

Abstract
Systemic anticancer chemotherapy is immunosuppressive and mostly induces nonimmunogenic tumor cell death. Here, we show that even in the absence of any adjuvant, tumor cells dying in response to anthracyclins can elicit an effective antitumor immune response that suppresses the growth of inoculated tumors or leads to the regression of established neoplasia. Although both antracyclins and mitomycin C induced apoptosis with caspase activation, only anthracyclin-induced immunogenic cell death was immunogenic. Caspase inhibition by Z-VAD-fmk or transfection with the baculovirus inhibitor p35 did not inhibit doxorubicin (DX)-induced cell death, yet suppressed the immunogenicity of dying tumor cells in several rodent models of neoplasia. Depletion of dendritic cells (DCs) or CD8+T cells abolished the immune response against DX-treated apoptotic tumor cells in vivo. Caspase inhibition suppressed the capacity of DX-killed cells to be phagocytosed by DCs, yet had no effect on their capacity to elicit DC maturation. Freshly excised tumors became immunogenic upon DX treatment in vitro, and intratumoral inoculation of DX could trigger the regression of established tumors in immunocompetent mice. These results delineate a procedure for the generation of cancer vaccines and the stimulation of anti-neoplastic immune responses in vivo.
The complete and permanent success of nonsurgical cancer therapy relies on the targeting of all tumor cells, including cancer stem cells, or theoretically on the direct removal of a fraction of the tumor accompanied by a “bystander effect” in which the immune system recognizes, attacks, and eradicates the remaining tumor cells, including cancer stem cells. Unfortunately, however, most widely used cytotoxic anticancer agents exert immunosuppressive side effects. Even worse, the preponderant type of cell death induced by chemotherapy is apoptosis, and apoptosis is frequently but not unanimously viewed as immunologically silent (leading to ignorance by the immune system) or even as tolerogenic (actively down-regulating the specific antitumor immune response; references 1 and 2). Thus, even after an initial therapeutic success, patients typically fail to mount a clinically relevant antitumor immune response and eventually succumb to tumor cell variants that escape from chemotherapy.
Although apoptosis is itself nonuniform with respect to the programmed signaling events responsible for cell death (3, 4), it is morphologically defined as a uniform type of cell death accompanied by chromatin condensation (pyknosis) and nuclear fragmentation (karyorhexis) occurring within an intact plasma membrane (5, 6). Therefore, it differs from necrosis in which the plasma membrane is destroyed early during the death process. In biochemical terms, apoptosis is frequently accompanied by the activation of a specific subset of cysteine kinases, the caspases (4). Because apoptosis is a physiological process that attains several millions of cells per second in the human body, it is inferred that this type of cell death occurs without emitting “danger signals” that would elicit a productive immune response (7). Apoptotic cells would hide away from immune recognition because they would be rapidly recognized and silently phagocytosed in an efficient manner. Thus, defects in the recognition/phagocytosis of apoptotic cells breach autotolerance and trigger autoimmune reactions (8–10). Moreover, apoptotic cells would mediate active immunosuppression by inhibiting the production of immunostimulatory cytokines (e.g., interleukin 1β by macrophages and interleukin 12 by DCs; references 11 and 12), by stimulating the production of immunosuppressive factors (e.g., transforming growth factor; reference 13), and/or by eliciting antigen-specific immune tolerance (14–16). In apparent contrast with this notion, however, DCs can capture apoptotic tumor cells in vitro and cross-present antigen derived from internalized dying cells on MHC class I molecules for recognition by CD8+ T cells in vivo, thus eliciting a productive immune response (2, 17–22), including in clinical trials (23). Systematic comparisons of the immunogenicity of apoptotic versus necrotic tumor cells have been inconclusive (24–27), however, suggesting that the nature of tumor cells and/or that the death-inducing stimulus would influence the experimental outcome (1).
Driven by these uncertainties and incognita, we decided to explore the possibility of inducing immunogenic tumor cell apoptosis by chemotherapy. Here, we show that anthracyclins are capable of eliciting immunogenic cell death in vitro, ex vivo, and in vivo. These data may have profound implications for the design of chemotherapeutic regimens with immunogenic properties.
Results
A mouse model of vaccination with apoptotic cancer cells
The colon carcinoma CT26 line was killed by treatment with doxorubicin (DX) or mitomycin C (MC) as determined by FACS analysis (Fig. 1 A), although the phenotypic manifestations of cell death differed. Both DX and MC induced caspase activation (Fig. 1 B) and chromatin condensation (Fig. 1, C and D), but only MC led to terminal desoxyribonucleotidyl transferase dUTP nick end labeling (TUNEL)-detectable DNA fragmentation (Fig. 1 C). When combined with DX, the broad spectrum caspase inhibitor Z-VAD-fmk (DXZ) delayed cell death (Fig. 1 A), reduced caspase activation (Fig. 1 B), and blunted chromatin condensation (Fig. 1 D). However, DXZ-treated cells manifested a strongly reduced clonogenic survival as compared with untreated controls, indicating that most of the cells underwent delayed caspase-independent death in vitro (Fig. 1 E) in accord with previous reports that DX can kill cells in the presence of caspase inhibitors (28, 29). Moreover, as compared with untreated controls, DX- and DXZ-treated cells generated tumors with a significant delay after inoculation into athymic nu/nu mice (Fig. 2 A). No tumor growth was found when DX-, DXZ-, or MC-treated cells were inoculated into syngenic, immunocompetent BALB/c mice (Fig. 2 B). To explore the putative immunogenicity of dead or dying cells, BALB/c mice were inoculated into one flank with live CT26 cells and, simultaneously, received an injection of DX-, DXZ-, or MC-treated or necrotic (frozen-thawed [F/T]) cells into the opposite flank. In this therapeutic regime, only DX-treated, but not DXZ-, MC-, or F/T-treated, cells reduced the frequency of tumors developing from live cells by ∼45% on day 30 and ∼30% on day 120 (Fig. 2 C). This degree of protection was obtained without the addition of any adjuvant. It was also independent of the presence of xenogenic antigens (FCS) in the culture media (not depicted). Animals not developing tumors after challenge with DX-treated CT26 cells (5 out of 15) were protected against a second inoculation of live CT 26 cells, but were not immune against the unrelated murine mammary adenocarcinoma TS/A (TSA) tumor (Fig. 2 D). Similarly, the isolated cases of animals not developing tumors after challenge with DXZ- or MC-treated CT26 cells (2 out of 28 and 1 out of 16, respectively) failed to develop tumors after rechallenge with live CT26 cells (not depicted), indicating that these treatments (DXZ and MC) could confer long-term antitumor immunity, albeit at a much lower level of efficacy as compared with DX-treated tumor cells. Splenic T cells from animals protected by inoculation of DX-treated cells could transfer efficient antitumor immunity when injected into naive recipients (Fig. 2 E). Of note, the efficacy of the tumor vaccination could be improved to 80% by one prophylactic injection of DX-treated cells before challenge with live tumor cells. Regardless of the immunization schedule, DX-treated cells did confer a higher degree of protection than DXZ-, MC-, or F/T-treated cells (Fig. 2 F). Moreover, DX-treated cells tended to be more immunogenic than CT26 cells dying after γ irradiation in the experimental setting of prophylactic inoculation of dead cells 1 wk before injection of live tumor cells into the opposite flank (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050915/DC1). Other anthracyclins than DX, such as daunorubicin and idarubicin, also induced immunogenic cell death (Fig. 2 G). Thus, dying tumor cells can elicit an effective antitumor immune response. We wondered whether the chemical nature of anthracyclins could be sufficient to explain the immunogenicity of DX-treated cells. Thus, CT26 cells were killed with MC and then treated with DX for 15 min in conditions that lead to DX incorporation into the cells at a level similar to that of cells killed by 24-h culture with DX (Fig. 3 A). However, the delayed treatment with DX did not ameliorate the poor immunogenicity of tumor cells killed by exposure to MC, as compared with cells killed with DX (Fig. 3 B). Thus, it is the mode of cell killing rather than the presence of DX itself that explains the immunogenicity of DX-treated tumor cells.
