

Abstract
Transfer of naive antigen-specific CD4+ T cells into lymphopenic mice that express an endogenous antigen as a systemic, secreted protein results in severe autoimmunity resembling graft-versus-host disease. T cells that respond to this endogenous antigen develop into effector cells that cause the disease. Recovery from this disease is associated with the subsequent generation of FoxP3+CD25+ regulatory cells in the periphery. Both pathogenic effector cells and protective regulatory cells develop from the same antigen-specific T cell population after activation, and their generation may occur in parallel or sequentially. Interleukin (IL)-2 plays a dual role in this systemic T cell reaction. In the absence of IL-2, the acute disease is mild because of reduced T cell effector function, but a chronic and progressive disease develops late and is associated with a failure to generate FoxP3+ regulatory T (T reg) cells in the periphery. Thus, a peripheral T cell reaction to a systemic antigen goes through a phase of effector cell–mediated pathology followed by T reg cell–mediated recovery, and both require the growth factor IL-2.
Two of the fundamental properties of the adaptive immune system are unresponsiveness to self-antigens (self-tolerance) and a steady state in terms of total lymphocyte numbers (homeostasis) (1–5). Disruption of self-tolerance and homeostasis has the potential of triggering pathologic immune reactions against a variety of self-antigens. The mechanisms that prevent such immune reactions are of great interest because defining these mechanisms and learning how to control systemic reactions against self-antigens may be useful for understanding and treating many types of diseases, not only autoimmune disorders. For instance, in graft-versus-host disease (GvHD), transplanted T cells react against host minor alloantigens that have many of the characteristics of self-antigens (such as constitutive and persistent expression in the absence of inflammation) (6, 7). In this scenario, disruption of homeostasis may be a major contributor to the fatality of the disease. Additionally, immunodeficiency or lymphopenia has been linked to autoimmunity in human disease and mouse models (8–10).
The goal of our experiments was to develop a monoclonal, antigen-specific model for a pathologic reaction to an endogenous systemic antigen that is presented like a self-antigen, i.e., continuously and in the absence of inflammatory stimuli. We hypothesized that, analogous to GvHD, tolerance can be broken by lymphopenia. We were especially interested in asking whether autoreactive T cells develop into pathogenic effector cells without any overt stimulation (other than encounter with endogenous antigen), whether they also have the ability to limit their own reactions, and what the mechanisms underlying this self-limitation are. With the recent interest in regulatory T (T reg) lymphocytes as a major control mechanism for immune responses (11–13), it has become important to determine if the conditions of antigen recognition that are needed to induce T reg cells are different from those that generate other T cell subsets. Many studies of antigen-specific T reg cells have focused on the development of so-called “natural” regulatory cells in the thymus, and only recently has the occurrence of peripherally generated T reg cells resembling the thymically derived subset been described using antigen delivery via an osmotic pump (14). However, little is known about the generation or functional significance of peripherally generated T reg cells (15).
In addition, we have explored the role of the cytokine IL-2 in the generation of effector and regulatory cells because of the recent realization that this cytokine has multiple, and often opposing, functions in immune regulation and because many immune therapies, especially for cancer and graft rejection, are based on administering or blocking IL-2 (16–19). IL-2 is known to be a potent inducer of T cell proliferation and differentiation into effector cells (20, 21). On the other hand, it is also required for the development of T reg lymphocytes in the thymus (22) and may promote apoptotic death of activated T cells (23, 24), thus contributing to termination of T cell responses. In fact, the lymphoproliferative and autoimmune phenotype of KO mice in which IL-2 production or signaling is disrupted indicates that the obligatory function of this cytokine may be in maintaining self-tolerance and homeostasis (25–28). To date, the regulatory function of IL-2 has been inferred largely from the phenotype of these germline KO mice. In such experimental systems, it is difficult to precisely define the role of IL-2 in the relative development of effector and T reg cells or to examine immune responses specific for known antigens.
To address these issues, we have exploited an experimental system in which naive CD4+ T cells with a single TCR specificity are transferred into mice expressing the cognate antigen as a systemic secreted protein (29). In this model, the antigen-recognizing T cells become functionally anergic and are eliminated, restoring homeostasis. We have modified this system by transferring the T cells into antigen-expressing mice on a lymphocyte-deficient (Rag−/−) background. Under these conditions, the systemic antigen triggers massive activation of the antigen-specific T cells, leading to fulminant autoimmunity that resembles acute GvHD. However, this acute phase is followed by a phase in which the mice recover, suggesting that the autoreactive T cells not only trigger autoimmunity but also eventually control the reaction. Because the entire reaction has been converted to the response of a monoclonal T cell population to a single antigen, we can define the T cell–intrinsic mechanisms that contribute to the establishment of tolerance. Our results show that recovery from the systemic reaction is associated with the de novo generation of CD25+ T reg cells. These T reg cells express high levels of FoxP3 mRNA and protein, and they develop faster from cells that have been preactivated in vitro. The T reg cells also develop in thymectomized animals, formally demonstrating that they are generated in the periphery. Absence of IL-2 results in less severe disease early, which is associated with reduced effector function of the T cells. In striking contrast, at late time points, in the absence of IL-2 no T reg cells are generated in the periphery, and the antigen-specific effector T cells undergo persistent expansion, accumulate in tissues, and cause severe injury. Thus, the same T cell population gives rise to both effector and T reg cells on recognition of the same antigen, and IL-2 is the critical cytokine for both phases of the response. The development of these two populations may occur sequentially or in parallel. The therapeutic implications of these results are discussed.
Results
A model of systemic T cell–mediated autoimmunity
The initial goal of this study was to define the mechanisms that limit or control autoimmunity and homeostasis in a setting where the only lymphocyte population that is present is reactive with an endogenous antigen that is expressed as a systemic protein. We hypothesized that transfer of T cells that are specific for this antigen into a lymphopenic host might result in autoimmunity. This hypothesis was spurred by the clinical correlation of immunodeficiency with autoimmunity (8, 9) and the finding that some autoimmune reactions in experimental animals are associated with lymphopenia (10). It was also supported by the notion that myeloablative or lymphocyte-depleting conditioning regimens used for bone marrow transplantation alter homeostasis and promote T cell responses to ubiquitously expressed antigens, resulting in GvHD (30).
To test this idea, we crossed transgenic mice that express a secreted form of OVA systemically (sOVA Tg) on to a Rag−/− background and adoptively transferred naive, monoclonal ovalbumin (OVA)-specific DO11.10 (DO11) CD4+ T cells (from DO11 Rag−/− mice) into these recipients. Within 1–2 wk the transfer recipients show clinical signs of acute disease (ruffled fur and hunching), lose weight, and begin to die (Fig. 1, A and B). Histopathologic examination of the skin shows massive infiltration of lymphocytes in the sOVA Tg Rag−/− recipients of DO11 cells (Fig. 1 C). The cellular infiltrates are associated with edema and congested vessels in the dermis and focal epidermal necrosis, which is characteristic of an acute graft-versus-host reaction (31).
Figure 1.
