

Abstract
Secretory leucoprotease inhibitor (SLPI) is a nonglycosylated protein produced by epithelial cells. In addition to its antiprotease activity, SLPI has been shown to exhibit antiinflammatory properties, including down-regulation of tumor necrosis factor α expression by lipopolysaccharide (LPS) in macrophages and inhibition of nuclear factor (NF)-κB activation in a rat model of acute lung injury. We have previously shown that SLPI can inhibit LPS-induced NF-κB activation in monocytic cells by inhibiting degradation of IκBα without affecting the LPS-induced phosphorylation and ubiquitination of IκBα. Here, we present evidence to show that upon incubation with peripheral blood monocytes (PBMs) and the U937 monocytic cell line, SLPI enters the cells, becoming rapidly localized to the cytoplasm and nucleus, and affects NF-κB activation by binding directly to NF-κB binding sites in a site-specific manner. SLPI can also prevent p65 interaction with the NF-κB consensus region at concentrations commensurate with the physiological nuclear levels of SLPI and p65. We also demonstrate the presence of SLPI in nuclear fractions of PBMs and alveolar macrophages from individuals with cystic fibrosis and community-acquired pneumonia. Therefore, SLPI inhibition of NF-κB activation is mediated, in part, by competitive binding to the NF-κB consensus-binding site.
Secretory leucoprotease inhibitor (SLPI) is an 11.7-kD nonglycosylated protein produced at mucosal surfaces, primarily the upper respiratory tract (1). SLPI forms inhibitory complexes with a variety of proteolytic enzymes, including neutrophil elastase, and therefore appears to be an important component of the antiprotease defense of the lung (2). The amino acid sequence of SLPI and the resulting nuclear magnetic resonance solution structure has revealed a protein composed of two highly homologous domains of 53 and 54 amino acids, 8 disulfide bridges in total, and a large number of positively charged residues (3).
Recently, it has been demonstrated that SLPI also possesses antiinflammatory, antiviral, and antibacterial activity (4). LPS hyporesponsive cells have been shown to transcribe SLPI and transfection of macrophages, with SLPI suppressing LPS-induced activation of NF-κB and production of nitric oxide and TNF-α by an unknown mechanism (5). In addition, IFN-γ can suppress expression of SLPI and restore LPS responsiveness to SLPI-producing cells (5). SLPI has also been shown to inhibit HIV infectivity of monocytes by blocking viral DNA synthesis by a mechanism that does not involve binding to HIV directly but is most likely due to interaction with the host cell (6). In a model of acute lung injury induced by intrapulmonary deposition of IgG immune complexes in rats, prior administration of SLPI attenuated pulmonary recruitment of neutrophils and decreased lung injury (7). In addition, prior administration of SLPI to these animals resulted in greatly reduced NF-κB activation in whole lung samples. Although, interestingly, down-regulation of NF-κB activation was not observed in alveolar macrophages (AMs) isolated by broncholaveolar lavage from these animals (7). Further investigation of the NF-κB regulatory proteins revealed that IκBβ degradation was prevented in animals pretreated with SLPI. These data suggest that the inhibitory effects of SLPI are selective for the signal transduction pathway leading to NF-κB activation. SLPI has been shown to inhibit LPS and lipoteichoic acid (LTA)-induced NF-κB activation in monocytic cells by preventing degradation of key regulatory proteins, such as IκBα and IκBβ (8, 9). However, SLPI does not prevent LPS-induced phosphorylation or polyubiquitination of IκBα and has no apparent inhibitory effect on peptidase activities associated with the 20S proteasome, which is responsible for degrading polyubiquitinated IκBα (8). An SLPI knockout mouse model has been demonstrated to develop septic shock after peritoneal administration of LPS, underlining the pivotal role played by SLPI in attenuating excessive inflammatory responses (10).
Our previous work showing inhibition of LPS-induced NF-κB activation in monocytic cells without affecting other features, such as IκBα phosphorylation and polyubiquitination, would indicate that SLPI might be mediating its effects internally after cell uptake (8). In this study, we show that SLPI is taken up into monocytes and becomes distributed within the cytoplasm and nucleus. We demonstrate that SLPI can bind DNA, and using chromatin immunoprecipitation (ChIP), we show that SLPI can bind to NF-κB regions within the IL-8 and TNF-α promoters but not the IL-10 promoter region, which contains no NF-κB binding site. We also show by ChIP that p65 binding to the NF-κB regions within the IL-8 and TNF-α promoters is inhibited in the presence of SLPI. In addition, we demonstrate that SLPI can bind specifically to labeled NF-κB consensus oligonucleotide sequence and diminish recombinant p65 binding. Finally, we demonstrate that SLPI is present in the nucleus of peripheral blood monocytes (PBMs) and AMs isolated from individuals with sepsis. Our results indicate that SLPI may attenuate inflammation in monocytes, in part, by binding NF-κB sites of proinflammatory immune response genes, thus preventing p65 binding and subsequent transactivation of these genes.
Results
SLPI uptake into monocytes
Cell binding and uptake studies.
U937 cells were incubated with fluorescently labeled SLPI, and the level of cell association was quantified using a spectrofluorimeter. A highly significant level of SLPI cell association was seen after just 1 h (Fig. 1). The level of cell association appeared to be time-dependent with increasing levels of cell association seen after longer incubations. A widely used tool for inhibiting transport processes is to apply a temperature block to cells. The results indicated that the binding and uptake of SLPI by U937 cells are significantly inhibited (P < 0.01) at a low temperature (Fig. 1). The role of endocytosis was assessed by pretreating cells with the endocytosis inhibitors (sodium azide, sodium fluoride, and antimycin A) that inhibit glycolysis and oxidative metabolism required for most endocytic processes. These inhibitors had no significant effect on SLPI cell association (not depicted).
Figure 1.

The level of cell association of SLPI-fluorescein with U937 cells. Cells were treated for 4 h with 10 μg/ml SLPI-fluorescein at 37 or 4°C. n = 3; mean ± SD; **, P < 0.01.
Intracellular distribution.
The cell-association studies provide little information on the fate of SLPI within the cells or on the degree of internalization versus binding. To this end, confocal laser scanning microscopy of the fluorescein-labeled protein was performed to determine the distribution of SLPI within cells. The distribution of SLPI within U937 cells is shown in Fig. 2 A, and the intracellular distribution of SLPI within PBMs is shown in Fig. 2 B. The uptake of SLPI into cells was rapid and widespread (Fig. 2, A and B). The protein is not simply bound to the cell membrane. It is internalized efficiently as evidenced by the high level of fluorescence within the cells. Fluorescein alone does not enter cells to a significant extent, and therefore, the probe is being carried into the cell by SLPI.
Figure 2.
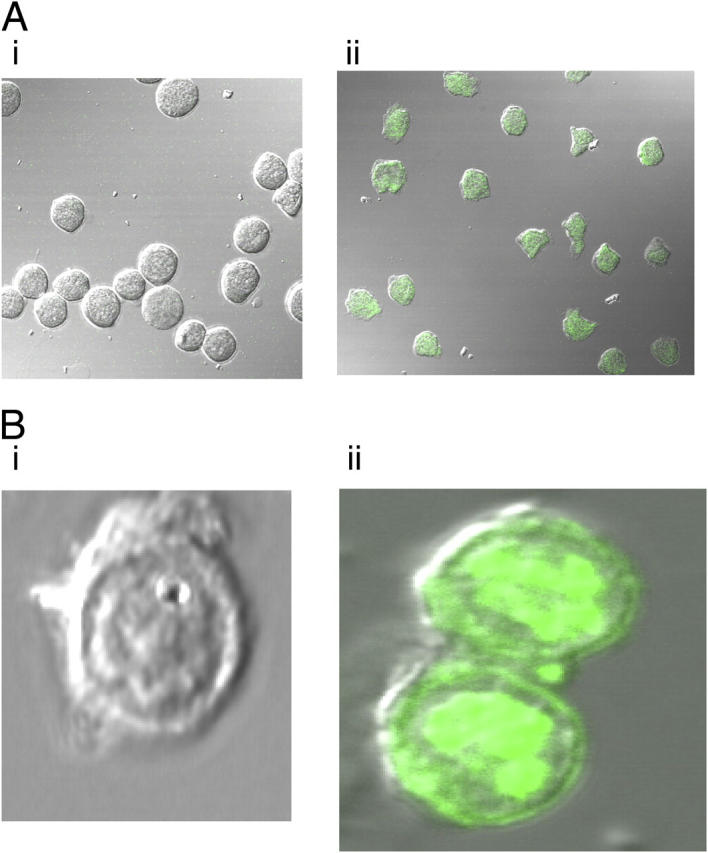
(A) Uptake of SLPI-fluorescein into U937 cells. 10 μg/ml SLPI-fluorescein was incubated with U937 cells for 1 h. (i) Untreated U937 cells. (ii) Treated U937 cells. (B) Uptake of SLPI-fluorescein into PBMs. 10 μg/ml SLPI-fluorescein was incubated with PBM for 1 h. (i) Untreated monocytes. (ii) Treated monocytes.