Figure 1.
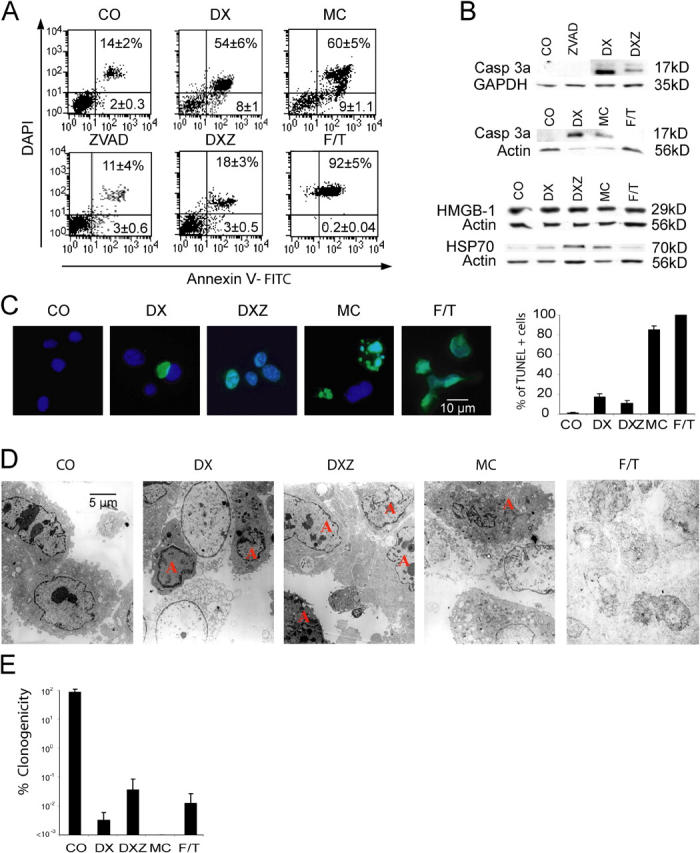
In vitro characterization of the different cell death modalities induced in CT26 cells. (A) FACS analysis of cells treated in vitro with the indicated agents as specified in Materials and methods. ZVAD, Z-VAD-fmk. Numbers indicate the percentage of cells (X ± SD, n = 5) in each quadrant. (B) Immunoblot analyses of cells treated as in A. Cellular extracts were subjected to the immunodetection of activated caspase 3 (Casp3a), HMGB-1, and HSP-70. (C) TUNEL staining (green) of cells counterstained with DAPI (blue). The percentage of TUNEL+ cells (X ± SEM, n = 3) was determined. (D) Transmission electron microscopy. Representative microphotographs are shown. Apoptotic cells showing chromatin condensation are labeled with an “A.” (E) Cells treated with the indicated agents as in A were washed and plated to determine the frequency of surviving clones, defining the control value of untreated cells as 100%. Results are representative of three independent experiments.
Figure 2.

Immunogenicity of the different cell death types. (A) Tumor evolution of DX- and DXZ-treated cells in nu/nu mice. Cells treated as in Fig. 1 were injected subcutaneously (3 × 106/mouse). (B) Tumor evolution of DX- and DXZ-treated cells in immunocompetent BALB/c. Note that MC- or F/T-treated mice also did not form tumors. (C) Immunogenic effect of DX-treated tumor cells. BALB/c mice were injected into one flank with live tumor cells and into the opposite flank with DX-, DXZ-, MC-, or F/T-treated cells, and the evolution of tumors was monitored. (D) Persistent, specific antitumor immunity elicited by DX-treated cells. Animals immunized with DX-treated CT26 cells that remain tumor-free after 120 d (see lower left panel in C) or age-matched control mice were challenged with CT126 cells or the unrelated TSA tumor cell line. (E) Adoptive transfer of antitumor immunity elicited by DX-treated cells. BALB/c mice were injected intravenously with 107 splenocytes from naive control animals or animals immunized with DX-treated CT26 cells that remain tumor-free (as in B and D) followed by subcutaneous injection of 5 × 105 tumor cells. (F) Effect of the timing of antitumor vaccination. CT26 cells treated as in Fig. 1 were injected at the same time as live tumor cells (simultaneous injection as in C) or were injected once 1 wk before challenge with live tumor cells (prophylactic immunization; n = 15 per group). (G) Replacement of DX by other anthracyclins. Animals were injected with CT26 cells treated with daunorubicin (DA) or idarubicin (ID), instead of DX, or PBS only (CO), and the growth of live CT26 cells simultaneously injected into the opposite flank was monitored as in C. All figures represent three to six independent experiments. *, P < 0.05 versus control (CO). Note that growth curves of tumors are only shown to the level at which ethical guidelines oblige to kill the animals.
Figure 3.

Contribution of DX to the immunogenic nature of apoptotic tumor cells. (A) DX incorporation into CT26 cells. Tumor cells were treated in the absence or presence of DX for 24 h as in Fig. 1, and the incorporation of DX into cells was measured by assessing the DX-specific red fluorescence in the FACS (left). Alternatively, CT26 cells were killed by prolonged culture in MC (as in Fig. 1) followed by the optional addition of DX during the last 15 min of culture, washing, and determination of the DX incorporation (right). (B) Immunogenic effect of DX in vivo. Cells treated as in A (DX, MC, or MC plus DX) were injected subcutaneously in a prophylactic setting (as in Fig. 2 F) 1 wk before challenge with life CT26 cells in the opposite flank followed by monitoring of tumor growth.
Immunogenicity of caspase-dependent, DX-induced cell death
As shown above, DX induced immunogenic tumor cell death. However, concomitant treatment with the broad-spectrum caspase inhibitor Z-VAD-fmk (DXZ) abolished the immunogenic potential of the treatment with DX (Fig. 2, C and F). Similar inhibitory effects were obtained when Z-VAD-fmk was replaced by a chemically related caspase inhibitor with a narrow spectrum of action, such as Z-DEVD-fmk (specific for caspase 3), whereas Z-VQID-CHO or Z-VDVAD-CHO, which are specific for caspases 6 and 2, respectively (not depicted), have no such effects. Z-VAD-fmk did not ameliorate the clonogenic survival of DX-treated CT26 cells (Fig. 1 E), nor did it reduce the incorporation of DX, which emits a red fluorescence, into the cell (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050915/DC1). To confirm that caspases rather than other proteases inhibited by Z-VAD-fmk (30) mark the difference between highly immunogenic (DX) and less immunogenic (DXZ) death, we stably transfected CT26 cells with the baculovirus caspase inhibitor p35, a protein that confers highly specific, irreversible caspase inhibition in vitro and in transgenic models (31, 32). Similarly to Z-VAD-fmk, p35 delayed DX-induced cell death (Fig. 4 A), yet did not restore the in vitro clonogenicity of DX-treated cells (Fig. 4 B). p35-expressing tumor cells failed to form tumors when treated in vitro with DX and inoculated into immunocompetent mice. However, as opposed to vector-only transfected DX-treated controls, p35-transfected DX-treated CT26 cells failed to elicit an antitumor immune response (Fig. 4, C and D). Animals protected against CT26 wild-type tumors also failed to develop tumors after inoculation of live p35-expressing CT26 cells, indicating that p35 did not interfere with the effector arm of the immune system (not depicted). Thus, two different protocols of caspase inhibition (pharmacological in the case of Z-VAD-fmk and genetic and cell-autonomous in the case of p35) had similar effects on the immunogenicity of DX-treated tumor cells.
Figure 4.

Inhibition of immunogenicity by the caspase inhibitor p35. (A) FACS analysis of CT26 cells transfected with vector only (Neo) or p35 cultured alone or in the presence of DX as in Fig. 1 A. Numbers indicate the percentage of cells (X ± SD, n = 5) in each quadrant. (B) Clonogenic survival of Neo or p35-transfected cells left untreated (100% values) or treated with DX. (C and D) Evolution of CT26 tumors in animals injected simultaneously with Neo-transfected, DX-treated, or p35-transfected DX-treated cells into the opposite flank. Note that only Neo-expressing DX-treated cells confer tumor immunity (P < 0.01).