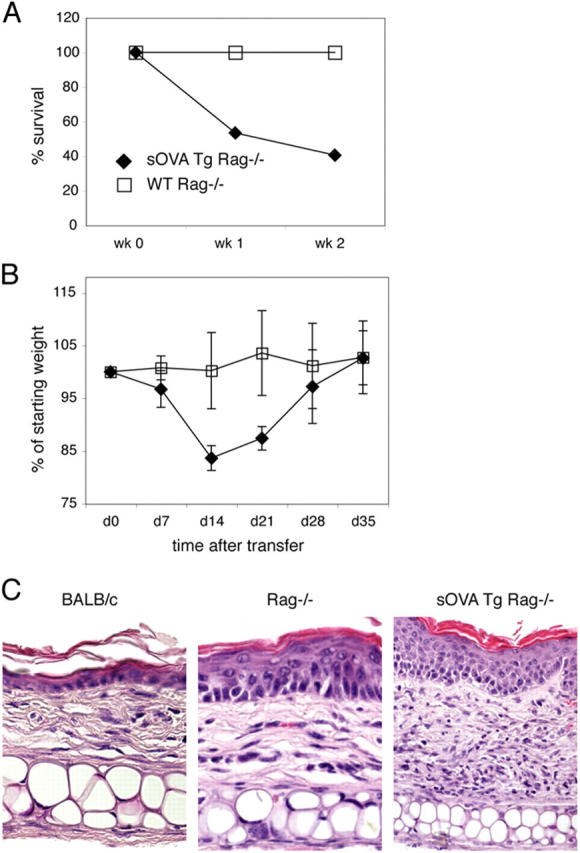
Lymphopenia promotes a systemic autoimmune reaction. 106 purified CD4+ DO11 Rag−/− T cells were transferred into sOVA Tg Rag−/− or nontransgenic Rag−/− mice on day 0. (A) Survival is plotted over time as the percentage of surviving mice from three experiments with four to seven mice per each group. (B) Body weight is plotted over time as the percentage of the starting weight before T cell transfer. Data are pooled from six to eight mice per group. In this analysis, only mice that survived were included. Values represent means ± SD. (C) Paraffin sections of the ear skin of BALB/c control, Rag−/−, and sOVA Tg Rag−/− mice 7 d after T cell transfer were stained with H&E.
One of the strengths of this experimental system is that a monoclonal T cell population reacting to a single antigen is responsible for the disease. It is therefore possible to examine these T cells and determine how their responses correlate with disease development. To do this, we assayed the recipients for the numbers and functions of the transferred DO11 T cells at different times after transfer. The controls we have used in these experiments are transfers of DO11 cells into Rag−/− recipients that do not produce OVA. In lymphopenic mice not expressing the cognate antigen, the transferred DO11 T cells undergo slow homeostatic proliferation with a fourfold increase in cell numbers between days 7 and 35 after transfer (Fig. 2, A and B), as has been previously described (32, 33). In the sOVA Tg Rag−/− recipients, the transferred DO11 cells undergo rapid proliferation and massive expansion, resulting in a >100-fold increase in cell numbers by day 7, and these numbers are maintained for up to 35 d (Fig. 2, A and B). Moreover, the T cells make large amounts of IL-2 and IFN-γ (Fig. 2, C and D). These reactions are in marked contrast to the responses seen in normal (lymphocyte-sufficient) sOVA Tg recipients, in which T cell expansion is modest (at least 10-fold less), transient (peaking at approximately day 4 and declining by days 7–10), and not accompanied by effector cytokine production, all of which are indicative of tolerance (29). Thus, the pathologic reactions and uncontrolled T cell activation in the antigen-expressing lymphopenic hosts suggest that tolerance has failed.
Figure 2.
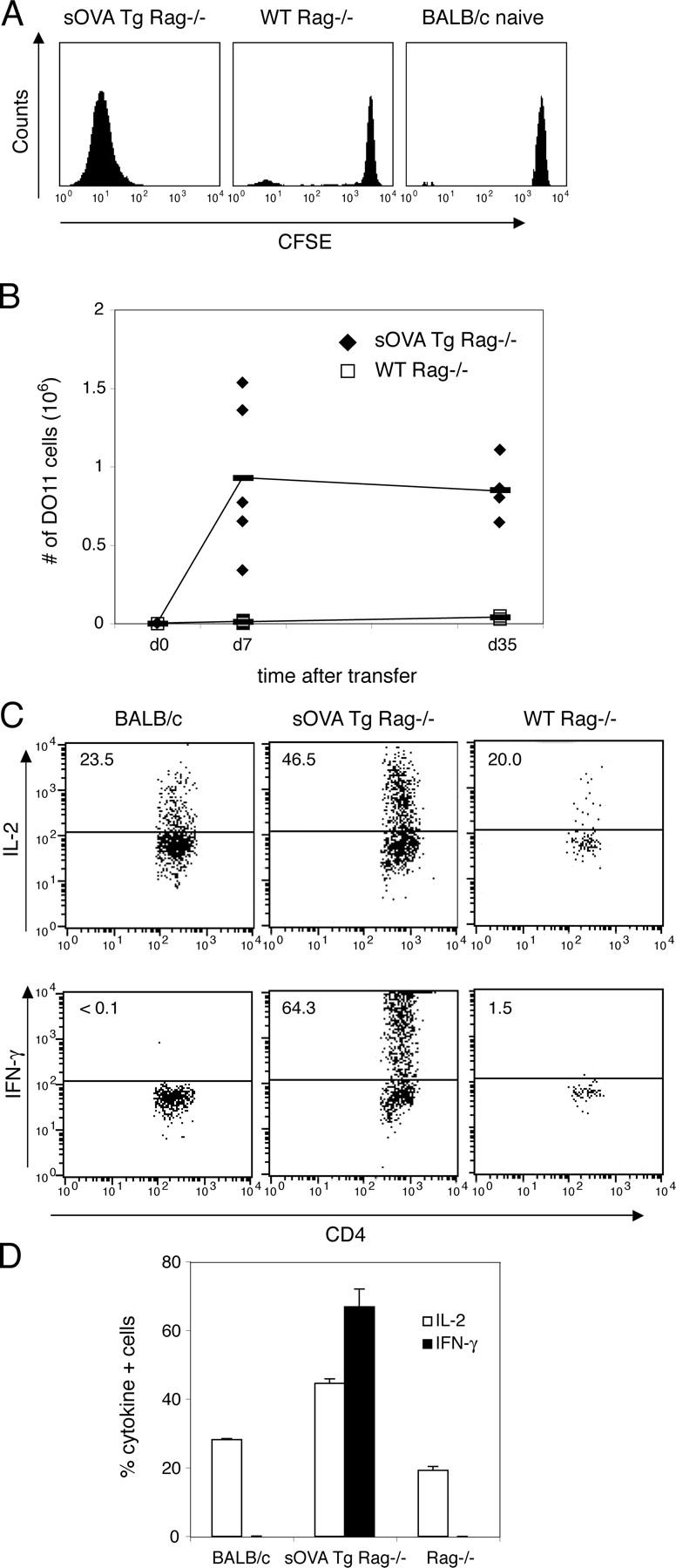
Expansion of antigen-recognizing T cells in lymphocyte-deficient recipients. 106 purified CD4+ DO11 Rag−/− cells labeled with CFSE were transferred into sOVA Tg Rag−/− mice, nontransgenic Rag−/− mice, or normal BALB/c recipients on day 0. (A) Peripheral lymph nodes were harvested 5 d after transfer, and the CFSE profile of gated KJ1-26+CD4+ cells was determined by flow cytometric analysis. (B) Peripheral lymph nodes were harvested at the indicated time points after transfer, and the total number of DO11 cells was determined by counting and a flow cytometric analysis of KJ1-26+CD4+ cells. Data are pooled from three experiments (n = 5, sOVA Tg Rag−/−; n = 9, Rag−/−). The horizontal lines indicate means. (C) Peripheral lymph node cells were harvested on day 4 after transfer, restimulated with OVA peptide and APCs, and stained for intracellular cytokines. All plots are gated on KJ1-26+CD4+ cells. Numbers refer to the percentage of cytokine-positive cells. Representative plots are shown from one experiment out of four with two mice per group. (D) Data are expressed as the percentage of cytokine-positive gated KJ1-26+CD4+ cells pooled from two mice per group from one representative experiment. Values represent means ± SD.