Nuclear import studies.
To confirm and visualize the degree of nuclear import of SLPI (Fig. 3, A and B, green), a nuclear counterstain propidium iodide (red) was used. The results are shown in Fig. 3 with colocalization represented by yellow on the confocal image. The degree of colocalization is also represented by a pixel intensity graph across one of the PBMs (Fig. 3 C).
Figure 3.
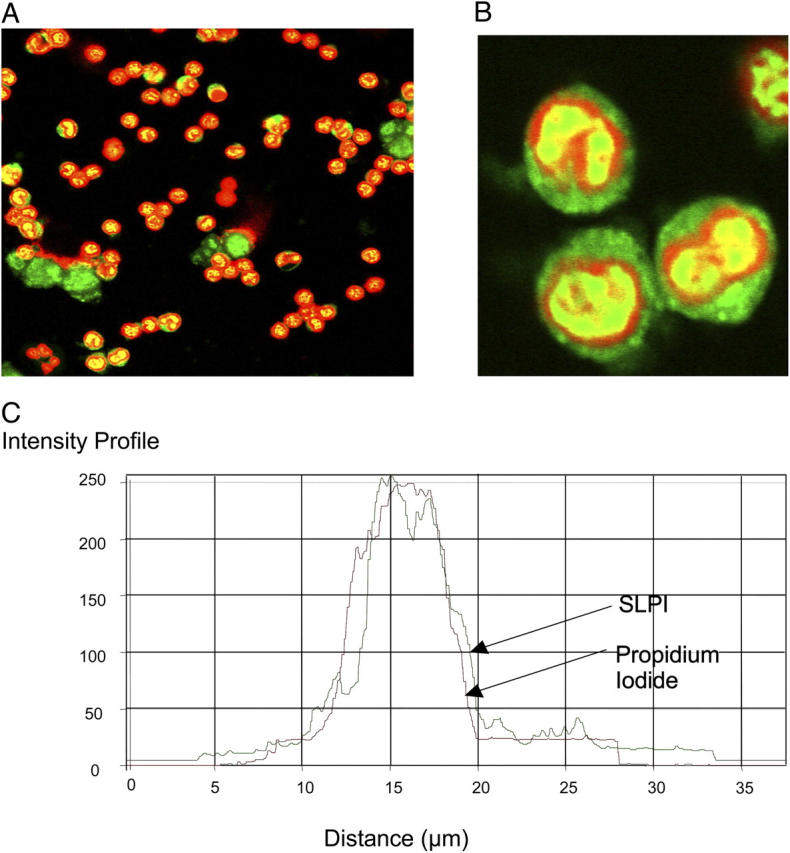
Nuclear import of SLPI-fluorescein in PBMs. 10 μg/ml SLPI-fluorescein (green) was incubated with monocytes for 1 h followed by counterstaining with propidium iodide. Red, nuclear stain. (A and B) Treated monocytes and (C) intensity profile. Red line, propidium iodide; green line, SLPI. (B) A magnification of the bottom left-hand quadrant of panel A.
Western analysis.
SLPI distribution in the cytoplasmic and nuclear compartments of U937 cells and PBMs was assessed by Western analysis. As shown in Fig. 4, SLPI was detected in the cytoplasmic and nuclear fractions of treated U937 cells (A) and PBMs (B). Western analysis of untreated U937 cells and PBMs revealed an absence of SLPI signal, indicating that both cells do not make appreciable quantities of SLPI that could be detected at this level. The purities of prepared extracts were confirmed by probing for cytoplasmic- and nuclear-specific proteins. As shown in Fig. 4 (A and B), GAPDH was detectable only in the cytoplasmic extract (ii), whereas Lamin B was only visible in the nuclear extract (iii). Equivalent amounts of GAPDH and Lamin B indicated equal loading for cytoplasmic and nuclear fractions, respectively.
Figure 4.

SLPI cytoplasmic and nuclear distribution in (A) U937 cells and (B) PBMs. Cells were incubated with 10 μg/ml SLPI for 1 h followed by isolation of cytoplasmic and nuclear fractions. Lane 1, untreated cytoplasmic fraction; lane 2, untreated nuclear fraction; lane 3, SLPI-treated cytoplasmic fraction; lane 4, SLPI-treated nuclear fraction subjected to Western blot and probed using antibodies to (i) SLPI, (ii) GAPDH, and (iii) Lamin B.
SLPI interaction with DNA
Binding to genomic DNA.
Due to SLPI's localization to the nucleus and its cationic charge, we considered the likelihood that SLPI could bind DNA directly. As shown in Fig. 5 A, SLPI was confirmed to bind to genomic DNA and, once bound, required up to 500 mM NaCl to be eluted. SLPI did not bind nonspecifically to the cellulose matrix (not depicted).
Figure 5.
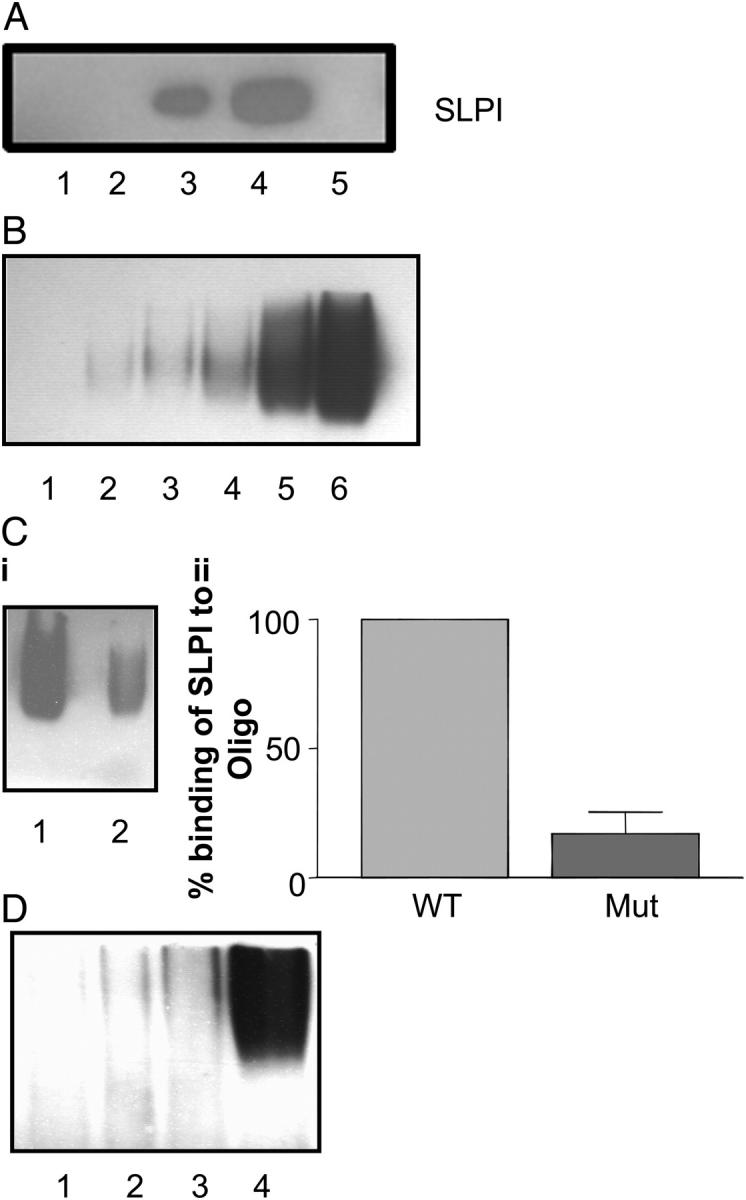
(A) SLPI binds to DNA. 2 μg SLPI was incubated with DNA-cellulose matrix, eluted in increasing amounts of NaCl, and subjected to Western blotting. Lane 1, flow through; lane 2, 150 mM NaCl elution; lane 3, 300 mM NaCl elution; lane 4, 500 mM NaCl elution; lane 5, 750 mM NaCl elution. (B) SLPI binding to biotin-labeled NF-κB consensus oligonucleotide. Various amounts of SLPI (1–50 ng) were incubated with labeled NF-κB oligonucleotide in the presence of poly(dI-dC·dI-dC): poly(dI-dC·dI-dC) and then electrophoresed on a 5% polyacrylamide gel. Lane 1, labeled oligonucleotide alone; lane 2, labeled oligonucleotide plus 1 ng SLPI; lane 3, labeled, oligonucleotide plus 2 ng SLPI; lane 4, labeled oligonucleotide plus 5 ng SLPI; lane 5, labeled oligonucleotide plus 10 ng SLPI; lane 6, labeled oligonucleotide plus 50 ng SLPI. (C, i) SLPI interaction with wild-type (WT) and mutant (Mut) NF-κB oligonucleotides. 100 ng SLPI was incubated with equal amounts of biotin-labeled wild-type NF-κB consensus oligonucleotide (lane 1) or mutant NF-κB oligonucleotide (lane 2). (ii) Densitometry graph of SLPI binding to wild-type versus mutant NF-κB oligonucleotide, representative of five experiments. (D) SLPI interaction with biotin-labeled GATA-1 (lane 1), Sp-1 (lane 2), CREB (lane 3), and AP-1 (lane 4) consensus oligonucleotides.