Critical role of DCs in the immune response elicited by dying tumor cells
In an attempt to understand why DX-induced cell death is immunogenic, we challenged DCs with dead or dying cells and measured their response. DX-treated CT26 cells were phagocytosed by all CD11c+/IAd+ DC subpopulations (CD11b+ B220+ DCs in Fig. 5 A and CD8α+ DCs not depicted) present in the spleen, whereas nonimmunogenic (live, DXZ, MC, or F/T) CT26 cells were neglected by DCs (Fig. 5, A and B). In contrast, several categories of dying (DX, DXZ, and F/T but not MC) tumor cells possessed the capacity of inducing DC maturation, as indicated by increased expression of the costimulatory molecules CD80 and CD86 as well as induction of MHC class II (Fig. 5 C). Soluble DX itself did not activate CD86 (Fig. 5 C) or CD80 (not depicted) expression in bone marrow–derived DCs when used at concentrations that cause apoptosis of a fraction of DCs (Fig. 5 D). Moreover, Z-VAD-fmk did not inhibit the DC-mediated phagocytosis of DX-treated CT26 cells (Fig. 5 E). Thus, the differential effect of MC-, DX-, and DXZ-treated cells on DCs cannot be due to the carryover of soluble DX or Z-VAD-fmk. DX-treated, but not DXZ-treated, CT26 cells elicited a cytotoxic T cell response after in vivo inoculation (Fig. 6 A). An intact cellular immune system was required for the DX-elicited immunogenic effect because athymic nu/nu mice failed to mount a productive antitumor immune response when challenged with DX-treated CT26 cells (Fig. 6 B). Similarly, the selective depletion of CD8+ cells, but not that of NK cells, by in vivo injection of specific mAbs abolished the antitumor immune response stimulated by DX-treated cells (Fig. 6 C). Thus, NK cells are dispensable for the antitumor immune response elicited by DX-treated cells, whereas CD8+ cells are required for this response. Additional data indicating the importance of CTLs were obtained in other tumor-relevant models. Injection of OVA-transfected B16F10 melanoma cells elicited the local recruitment of OVA-specific CD8+ T cells, which recognize the Kb/SIINFEKL tetramer, into (as well as the production of IFN-γ by cells from) the draining lymph node of syngenic C57BL/6 mice, provided that the injected cells were pretreated with DX (Fig. 7, A–C). However, this response was blunted when the inoculum had been treated with DXZ or MC, or when the DX-treated cells were rendered necrotic by freeze-thawing (Fig. 7, A–C). To determine the impact of CD11c+ DCs on the immune response in vivo, diphteria toxin was injected into transgenic C57BL/6 mice expressing the diphteria toxin receptor specifically in DCs (under the control of the CD11c promoter; reference 33). Vehicle-injected control animals recruited CD8+ T cells with a TCR specific for the OVA-derived Kb/SIINFEKL epitope into the draining lymph node, but DC-depleted animals failed to do so in response to injection of apoptotic OVA-transfected B16F10 cells (Fig. 7 D). These data confirm that DX-treated cells are particularly effective in stimulating a cytotoxic immune response and that this response is mediated by DCs.
Figure 5.
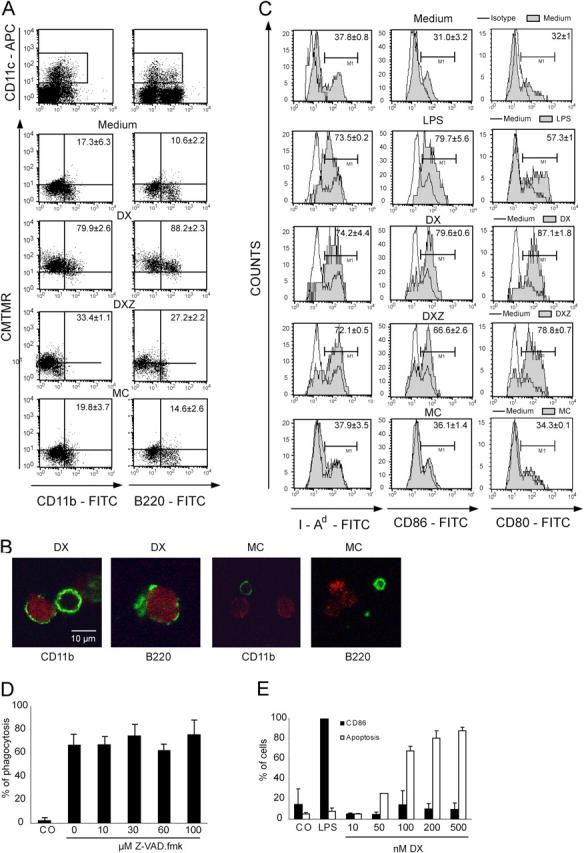
Effect of dying tumor cells on DCs. (A and B) In vitro phagocytosis of the DX-, DXZ-, or MC-treated cells (stained with CMTMR) by spleen DCs from Flt3L-injected mice. Representative FACS diagrams are depicted in A, and confocal images of dying tumor cells (red) phagocytosed by purified DC subsets (green) are shown in B. (C) DC maturation of splenic DCs induced by LPS (positive control) and dying or dead CT26 cells. Percentage values in A and C are means of three independent determinations ± SD. (D) Failure of Z-VAD-fmk to inhibit the phagocytosis of DX-treated CT26 cells by DCs. DCs generated as in D were incubated for 90 min with a twofold excess of DX-treated CT26 cells in the presence of the indicated concentrations of Z-VAD-fmk, and the percentage of DCs containing dying or dead CT26 cells was determined as in A. (E) Failure of DX to activate DCs. Bone marrow–derived DCs were activated with LPS as a positive control or with the indicated concentration of DX, and the frequency of apoptotic cells (annexin V+) and CD86+ cells was determined by immunofluorescence and cytofluorometric analysis. Percentage values in A and C–E are means of three independent determinations ± SD.
Figure 6.

Characterization of the immune effectors induced by dying tumor cells. (A) CTL response of DX- and DXZ-treated cells in immunized mice. Splenic T cells from animals vaccinated with PBS only (CO), DX-treated CT26 cells, or DXZ-treated CT26 cells were tested for their capacity to lyse syngenic cells pulsed with the immunodominant H2Ld- restricted CT26-derived peptide SPSYVYHQF. Representative CTL responses from individual mice are shown on the left, and mean values are shown on the right. R and NR refer to responders and nonresponders, respectively. *, P < 0.01 versus control (CO). (B) Failure of nude mice to mount an immune response against DX-treated CT26 cells. Wild-type or nu/nu BALB/c mice were inoculated with DX-treated CT26 cells 1 wk before the injection of live CT26 cells into the opposite flank, and tumor-free survival was monitored. (C) Requirement of CD8+ but not NK cells for the antitumor immune response. Wild-type BALB/c mice received intraperitoneal injections of depleting antibodies specific for CD8 or for the NK marker asialo-GM1 3–4 d before challenge with dying (DX-treated) and live (untreated) CT26 cells into opposite flanks at 1 wk of interval.
Figure 7.

Contribution of specific CTLs and DCs to the immune response against DX-treated tumor cells. (A) Percentages of CD8+ lymph node cells expressing TCR that interact with the OVA-derived peptide SIINFEKL presented by H-2Kb 5 d after challenge with either peptide plus adjuvant (P+A) or with DX, DX F/T (cells treated with DX and then F/T), DXZ-, MC-, and F/T-treated B16-OVA cells (three experiments). (B) Absolute number of specific CD8+ cells per lymph node determined as in A. Values are X ± SD (n = 3). (C) IFN-γ production as measured after in vitro culture with SIINFEKL in the same experiment. Control values are <50 pg/ml. (D) Effect of the depletion of DCs on the accumulation of specific T cells. Transgenic mice specifically expressing the diphteria toxin (DT) receptor in DCs were pretreated with PBS alone or a dose of diphteria toxin that depletes DCs. The mice were then challenged with DX-treated B16-OVA cells into the food pad. Draining lymph nodes were recovered 48 h later and stained for simultaneous detection of CD8 and OVA peptide–specific T cell receptors. Representative FACS pictograms are shown and values are X ± SD (n = 3).