Role of T reg cells in recovery from disease
Despite this acute systemic disease after transfer of DO11 cells into sOVA Tg Rag−/− recipients, there is little or no additional lethality after 2–3 wk subsequent to transfer, and the surviving mice begin to gain weight and recover (Fig. 1 B). Because the DO11 cells are still present in large numbers even as the mice are recovering, we hypothesized that recovery may be because of the generation of T reg cells. Because CD25+CD4+FoxP3+ T cells are not detectable in DO11 Rag−/− animals (see Fig. 4 and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050855/DC1) and therefore could not be present in the transferred T cells, any T reg cells that develop must be generated from the transferred naive DO11 cells. As illustrated in a representative series of FACS plots in Fig. 3 A and summarized in Fig. 3 B, within 4 d after transfer most of the DO11 cells express moderate to high levels of CD25, presumably as a result of activation by the antigen. CD25 expression gradually declines over 2 wk. However, at day 30–35 (Fig. 3, A and D), a distinct new population of CD25high cells has clearly emerged, with most of these expressing high levels of CD62L (Fig. 3 D). This CD25highCD62L+ population can be detected as early as day 14 and gradually increases as a percentage of the T cells. The CD25high T cells do not produce any IL-2, IFN-γ, or IL-4, suggesting that they may be T reg cells in which cytokine production has been suppressed (Fig. 3 C).
Figure 4.

T reg cells develop more rapidly from activated than from naive DO11 cells. Purified CD4+ DO11 Rag−/− cells were stimulated on mitomycin C–treated splenocytes in the presence of 1 μg/ml OVA peptide for 4 d. 106 in vitro–activated DO11 Rag−/− or naive DO11 Rag−/− cells were transferred into sOVA Tg Rag−/− on day 0. (A) FoxP3 expression of naive and in vitro–activated DO11 Rag−/− cells before transfer. (B) Splenocytes were harvested 4, 10, and 30 d after transfer. FoxP3 expression of gated KJ1-26+CD4+ cells was measured by flow cytometric analysis. Splenocytes were restimulated with OVA peptide and APCs and stained for IFN-γ on days 4, 10, and 30 after transfer. All plots are gated on KJ1-26+CD4+ cells. Representative plots are shown from three experiments with two mice per group.
Figure 3.
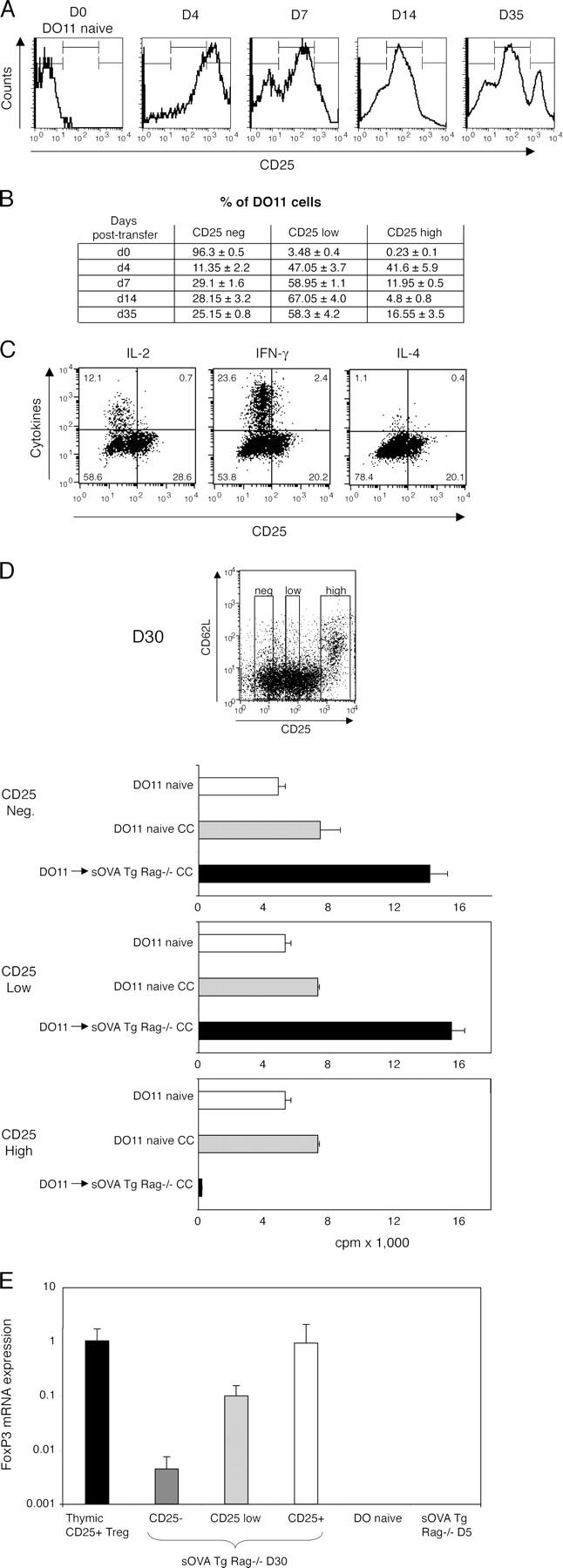
Peripheral development of effector cells and T reg cells. 106 CD4+-purified DO11 Rag−/− cells were transferred into sOVA Tg Rag−/− mice on day 0. (A) Peripheral lymph nodes were harvested at the indicated time points after transfer and analyzed for expression of KJ1-26+CD4+CD25+ cells by flow cytometry. Histograms show CD25 expression gated on KJ1-26+CD4+ cells from one representative mouse out of three experiments. (B) The table summarizes data pooled from two mice per group, based on gates shown in A. (C) Lymph node cells were harvested 30 d after transfer, restimulated with OVA peptide and APCs, and stained for intracellular cytokines. CD25 staining was done before permeabilization. All plots are gated on KJ1-26+CD4+ cells. Representative plots are shown from one experiment out of four with two mice per group. (D) Peripheral lymph node cells were harvested on day 30 after transfer and sorted for KJ1-26+CD4+ and negative (Neg.), intermediate (Low), or high (High) levels of CD25 expression as indicated in the FACS plot. The cells were co-cultured (CC) with sorted naive KJ1-26+CD4+CD25− cells from DO11 Rag−/− mice, and [3H]thymidine incorporation was assayed on day 3. (E) Lymph node and spleen cells from sOVA Tg Rag−/− mice at day 30 after transfer were sorted on KJ1-26+CD4+CD25 negative (−), low, and high (+) expression and analyzed for FoxP3 mRNA by real-time fluorogenic RT-PCR. Thymic- generated KJ1-26+CD4+CD25+ cells from DO11 × RIP-mOVA mice were included as a positive control; naive DO11 Rag−/− and KJ1-26+CD4+ cells recovered from sOVA Tg Rag−/− mice on day 5 were included as negative controls. Data are pooled from two experiments. Values in D and E represent means ± SD.