SLPI binding to NF-κB oligonucleotide.
Due to SLPI's ability to inhibit LPS- and LTA-induced NF-κB activation, and with the new information presented above that SLPI can localize to the nucleus and bind DNA, we considered the possibility that SLPI might bind directly to NF-κB binding sites. In Fig. 5 B, we demonstrate increased SLPI binding to wild-type biotinylated NF-κB consensus oligonucleotide in a dose-dependant manner. To test for the specificity of binding, a fixed amount of SLPI (100 ng) was incubated with wild-type biotinylated NF-κB consensus oligonucleotide and mutant NF-κB oligonucleotide, which contains a single base change rendering it unable to bind NF-κB. We demonstrate in Fig. 5 C (i and ii) that SLPI can bind with much greater affinity to wild-type NF-κB consensus oligonucleotide compared with binding to the mutant oligonucleotide. This result indicates SLPI's preference for binding to NF-κB sites, corresponding with its ability to inhibit NF-κB activation in activated U937 cells (8, 9). SLPI interaction with other consensus transcription factor binding sites was also assessed. Although there was no binding of SLPI to GATA-1, Sp-1, or CREB consensus oligonucleotides, strong binding of SLPI to AP-1 probe was observed (Fig. 5 D), indicating that SLPI may also bind sites other than NF-κB.
Competition studies.
To assess the ability of SLPI to compete for binding to NF-κB DNA sites compared with p65, we incubated a fixed amount of recombinant p65 (100 ng) with various concentrations of SLPI (1–2,000 ng). p65–DNA complexes were detected by electromobility shift assay (EMSA). In the presence of increasing SLPI concentrations (10–2,000 ng), the intensity of the p65–NF-κB DNA complex signal decreased (Fig. 6 A). At concentrations of 1,000 and 2,000 ng, SLPI markedly suppressed the binding of 100 ng p65 to NF-κB consensus oligonucleotide (Fig. 6 A, lanes 5 and 6). In these lanes, increased SLPI–oligonucleotide complexes could also be observed. A densitometry curve for SLPI inhibition of p65 binding to NF-κB oligonucleotide is shown in Fig. 6 B. In addition, we also demonstrated that SLPI could prevent recombinant p65 binding to NF-κB oligonucleotide in a p65 activity ELISA (Fig. 6 C, i), with similar results to those shown in panels A and B, as well as displace p65 from binding to the NF-κB oligonucleotide (Fig. 6 C, ii).
Figure 6.

(A) Increasing SLPI concentration prevents p65 binding to the NF-κB site. EMSA: 10–2,000 ng SLPI was incubated with NF-κB consensus binding site for 15 min followed by incubation with fixed amounts of p65 (100 ng) for an additional 20 min. Lane 1, p65 plus NF-κB DNA; lane 2, 10 ng SLPI plus p65 plus NF-κB DNA; lane 3: 100 ng SLPI plus p65 plus NF-κB DNA; lane 4, 500 ng SLPI plus p65 plus NF-κB DNA; lane 5, 1,000 ng SLPI plus p65 plus NF-κB DNA; lane 6, 2,000 ng SLPI plus p65 plus NF-κB DNA. P65–oligonucleotide and SLPI–oligonucleotide complexes are shown. (B) Densitometric evaluation of SLPI inhibition of p65 binding to NF-κB consensus oligonucleotide. (C, i) Increasing SLPI concentration prevents p65 binding to NF-κB site. p65 activity ELISA: 10–1,000 ng SLPI was incubated in wells containing the NF-κB consensus oligonucleotide for 15 min followed by incubation with fixed amounts of p65 (10 ng). p65 binding was detected by anti-p65 antibody/peroxidase-linked secondary antibody. (ii) SLPI displacement of p65 binding to NF-κB oligonucleotide. (D) p65 nuclear localization in U937 cells treated with (i) LPS or (ii) SLPI/LPS. U937 cells were incubated with 1 μg/ml LPS or 10 μg/ml SLPI for 1 h followed by LPS. Nuclear fractions were prepared and probed for NF-κB p65 over time. Lane 1, control; lane 2, 30 min; lane 3, 60 min; lane 4, 120 min. (E) SLPI and p65 concentrations in 106 cells. Nuclear extracts from SLPI-incubated U937 cells were subjected to Western blot, and SLPI and p65 levels were estimated by densitometry using SLPI and p65 standards. p65 = 5.61 ± 0.47 ng; SLPI = 194 ± 3.03 ng.
p65/SLPI nuclear localization and concentration in activated U937 cells.
To determine the physiological relevance of this effect, we performed two experiments to ascertain: (a) whether p65 still localized to the nucleus after activation with LPS in the presence of SLPI and (2) the concentration of SLPI and p65 in the nuclear compartment of U937 cells after incubation with SLPI and LPS. As shown in Fig. 6 D, p65 localized to the nucleus of U937 cells within 30 min after stimulation with LPS. Likewise, the kinetics of p65 localization to the nucleus was very similar in the presence of SLPI/LPS, indicating that inhibition of NF-κB activation in these cells was not due to exclusion of p65 from the nucleus. p65 and SLPI levels in the nucleus of activated U937 cells were determined by densitometric analysis of nuclear fractions separated by SDS-PAGE and blotted for the respective protein. Fig. 6 E reveals that the amount of p65 in the nucleus of activated U937 cells was 5.61 ± 0.47 ng compared with the amount of SLPI in the same fraction, which was 194 ± 3.03 ng. This result revealed that SLPI levels were ∼35-fold higher than p65, indicating that SLPI could inhibit p65 binding to NF-κB sites in the genome according to the results obtained in Fig. 6 (A and B).
SLPI interaction with NF-κB sites in the genome.
Because SLPI is distributed in the nucleus of monocytic cells and interacts with NF-κB sites, we tested to see whether SLPI could interact with an endogenous NF-κB–rich promoter region. To this end, we investigated the binding of SLPI to the IL-8 and TNF-α promoters, which contain multiple NF-κB binding sites, and the IL-10 promoter, which contains no NF-κB sites, in U937 cells. U937 cells treated with or without SLPI were fixed with formaldehyde and subjected to ChIP analysis as outlined in Materials and methods. As shown in Fig. 7, SLPI could bind to the IL-8 and TNF-α promoters, but did not interact with the IL-10 or GAPDH promoters. In contrast, p65 binding to the TNF-α and IL-8 promoters was observed in cells treated with LPS alone (as expected), but not in those cells preincubated with SLPI (Fig. 7).
Figure 7.