DX-elicited apoptosis is immunogenic in several tumor models
One single subcutaneous injection of DX-treated CT26 cells conferred a strong protection against the development of pulmonary metastases induced by simultaneous intravenous injection of tumor cells (Fig. 8 A). In this system, MC-treated and F/T cells were inefficient. Freeze-thawing of DX-treated CT26 cells (DX F/T) annihilated their immunogenic effect, and coincubation with Z-VAD-fmk led to a partial reduction of the DX effect (Fig. 8 A). The anti-metastatic effect was complete when DX-treated CT26 cells were injected twice before intravenous challenge with untreated CT26 cells (Fig. 8 B). In a further round of experiments, we determined whether the observed tumor vaccination effects were a general phenomenon. When replacing CT26 cells with B16F10 cells, we found that this melanoma cell line was immunogenic if DX treated and injected into C57BL/6 mice (not depicted). B16F10A2/gp100 melanoma cells present peptides from the human gp100 tumor antigen in the context of HLA class I A2 (34). Again, in vitro treatment with DX followed by subcutaneous injection of the cells was able to induce a significant protection against later challenge with live B16F10A2/gp100 tumor cells in “humanized” mice expressing an HLA.A2 transgene (Fig. 8 C; references 34 and 35). As a side observation, such humanized mice were largely deficient for NKT cells (NK1.1+ TCRα/β+ CD4+; Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050915/DC1), suggesting that NKT cells are not required for the antitumor immune response elicited by DX-treated tumor cells. Thus, DX-treated dying tumor cells turned out to be immunogenic on a human MHC background. Furthermore, inbred BDIX rats inoculated with the syngenic PROb colon carcinoma (36) could be protected by injection of DX-treated tumor cells, and this effect was again suppressed by simultaneous pretreatment of the tumor cells with DX and Z-VAD-fmk (Fig. 8 D). In conclusion, this protocol of cancer immunization is applicable to a series of distinct tumors, species, and MHC backgrounds.
Figure 8.

Antitumor vaccination with DX-treated melanoma or colon carcinoma cells in distinct rodent models. (A) Percentage of metastasis-free mice observed after intravenous injection of live CT26 cells into animals injected simultaneously with vehicle only (CO) or dying tumor cells generated as in Fig. 1. Untreated controls developed 46 ± 22 metastases (X ± SD, n = 46). (B) Effect of two immunizations with DX-treated CT26 cells 14 and 7 d before intravenous injection of live CT26 cells. Data in A and B have been pooled from three independent experiments. *, P < 0.001 versus control (CO). (C) DX-treated B16A2 cells were injected twice into mice transgenic for HLA.A2 1 and 2 wk before challenge with live B16A2 cells into the opposite flank. *, P < 0.05 versus control (CO). (D) The protocol of simultaneous subcutaneous injection of dying (DX- or DXZ-treated) tumor cells was applied to BDIX rats challenged with PROb cells. *, P < 0.001 versus control (CO). This experiment has been repeated three times, yielding similar results.
Ex vivo and in vivo induction of immunogenic apoptosis
We investigated whether it would be required to immunize animals with dying cells cultured as an established cell line or whether it would be feasible to take advantage of resected tumors (as this would be the case in human cancer patients). CT26 tumors were generated in BALB/c mice and then surgically removed and enzymatically dissociated optionally after a step of cryostorage in DMSO-containing medium, treated with DX in vitro, and used as antitumor vaccine (Fig. 9 A). This ex vivo removal/in vitro treatment elicited an efficient antitumor immune response if DX was added during the in vitro incubation, although cryostored cells were less efficient than freshly recovered tumor cells. The omission of DX curtailed the efficacy of the vaccine (Fig. 9 A). As an alternative to this ex vivo/in vitro protocol of vaccine generation, we injected DX into palpable (>125 mm3) subcutaneous CT26 tumors established in BALB/c mice. Using this protocol of intratumoral as opposed to intravenous (not depicted) chemotherapy, we achieved stable disease or complete tumor regression in 16 out of 40 (40%) of the animals (Fig. 9 B). We have no explanation why some animals showed no therapeutic effect, others demonstrated a delay in tumor growth, and yet others were completely cured. One possible explanation for this heterogeneity is a variation in the local infiltration of the tumor that escapes macroscopic inspection. When DX was injected in combination with Z-VAD-fmk or when DX was replaced by MC, we only observed a delay in tumor growth with no stable disease or tumor regression (n = 15; Fig. 9 B). Rechallenge of animals that manifested a complete therapeutic response with DX-treated CT26 cells failed to produce tumors (Fig. 9 C), indicating that this therapeutic regimen had induced a persistent antitumor immunity. Importantly, DX injection into tumors failed to yield a therapeutic response (beyond a delay in tumor growth by several days) in nu/nu mice (Fig. 9 D), indicating that the immune system participated in the beneficial effects of DX injection as observed in immunocompetent mice (Fig. 9 B). When we investigated the effect of intratumoral DX injection into tumors of BDIX rats carrying PROb cancers, we found again that the therapeutic effect of DX injection was only observed in immunocompetent rats (not in athymic animals) and that its effect was abolished by coinjection of Z-VAD-fmk (Fig. 9 E). Local injection of MC caused a similar delay in PROb tumor growth in immunocompetent and athymic rats (Fig. 9 E). These findings corroborate the notion that DX causes immunogenic tumor cell death, whereas MC causes a nonimmunogenic (or less immunogenic) type of cell death in vivo.
Figure 9.

Ex vivo and in vivo induction of immunogenic cell death. (A) Evolution of tumors in mice vaccinated with cells from freshly resected or cryopreserved CT26 tumors, which were left untreated (CO) or treated with DX. (B) Effect of intratumoral chemotherapy on the growth of CT26 colon cancer cells inoculated into immunocompetent BALB/c mice. BALB/c mice previously inoculated with live CT26 cells received intratumoral injection (arrows) of DX, DXZ, or MC as soon as they were palpable ∼14 d after inoculation of tumor cells, and the tumor growth was monitored. Although some DX-treated animals showed a normal tumor growth (n = 12) or a simple delay in tumor growth (n = 12), other animals exhibited stable disease (n = 7) or complete tumor regression (n = 9). (C) Rechallenge of animals cured by intratumoral injection of DX (as in B) or age-matched control (CO) mice with live CT26 cells. Similar results were obtained in three independent experiments. (D) Failure of intratumoral DX injections to confer stable antitumor effects in nu/nu BALB/c mice carrying CT26 tumors. The experiment was performed as in B except that the animals were immunodeficient. (E) Intratumoral injections in the PROb rat colon carcinoma model. Immunocompetent or nu/nu BDIX rats carrying PROb tumors received intratumoral injections of DX, DXZ, or MC, and tumor growth was monitored.