To ask if the CD25high cells have suppressive activity, we purified DO11 cells 30 d after transfer, separated them into CD25-negative, -low, and -high fractions by cell sorting, and tested the suppressive ability of each fraction in co-culture assays with naive DO11 cells. As shown in Fig. 3 D, the CD25high cells isolated 30 d after transfer have potent suppressive activity in vitro. In addition, DO11 cells recovered 4 wk after transfer into sOVA Tg Rag−/− recipients are unable to cause disease when transferred into another set of sOVA Tg Rag−/− mice (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050855/DC1). This result further supports the idea that T reg cells have developed as a result of prolonged exposure to the antigen. We have found that both CD25neg/low and CD25high cells are nonpathogenic on transfer into a second antigen-expressing recipient, presumably because even the CD25neg/low cells rapidly develop into CD25high cells on repeat encounter with the antigen (Fig. S2).
Expression of FoxP3 mRNA and protein has been demonstrated to be a functionally relevant marker for CD4+CD25+ T reg cells (34, 35). Indeed, FoxP3 mRNA expression is high in CD25high DO11 Rag−/− cells isolated from sOVA Tg Rag−/− recipients 30 d after transfer (Fig. 3 E). Furthermore, FoxP3 mRNA levels are similar to those in thymically generated CD4+CD25+ cells from DO11 × RiP-mOVA mice (29, 36). Surprisingly, FoxP3 expression can also be detected in CD25neg and CD25low cells 30 d after transfer, although it is lower than in the CD25high regulatory population. In contrast, DO11 cells that are isolated from sOVA Tg Rag−/− recipients early (i.e., on day 5 after transfer) or naive DO11 Rag−/− cells do not express detectable FoxP3 mRNA. Thus, FoxP3 expression may increase progressively after chronic antigen recognition and is highest in cells that have the functional characteristics of T reg cells.
It is theoretically possible that the adoptively transferred T cells migrate to the thymus and this is where the T reg cells are generated. To test this, we examined T reg cell generation in thymectomized antigen-expressing recipients. Because of the small thymus, it is not possible to reliably thymectomize the sOVA Tg Rag−/− mice. We therefore set up another system in which intact sOVA Tg mice were thymectomized, lethally irradiated, and transplanted with Rag−/− bone marrow, again providing lymphopenic antigen-expressing mice as recipients for transferred DO11 Rag−/− cells. These recipients undergo similar clinical deterioration with massive weight loss and subsequent recovery as do sOVA Tg Rag−/− mice (unpublished data). If DO11 cells are transferred into these sOVA Tg bone marrow chimeras, a CD25high population develops as it does in sOVA Tg Rag−/− recipients. The CD25high cells show regulatory function in vitro, thus demonstrating that the development of regulatory cells occurs in the periphery independently of thymic selection (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050855/DC1).
Kinetics of development of T reg cells from naive and activated T cells
To dissect the differentiation pathway of the T reg cells, we first examined the time it took to generate these cells without or with a cycle of prior activation. DO11 Rag−/− cells were primed for 4 d with antigen and APCs in vitro. At this time, all the T cells had cycled and up-regulated activation markers such as CD25 (unpublished data). We have previously shown that T cells primed in this way develop into effector and memory cells on transfer into BALB/c mice (37). When these activated DO11 cells are transferred into sOVA Tg Rag−/− mice, a population of CD25highFoxP3+ T reg cells develops within 4 d and the numbers of these increase progressively (Fig. 4). Transfer of naive cells does not generate CD25highFoxP3+ T reg cells until after days 10–12 (Figs. 3 and 4). Co-culture assays also show that CD25high DO11 cells recovered 10–12 d after transfer of activated cells have suppressive activity but not after transfer of naive DO11 cells (unpublished data). Consistent with these accelerated kinetics of T reg cell development from primed cells, sOVA Tg Rag−/− mice that have been transferred with in vitro–activated cells do clinically better than recipients transferred with naive cells, and in five independent experiments with a total of 12 mice per group, no mortality has been observed after transfer of in vitro–activated cells (unpublished data). Thus, T reg cells appear more rapidly if the T cells have been activated before encounter with systemic antigen.
To further define a possible relationship between effector T cells and T reg cells, we analyzed the DO11 cells for intracellular IFN-γ production as a marker of effector cells at the same time points as for FoxP3 expression. These experiments showed that IFN-γ+ cells appear after 4 d subsequent to transfer of naive or activated DO11 T cells, increase through days 10–12, and decrease by day 30. (Fig. 4 C). The finding that there is a progressive increase in T reg cells and decrease in IFN-γ–producing cells may suggest that the two populations arise independently (and even reciprocally). Consistent with this possibility, we have not found any cells that simultaneously express IFN-γ and FoxP3 protein at day 30 by two-color stains (unpublished data). However, it is also possible that the development of the T reg cell phenotype actively extinguishes the production of effector cytokines, thus creating the impression that these are nonoverlapping populations.
Role of IL-2 in the systemic T cell reaction
Having established a system in which a single antigen generated effector and regulatory cells from one monoclonal antigen-specific T cell population, we were interested in asking what stimuli might be responsible for generating these two cell types. We have focused on the role of IL-2 because of the demonstrated ability of this cytokine to both stimulate and terminate T cell responses and the importance of therapeutic approaches targeting IL-2 or its receptor. IL-2 has been implicated in the thymic generation of T reg cells, but its role in peripheral T reg cell generation is unknown (38). For these experiments, we transferred either WT DO11 cells (from DO11 Rag−/− mice) into WT sOVA Tg Rag−/− recipients or IL-2–deficient DO11 cells (from DO11 Rag−/− IL-2−/− mice) into antigen-expressing recipients that also lacked IL-2 (sOVA Tg Rag−/− IL-2−/−). Thus, in these experiments, IL-2 could be supplied either by the transferred T cells or by another nonlymphocytic cell population in the host, such as DCs (39), or it was completely absent. Importantly, by crossing the sOVA Tg IL-2−/− and the DO11 IL-2−/− mice with Rag−/− mice, neither donor nor recipient develops the autoimmune and lymphoproliferative disease that develops in conventional IL-2 KOs (25, 26). Furthermore, DO11 Rag−/− WT and DO11 Rag−/− IL-2−/− cells have a similar naive phenotype (Fig. S1), and similar cell numbers can be recovered from the thymus and peripheral lymphoid organs (unpublished data).
Inspection of the transfer recipients showed that, in the first 1–2 wk after cell transfer, the IL-2−/− recipients of IL2−/− DO11 Rag−/− cells appear healthy and there is less weight loss than when IL-2–producing DO11 Rag−/− cells are transferred into WT sOVA Tg recipients (Fig. 5 A). In addition, the clinical score and the epidermal damage in the skin at day 10 are also less in the absence of IL-2 (Fig. 5 B). Strikingly, WT mice that survive the acute reaction begin to gain weight and recover by clinical score, and by day 30 there is less dermal inflammation in the skin by histopathology (Fig. 5, A and C). In contrast, when IL-2 is absent, the weight loss is progressive (Fig. 5 A), the mice do not recover by clinical score, and by day 30 the skin is severely inflamed and the mice have almost complete alopecia (Fig. 5 C). Thus, the absence of IL-2 leads to a persistent and progressive pathologic reaction resembling chronic GvHD (40).
Figure 5.

Delayed pathology in the absence of IL-2. 106 purified CD4+ DO11 Rag−/− cells (DOWT) were transferred into sOVA Tg Rag−/− recipients or DO11 Rag−/− IL-2−/− (DOIL-2−/−) cells were transferred into sOVA Tg Rag−/− IL-2−/− mice. (A) Survival is plotted over time as the percentage of surviving mice from five experiments with a total of 19–37 mice/group. Body weight is plotted over time as the percentage of the starting weight before T cell transfer. Data are pooled from three individual experiments with 8–13 mice/group. *, P ≤ 0.05 by t test. (B and C) Paraffin sections of ear skin of sOVA Tg Rag−/− and sOVA Tg Rag−/− IL-2−/− mice 10 (B) and 30 (C) d after T cell transfer were stained with H&E. Frozen sections were stained with anti-CD4 or KJ1-26. One representative section from each group is shown. Clinical scores calculated as described in Materials and methods based on eight parameters (ear swelling, eye swelling, foot swelling, hunching, movement, ruffled fur, alopecia, and scaling) are shown from sOVA Tg Rag−/− and sOVA Rag−/−IL-2−/− mice at days 10 and 30. Values in B and C represent means ± SD.