Interaction of SLPI with NF-κB sites in genomic DNA. U937 cells were incubated with or without 10 μg/ml SLPI for 1 h and then 1 μg/ml LPS for 1 h, and DNA was isolated by ChIP. U937 cells were incubated with anti-SLPI IgG, anti-p65 IgG, anti-acetyl histone H3 (positive control), or no antibody (negative control). DNA interacting with SLPI was isolated and subjected to PCR for detection of IL-8 promoter region (A), TNF-α promoter region (B), IL-10 promoter region (C), and GAPDH promoter (D). Lanes 1, 3, 5, and 7, cells incubated minus SLPI; lanes 2, 4, 6, and 8, cells incubated with SLPI; lanes 1 and 2, no antibody immunoprecipitation; lanes 3 and 4, SLPI antibody immunoprecipitation; lanes 5 and 6, p65 antibody immunoprecipitation (A and B only); lanes 7 and 8, acetyl histone H3 antibody immunoprecipitation.
Effect of SLPI on LPS-induced cytokine production.
To determine the effect of SLPI on LPS-induced cytokine production, U937 cells were differentiated to a macrophage lineage, and LPS-induced TNF-α, IL-8, and IL-10 production in the presence of SLPI was assessed. As can be seen in Fig. 8, SLPI decreased LPS-induced TNF-α (LPS:1,420 ± 26.8 pg/ml vs. SLPI/LPS: 914.3 ± 47.1 pg/ml) and IL-8 production (LPS: 599.2 ± 15.4 pg/ml vs. SLPI/LPS: 340.2 ± 4.0 pg/ml), but not IL-10 (LPS: 219.7 ± 0.8 pg/ml vs. SLPI/LPS: 223.2 ± 4.3 pg/ml).
Figure 8.
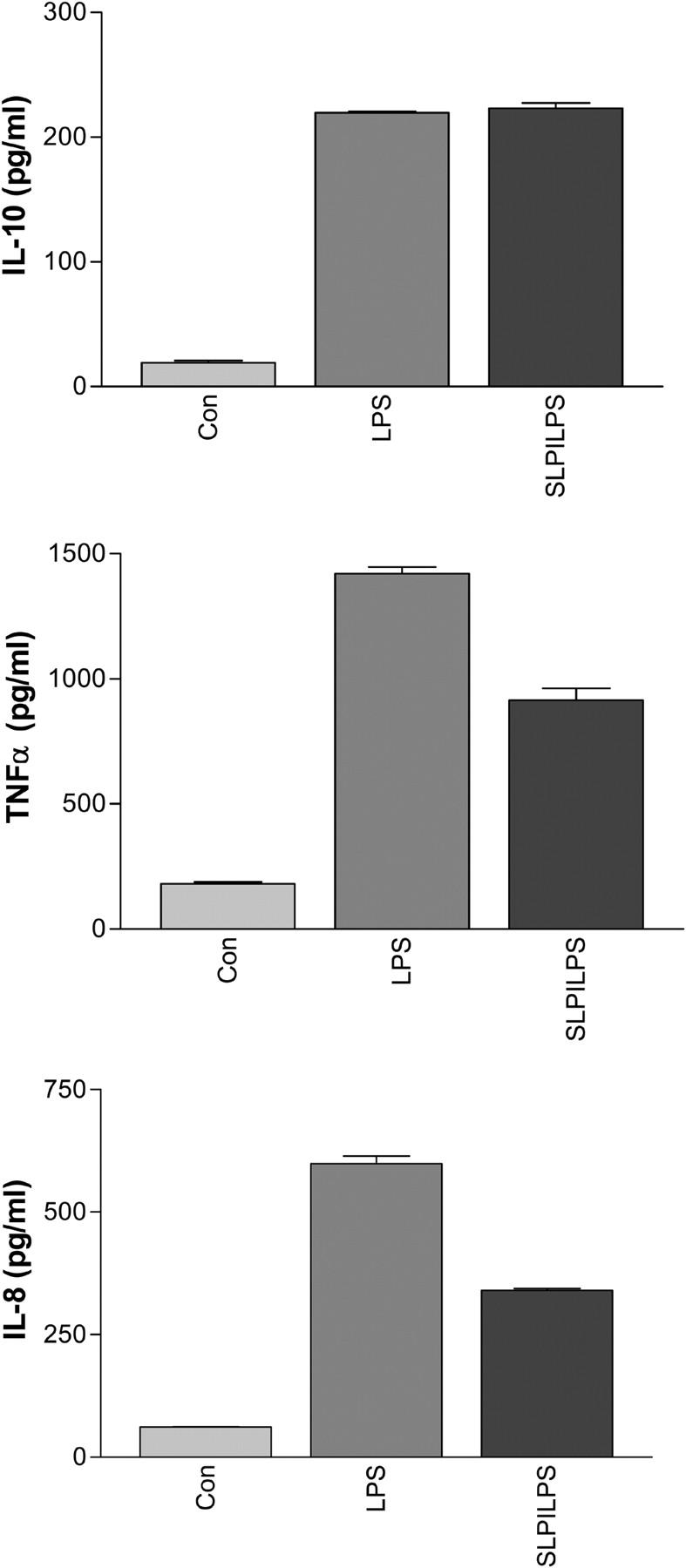
Effect of SLPI on LPS-induced TNF-α (A), IL-8 (B), and IL-10 (C) protein production by U937 macrophages.
Presence of SLPI in PBM and AM nuclear fractions.
AMs and PBMs were isolated from six individuals with pulmonary sepsis—three with cystic fibrosis (ΔF508 homozygous) and three with community-acquired pneumonia—as well as from three healthy controls (AMs and PBMs). Nuclear fractions were prepared from both cell populations, and SLPI was measured by ELISA. SLPI was shown to be present in AM (17,897 ± 4,508 pg/106 cells) and PBM (133.9 ± 34.4 pg/106 cells) nuclear fractions from sepsis patients (Fig. 9). SLPI was also present in control AMs (13,499 ± 3,768 pg/106 cells) but could not be detected in control PBMs (Fig. 9).
Figure 9.

Measurement of SLPI in the nucleus of AMs and PBMs. Nuclear fractions were prepared from 106 AMs or PBMs from individuals with sepsis (cystic fibrosis and community-acquired pneumonia) and healthy controls. SLPI levels were determined by ELISA. Con, control; Sep, sepsis.
Discussion
Previous studies have demonstrated SLPI's ability to inhibit LPS-induced NF-κB activation in monocytes and macrophages as well as prevent TNF-α, nitric oxide, and matrix metalloprotease production (5, 11). In this study, we show that SLPI can enter monocytes rapidly in a temperature-dependent process, becoming localized to the cytoplasm and nucleus. The ability of SLPI to enter the nucleus is particularly interesting due to the cationic nature of this protein and its potential to bind negatively charged regions of DNA. We show that SLPI can bind DNA and NF-κB sites in a specific manner. In addition, we show that SLPI can compete with NF-κB (p65) for binding to the NF-κB DNA site, thereby providing a mechanism by which SLPI can prevent LPS- and LTA-induced NF-κB activation in monocytes. Using ChIP technology, we also prove that SLPI binds the NF-κB–rich region of the IL-8 promoter, but not the IL-10 promoter, which possesses no NF-κB binding sites. We also showed by ChIP that p65 binding was inhibited in the presence of SLPI. Finally, we demonstrate that SLPI is present in in vivo PBM and AM nuclear fractions.
Other proteins have been identified that are rapidly and efficiently internalized by cells, e.g., Drosophilia homeotic transcription protein antennapedia (Antp) and the human immunodeficiency virus I transcriptional activator (HIV-TAT), due to the presence of small regions called protein transduction domains in their structure (12). The common feature of these protein transduction domains is the high content of lysine and arginine amino acids (13). A great deal of debate still continues about the exact mechanism of protein transduction into cells. Initial studies conducted on Antp and HIV-TAT indicated a nonendocytic uptake process for these arginine-rich peptides, but it has recently been suggested that the mechanism of uptake is very much protein-dependent. A number of models have been devised to explain the ability of basic, hydrophilic peptides to translocate across cell membranes, including the involvement of hydrogen bonding and membrane potential in the translocation process (14). Therefore, it might be the arginine-rich nature of SLPI that enables it to interact with the predominantly negatively charged cell membrane and be internalized. Indeed, it could be hypothesized that SLPI contains its own unique protein transduction domain. There is recent evidence to show that SLPI can bind to the cell surface protein annexin II, which is present on the cell surface of macrophages, U937 cells, and PBMs (15, 16, 17).
