical reading of this manuscript.
The authors have no conflicting financial interests.
Abbreviations used: BCG, bacillus of Calmette and Guerin; BMDCs, BM-derived DCs; BMM, BM-derived macrophages; DKO, double KO; MOI, multiplicity of infection; TLR, Toll-like receptor.
A. Bafica and C.A. Scanga contributed equally to this work.
References
- 1.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 2.Iwasaki, A., and R. Medzhitov. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5:987–995. [DOI] [PubMed] [Google Scholar]
- 3.Krutzik, S.R., and R.L. Modlin. 2004. The role of Toll-like receptors in combating mycobacteria. Semin. Immunol. 16:35–41. [DOI] [PubMed] [Google Scholar]
- 4.Quesniaux, V., C. Fremond, M. Jacobs, S. Parida, D. Nicolle, V. Yeremeev, F. Bihl, F. Erard, T. Botha, M. Drennan, et al. 2004. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6:946–959. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. The World Health Report 2004. Geneva. 32 pp.
- 6.Caruso, A.M., N. Serbina, E. Klein, K. Triebold, B.R. Bloom, and J.L. Flynn. 1999. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J. Immunol. 162:5407–5416. [PubMed] [Google Scholar]
- 7.Scanga, C.A., V.P. Mohan, K. Yu, H. Joseph, K. Tanaka, J. Chan, and J.L. Flynn. 2000. Depletion of CD4+ T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon γ and nitric oxide synthase 2. J. Exp. Med. 192:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper, A.M., A.D. Roberts, E.R. Rhoades, J.E. Callahan, D.M. Getzy, and I.M. Orme. 1995. The role of interleukin-12 in acquired immunity to Mycobacterium tuberculosis infection. Immunology. 84:423–432. [PMC free article] [PubMed] [Google Scholar]
- 9.Flynn, J.L., M.M. Goldstein, K.J. Triebold, J. Sypek, S. Wolfand, and B.R. Bloom. 1995. IL-12 increases resistance of BALB/c mice to Mycobacterium tuberculosis infection. J. Immunol. 155:2515–2524. [PubMed] [Google Scholar]
- 10.Flynn, J.L., M.M. Goldstein, J. Chan, K.J. Triebold, K. Pfeffer, C.J. Lowenstein, R. Schreiber, T.W. Mak, and R.R. Bloom. 1995. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 2:561–572. [DOI] [PubMed] [Google Scholar]
- 11.Bean, A.G., D.R. Roach, H. Briscoe, M.P. France, H. Korner, J.D. Sedgwick, and W.J. Britton. 1999. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 162:3504–3511. [PubMed] [Google Scholar]
- 12.Cooper, A.M., D.K. Dalton, T.A. Stewart, J.P. Griffin, D.G. Russell, and I.M. Orme. 1993. Disseminated tuberculosis in interferon γ gene-disrupted mice. J. Exp. Med. 178:2243–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flynn, J.L., J. Chan, K.J. Triebold, D.K. Dalton, T.A. Stewart, and B.R. Bloom. 1993. An essential role for interferon γ in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178:2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casanova, J.L., and L. Abel. 2002. Genetic dissection of immunity to mycobacteria: the human model. Annu. Rev. Immunol. 20:581–620. [DOI] [PubMed] [Google Scholar]
- 15.Feng, C.G., D. Jankovic, M. Kullberg, A. Cheever, C.A. Scanga, S. Hieny, P. Caspar, G.S. Yap, and A. Sher. 2005. Maintenance of pulmonary Th1 effector function in chronic tuberculosis requires persistent IL-12 production. J. Immunol. 174:4185–4192. [DOI] [PubMed] [Google Scholar]
- 16.Thoma-Uszynski, S., S. Stenger, O. Takeuchi, M.T. Ochoa, M. Engele, P.A. Sieling, P.F. Barnes, M. Rollinghoff, P.L. Bolcskei, M. Wagner, et al. 2001. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 291:1544–1547. [DOI] [PubMed] [Google Scholar]