To correlate the disease phenotype with the T cell response, we examined the status of the DO11 T cells in the lymphoid organs of the transfer recipients. By day 10 after transfer the numbers of cells recovered from mice lacking IL-2 are about twofold more than from WT mice, and this difference is even greater at day 30 (Fig. 6 A). If the transferred DO11 cells lack IL-2, fewer cells produce IFN-γ than do WT cells (WT, 0.58 × 106 vs. KO, 0.37 × 106; Fig. 6 B). However, on day 30, even though the percentage of IFN-γ–producing DO11 cells is lower in the absence of IL-2, the total numbers in lymphoid tissues are about the same as in the presence of IL-2 (WT, 0.23 × 106 vs. KO, 0.27 × 106; Fig. 6 B). We have not detected IL-4–producing DO11 cells in the absence of IL-2, suggesting that the reduced pathology early and the chronic disease late are not attributable to an imbalanced Th2 cell reaction. Thus, systemic antigen recognition in the absence of IL-2 has two effects: early in the course, the inflammatory phenotype is ameliorated, whereas late after adoptive transfer it leads to persistent pathology and greatly increased numbers of the antigen-recognizing T cells.
Figure 6.

T cell expansion is not controlled in the absence of IL-2. 106 DO11 Rag−/− cells (DOWT) were transferred into sOVA Tg Rag−/− recipients, or DO11 Rag−/− IL-2−/− (DOIL-2−/−) cells were transferred into sOVA Tg Rag−/− IL-2−/− mice. (A) Peripheral lymph node cells harvested on days 10 and 30 were gated on KJ1-26+CD4+, and the total number of DO11 cells was determined by counting and flow cytometry and normalized to the number of DO11 cells recovered from sOVA Tg Rag−/− recipients on day 10 in each experiment. Data are pooled from two experiments. (B) The cells were restimulated with OVA peptide and APCs and stained for intracellular cytokines. Each bar represents the mean percentage of cytokine-positive cells in three to six mice per group. Values in A and B represent means ± SD. (C) Peripheral lymph node cells harvested on day 30 were stained with KJ1-26, anti-CD4, anti-CD25, and Annexin V and analyzed by flow cytometry. Plots are gated on KJ1-26+CD4+CD25neg/low or CD25high cells from one representative mouse per group. Naive DO11 Rag−/− WT and DO11 Rag−/− IL-2−/− cells were included as controls.
One possible explanation for the increased cell numbers in mice transferred with IL-2−/− DO11 Rag−/− cells could be a resistance of the T cells to activation-induced cell death (23, 24). To test this hypothesis, we assayed sOVA Tg Rag−/− mice transferred with DO11 Rag−/− cells and sOVA Tg Rag−/− IL-2−/− recipients transferred with DO11 Rag−/− IL-2−/− cells for the number of apoptotic cells 30 d after transfer. As shown in Fig. 6 C, T cells in both the sOVA Tg Rag−/− and sOVA Tg Rag−/− IL-2−/− recipients have a comparable fraction of Annexin V binding cells, an early marker of apoptosis. Naive DO11 Rag−/− and DO11 Rag−/− IL-2−/− cells show a similar and very low number of apoptotic cells by this assay. Thus, the increased cell number in the absence of IL-2 cannot be explained by a reduced frequency of apoptotic cells.
Role of IL-2 in the development of T reg cells
Because peripherally generated T reg cells are likely involved in recovery from disease (Fig. 3), it follows that in the absence of IL-2 failure to generate these T cells may be responsible for the delayed, chronic reaction. To address this, we examined sOVA Tg Rag−/− IL-2−/− mice that had been transferred with IL-2−/− DO11 cells for the numbers of CD25+ T cells. As shown in Fig. 7 A, at day 30 after transfer few cells expressing CD25 can be detected but no distinct CD25high population can be found if the DO11 cells are from IL-2−/− mice. This experimental system also allows us to establish the source of the cytokine (transferred T cells or host) required for the generation of T reg cells. CD25high cells develop from WT DO11 cells regardless of whether or not the host produces IL-2, but CD25high cells do not develop if the T cells cannot produce IL-2 (Fig. 7 A). Thus, IL-2 production from cells other than lymphocytes (e.g., DCs) is not sufficient for peripheral generation of CD25+ T reg cells.
Figure 7.
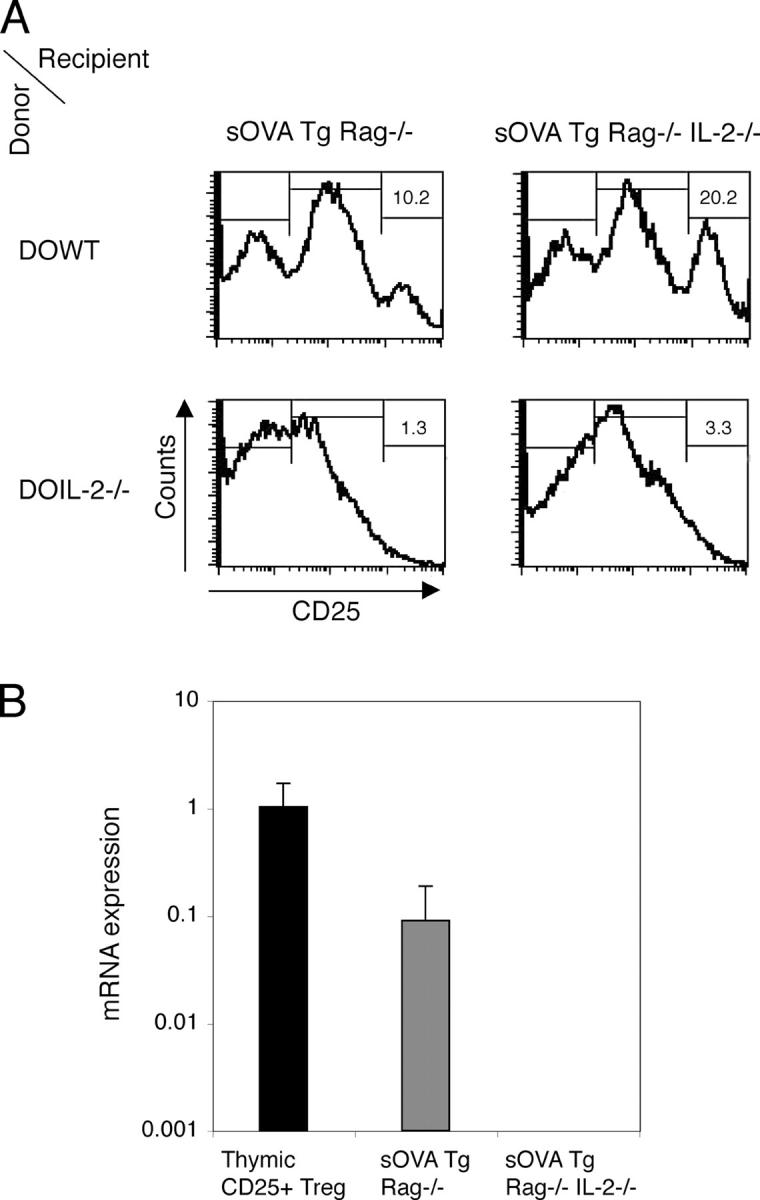
IL-2 is crucial for peripheral generation of CD25+ cells. 106 purified CD4+ DO11 Rag−/− cells (DOWT) or DO11 Rag−/− IL-2−/− (DOIL-2−/−) cells were transferred either into sOVA Tg Rag−/− recipients or into sOVA Tg Rag−/− IL-2−/− mice. (A) Peripheral lymph node cells were harvested on day 30. CD25 expression on gated KJ1-26+CD4+ cells was analyzed by flow cytometry. Numbers refer to the percentages of CD25high cells. (B) Lymph node cells of sOVA Tg Rag−/− recipients transferred with DO11 Rag−/− cells or sOVA Tg Rag−/− IL-2−/− mice transferred with DO11 Rag−/− IL-2−/− cells were sorted for KJ1-26+CD4+ cells on day 30 after transfer and analyzed for FoxP3 mRNA by real-time fluorogenic RT-PCR. Thymic-generated KJ1-26+CD4+CD25+ cells from DO11 × RIP-mOVA mice were included as a positive control. Data are pooled from two experiments. Values represent means ± SD.