Like most of the cationic proteins of the respiratory tract, SLPI possesses antibacterial activity. However, it is clear that many of these proteins can also exert proinflammatory or antiinflammatory effects. The neutrophil-derived peptide LL-37 can induce IL-8 expression in monocytes via ERK and p38-dependent pathways (18) similar to α defensins, which can also induce IL-8 expression by epithelial cells (19). In contrast, lactoferrin, which is one of the most abundant cationic proteins of the airways, can inhibit LPS-induced IL-8 expression by epithelial cells. Similar to the data presented in this study, lactoferrin has also been shown to enter cells becoming localized to the nucleus (20). Lactoferrin's interaction with DNA has also been verified, although its ability to up-regulate AP-1 activity in fibroblasts is not due to direct binding and transactivation of the AP-1 promoter site (21). Another peptide, PR-39, has also been shown to enter cells and inhibit TNF-α–induced effects by binding directly to subunits of the proteasome, thus affecting degradation of polyubiquitinated, phosphorylated IκBα (22). We have previously shown that SLPI can prevent degradation of IκBα and β and subsequently inhibit NF-κB activity without affecting IκBα phosphorylation and ubiquitination (8). But in this study, we have demonstrated that p65 can still translocate to the nucleus in SLPI-treated cells. Therefore, how does SLPI prevent IκBα degradation and at the same time allow p65 entry to the nucleus? These diverging effects may occur as a result of SLPI's multifunctional characteristics of size, charge, and protease inhibition activity as we will now explain. Due to SLPI's inhibition of p65, it might be expected that SLPI would be proapoptotic. However, we have generated some data to show that instead of increasing TNF-α–induced caspase-3 activation in U937 cells, SLPI in fact decreases TNF-α–induced caspase-3 activity (not depicted). It is possible that SLPI is interfering with apoptotic pathways in the cell, most of which are controlled by proteases, particularly caspases, and that SLPI might be inhibiting one of these proteases directly. Interestingly, caspase-3 has been shown previously to cleave IκBα (23). This raises the possibility that SLPI may inhibit caspase-3–mediated IκBα cleavage and that our previous finding, demonstrating SLPI inhibition of LPS-induced IκBα degradation (8), might be due in part to SLPI's antiapoptotic effect as opposed to any direct effect on the ubiquitin–proteasome pathway. Therefore, SLPI inhibition of caspase activity may lead to accumulation of a certain amount of phosphorylated, polyubiquitinated IκBα, whereas a certain amount of IκBα may still be degraded by the proteasome, allowing release of p65 from the NFκB–IκBα complex. Our current findings, demonstrating SLPI binding to NF-κB DNA sites, might be antiprotease-independent and occur as a result of SLPI's relatively small size (allowing it to passively diffuse into the nucleus) and its positive charge (allowing it to bind to negatively charged DNA, albeit in a specific manner).
Studies in SLPI knockout mice have demonstrated that these develop septic shock when challenged peritoneally with LPS (10). This was explained in part by increased activity of the transcription factors NF-κB and C/EBP-β in macrophages from SLPI−/− mice compared with SLPI+/+ mice. These findings lend support to our own observations that SLPI can inhibit NF-κB activity by interacting directly with NF-κB binding sites and subsequently inhibiting p65 binding. We have also evaluated SLPI binding to other transcription factor binding sites, including GATA-1, Sp-1, CREB, and AP-1. Although we did not observe binding of SLPI to GATA-1, Sp-1, and CREB, we did observe SLPI binding to the AP-1 transcription factor binding site. AP-1 is involved in a number of processes, including inflammation and cell proliferation, and future studies will be aimed at elucidating the AP-1–dependent effects of SLPI. It is possible that some of the effects that we have seen on TNF-α and IL-8 production in this study (Fig. 8) are due in part to an effect of SLPI on AP-1 activity. However, it has been shown previously that LPS-induced IL-8 production from macrophages is dependent on NF-κB activity (24). To determine further the specific effects of SLPI on AP-1 activity as compared with NF-κB, an approach using siRNA to p65 or ATF-2/c-jun (subunits of AP-1) and the subsequent effect of SLPI on LPS-stimulated cytokine production in siRNA-transfected U937 cells will provide more definitive data.
Other studies in support of our findings in this study have demonstrated that cyclin D1 gene expression is increased in an SLPI knockout cell line compared with the parental cell line and that this activity is due in part to SLPI regulation of the cyclin D1 promoter (25). Interestingly, NF-κB is a transactivator of cyclin D1 gene expression, suggesting that SLPI may also influence the regulation of this gene by inhibiting NF-κB binding. In addition, one of the few augmentation trials involving the administration of SLPI to the airways of individuals with cystic fibrosis resulted in decreased IL-8 levels in the lungs of treated individuals (26). This correlates well with our findings in this study in which we have shown that SLPI can bind to the NF-κB–rich promoter region of the IL-8 gene in monocytes and, in so doing, may inhibit NF-κB activation of the IL-8 gene and subsequent IL-8 protein production. Another previous study has shown that SLPI can prevent LPS-induced TNF-α production in macrophages, and we have demonstrated here that SLPI can bind to the NF-κB–rich TNF-α promoter (5). We also investigated SLPI binding to the IL-10 promoter and demonstrated an absence of SLPI binding to the promoter region of this gene. However, it has been shown previously that IL-10 expression is regulated by MAP kinases and Sp-1 (27, 28), but not NF-κB (29).
Our finding that SLPI is present in the nucleus of human PBMs and AMs also lends support to our observation that SLPI can enter the nucleus of monocytes and macrophages. We have demonstrated differences in SLPI concentrations in the nucleus of both cell populations, which probably reflect the SLPI concentration in the surrounding environment of these cell types. Although SLPI levels can exceed 100 ng/ml in serum, saliva levels of SLPI are in the range of 4–24 μg/ml and lung epithelial lining fluid levels of SLPI are as high as 10 μg/ml (30–32). This information and our findings in this study would suggest that SLPI may exert its greatest anti–NF-κB effects in the respiratory/upper respiratory tract in vivo. Therefore, the use of SLPI as an augmentation therapy directed toward inhibiting NF-κB would necessitate augmentation levels that would reach 10 μg/ml and higher to have an effect.
In conclusion, we have demonstrated that SLPI can enter monocytic cells and become localized to the nucleus. We have also shown that SLPI can bind an NF-κB consensus sequence as well as the promoter region of the IL-8 gene in monocytes and is capable of inhibiting p65 binding to the NF-κB DNA binding site. Therefore, our results may go some way toward explaining SLPI's immunomodulatory/antiinflammatory role in suppressing NF-κB–induced responses in monocytes.
MATERIALS AND METHODS
All experiments were repeated at least three times.
SLPI intracellular localization
SLPI labeling.
SLPI (R&D Systems) was fluorescently labeled using a dye reagent (Fluorescein-EX; Invitrogen) that has a reactive succinimidyl ester moiety, which reacts with the primary amines of the protein to form a stable SLPI-fluorescein conjugate. Free dye was removed using a Spin-OUT column (Geno Technology) with a molecular weight cut-off of 6,000. The yield after purification was quantified using a Bio-Rad Laboratories protein assay, and the degree of labeling was determined by UV absorbance.
Cell culture.
U937 cells were cultured routinely in RPMI containing 10% fetal calf serum. PBMs were isolated from the blood of healthy volunteers using Ficoll-Paque separation from which the mononuclear cell layer was removed and washed twice in RPMI. Monocytes were positively selected using anti-CD14–coated magnetic beads (Miltenyi Biotec) and resuspended in RPMI/10% fetal calf serum.
Cellular binding and uptake assays.
Before treatment, the medium was removed from U937 cells and replaced with fresh medium. The cells were seeded onto 24-well plates (Nunc), and SLPI-fluorescein was added to each well and incubated at 37°C under 5% CO2. After incubation, the cells were washed three times with PBS and incubated with 1 ml Triton X-100 (1% wt/vol) for 30 min at 37°C. Cell-associated fluorescence was determined at excitation and emission wavelengths of 494 and 518 nm, respectively, on a spectrofluorimeter (PerkinElmer) and expressed as fluorescence/mg protein (Bio-Rad Laboratories protein assay kit).