Discussion
Apoptosis is often considered as an immunologically silent or even tolerogenic death modality (8–16, 37). Challenging this viewpoint, it has been shown that gemcitabine-induced apoptosis of hemagglutinin-expressing AB1 mesothelioma cells can prime CD8 T cells in vivo, thus eliciting an effective antitumor immune response (38). Similarly, local γ irradiation of OVA-transfected B16F10 tumors can increase both the generation of antitumor immune effector cells and their trafficking to the tumor site (39). The present data suggest that some but not all regimens of chemotherapy-induced apoptosis can elicit an effective antitumor immune response. Although anthracyclins (and in particular DX, which we used as a model agent) elicited immunogenic apoptosis in vitro (Figs. 2–4 and 6–8), ex vivo (Fig. 9 A) and in vivo (Fig. 9, B–E) MC (Figs. 2, 3, 5 C, 6 A, 7 A, 8 A, and 9 B) was relatively inefficient in eliciting immunogenic cell death. The incorporation of DX a posteriori into MC-killed cells could not substitute for the DX treatment ab initio (Fig. 3), underscoring the contribution of the host cell (rather than the chemical nature of anthracyclins) to the immunogenic phenotype. Preparations of DX- or MC-treated cells that contained a similar percentage of dying cells (Fig. 1 A) were injected, and the morphological aspect of cell death elicited by DX and MC was similar (Fig. 1, C and D). Both were accompanied by caspase activation (Fig. 1 B) without any difference in the expression of some major endogenous adjuvants, such as HSP70 and HMGB-1 (Fig. 1 B), and the only difference resided in the degree of DNA fragmentation, which was more advanced with MC (Fig. 1 C). Moreover, DCs phagocytosed MC-treated cells less efficiently than DX-treated cells (Fig. 5 A), thus underscoring a hitherto unsuspected heterogeneity in apoptotic cells with respect to their uptake by DCs (40). Importantly, inhibition of caspase activation by Z-VAD-fmk or transfected p35 greatly reduced the immunogenic potential of DX-treated cells, an effect that was observed in a variety of different tumor models in vitro and in vivo (Figs. 2–9) and also affected DC-mediated recognition and phagocytosis (Fig. 5 A). However, caspase activation is not sufficient to elicit an immunogenic cell death because MC-treated cells, which do exhibit caspase activation (Fig. 1 B), are inefficient antitumor vaccines (Fig. 2). Thus, a specific combination of factors, caspase activation, and a yet-to-be defined antigenic property of anthracyclin-treated cells have to cooperate to elicit an antitumor immune response. Disruption of the membrane integrity of DX-treated tumor cells by freeze-thawing is sufficient to abolish their immunogenic property (Figs. 2 F, 7, A–C, and 8 A), meaning that intact apoptotic cells or bodies are required for optimal antigenicity. Induction of necrotic cell death is usually achieved by several cycles (usually three or more) of freeze-thawing, whereas we performed one single freeze-thaw cycle, a procedure that avoids a gross liquefaction of cellular structures. Thus, in contrast with other reports (41, 42), freeze thawing as performed here did not result in the uptake of cells by DCs (Fig. 5 A) nor in immunogenic cell death (Fig. 2 C). Regardless of these mechanistic details, it appears that the particular protocol of apoptosis induction determines the immunogenicity of cell death. Apparently, two different subcategories of apoptosis exist, one that is highly immunogenic and another that is comparatively less immunogenic.
DX-treated dying tumor cells are efficiently phagocytosed by myeloid and plasmacytoid DCs (Fig. 5, A and B) and are highly efficient in eliciting antigen-specific CTLs (Figs. 6 A and 7), which are required for the antitumor immune response (Fig. 6, B and C). In contrast, NK cells (Fig. 6 C) and NKT cells (Fig. S3) are not stringently required for mounting an antitumor immune response, at least in this experimental setting. Thus, in contrast to other experimental systems, such as viral infection (43), NK cells are dispensable for the priming of DCs by DX-treated tumor cells. Of note, intratumoral DX injection could elicit a chemotherapeutic response whose positive outcome requires an intact cellular immune system (Fig. 9, D and E). Again, these properties (phagocytosis of DX-treated cells by DCs, induction of antigen-specific CTLs, and elicitation of an immune response by intratumoral injection of DX) were largely reduced by simultaneous treatment with the caspase inhibitor Z-VAD-fmk, correlating with a reduced immunogenicity. To our knowledge, this is the first report indicating that caspase activation is broadly required for the elicitation of immunogenic tumor cell death. Previous reports have described that caspase activation rather reduces the immunogenic potential of tumor cells. Thus, a PROb clone (REGb) that fails to activate caspases is much more immunogenic than the parental PROb line (44). Similarly, a PROb clone manipulated to stably express an anti-sense cytochrome c cDNA undergoes atypical apoptosis without caspase activation, yet has an increased immunogenic potential (27). As another example, caspases have been documented to destroy immunodominant epitopes, thus blunting the immune response against lymphoma cells (45). In NOD mice, which are prone to develop autoimmune diabetes, systemic caspase inhibition reduced the priming of T cells by β cells undergoing spontaneous apoptosis (46) and, conversely, local caspase inhibition abolished the tolerogenic effect of β cell apoptosis induced by streptozotocin (37). The data presented here, which address the importance of caspase activation in a direct fashion, suggest that caspase activation ameliorates the antigenic properties of dying tumor cells. This points to an interesting problem. The expression of caspase inhibitors has negative prognostic value for some tumors (47–49). It is possible that failure to activate caspases not only increases the resistance of tumor cells to chemotherapeutic agents, in a cell-autonomous fashion, but also reduces the probability of immunogenic death, thus blunting the host's antitumor defense system.
It is important to underscore the fact that the immunogenic effects of anthracyclin-treated tumor cells were observed in the absence of any adjuvant or costimulus. Hence, coapplication of adjuvants might be expected to improve the efficacy of the vaccination schedule. As a fascinating possibility, our data suggest a strategy of inducing immunogenic cell death in established tumors either ex vivo or in vivo. Whether such a simple strategy might be efficient in treating patients has to be addressed in the future.
MATERIALS AND METHODS
Cell lines and cell death induction.
CT26, PROb, B16F10, B16/F10.9-OVA, and B16F10A2/gp100 (B16F10 transfected with gp100 and HLA A2.1 and selected in 50 μg/ml hygromicin and G418) cells were cultured at 37°C under 5% CO2 in RPMI 1640 medium supplemented with 10% FCS, penicillin, streptomycin, 1 mM pyruvate, and 10 mM Hepes in the presence of DX for 24 h (25 μM for CT26, 30 μM for PROb, and 2.5 μM for B16F10 and its derivatives), 5 μM daunorubicin for 24 h (GE Healthcare), 1 μM idarubicin for 24 h (Aventis), 30 μM MC for 48 h (Sanofi-Synthelabo), and/or 100 μM zVAD-fmk for 24 h (Bachem). Necrosis was induced by one freeze-thaw cycle in liquid N2 and a 37°C water bath. CT26 cells were stably transfected with vector only (Neo) or with a pcDNA3.1 vector encoding p35 (31, 32). In one experiment, 100 μM DX was added for 15 min to cells cultured in MC for 2 d. The incorporation of DX into washed (three times in PBS) cells was measured by assessing the DX-specific red fluorescence in a FACSVantage (Becton Dickinson) equipped with an argon laser.
Cell death assays.
Cells were trypsinized and subjected to cytofluorometric analysis with a FACSVantage after staining with 2.5 μM DAPI for 10 min (Invitrogen) for determination of cell viability, and annexin V was conjugated with fluorescein isothiocyanate (Bender Medsystems) for the assessment of phosphatidylserine exposure (50, 51). TUNEL assays were performed on cells (let to adhere for 1 h in PBS on polylysine-coated slides [O. Kindler GmbH] and fixed with 4% paraformaldehyde for 30 min) using the In situ Cell Death Detection Kit (Roche) and a fluorescence microscope (IRE2; Leica) equipped with a camera (DC300F; Leica). For electron microscopy, cells (fixed for 1 h at 4°C in 2.5% glutaraldehyde in phosphate buffer, pH 7.4, washed, and fixed again in 2% osmium tetroxide and then embedded in Epon) and ultrathin sections (80 nm) were stained with uranyl acetate and lead citrate and examined with an electron microscope (902; Leo) at 80 kV.
Immunoblot analyses.
Cells were washed with cold PBS at 4°C and lysed in a buffer containing 50 mM Tris HCl, pH 6.8, 10% glycerol, and 2% SDS. Primary antibodies detecting activated caspase 3 (dilution 1/1,000; Cell Signaling Technology), HSP70 (dilution 1/1,000; Stratagene) or 2 μg/ml HMGB-1 (BD Biosciences) were revealed with the appropriate horseradish peroxidase–labeled secondary antibody (Southern Biotechnology Associates, Inc.) and detected by enhanced chemiluminescence (Pierce Chemical Co.). Anti-actin or anti-GAPDH (Chemicon) was used to control equal loading.
Antitumor vaccination and assessment of tumor growth in vivo.