IL-2 has been implicated in the generation and/or the function of thymic T reg cells (38, 41). We have shown that there are no CD25high T cells generated if the T cells are IL-2 deficient (Fig. 7 A). However, it could be possible that CD25negative T reg cells exist but are not functional. Because FoxP3 mRNA expression is an accepted marker for T reg cells, we sorted DO11 Rag−/− or DO11 Rag−/− IL-2−/− cells 30 d after transfer into sOVA Tg Rag−/− WT or sOVA Tg Rag−/− IL-2−/− recipients, respectively, and assayed the cells for FoxP3 mRNA. As shown in Fig. 7 B, FoxP3 mRNA expression is detectable only in the WT and not in the IL-2–deficient DO11 T cells, indicating that IL-2 is critical for the generation of FoxP3-positive T reg cells in the periphery.
Discussion
Lymphocytes that respond to an antigen have the potential of developing into several functionally and phenotypically different populations. CD4+ T cells can differentiate into effector cells (including Th1 and Th2 cell subsets), memory cells, and regulatory cells. A major question in immunology is how these fates are determined, and, more specifically, what is the influence of the nature of the antigenic stimulus and the conditions of stimulation. This question has been addressed in great detail for Th1/Th2 cell differentiation but is largely unanswered for regulatory cells. We were especially interested in asking whether an endogenous ubiquitously expressed antigen is capable of inducing both effector and T reg cells and whether there are distinct requirements for these cell populations. The importance of this question is that the choice between self-tolerance and autoimmunity may be determined, to a substantial extent, by the balance between effectors and regulators, and learning how to manipulate this balance has obvious therapeutic relevance.
To address this question, we have developed an experimental system in which CD4+ T cells specific for the protein OVA are exposed to OVA expressed as a secreted, systemic antigen in a lymphopenic host. In these lymphopenic antigen-expressing mice, the antigen-recognizing T cells do not become tolerant. Instead, the T cells expand massively, rapidly differentiate into IFN-γ–producing effector cells, and cause a severe immune reaction (Figs. 1 and 2). Some of the effector cells that develop in the lymphopenic recipients infiltrate the skin, where they cause severe epidermal necrosis and lesions resembling acute GvHD. In many respects, this sequence of events is similar to what one sees in GvHD when transferred T cells react against host alloantigens (6, 7). This reaction contrasts sharply with the T cell response seen in antigen-expressing intact (lymphocyte-sufficient) hosts in which the T cells become tolerant by a combination of anergy and deletion (29).
Despite the severe pathology induced by antigen recognition in lymphopenic recipients, within 2–3 weeks the surviving mice begin to recover. Recovery is associated with the development of CD25+ T cells with the phenotypic and functional properties of T reg cells. These cells are CD25high, express high levels of the FoxP3 transcription factor, and suppress T cell responses in co-cultures (Fig. 3). The appearance of these cells also correlates with an inability of the DO11 T cells to cause disease on retransfer into a new antigen-expressing recipient (Fig. S2). The role of T reg cells in controlling a variety of immune reactions is well established (11, 12, 42). It is generally accepted that the majority of CD4+CD25+ T reg cells are generated by self-antigen recognition in the thymus (15). It is known that T cells with suppressive function, called Th3 or Tr1 cells, can be induced from mature CD4+ T cells by special culture conditions in vitro (43–47), and one report has demonstrated that chronic exposure of mature CD4+ T cells to their cognate antigen delivered via an osmotic pump can induce CD4+CD25+ T reg cells in the periphery with the same properties as thymically derived T reg cells (14). We have set up a system in which T reg cells are induced by an endogenously produced antigen that has the properties of a self-antigen. The T reg cells generated in our system develop even in thymectomized mice (Fig. S3), formally demonstrating that they arise in peripheral tissues. Also, even though most of our experiments have been done with Rag−/− recipients, we obtained similar results in a model using irradiation and bone marrow reconstitution (Fig. S3), further emphasizing the relevance to bone marrow transplantation therapy and GvHD in humans, which, by nature, is a highly controlled setting.
The question remains as to what role peripherally generated T reg cells play in tolerance maintenance in normal (lymphocyte-sufficient) hosts. It is possible that under normal circumstances, thymic-derived T reg cells are sufficient to maintain self-tolerance. We have found that transfer of DO11 cells into lymphosufficient sOVA Tg mice leads to profound deletion with few anergic cells remaining (29). Under these circumstances, the transferred T cells may simply not encounter antigen for long enough to develop into T reg cells. T reg cells may only be generated in the periphery if other defects occur, such as breakdown of lymphocyte homeostasis. Also, in lymphopenic hosts the transferred T cells do not have to compete with endogenous cells for the growth factors and other stimuli that may be needed for T reg cell generation.
An important question emerges from these findings: do the T reg cells arise from effector cells or are these parallel differentiation pathways? Our experiments demonstrate that T reg cells develop more rapidly from cells that have been stimulated with antigen ex vivo than from naive T cells (Fig. 4), suggesting that T reg cells can develop from activated cells. Furthermore, T reg cells develop faster from CD25neg/low cells that have been exposed to the antigen for a month than from naive T cells (Fig. S2). By costaining for intracellular FoxP3 and IFN-γ, we have found no overlap between FoxP3+ and IFN-γ+ populations. These data allow two possible interpretations: (a) T reg and effector cells develop in parallel and independently from activated cells that have not yet undergone effector differentiation or (b) T reg cells develop from effector cells, but IFN-γ production is extinguished when the cells acquire the T reg cell phenotype. Future experiments are directed at distinguishing these possibilities. Importantly, the development of pathogenic effectors and protective regulatory cells occurs in the same animal, and, therefore, in the presence of the same innate immune responses, costimulators, or other overt stimuli. It is conceivable that different populations of APCs induce effectors and regulatory cells from the naive T lymphocytes, and it is also possible that the effector T cells themselves alter antigen presentation by interacting with APCs, which subsequently leads to preferential generation of T reg cells. Identifying the stimuli that determine the balance between effector and T reg cells may provide clues to the control of a variety of T cell–dependent immune responses.