To determine the mechanism of SLPI–cell interaction, incubations were performed at 4°C and in the presence of endocytosis inhibitors (0.1% wt/vol sodium azide, 10 mM sodium fluoride, and 1 μg/ml antimycin A).
Confocal laser scanning microscopy.
106/ml U937 cells or PBMs were incubated with 10 μg/ml SLPI-fluorescein for various time periods (10 min to 4 h) followed by washing three times in PBS. 10,000 cells were then cytospun onto glass microscope slides and air-dried. The cytospins were incubated for 5 min in 1% wt/vol paraformaldehyde followed by washing three times in PBS. Finally, some cytospins were incubated with 0.5 μM propidium iodide for 30 min in the dark and washed three times in PBS. Fluorescence-free glycerol-based mounting medium was added before covering the slide with a coverslip for visualization. The cells were viewed using an upright confocal microscope (LSM510 Axioplan 2; Carl Zeiss MicroImaging, Inc.).
Western blotting
Cytoplasmic and nuclear fractions from 10 μg/ml of untreated and SLPI-treated monocytes were prepared as described previously (8). The purity of the fractions were tested by separating equal amounts of protein (10 μg) on 10% SDS-PAGE and followed by transfer to nitrocellulose. Western analysis was performed using antibodies to GAPDH (1:500 for cytoplasmic detection) and Lamin B (1:100 for nuclear detection) in 5% nonfat skimmed milk and 1% BSA in PBST overnight at 4°C. All antibodies were from Santa Cruz Biotechnology, Inc. Antibody–antigen complexes were detected using appropriate horseradish peroxidase–labeled secondary antibodies, and blots were developed using supersignal chemiluminescent reagent (Pierce Chemical Co.). For detection of SLPI, 10 μg of cytoplasmic and nuclear fractions were separated on 15% SDS-PAGE, transferred to nitrocellulose, and detected using a rabbit anti–SLPI IgG (1:1,000) incubated overnight in 0.2% I-block (Tropix) in PBST. Antibody–antigen complex was detected using anti–rabbit horseradish peroxidase for 1 h in 0.2% I-block (Tropix) in PBST, and the blot was developed as described above.
Activation of U937 cells by LPS.
106/ml U937 cells were incubated alone, in the presence of 1 μg/ml LPS (Escherichia coli B111:B4; Sigma-Aldrich), or preincubated with 10 μg/ml SLPI followed by 1 μg/ml LPS for 120 min as described previously (8, 9). Nuclear fractions were prepared and equal amounts of protein (20 μg) were separated by 15% SDS-PAGE. After transfer to nitrocellulose, the blot was blocked in 5% nonfat skimmed milk and 1% BSA and then incubated with mouse anti–p65 IgG overnight at 4°C. The rest of the blotting procedure was performed as described above. SLPI was also detected in the same samples (as described above), and levels of SLPI and p65 were determined by densitometry using the GeneGenius Gel Documentation and Analysis System (Syngene) as well as Syngene GeneSnap and GeneTools software. Comparison of p65 and SLPI in U937 nuclear fractions was made to fixed standards of recombinant SLPI and p65 separated on the same gels as the nuclear fractions.
DNA binding studies
Genomic DNA binding.
To test SLPI binding to DNA, SLPI was incubated with 4 mg of the bovine thymus genomic DNA–cellulose conjugate (Sigma-Aldrich), or cellulose-type 50 (Sigma-Aldrich) was incubated with 2 μg SLPI at 4°C for 1 h in 20 mM Tris, pH 7.5, 100 mM KCl, 10% glycerol, 1 mM EDTA, 1 mM DTT, 1 mg/ml BSA, 0.2% Triton X-100, and protease inhibitor cocktail. The beads were thoroughly washed with 20 mM Tris, pH 7.5, 110 mM potassium acetate, 10% glycerol, 1 mM EDTA, 1 mg/ml BSA, and 0.2% Triton X-100. The beads were then equilibrated in 20 mM Tris, pH 7.5, and elution was performed sequentially in PBS with increasing concentrations of NaCl (from 150 to 750 mM). The eluate was centrifuged at 960 g for 1 min, and the supernatant was used for analysis. For Western blot analysis, proteins were resolved on a 15% SDS-PAGE and transferred to a nitrocellulose membrane. SLPI was detected by Western blot as indicated above.
EMSA.
EMSA was performed as described previously with some modifications. Various concentrations of SLPI (1–2,000 ng) were incubated with double-stranded wild-type biotinylated NF-κB consensus oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGGC-3′; MWG Biotech) alone or in the presence of 100 ng of recombinant p65 (Active Motif) for 30 min at room temperature in binding buffer (4% [vol/vol] glycerol, 1 mM EDTA, 10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM DTT, and 0.1 mg/ml nuclease-free BSA) and 2 μg poly (dI-dC·dI-dC):poly(dI-dC·dI-dC): (Sigma-Aldrich). In some experiments, double-stranded mutant biotinylated NF-κB oligonucleotide (5′-AGTTGAGGCGACTTTCCCAGGC-3′; MWG Biotech) was added to test the specificity of SLPI binding. Reaction mixtures were electrophoresed on native 5% (for detection of NF-κB–DNA complexes) or 15% (for detection of SLPI–DNA complexes) polyacrylamide gels. The gels were then transferred to nitrocellulose in 1X TBE for 30 min at 380 mA, 100 V, and then cross-linked on a UV transilluminator for 10 min. Nitrocellulose membrane was used in preference to nylon as SLPI–NF-κB probe complexes could not be detected on nylon membrane, most likely due to the inability of the positively charged SLPI molecule to bind to the positively charged nylon membrane. Detection of SLPI–DNA or p65–DNA complexes was performed using the Chemiluminescent Nucleic Acid Detection Module (Pierce Chemical Co.). In brief, after cross-linking, the blot was incubated in blocking buffer for 1 h at 37°C. Streptavidin-peroxidase was added for 15 min in blocking buffer at room temperature, and the blot was then washed six times in wash buffer. The blot was incubated for 5 min in equilibration buffer and developed with the chemiluminescent reagents provided with the kit. SLPI inhibition of p65 binding was determined by densitometry using the GeneGenius Gel Documentation and Analysis System (Syngene) as well as Syngene GeneSnap and GeneTools software.
SLPI binding to other consensus oligonucleotide sequences.
SLPI binding to other transcription factor consensus oligonucleotide binding sites was also tested as described above using 100 ng SLPI and the following biotin-labeled double-stranded oligonucleotides: CGCTTGATGAGTCAGCCGGAA (AP-1), AATTGGACCGTATCTCCC (GATA), ATTCGATCGGGCGGGGCGAG (Sp-1), and AGAGATTGCCTGACGTCAGACAGCTAG (CREB).
NF-κB activity ELISA.
The effect of SLPI on p65 binding activity was determined using the TransAM NF-κB ELISA (Active Motif). Initially, 10–1,000 ng SLPI was incubated in 30 μl binding buffer (+DTT) and added to wells containing the NF-κB consensus oligonucleotide for 15 min at room temperature with rotation. 10 ng of recombinant p65 (Active Motif) was added to each well in 10 μl of complete lysis buffer, and incubation was continued for an additional hour. After washing, 100 μl anti-p65 antibody was added to each well for 1 h without agitation followed by additional washing and incubation with peroxidase-labeled secondary antibody without agitation. After the final wash, 100 μl developer solution was added to each well for 5 min followed by 100 μl stop solution and then read at 450/650 nm on a plate reader. In separate experiments to examine SLPI's ability to displace p65 from the NF-κB oligonucleotide, 10 ng p65 was incubated in 30 μl binding buffer (+DTT) and added to wells containing the NF-κB consensus oligonucleotide for 15 min at room temperature with rotation. 100 or 1,000 ng SLPI was added to each well in 10 μl of complete lysis buffer, and incubation was continued for an additional hour. Incubation with anti-p65 antibody/secondary antibody and development were performed as described above.