- 17.Jones, B.W., T.K. Means, K.A. Heldwein, M.A. Keen, P.J. Hill, J.T. Belisle, and M.J. Fenton. 2001. Different Toll-like receptor agonists induce distinct macrophage responses. J. Leukoc. Biol. 69:1036–1044. [PubMed] [Google Scholar]
- 18.Abel, B., N. Thieblemont, V.J.F. Quesniaux, N. Brown, J. Mpagi, K. Miyake, F. Bihl, and B. Ryffel. 2002. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J. Immunol. 169:3155–3162. [DOI] [PubMed] [Google Scholar]
- 19.Feng, C.G., C.A. Scanga, C.M. Collazo-Custodio, A.W. Cheever, S. Hieny, P. Caspar, and A. Sher. 2003. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J. Immunol. 171:4758–4764. [DOI] [PubMed] [Google Scholar]
- 20.Scanga, C.A., A. Bafica, C.G. Feng, A.W. Cheever, S. Hieny, and A. Sher. 2004. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect. Immun. 72:2400–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fremond, C.M., V. Yeremeev, D.M. Nicolle, M. Jacobs, V.F. Quesniaux, and B. Ryffel. 2004. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J. Clin. Invest. 114:1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi, S., C. Nathan, D. Schnappinger, J. Drenkow, M. Fuortes, E. Block, A. Ding, T.R. Gingeras, G. Schoolnik, S. Akira, et al. 2003. MyD88 primes macrophages for full-scale activation by interferon-γ yet mediates few responses to Mycobacterium tuberculosis. J. Exp. Med. 198:987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiling, N., C. Holscher, A. Fehrenbach, S. Kroger, C.J. Kirschning, S. Goyert, and S. Ehlers. 2002. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J. Immunol. 169:3480–3484. [DOI] [PubMed] [Google Scholar]
- 24.Kamath, A.B., J. Alt, H. Debbabi, and S.M. Behar. 2003. Toll-like receptor 4-defective C3H/HeJ mice are not more susceptible than other C3H substrains to infection with Mycobacterium tuberculosis. Infect. Immun. 71:4112–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugawara, I., H. Yamada, C. Li, S. Mizuno, O. Takeuchi, and S. Akira. 2003. Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol. Immunol. 47:327–336. [DOI] [PubMed] [Google Scholar]
- 26.Sugawara, I., H. Yamada, S. Hua, and S. Mizuno. 2001. Role of interleukin (IL)-1 type 1 receptor in mycobacterial infection. Microbiol. Immunol. 45:743–750. [DOI] [PubMed] [Google Scholar]
- 27.Kinjo, Y., K. Kawakami, K. Uezu, S. Yara, K. Miyagi, Y. Koguchi, T. Hoshino, M. Okamoto, Y. Kawase, K. Yokota, et al. 2002. Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: a comparative study with IL-12p40. J. Immunol. 169:323–329. [DOI] [PubMed] [Google Scholar]
- 28.Drennan, M.B., D. Nicolle, V.J. Quesniaux, M. Jacobs, N. Allie, J. Mpagi, C. Fremond, H. Wagner, C. Kirschning, and B. Ryffel. 2004. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am. J. Pathol. 164:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Means, T.K., B.W. Jones, A.B. Schromm, B.A. Shurtleff, J.A. Smith, J. Keane, D.T. Golenbock, S.N. Vogel, and M.J. Fenton. 2001. Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis-induced macrophage responses. J. Immunol. 166:4074–4082. [DOI] [PubMed] [Google Scholar]
- 30.Underhill, D.M., A. Ozinsky, K.D. Smith, and A. Aderem. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA. 96:14459–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krieg, A.M., A.K. Yi, S. Matson, T.J. Waldschmidt, G.A. Bishop, R. Teasdale, G.A. Koretzky, and D.M. Klinman. 1995. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 374:546–549. [DOI] [PubMed] [Google Scholar]
- 32.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9–mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tokunaga, T., H. Yamamoto, S. Shimada, H. Abe, T. Fukuda, Y. Fujisawa, Y. Furutani, O. Yano, T. Kataoka, T. Sudo, et al. 1984. Antitumor activity of deoxyribonucleic acid fraction from Mycobacterium bovis BCG. I. Isolation, physicochemical characterization, and antitumor activity. J. Natl. Cancer Inst. 72:955–962. [PubMed] [Google Scholar]