IL-2 has multiple and often opposing functions in immune regulation (48, 49). Although IL-2 is a powerful T cell growth factor in vitro, the analysis of KO mice lacking IL-2 or IL-2 receptors has suggested that the major obligatory function of IL-2 may be to control immune responses (25–28). Studies with KO mice indicate that IL-2 is required for thymic generation of T reg cells (22), but it may also play a role in the function of T reg cells (41, 50). Thus, IL-2 is clearly important for controlling immune responses; it is, however, unclear from the KO mouse data whether IL-2 also plays an obligatory role in stimulating immune responses or if its absence can be compensated for by other cytokines in vivo. Our experiments formally prove the dual functions of IL-2 in a model of antigen-specific systemic immune disease. The absence of IL-2 leads to a mild acute disease early, with less pathology and no lethality (Fig. 5). In the absence of IL-2, DO11 cells produce less IFN-γ (Fig. 6 B), and the total number of IFN-γ–producing cells is also reduced. These data suggest that the worse clinical disease in the WT setting early is mediated by cytokines that may include IFN-γ. It is conceivable that IL-2 in the WT recipients also promotes the production of other proinflammatory Th1 cytokines such as TNF-α, or IL-2 may activate other cell populations such as NK cells that may contribute to the phenotype.
In contrast to the early amelioration of disease, the absence of IL-2 leads to a failure to recover from the disease and the development of a chronic reaction (Fig. 5). This failure to recover seems to be dependent on the massive infiltration of lymphocytes in the periphery, as shown by skin histology and clinical score (Fig. 5 C), and is associated with a failure to generate CD25+ T cells (Fig. 7). Our results demonstrate that IL-2 is required for T reg cell generation in the periphery. The lack of FoxP3-expressing cells in the absence of IL-2 indicates that T reg cells do not develop in the absence of IL-2, rather than being present but nonfunctional. This is in contrast to a previous study that showed normal FoxP3 expression in thymocytes of IL-2–deficient mice (51). The absolute dependence on IL-2 in our system could therefore be one of the differences between thymically and peripherally generated T reg cells. Our experiments also show that T cells, not nonlymphocyte populations such as DCs, must produce the IL-2 in order to develop into T reg cells (Fig. 7). When the development of T reg cells fails because of an inability to produce IL-2, the antigen-stimulated T cells continue to expand (Fig. 6). However, we cannot differentiate whether the IL-2 has to be produced by the T cells developing into T reg cells in an autocrine fashion or whether paracrine IL-2 that could be provided by CD25low effector cells is sufficient for T reg cell development, as has been suggested by others (51, 52).
The analysis of systemic immune reactions, as in autoimmunity and GvHD, are important for the development of clinical therapies for these immunologic diseases. Among the many treatments that have been tried in such diseases, one targeted therapy that has received some attention recently is cytokine antagonism, particularly blocking the IL-2/IL-2R pathway (18, 19, 53). Our results clearly raise caution about the usefulness and timing of IL-2 blockade for controlling pathologic immune reactions. It may be that an acute blockade of IL-2 action will result in reduced T cell expansion and reduced development of effector cells, which may ameliorate pathologic immune reactions. This is consistent with several clinical studies in which blockade of the IL-2/IL-2R pathway leads to reduced incidence of acute GvHD (54–56). However, prolonged inhibition of IL-2 may promote late, uncontrolled immune responses and predisposition to persistent chronic GvHD. In fact, a recent report has also suggested that IL-2R blockade in conjunction with corticosteroids after bone marrow transplantation leads to increased relapse- and nonrelapse-related mortality compared with corticosteroids alone (57). Our data suggest that IL-2R blockade at late time points after bone marrow transplantation, in particular using myeloablative or T cell–depleting regimens, may have detrimental effects, because this strategy will also inhibit the subsequent development of regulatory cells and may therefore reduce the potential of endogenous control and restoration of homeostasis. The idea that IL-2 antagonism may be beneficial early and harmful late is applicable to other pathologic immune responses, as in graft rejection and even autoimmune diseases. The timing of such treatment may, therefore, be the key to its successful clinical application.
MATERIALS AND METHODS
Mice
All experimental mice were used at 6–12 wk of age. All mice were age and sex matched ±2 wk. BALB/c mice were purchased from Charles River Laboratories. Transgenic mice expressing the DO11.10 TCR (DO11), specific for the chicken OVA peptide (OVA323-339) in the context of the MHC class II molecule I-Ad, were obtained from K. Murphy (Washington University, St. Louis, MO). All experiments were performed using DO11 cells that had been crossed either onto a Rag1−/− or Rag2−/− background. For some experiments, DO11 Rag2−/− mice were bred onto an IL-2−/− background. DO11 × Rip-mOVA mice have been described previously (36). sOVA Tg on a BALB/c background, expressing a soluble form of OVA in the serum under control of the Metallothionein promoter I, and PCR typing for OVA expression have been described previously (unpublished data) (29). sOVA Tg mice were bred onto a Rag2−/− background or onto a Rag2−/− IL-2−/− background. Animals that died or had to be killed were excluded from all analyses except survival curves. Statistical analysis comparing weight between groups was performed using two-tailed Student's t test, assuming equal variances.
Clinical scoring was done using the following parameters: ear swelling, as measured by microcaliper in millimeters (<0.25 = 0, <0.3 = 0.25, <0.35 = 0.5, <0.4 = 0.75, <0.45 = 1, <0.5 = 1.25, and >0.5 = 1.5 points, respectively); eye swelling (no, moderate, and severe = 0, 0.5, and 1 points, respectively); foot swelling (no and yes = 0 and 1 points, respectively); ruffled fur (no and yes = 0 and 1 points, respectively); hunching (no, moderate, and severe = 0, 0.5, and 1 points, respectively); movement (normal, moderately decreased, and severely decreased = 0, 0.5, and 1 points, respectively); alopecia (<1 cm = 1, <2 cm = 2, and >2 cm = 3 points, respectively); and scaling (0.3 points per lesion on tail, paws, and ears).
In some experiments, the thymus of sOVA Tg mice was removed by suction, and the mice were lethally irradiated (900 rad) and transplanted with Rag−/− bone marrow. Radioresistant CD4+ cells were depleted with GK1.5 antibody before adoptive transfer of DO11 Rag−/− T cells.
All mice were bred and maintained in our pathogen-free facility in accordance with the guidelines of the Laboratory Animal Resource Center of the University of California, San Francisco. All experiments were conducted with the approval of the Committee on Animal Research of the University of California, San Francisco.
Antibodies and flow cytometry
Lymph node cells and splenocytes were stained with the clonotypic antibodies KJ1-26 (Caltag Laboratories), anti-CD4 (GK1.5, H129.19, and RM4-5), anti-CD25 (PC61 and 7AD), anti-CD62L (MEL-14), Annexin V, and FoxP3 (eBioscience). All antibodies were obtained from BD Biosciences unless otherwise stated. Antibodies were used as FITC, PE, PE–Cy7, PE–Texas red, allophycocyanin, or peridin chlorophyll protein conjugates. Fc-block (anti-CD16/CD32) was added before staining. Flow cytometric analyses were done on a FACSCalibur with Cellquest Software (both from Becton Dickinson). Percentages were rounded up after the first decimal place. Cells were sorted with a cell sorter (MoFlo; DakoCytomation). For intracellular cytokine staining, DO11 T cells recovered from peripheral lymph nodes or spleens of transfer recipients were restimulated on mitomycin C–treated BALB/c splenocytes for 14 h in the presence of 1 μg/ml OVA peptide. 10 μg/ml Brefeldin A (Epicentre) was added for the final 2 h of stimulation. Cells were stained for the intracellular cytokines IL-2, IFN-γ, and IL-4, with appropriate isotype controls, and were analyzed by flow cytometry.