ChIP
ChIP was performed using an EZ ChIP kit (Upstate Biotechnology). U937 monocytes were pretreated with or without 10 μg/ml SLPI for 1 h and incubated for an additional hour with 1 μg/ml LPS. After culture, cells were cross-linked in the cell culture plates by adding 37% formaldehyde to the culture medium to a final concentration of 1% and incubating at room temperature for 10 min. The cross-linking reaction was stopped by the addition of 0.125 M glycine for 5 min. Cells were washed with ice-cold PBS containing protease inhibitors and lysed in SDS lysis buffer containing protease inhibitors. Samples were sonicated on ice to an average size of 200–1,000 bp and centrifuged for 10 min at 14,000 rpm at 4°C to remove insoluble material. The supernatant was diluted 10-fold in ChIP dilution buffer containing protease inhibitors. Chromatin was precleared with salmon sperm DNA/protein G agarose for 1 h at 4°C, and 1% of this solution was saved (input chromatin) and processed with the eluted immunoprecipitates beginning with the cross-link reversal step. Samples were incubated overnight at 4°C with 5 μg goat anti–SLPI antibody, 5 μg anti-p65 antibody (sc-109x; Santa Cruz Biotechnology, Inc.), 10 μg anti-acetyl histone H3 (positive control; Upstate Biotechnology), or no antibody (negative control) with rotation. Immune complexes were collected by incubating samples with salmon sperm DNA/protein G agarose for 1 h at 4°C with rotation. The beads were pelleted by brief centrifugation and washed as per the manufacturer's instructions with low salt complex wash buffer, high salt complex wash buffer, LiCl immune complex wash buffer, and TE buffer, pH 8.0. The immunocomplexes were eluted from the agarose beads by adding 200 μl elution buffer (1% SDS and 0.1 M NaHCO3), vortexing, and rotating at room temperature for 30 min. Input and immunoprecipitated chromatin were incubated with 200 mM NaCl at 65°C for 4 h to reverse DNA–protein cross-links. Samples were RNase-treated for 30 min at 37°C. 10 mM EDTA, 40 mM Tris-HCL, pH 6.5, and 20 μg proteinase K (Sigma-Aldrich) were then added to chromatin and incubated at 45°C for 1 h. Input DNA was recovered by phenol/chloroform extraction and ethanol precipitation. Immunoprecipitated DNA was purified as per the manufacturer's instructions (Upstate Biotechnology). Promoter sequences were detected in input and immunoprecipitated DNA by PCR using specific primers: IL-8 (forward: GGGCCATCAGTTGCAAATC; reverse: TTCCTTCCGGTGGTTTCTTC); IL-10 (forward: AGAGGAAAGTAAGGGACCTCC; reverse: AGACTGGCTTCCTACAGGACA); TNF-α (forward: CCCTCCAGTTCTAGTTCTATC; reverse: GGGGAAAGAATCATTCAACCA), and GAPDH (forward: TACTAGCGGTTTTACGGGCG; reverse: TCGAACAGGAGGAGCAGAGAGCGA).
Effect of SLPI on LPS-induced cytokine production
To assess the effect of SLPI on LPS-induced TNF-α, IL-8, and IL-10 protein production, U937 cells were differentiated to a macrophage phenotype by incubating with 100 ng/ml PMA for 48 h. The adhered cells were then washed and incubated for an additional 3 d in fresh medium, washed again, and resuspended in fresh medium. The cells were then incubated for an additional 24 h in medium alone or with 100 ng/ml LPS, or they were preincubated with 10 μg/ml SLPI for 1 h followed by LPS for 24 h. TNF-α, IL-8, and IL-10 levels were measured in the cell supernatant using commercially available ELISA kits (R&D Systems).
SLPI nuclear localization in vivo
To determine whether SLPI could be detected in the nucleus of in vivo samples, PBMs were isolated from six individuals with pulmonary sepsis—three with cystic fibrosis and three with community-acquired pneumonia. PBMs were also isolated from three healthy controls. The cystic fibrosis patients were homozygous for the ΔF508 CFTR mutation. In addition, AMs were isolated from the same individuals. In brief, bronchoalveolar lavage fluid was collected from each individual and centrifuged at 3,000 rpm for 10 min and washed twice in RPMI medium containing 10% fetal calf serum. Macrophages were adhered to six-well tissue culture plates and cultured for 1 h followed by washing in medium. Adhered macrophages were scraped from the wells in PBS and counted using trypan blue.
Cytoplasmic and nuclear extracts were prepared from the isolated PBM and AM samples as described above in preparation for SLPI analysis by ELISA.
SLPI ELISA.
Immunlon-2 96-well plates were coated with rabbit anti–human SLPI antibody overnight at 4°C in Voller's buffer (1:1,000). The plates were washed with PBST, and SLPI standards (2,000–31.25 pg/ml) were applied to the plate in 100-μl aliquots. The monocyte and AM nuclear samples were made up to 200 μl in PBST, diluted 1:2 across the plate, and left at room temperature for 2 h. The plates were then washed as before, and biotinylated anti–human SLPI antibody (R&D Systems) diluted 1:100 in PBST was loaded onto the plate in 100-μl aliquots and left at room temperature for 2 h. Finally, streptavidin-peroxidase (Zymed Laboratories) diluted 1:2,500 in PBST was pipetted onto the plates in 100-μl aliquots and left at room temperature for 30 min. After a final wash, 100 μl ABTS (Zymed Laboratories) was added to each well, and the color was left to develop. Absorbance values at 405 nm were converted to actual SLPI concentrations using GraphPad Prism software (GraphPad Software).
Acknowledgments
We wish to thank Dr. Stephen Smith (Department of Medical Microbiology, Trinity College Dublin) for technical assistance in the drafting of the manuscript.
We wish to acknowledge funding from the Alpha One Foundation (to C.C. Taggart), the Health Research Board of Ireland (RP/198/2002 to C.C. Taggart), Science Foundation Ireland (04/BR/B0640 to C.C. Taggart and SC/2004/B0419 to S.-A. Cryan), the Research Committee of the Royal College of Surgeons in Ireland (13/02 to C.C. Taggart), the Programme for Research in Third Level Institutes administered by Higher Education Authority of Ireland (to N.G. McElvaney), the Cystic Fibrosis Association of Ireland (to C.C. Taggart), the CF Research Trust, and the CF Hope Source.
The authors have no conflicting financial interests.
Abbreviations used: AM, alveolar macrophage; ChIP, chromatin immunoprecipitation; EMSA, electromobility shift assay; LTA, lipoteichoic acid; PBM, peripheral blood monocyte; SLPI, secretory leucoprotease inhibitor.