- 34.Ronaghy, A., B.J. Prakken, K. Takabayashi, G.S. Firestein, D. Boyle, N.J. Zvailfler, S.T. Roord, S. Albani, D.A. Carson, and E. Raz. 2002. Immunostimulatory DNA sequences influence the course of adjuvant arthritis. J. Immunol. 168:51–56. [DOI] [PubMed] [Google Scholar]
- 35.Krieg, A.M. 2001. CpG motifs in bacterial DNA and their immune effects. 2002. Annu. Rev. Immunol. 20:709–760. [DOI] [PubMed] [Google Scholar]
- 36.Tokunaga, T., T. Yamamoto, and S. Yamamoto. 1999. How BCG led to the discovery of immunostimulatory DNA. Jpn. J. Infect. Dis. 52:1–11. [PubMed] [Google Scholar]
- 37.Yamamoto, S., T. Yamamoto, S. Shimada, E. Kuramoto, O. Yano, T. Kataoka, and T. Tokunaga. 1992. DNA from bacteria, but not from vertebrates, induces interferons, activates natural killer cells and inhibits tumor growth. Microbiol. Immunol. 36:983–997. [DOI] [PubMed] [Google Scholar]
- 38.Ahmad-Nejad, P., H. Hacker, M. Rutz, S. Bauer, R.M. Vabulas, and H. Wagner. 2002. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 32:1958–1968. [DOI] [PubMed] [Google Scholar]
- 39.Latz, E., A. Schoenemeyer, A. Visintin, K.A. Fitzgerald, B.G. Monks, C.F. Knetter, E. Lien, N.J. Nilsen, T. Espevik, and D.T. Golenbock. 2004. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 5:190–198. [DOI] [PubMed] [Google Scholar]
- 40.Leifer, C.A., M.N. Kennedy, A. Mazzoni, C. Lee, M.J. Kruhlak, and D.M. Segal. 2004. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J. Immunol. 173:1179–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalis, C., M. Gumenscheimer, N. Freudenberg, S. Tchaptchet, G. Fejer, A. Heit, S. Akira, C. Galanos, and M.A. Freudenberg. 2005. Requirement for TLR9 in the immunomodulatory activity of Propionibacterium acnes. J. Immunol. 174:4295–4300. [DOI] [PubMed] [Google Scholar]
- 42.Rutz, M., J. Metzger, T. Gellert, P. Luppa, G.B. Lipford, H. Wagner, and S. Bauer. 2004. Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur. J. Immunol. 34:2541–2550. [DOI] [PubMed] [Google Scholar]
- 43.Jang, S., S. Uematsu, S. Akira, and P. Salgame. 2004. IL-6 and IL-10 induction from dendritic cells in response to Mycobacterium tuberculosis is predominantly dependent on TLR2-mediated recognition. J. Immunol. 173:3392–3397. [DOI] [PubMed] [Google Scholar]
- 44.Fulton, S.A., J.M. Johnsen, S.F. Wolf, D.S. Sieburth, and W.H. Boom. 1996. Interleukin-12 production by human monocytes infected with Mycobacterium tuberculosis: role of phagocytosis. Infect. Immun. 64:2523–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi, S., A. Blumenthal, C.M. Hickey, S. Gandotra, D. Levy, and S. Ehrt. 2005. Expression of many immunologically important genes in Mycobacterium tuberculosis-infected macrophages is independent of both TLR2 and TLR4 but dependent on IFN-{alpha}{beta} receptor and STAT1. J. Immunol. 175:3318–3328. [DOI] [PubMed] [Google Scholar]
- 46.Napolitani, G., A. Rinaldi, F. Bertoni, F. Sallusto, and A. Lanzavecchia. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745. [DOI] [PubMed] [Google Scholar]