All animals were maintained in specific pathogen-free conditions, and all experiments followed the Federation of European Laboratory Animal Science Association guidelines. 3 × 106 treated CT26 cells were inoculated subcutaneously in 200 μl PBS into 6-wk-old female BALB/c mice (Charles River Laboratories) into the lower flank, whereas 5 × 105 untreated control cells were inoculated into the contralateral flank (52). PROb cells (3 × 106 treated cells and 106 control cells) were injected subcutaneously into the lower flanks of syngeneic 2-mo-old BDIX rats (Charles River Laboratories), and B16A2 and B16F10 (1.8 × 105 treated cells and 3 × 104 control cells) were injected into 6-wk-old female HHD2 transgenic mice (bred in the Institut Gustave Roussy [IGR] animal facility) and C57BL/6 mice (Charles River Laboratories), respectively. For the tumorigenicity assay, 3 × 106 treated or untreated CT26 cells were injected subcutaneously into nu/nu mice (IGR animal facility). To assess the specificity of the immune response against CT26, we injected either 5 × 105 or 5 × 106 of CT26 (for the mice immunized in a standard protocol or vaccination protocol, respectively) or 106 TSA cells. Tumors of the control side were evaluated weekly using a caliper. Animals bearing tumors in excess of 20–25% of the body mass were killed. In one series of experiments, splenocytes from naive or tumor-protected BALB/c mice (i.e., mice that remain tumor-free for at least 60 d after subcutaneous challenge with apoptotic and live CT26 cells into the right and left flanks, respectively) were injected intravenously (107 cells) into 6-wk-old female BALB/c mice, which received 5 × 105 CT26 cells subcutaneously 1 d later. Lung metastases were generated by injecting 105 CT26 cells into the tail vein simultaneously with a subcutaneous injection of 3 × 106 treated cells. Mice were injected with Indian ink 20 d later, killed, and lung metastases were counted with a loupe by three independent investigators in a blind fashion. For vaccination with primary tumors, subcutaneous tumors were dissected and dissociated with 1.6 mg/ml collagenase and 350 U/ml DNase for 20 min at 37°C, treated with 20 μM DX for 24 h, and then injected subcutaneously with 3 × 106 cells. Alternatively, the tumors were frozen (in FCS with 10% DMSO) as ∼1-mm3 cubes at −80°C and dissociated and treated the same way after thawing. In a series of experiments, BALB/c (wild-type or nu/nu) carrying palpable CT26 tumors or rats (BDIX or nude) carrying PROb tumors (implanted 14 d before by injection of 106 tumor cells) received a single intratumoral injection of MC (100 μl of a 3-mM solution), DX (20 μl of a 10-mM solution), and/or Z-VAD.fmk (10 μl of a 100-mM solution). Depletion of CD8+ or NK cells was achieved by intraperitoneal injection of 250 μg anti-CD8 mAb (obtained from the rat anti–mouse hybridoma H35.17.2; American Type Culture Collection) or anti-asialo GM1 mAb (Wako Bioproducts), respectively, 4 d before challenge with dying (DX-treated) tumor cells and 3 d before injection of live tumor cells (inoculated 1 wk after that of the DX-treated cells).
Assessment of specific T cell responses.
The CTL response against the immunodominant CT26-specific AH1 H2Ld-restricted peptide (SPSYVYHQF; Neosystem; reference 53) was determined. Splenocytes were isolated from immunized mice 10 d after injection of apoptotic cells and restimulated in vitro for 5 d with or without 5 μg/ml AH1 in the presence of syngeneic irradiated naive spleen cells. The cytotoxic activity was determined during a classic 5-h in vitro 51Cr-release assay using 51Cr-P815 tumor cells loaded with 50 μM AH1 peptide as target cells (52). The percentage of specific lysis was calculated as 100 × (experimental release − spontaneous release)/(maximal release − spontaneous release). Maximum release was obtained by adding 10% Triton X-405 to target cells, and spontaneous release was determined by incubating target cells in medium alone. 5–6-wk-old female C57BL/6 mice (H-2b) were injected into both footpads with PBS and 50 μg OVA peptide (H-2Kb-restricted, SIINFEKL, amino acids 257–264; Eurogentec) mixed with incomplete Freund adjuvant (Sigma-Aldrich) or 106 B16/F10.9-OVA cells. Animals were killed 5 d after footpad immunization, and popliteal and inguinal lymph nodes were removed and processed individually. A single cell suspension was obtained by homogenizing and filtering the organ through a sterile cell strainer (70 μm; Becton Dickinson). For FACS analysis, 2 × 106 cells were stained at room temperature with a tetramer-OVA-PE (Beckman Coulter) and then stained with an anti–CD3-FITC and an anti–CD8-APC mAb (Becton Dickinson). Cells were analyzed using a FACSCalibur cytometer (Becton Dickinson) with CELLQuest Pro software. Alternatively, 105 cells were cultured in complete culture medium containing 2% of mouse serum in the presence or absence of 40 μg/ml SIINFEKL peptide. 3 d later, the supernatants were harvested and IFN-γ secretion was determined by ELISA (OptEIA ELISA Kit; BD Biosciences). In one series of experiments, CD11c-GFP DT Kb mice (33) were injected intraperitoneally with 100 ng diphteric toxin (or PBS as a vehicle control) 48 h before the injection of DX-treated B16/F10.9-OVA cells into the food pad.
Priming and isolation of DCs.
Mice were injected intraperitoneally daily with 10 μg of Flt3L (Immunex) for 10 consecutive days. Splenic cells were coincubated with 1 μM CytoTracker Red– (CMTMR; Invitrogen) prestained apoptotic or necrotic CT26 cells at a ratio of 1:1 or 5:1 for 2 h for the phagocytosis experiments or 24 h for maturation experiments. Cells were then analyzed by FACS with FITC-conjugated antibodies (Becton Dickinson) specific for B220, CD11b, CD8α, MHC class II, CD40, CD80, or CD86 molecules, as well as an APC-conjugated anti-CD11c antibody. Alternatively, CD11c+ splenocytes were purified using immunomagnetic beads (Miltenyi Biotec) coincubated for 2 h with the CMTMR-labeled CT26 cells at a ratio of 1:1. The cells were then stained with B220, CD11b, and CD8α (BD Biosciences), as well as a goat anti–mouse antibody conjugated with Alexa Fluor 488 (Invitrogen). Confocal microscopy was performed with a microscope (LSM 510; Carl Zeiss MicroImaging, Inc.) equipped with a 63X objective. For the generation of bone marrow–derived DCs, bone marrow cells were flushed from the tibias and femurs of C57BL/6 mice with RPMI 1640 medium supplemented with 10% heat-inactivated FBS (Invitrogen), 2 mM l-glutamine, 50 μM 2-ME (Sigma-Aldrich), 10 mM Hepes buffer, pH 7.4 (Invitrogen), and 50 μg/ml penicillin/streptomycin (Invitrogen). After one centrifugation, bone marrow cells were resuspended in Tris-ammonium chloride for 2 min to lyse red blood cells. After one more centrifugation, bone marrow cells were cultured for 8 d at 106 cells/ml in culture medium supplemented with 100 ng/ml recombinant mouse FLT3 liter (R&D Systems) in six-well plates (Costar Corning).
Statistical analyses.
Data are presented as arithmetic means ± SD or percentages. All statistical analyses were performed using JMP software (SAS Institute Inc.). The Student's t test was used to compare continuous variables (comparison of tumor growth), the Chi square test was used for nonparametrical variables (comparison of animal cohorts), and log-rank tests were used for Kaplan Meier curves. For all tests, the statistical significance level was set at 0.05.
Online supplemental material.
Fig. S1 shows the immunogenicity of γ-irradiated apoptotic CT26 cells in vivo. Mice were immunized by a prophylactic injection of γ-irradiated CT26 cells before challenge with live tumor cells. Fig. S2 is a cytofluorometric analysis of CT26 cells treated with PBS, DX alone (DX), or DX and z.VAD-fmk (DZ) for 24 h. Fig. S3 shows the absence of NKT cells in HHD2 transgenic mice. Splenic cells were obtained from both 6-wk-old female C57BL/6 and HHD2 transgenic mice and then analyzed by cytofluorometry with antibodies against CD4, NK1.1, and TCR αβ. Figs. S1–S3 are available at http://www.jem.org/cgi/content/full/jem.20050915/DC1.