References
- 1.Abe, T., N. Kobayashi, K. Yoshimura, B.C. Trapnell, H. Kim, R.C. Hubbard, M.T. Brewer, R.C. Thompson, and R.G. Crystal. 1991. Expression of the secretory leukoprotease inhibitor gene in epithelial cells. J. Clin. Invest. 87:2207–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gauthier, F., U. Fryksmark, K. Ohlsson, and J.G. Bieth. 1982. Kinetics of the inhibition of leukocyte elastase by the bronchial inhibitor. Biochim. Biophys. Acta. 700:178–183. [DOI] [PubMed] [Google Scholar]
- 3.Seemuller, U., M. Arnhold, H. Fritz, K. Wiedenmann, W. Machleidt, R. Heinzel, H. Applehans, H.G. Gassen, and F. Lottspeich. 1986. The acid-stable proteinase inhibitor of human mucous secretions (HUSI-I, antileukoprotease). Complete amino acid sequence as revealed by protein and cDNA sequencing and structural homology to whey proteins and Red Sea turtle proteinase inhibitor. FEBS Lett. 199:43–48. [DOI] [PubMed] [Google Scholar]
- 4.Hiemstra, P.S., B.A. Fernie-King, J. McMichael, P.J. Lachmann, and J.M. Sallenave. 2004. Antimicrobial peptides: mediators of innate immunity as templates for the development of novel anti-infective and immune therapeutics. Curr. Pharm. Des. 10:2891–2905. [DOI] [PubMed] [Google Scholar]
- 5.Jin, F.Y., C. Nathan, D. Radzioch, and A. Ding. 1997. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell. 88:417–426. [DOI] [PubMed] [Google Scholar]
- 6.McNeely, T.B., D.C. Shugars, M. Rosendahl, C. Tucker, S.P. Eisenberg, and S.M. Wahl. 1997. Inhibition of human immunodeficiency virus type 1 infectivity by secretory leukocyte protease inhibitor occurs prior to viral reverse transcription. Blood. 90:1141–1149. [PubMed] [Google Scholar]
- 7.Lentsch, A.B., J.A. Jordan, B.J. Czermak, K.M. Diehl, E.M. Younkin, V. Sarma, and P.A. Ward. 1999. Inhibition of NF-kappaB activation and augmentation of IkappaBbeta by secretory leukocyte protease inhibitor during lung inflammation. Am. J. Pathol. 154:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taggart, C.C., C.M. Greene, N.G. McElvaney, and S.J. O'Neill. 2002. Secretory leucoprotease inhibitor prevents lipopolysaccharide-induced IkappaBalpha degradation without affecting phosphorylation or ubiquitination. J. Biol. Chem. 277:33648–33653. [DOI] [PubMed] [Google Scholar]
- 9.Greene, C.M., N.G. McElvaney, S.J. O'Neill, and C.C. Taggart. 2004. Secretory leucoprotease inhibitor impairs Toll-like receptor 2- and 4-mediated responses in monocytic cells. Infect. Immun. 72:3684–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura, A., Y. Mori, K. Hagiwara, T. Suzuki, T. Sakakibara, T. Kikuchi, T. Igarashi, M. Ebina, T. Abe, J. Miyazaki, et al. 2003. Increased susceptibility to LPS-induced endotoxin shock in secretory leukoprotease inhibitor (SLPI)-deficient mice. J. Exp. Med. 197:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang, Y., D.L. DeWitt, T.B. McNeely, S.M. Wahl, and L.M. Wahl. 1997. Secretory leukocyte protease inhibitor suppresses the production of monocyte prostaglandin H synthase-2, prostaglandin E2, and matrix metalloproteinases. J. Clin. Invest. 99:894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki, T., S. Futaki, M. Niwa, S. Tanaka, K. Ueda, and Y. Sugiura. 2002. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 277:2437–2443. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell, D.J., D.T. Kim, L. Steinman, C.G. Fathman, and J.B. Rothbard. 2000. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 56:318–325. [DOI] [PubMed] [Google Scholar]
- 14.Gupta, B., T.S. Levchenko, and V.P. Torchilin. 2005. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev. 57:637–651. [DOI] [PubMed] [Google Scholar]
- 15.Ma, G., T. Greenwell-Wild, K. Lei, W. Jin, J. Swisher, N. Hardegen, C.T. Wild, and S.M. Wahl. 2004. Secretory leukocyte protease inhibitor binds to annexin II, a cofactor for macrophage HIV-1 infection. J. Exp. Med. 200:1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olwill, S.A., H. McGlynn, W.S. Gilmore, and H.D. Alexander. 2005. Annexin II cell surface and mRNA expression in human acute myeloid leukaemia cell lines. Thromb. Res. 115:109–114. [DOI] [PubMed] [Google Scholar]
- 17.Brownstein, C., A.B. Deora, A.T. Jacovina, R. Weintraub, M. Gertler, K.M. Khan, D.J. Falcone, and K.A. Hajjar. 2004. Annexin II mediates plasminogen-dependent matrix invasion by human monocytes: enhanced expression by macrophages. Blood. 103:317–324. [DOI] [PubMed] [Google Scholar]
- 18.Bowdish, D.M., D.J. Davidson, D.P. Speert, and R.E. Hancock. 2004. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J. Immunol. 172:3758–3765. [DOI] [PubMed] [Google Scholar]
- 19.Van Wetering, S., S.P. Mannesse-Lazeroms, M.A. Van Sterkenburg, M.R. Daha, J.H. Dijkman, and P.S. Hiemstra. 1997. Effect of defensins on interleukin-8 synthesis in airway epithelial cells. Am. J. Physiol. 272:L888–L896. [DOI] [PubMed] [Google Scholar]
- 20.He, J., and P. Furmanski. 1995. Sequence specificity and transcriptional activation in the binding of lactoferrin to DNA. Nature. 373:721–724. [DOI] [PubMed] [Google Scholar]
- 21.Oh, S.M., D.H. Hahm, I.H. Kim, and S.Y. Choi. 2001. Human neutrophil lactoferrin trans-activates the matrix metalloproteinase 1 gene through stress-activated MAPK signaling modules. J. Biol. Chem. 276:42575–42579. [DOI] [PubMed] [Google Scholar]
- 22.Gao, Y., S. Lecker, M.J. Post, A.J. Hietaranta, J. Li, R. Volk, M. Li, K. Sato, A.K. Saluja, M.L. Steer, et al. 2000. Inhibition of ubiquitin-proteasome pathway-mediated I kappa B alpha degradation by a naturally occurring antibacterial peptide. J. Clin. Invest. 106:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barkett, M., D. Xue, H.R. Horvitz, and T.D. Gilmore. 1997. Phosphorylation of IkappaB-alpha inhibits its cleavage by caspase CPP32 in vitro. J. Biol. Chem. 272:29419–29422. [DOI] [PubMed] [Google Scholar]
- 24.Koch, A., M. Giembycz, R.G. Stirling, S. Lim, I. Adcock, K. Wassermann, E. Erdmann, and K.F. Chung. 2004. Effect of smoking on MAP kinase-induced modulation of IL-8 in human alveolar macrophages. Eur. Respir. J. 23:805–812. [DOI] [PubMed] [Google Scholar]
- 25.Zhang, D., R.C. Simmen, F.J. Michel, G. Zhao, D. Vale-Cruz, and F.A. Simmen. 2002. Secretory leukocyte protease inhibitor mediates proliferation of human endometrial epithelial cells by positive and negative regulation of growth-associated genes. J. Biol. Chem. 277:29999–30009. [DOI] [PubMed] [Google Scholar]
- 26.McElvaney, N.G., H. Nakamura, P. Birrer, C.A. Hebert, W.L. Wong, M. Alphonso, J.B. Baker, M.A. Catalano, and R.G. Crystal. 1992. Modulation of airway inflammation in cystic fibrosis. In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J. Clin. Invest. 90:1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim, W., W. Ma, K. Gee, S. Aucoin, D. Nandan, F. Diaz-Mitoma, M. Kozlowski, and A. Kumar. 2002. Distinct role of p38 and c-Jun N-terminal kinases in IL-10-dependent and IL-10-independent regulation of the costimulatory molecule B7.2 in lipopolysaccharide-stimulated human monocytic cells. J. Immunol. 168:1759–1769. [DOI] [PubMed] [Google Scholar]
- 28.Brightbill, H.D., S.E. Plevy, R.L. Modlin, and S.T. Smale. 2000. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J. Immunol. 164:1940–1951. [DOI] [PubMed] [Google Scholar]
- 29.Bondeson, J., K.A. Browne, F.M. Brennan, B.M. Foxwell, and M. Feldman. 1999. Selective regulation of cytokine induction by adenoviral gene transfer of IkappaBalpha into human macrophages: lipopolysaccharide-induced, but not zymosan-induced, proinflammatory cytokines are inhibited, but IL-10 is nuclear factor-kappaB independent. J. Immunol. 162:2939–2945. [PubMed] [Google Scholar]
- 30.Grobmyer, S.R., P.S. Barie, C.F. Nathan, M. Fuortes, E. Lin, S.F. Lowry, C.D. Wright, M.J. Weyant, L. Hydo, F. Reeves, et al. 2000. Secretory leukocyte protease inhibitor, an inhibitor of neutrophil activation, is elevated in serum in human sepsis and experimental endotoxemia. Crit. Care Med. 28:1276–1282. [DOI] [PubMed] [Google Scholar]
- 31.McNeely, T.B., M. Dealy, D.J. Dripps, J.M. Orenstein, S.P. Eisenberg, and S.M. Wahl. 1995. Secretory leukocyte protease inhibitor: a human saliva protein exhibiting anti-human immunodeficiency virus 1 activity in vitro. J. Clin. Invest. 96:456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taggart, C.C., G.J. Lowe, C.M. Greene, A.T. Mulgrew, S.J. O'Neill, R.L. Levine, and N.G. McElvaney. 2001. Cathepsin B, L, and S cleave and inactivate secretory leucoprotease inhibitor. J. Biol. Chem. 276:33345–33352. [DOI] [PubMed] [Google Scholar]