- 48.Coban, C., K.J. Ishii, T. Kawai, H. Hemmi, S. Sato, S. Uematsu, M. Yamamoto, O. Takeuchi, S. Itagaki, N. Kumar, et al. 2005. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gautier, G., M. Humbert, F. Deauvieau, M. Scuiller, J. Hiscott, E.E. Bates, G. Trinchieri, C. Caux, and P. Garrone. 2005. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor–induced interleukin-12p70 secretion by dendritic cells. J. Exp. Med. 201:1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicolle, D., C. Fremond, X. Pichon, A. Bouchot, I. Maillet, B. Ryffel, and V.J. Quesniaux. 2004. Long-term control of Mycobacterium bovis BCG infection in the absence of Toll-like receptors (TLRs): investigation of TLR2-, TLR6-, or TLR2-TLR4-deficient mice. Infect. Immun. 72:6994–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang, T.J., and G.T. Chae. 2001. Detection of Toll-like receptor 2 (TLR2) mutation in the lepromatous leprosy patients. FEMS Immunol. Med. Microbiol. 31:53–58. [DOI] [PubMed] [Google Scholar]
- 52.Ogus, A.C., B. Yoldas, T. Ozdemir, A. Uguz, S. Olcen, I. Keser, M. Coskun, A. Cilli, and O. Yegin. 2004. The Arg753GLn polymorphism of the human toll-like receptor 2 gene in tuberculosis disease. Eur. Respir. J. 23:219–223. [DOI] [PubMed] [Google Scholar]
- 53.Ben-Ali, M., M.R. Barbouche, S. Bousnina, A. Chabbou, and K. Dellagi. 2004. Toll-like receptor 2 Arg677Trp polymorphism is associated with susceptibility to tuberculosis in Tunisian patients. Clin. Diagn. Lab. Immunol. 11:625–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lazarus, R., W.T. Klimecki, B.A. Raby, D. Vercelli, L.J. Palmer, D.J. Kwiatkowski, E.K. Silverman, F. Martinez, and S.T. Weiss. 2003. Single-nucleotide polymorphisms in the Toll-like receptor 9 gene (TLR9): frequencies, pairwise linkage disequilibrium, and haplotypes in three U.S. ethnic groups and exploratory case-control disease association studies. Genomics. 81:85–91. [DOI] [PubMed] [Google Scholar]
- 55.Medina, E., and R.J. North. 1998. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology. 93:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacMicking, J.D., R.J. North, R. LaCourse, J.S. Mudgett, S.K. Shah, and C.F. Nathan. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA. 94:5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flynn, J.L., M.M. Goldstein, K.J. Triebold, B. Koller, and B.R. Bloom. 1992. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA. 89:12013–12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Behar, S.M., C.C. Dascher, M.J. Grusby, C.R. Wang, and M.B. Brenner. 1999. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J. Exp. Med. 189:1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D'Souza, C.D., A.M. Cooper, A.A. Frank, S. Ehlers, J. Turner, A. Bendelac, and I.M. Orme. 2000. A novel nonclassic beta2-microglobulin-restricted mechanism influencing early lymphocyte accumulation and subsequent resistance to tuberculosis in the lung. Am. J. Respir. Cell Mol. Biol. 23:188–193. [DOI] [PubMed] [Google Scholar]
- 60.Lutz, M.B., N. Kukutsch, A.L. Ogilvie, S. Rossner, F. Koch, N. Romani, and G. Schuler. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 223:77–92. [DOI] [PubMed] [Google Scholar]
- 61.Rothfuchs, A.G., D. Gigliotti, K. Palmblad, U. Andersson, H. Wigzell, and M.E. Rottenberg. 2001. IFN-alpha/beta-dependent, IFN-gamma secretion by bone marrow-derived macrophages controls an intracellular bacterial infection. J. Immunol. 167:6453–6461. [DOI] [PubMed] [Google Scholar]
- 62.Bafica, A., C.A. Scanga, M.L. Schito, S. Hieny, and A. Sher. 2003. Cutting edge: in vivo induction of integrated HIV-1 expression by mycobacteria is critically dependent on Toll-like receptor 2. J. Immunol. 171:1123–1127. [DOI] [PubMed] [Google Scholar]
- 63.Bafica, A., C.A. Scanga, C. Serhan, F. Machado, S. White, A. Sher, and J. Aliberti. 2005. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J. Clin. Invest. 115:1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]