Acknowledgments
We thank Drs. Mathew Albert (Pasteur Institute, Paris, France) for helpful discussion, François Lemonnier (Pasteur Institute) for providing transgenic mice, Abdelali Jalil (Institut Gustave Roussy, Villejuif, France) for technical support, and Guy Salvesen (Burnham Institute, La Jolla, CA) for the p35 plasmid.
G. Kroemer is supported by a special grant from Ligue Nationale contre le cancer as well as grants from Agence Nationale pour la Recherche sur le SIDA, European Community (Active p53, RIGHT), and Sidaction. N. Casares and A. Tesniere have fellowships from Fondation pour la Recherche Médicale, and M.O. Pequignot has a fellowship from Institut Gustave Roussy. L Zitvogel is supported by the EU grants Allosem and DC-THERA.
The authors have no conflicting financial interests.
Abbreviations used: DX, doxorubicin; DXZ, DX combined with Z-VAD-fmk; F/T, frozen-thawed; MC, mitomycin C; TSA, murine mammary adenocarcinoma TS/A; TUNEL, terminal desoxyribonucleotidyl transferase dUTP nick end labeling.
N. Casares and M.O. Pequignot contributed equally to this work.
References
- 1.Zitvogel, L., N. Casares, M. Pequignot, M.L. Albert, and G. Kroemer. 2004. The immune response against dying tumor cells. Adv. Immunol. 84:131–179. [DOI] [PubMed] [Google Scholar]
- 2.Steinman, R.M., and I. Mellman. 2004. Immunotherapy: bewitched, bothered, and bewildered no more. Science. 305:197–200. [DOI] [PubMed] [Google Scholar]
- 3.Ferri, K.F., and G.K. Kroemer. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3:E255–E263. [DOI] [PubMed] [Google Scholar]
- 4.Igney, F.H., and P.H. Krammer. 2002. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer. 2:277–288. [DOI] [PubMed] [Google Scholar]
- 5.Kerr, J.F.R., A.H. Wyllie, and A.R. Currie. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 26:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer, G., and S.J. Martin. 2005. Caspase-independent cell death. Nat. Med. 11:725–730. [DOI] [PubMed] [Google Scholar]
- 7.Matzinger, P. 2002. The danger model: a renewed sense of self. Science. 296:301–305. [DOI] [PubMed] [Google Scholar]
- 8.Hanayama, R., M. Tanaka, K. Miyasaka, K. Aozasa, M. Koike, Y. Uchiyama, and S. Nagata. 2004. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 304:1147–1150. [DOI] [PubMed] [Google Scholar]
- 9.Asano, K., M. Miwa, K. Miwa, R. Hanayama, H. Nagase, S. Nagata, and M. Tanaka. 2004. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J. Exp. Med. 200:459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lauber, K., S.G. Blumenthal, M. Waibel, and S. Wesselborg. 2004. Clearance of apoptotic cells: getting rid of the corpses. Mol. Cell. 14:277–287. [DOI] [PubMed] [Google Scholar]
- 11.Fadok, V.A., D.L. Bratton, and P.M. Henson. 2001. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J. Clin. Invest. 108:957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim, S., K.B. Elkon, and X. Ma. 2004. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 21:643–653. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann, P.R., J.A. Kench, A. Vondracek, E. Kruk, D.L. Daleke, M. Jordan, P. Marrack, P.M. Henson, and V.A. Fadok. 2005. Interaction between phosphatidylserine and the phosphatidylserine receptor inhibits immune responses in vivo. J. Immunol. 174:1393–1404. [DOI] [PubMed] [Google Scholar]
- 14.Steinman, R.M., S. Turley, I. Mellman, and K. Inaba. 2000. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 191:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu, K., T. Iyoda, M. Saternus, Y. Kimura, K. Inaba, and R.M. Steinman. 2002. Immune tolerance after delivery of dying cells to dendritic cells in situ. J. Exp. Med. 196:1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferguson, T.A., J. Herndon, B. Elzey, T.S. Griffith, S. Schoenberger, and D.R. Green. 2002. Uptake of apoptotic antigen-coupled cells by lymphoid dendritic cells and cross-priming of CD8+ T cells produce active immune unresponsiveness. J. Immunol. 168:5589–5595. [DOI] [PubMed] [Google Scholar]
- 17.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 18.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 392:86–89. [DOI] [PubMed] [Google Scholar]
- 19.Russo, V., S. Tanzarella, P. Dalerba, D. Rigatti, P. Rovere, A. Villa, C. Bordignon, and C. Traversari. 2000. Dendritic cells acquire the MAGE-3 human tumor antigen from apoptotic cells and induce a class I-restricted T cell response. Proc. Natl. Acad. Sci. USA. 97:2185–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyoda, T., S. Shimoyama, K. Liu, Y. Omatsu, Y. Akiyama, Y. Maeda, K. Takahara, R.M. Steinman, and K. Inaba. 2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maranon, C., J.F. Desoutter, G. Hoeffel, W. Cohen, D. Hanau, and A. Hosmalin. 2004. Dendritic cells cross-present HIV antigens from live as well as apoptotic infected CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 101:6092–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blachere, N.E., R.B. Darnell, and M.L. Albert. 2005. Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol. 3:e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirschowitz, E.A., T. Foody, R. Kryscio, L. Dickson, J. Sturgill, and J. Yannelli. 2004. Autologous dendritic cell vaccines for non-small-cell lung cancer. J. Clin. Oncol. 22:2808–2815. [DOI] [PubMed] [Google Scholar]
- 24.Strome, S.E., S. Voss, R. Wilcox, T.L. Wakefield, K. Tamada, D. Flies, A. Chapoval, J. Lu, J.L. Kasperbauer, D. Padley, et al. 2002. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res. 62:1884–1889. [PubMed] [Google Scholar]
- 25.Scheffer, S.R., H. Nave, F. Korangy, K. Schlote, R. Pabst, E.M. Jaffee, M.P. Manns, and T.F. Greten. 2003. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int. J. Cancer. 103:205–211. [DOI] [PubMed] [Google Scholar]
- 26.Bartholomae, W.C., F.H. Rininsland, J.C. Eisenberg, B.O. Boehm, P.V. Lehmann, and M. Tary-Lehmann. 2004. T cell immunity induced by live, necrotic, and apoptotic tumor cells. J. Immunol. 173:1012–1022. [DOI] [PubMed] [Google Scholar]
- 27.Schmitt, E., A. Parcellier, F. Ghiringhelli, N. Casares, S. Gurbuxani, N. Droin, A. Hamai, M. Pequignot, A. Hamman, M. Moutet, et al. 2004. Increased immunogenicity of colon cancer cells by selective depletion of cytochrome c. Cancer Res. 64:2705–2711. [DOI] [PubMed] [Google Scholar]
- 28.Hopkins-Donaldson, S., P. Yan, K.B. Bourloud, A. Muhlethaler, J.L. Bodmer, and N. Gross. 2002. Doxorubicin-induced death in neuroblastoma does not involve death receptors in S-type cells and is caspase-independent in N-type cells. Oncogene. 21:6132–6137. [DOI] [PubMed] [Google Scholar]
- 29.Carter, B.Z., S.M. Kornblau, T. Tsao, R.Y. Wang, W.D. Schober, M. Milella, H.G. Sung, J.C. Reed, and M. Andreeff. 2003. Caspase-independent cell death in AML: caspase inhibition in vitro with pan-caspase inhibitors or in vivo by XIAP or Survivin does not affect cell survival or prognosis. Blood. 102:4179–4186. [DOI] [PubMed] [Google Scholar]
- 30.Rozman-Pungercar, J., N. Kopitar-Jerala, M. Bogyo, D. Turk, O. Vasiljeva, I. Stefe, P. Vandenabeele, D. Bromme, V. Puizdar, M. Fonovic, et al. 2003. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 10:881–888. [DOI] [PubMed] [Google Scholar]
- 31.Stennicke, H.R., C.A. Ryan, and G.S. Salvesen. 2002. Reprieval from execution: the molecular basis of caspase inhibition. Trends Biochem. Sci. 27:94–101. [DOI] [PubMed] [Google Scholar]
- 32.Date, T., Z. Luo, M. Yamakawa, A.J. Belanger, A. Scaria, S.H. Cheng, R.J. Gregory, S. Mochizuki, and C. Jiang. 2003. Myocardial expression of baculoviral p35 alleviates doxorubicin-induced cardiomyopathy in rats. Hum. Gene Ther. 14:947–957. [DOI] [PubMed] [Google Scholar]
- 33.Jung, S., D. Unutmaz, P. Wong, G. Sano, K. De los Santos, T. Sparwasser, S. Wu, S. Vuthoori, K. Ko, F. Zavala, et al. 2002. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 17:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mullins, D.W., T.N. Bullock, T.A. Colella, V.V. Robila, and V.H. Engelhard. 2001. Immune responses to the HLA-A*0201-restricted epitopes of tyrosinase and glycoprotein 100 enable control of melanoma outgrowth in HLA-A*0201-transgenic mice. J. Immunol. 167:4853–4860. [DOI] [PubMed] [Google Scholar]
- 35.Shirai, M., T. Arichi, M. Nishioka, T. Nomura, K. Ikeda, K. Kawanishi, V.H. Engelhard, S.M. Feinstone, and J.A. Berzofsky. 1995. CTL responses of HLA-A2.1-transgenic mice specific for hepatitis C viral peptides predict epitopes for CTL of humans carrying HLA-A2.1. J. Immunol. 154:2733–2742. [PubMed] [Google Scholar]
- 36.Caignard, A., H. Pelletier, and F. Martin. 1988. Specificity of the immune response leading to protection or enhancement by regressive and progressive variants of a rat colon carcinoma. Int. J. Cancer. 42:883–886. [DOI] [PubMed] [Google Scholar]
- 37.Hugues, S., E. Mougneau, W. Ferlin, D. Jeske, P. Hofman, D. Homann, L. Beaun, C. Schrike, M. Von Herrath, A. Lehuen, and N. Glaichenhaus. 2002. Tolerance to islet antigens and prevention from diabetes induced by limited apoptosis of pancreatic beta cells. Immunity. 16:169–181. [DOI] [PubMed] [Google Scholar]
- 38.Nowak, A.K., R.A. Lake, A.L. Marzo, B. Scott, W.R. Heath, E.J. Collins, J.A. Frelinger, and B.W. Robinson. 2003. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J. Immunol. 170:4905–4913. [DOI] [PubMed] [Google Scholar]
- 39.Lugade, A.A., J.P. Moran, S.A. Gerber, R.C. Rose, J.G. Frelinger, and E.M. Lord. 2005. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 174:7516–7523. [DOI] [PubMed] [Google Scholar]
- 40.Demaria, S., F.R. Santori, B. Ng, L. Liebes, S.C. Formenti, and S. Vukmanovic. 2005. Select forms of tumor cell apoptosis induce dendritic cell maturation. J. Leukoc. Biol. 77:361–368. [DOI] [PubMed] [Google Scholar]
- 41.Herr, W., E. Ranieri, W. Olson, H. Zarour, L. Gesualdo, and W.J. Storkus. 2000. Mature dendritic cells pulsed with freeze-thaw cell lysates define an effective in vitro vaccine designed to elicit EBV-specific CD4(+) and CD8(+) T lymphocyte responses. Blood. 96:1857–1864. [PubMed] [Google Scholar]
- 42.Gitlitz, B.J., A.S. Belldegrun, A. Zisman, D.H. Chao, A.J. Pantuck, A. Hinkel, P. Mulders, N. Moldawer, C.L. Tso, and R.A. Figlin. 2003. A pilot trial of tumor lysate-loaded dendritic cells for the treatment of metastatic renal cell carcinoma. J. Immunother. 26:412–419. [DOI] [PubMed] [Google Scholar]
- 43.Andoniou, C.E., S.L. van Dommelen, V. Voigt, D.M. Andrews, G. Brizard, C. Asselin-Paturel, T. Delale, K.J. Stacey, G. Trinchieri, and M.A. Degli-Esposti. 2005. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 6:1011–1019. [DOI] [PubMed] [Google Scholar]
- 44.Larmonier, N., C. Billerey, C. Rebe, A. Parcellier, M. Moutet, A. Fromentini, G. Kroemer, C. Garrido, E. Solary, F. Martin, and B. Bonnotte. 2002. An atypical caspase-independent death pathway for an immunogenic cancer cell line. Oncogene. 21:6091–6100. [DOI] [PubMed] [Google Scholar]
- 45.Castiglioni, P., A. Martin-Fontecha, G. Milan, V. Tomajer, F. Magni, J. Michaelsson, C. Rugarli, A. Rosato, and M. Bellone. 2002. Apoptosis-dependent subversion of the T-lymphocyte epitope hierarchy in lymphoma cells. Cancer Res. 62:1116–1122. [PubMed] [Google Scholar]
- 46.Turley, S., L. Poirot, M. Hattori, C. Benoist, and D. Mathis. 2003. Physiological β cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J. Exp. Med. 198:1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamm, I., S.M. Kornblau, H. Segall, S. Krajewski, K. Welsh, S. Kitada, D.A. Scudiero, G. Tudor, Y.H. Qui, A. Monks, et al. 2000. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin. Cancer Res. 6:1796–1803. [PubMed] [Google Scholar]
- 48.Ferreira, C.G., P. van der Valk, S.W. Span, I. Ludwig, E.F. Smit, F.A. Kruyt, H.M. Pinedo, H. van Tinteren, and G. Giaccone. 2001. Expression of X-linked inhibitor of apoptosis as a novel prognostic marker in radically resected non-small cell lung cancer patients. Clin. Cancer Res. 7:2468–2474. [PubMed] [Google Scholar]
- 49.Muris, J.J., S.A. Cillessen, W. Vos, I.S. van Houdt, J.A. Kummer, J. van Krieken, N.M. Jiwa, P.M. Jansen, H.C. Kluin-Nelemans, G.J. Ossenkoppele, C. Gundy, C.J. Meijer, and J.J. Oudejans. 2005. Immunohistochemical profiling of caspase signaling pathways predicts clinical response to chemotherapy in primary nodal diffuse large B-cell lymphomas. Blood. 105:2916–2923. [DOI] [PubMed] [Google Scholar]
- 50.Castedo, M., T. Roumier, J. Blanco, K.F. Ferri, J. Barretina, K. Andreau, J.-L. Perfettini, A. Armendola, R. Nardacci, P. LeDuc, et al. 2002. Sequential involvement of Cdk1, mTOR and p53 in apoptosis induced by the human immunodeficiency virus-1 envelope. EMBO J. 21:4070–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castedo, M., T. Hirsch, S.A. Susin, N. Zamzami, P. Marchetti, A. Macho, and G. Kroemer. 1996. Sequential acquisition of mitochondrial and plasma membrane alterations during early lymphocyte apoptosis. J. Immunol. 157:512–521. [PubMed] [Google Scholar]
- 52.Casares, N., J.J. Lasarte, A.L. de Cerio, P. Sarobe, M. Ruiz, I. Melero, J. Prieto, and F. Borras-Cuesta. 2001. Immunization with a tumor-associated CTL epitope plus a tumor-related or unrelated Th1 helper peptide elicits protective CTL immunity. Eur. J. Immunol. 31:1780–1789. [DOI] [PubMed] [Google Scholar]
- 53.Huang, A.Y., P.H. Gulden, A.S. Woods, M.C. Thomas, C.D. Tong, W. Wang, V.H. Engelhard, G. Pasternack, R. Cotter, D. Hunt, et al. 1996. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc. Natl. Acad. Sci. USA. 93:9730–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]