Cell preparations, purifications, and adoptive transfer
CD4+ cells for adoptive transfer were purified from spleens and lymph nodes using Dynabeads according to the manufacturer's protocol (Dynal). CD4+ purified DO11 Rag−/− or DO11 Rag−/− IL-2−/− cells were adoptively transferred into sOVA Tg Rag−/− or sOVA Tg Rag−/− IL-2−/− recipients by tail vein injection. For in vitro activation, CD4+ cells were labeled with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen) at 10 × 106 cells/ml for 10 min at 37°C.
In vitro proliferation and suppressor assays
For co-culture assays, 5,000 sorted KJ1-26+CD4+CD25− T cells from DO11 Rag−/− mice were used as responders and stimulated on 25,000 APCs with 1 μg/ml OVA in 200 μl of RPMI 1640 with 10% FCS in 96-well plates (Costar). KJ1-26+CD4+ cells from sOVA Tg Rag−/− recipients that were harvested at the time points indicated in the figures and sorted on the basis of high, intermediate, or no CD25 expression were used as suppressors. The ratio of responder/suppressor cells remained constant at 1:1. 1 μCi/well [3H]thymidine was added during the final 16 h of culture, and incorporation was measured by scintillation counting at the time points indicated in the figures.
For in vitro activation, 0.25 × 106 CD4+-purified and CFSE-labeled DO11 Rag−/− cells were stimulated on 2.5 × 106 mitomycin C–treated splenocytes in the presence of 1 μg/ml OVA peptide for 4 d. Cells were ficolled and rested overnight in 1 ng/ml IL-2 before injection.
Histology and immunohistochemistry
Tissues were fixed in 10% neutral buffered formalin and embedded in paraffin. 5-μm sections were cut and stained with hematoxylin and eosin (H&E). For immunohistochemistry, tissues were immersed in OCT (TissueTek), flash frozen, cut into 5-μm sections, and stained with rat anti-CD4–Biotin (GK1.5; BD Biosciences) or biotinylated KJ1-26 and subsequently stained with streptavidin–horseradish peroxidase (BD Biosciences). Visualization was done with 3,3′-diaminobenzidine (Sigma-Aldrich).
Real-time RT-PCR
Quantitative RT-PCR was performed using real-time fluorogenic 5′-nuclease PCR on a sequence detection system (ABI Prism 7700; PE Biosystems) according to the manufacturer's instructions (TaqMan; Perkin Elmer). Total RNA was isolated using the Absolutely RNA RT-PCR Miniprep Kit (Stratagene). To avoid contamination with DNA, samples were treated with DNase (Ambion) before amplification and reverse transcribed using Superscript II Kit for RT-PCR (Invitrogen). Primer and probe sequences for FoxP3 and HPRT were used as published (58, 59). FoxP3 transcript expression was normalized to HPRT abundance.
Online supplemental material
Fig. S1 demonstrates that DO11 Rag−/− WT and DO11 Rag−/− IL-2−/− lymphocytes have a naive phenotype by staining for CD25, CD69, and CD62L expression. Fig. S2 shows that CD25neg/low DO11 T cells recovered 30 d after transfer into sOVA Tg Rag−/− do not cause disease and rapidly develop a population of CD25high cells on transfer into a new sOVA Tg Rag−/− host. Fig. S3 demonstrates the development of T reg cells in thymectomized sOVA Tg WT mice that have been lethally irradiated and transplanted with Rag−/− bone marrow before transfer of DO11 Rag−/− cells. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050855/DC1).
Acknowledgments
We are indebted to S. Jiang and C. McArthur for expert cell sorting and C. Benitez for mouse typing. We thank Dr. H. Sanchez for assistance with microscopy and digital photography. We thank the members of the Abbas and Bluestone labs for helpful discussions and Dr. M. Krummel for critical comments on the manuscript. We thank Drs. Qizhi Tang and Helene Bour-Jordan for assistance with thymectomy.
This work was supported by grants PO1 AI35297 and RO1 AI64677 from the National Institutes of Health (to A.K. Abbas) and by fellowships KN 533/1-1 and LO 808/1-1 from the Deutsche Forschungsgemeinschaft (to B. Knoechel and J. Lohr, respectively).
The authors declare that they have no competing financial interests.
Abbreviations used: CFSE, carboxyfluorescein diacetate succinimidyl ester; GvHD, graft-versus-host disease; H&E, hematoxylin and eosin.
B. Knoechel and J. Lohr contributed equally to this work.
References
- 1.Tanchot, C., D.L. Barber, L. Chiodetti, and R.H. Schwartz. 2001. Adaptive tolerance of CD4+ T cells in vivo: multiple thresholds in response to a constant level of antigen presentation. J. Immunol. 167:2030–2039. [DOI] [PubMed] [Google Scholar]
- 2.Jameson, S.C. 2002. Maintaining the norm: T-cell homeostasis. Nat. Rev. Immunol. 2:547–556. [DOI] [PubMed] [Google Scholar]
- 3.Walker, L.S., and A.K. Abbas. 2002. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2:11–19. [DOI] [PubMed] [Google Scholar]
- 4.Grossman, Z., B. Min, M. Meier-Schellersheim, and W.E. Paul. 2004. Concomitant regulation of T-cell activation and homeostasis. Nat. Rev. Immunol. 4:387–395. [DOI] [PubMed] [Google Scholar]
- 5.Goldrath, A.W. 2002. Maintaining the status quo: T-cell homeostasis. Microbes Infect. 4:539–545. [DOI] [PubMed] [Google Scholar]
- 6.Deeg, H.J., and R. Storb. 1984. Graft-versus-host disease: pathophysiological and clinical aspects. Annu. Rev. Med. 35:11–24. [DOI] [PubMed] [Google Scholar]
- 7.Klingebiel, T., and P.G. Schlegel. 1998. GVHD: overview on pathophysiology, incidence, clinical and biological features. Bone Marrow Transplant. 21:S45–S49. [PubMed] [Google Scholar]
- 8.Arkwright, P.D., M. Abinun, and A.J. Cant. 2002. Autoimmunity in human primary immunodeficiency diseases. Blood. 99:2694–2702. [DOI] [PubMed] [Google Scholar]
- 9.Etzioni, A. 2003. Immune deficiency and autoimmunity. Autoimmun. Rev. 2:364–369. [DOI] [PubMed] [Google Scholar]
- 10.King, C., A. Ilic, K. Koelsch, and N. Sarvetnick. 2004. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 117:265–277. [DOI] [PubMed] [Google Scholar]
- 11.Maloy, K.J., and F. Powrie. 2001. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2:816–822. [DOI] [PubMed] [Google Scholar]
- 12.Sakaguchi, S. 2004. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22:531–562. [DOI] [PubMed] [Google Scholar]
- 13.Shevach, E.M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 14.Apostolou, I., and H. von Boehmer. 2004. In vivo instruction of suppressor commitment in naive T cells. J. Exp. Med. 199:1401–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bluestone, J.A., and A.K. Abbas. 2003. Natural versus adaptive regulatory T cells. Nat. Rev. Immunol. 3:253–257. [DOI] [PubMed] [Google Scholar]
- 16.Rosenberg, S.A., M.T. Lotze, L.M. Muul, S. Leitman, A.E. Chang, S.E. Ettinghausen, Y.L. Matory, J.M. Skibber, E. Shiloni, J.T. Vetto, et al. 1985. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 313:1485–1492. [DOI] [PubMed] [Google Scholar]
- 17.Kovacs, J.A., S. Vogel, J.M. Albert, J. Falloon, R.T. Davey Jr., R.E. Walker, M.A. Polis, K. Spooner, J.A. Metcalf, M. Baseler, et al. 1996. Controlled trial of interleukin-2 infusions in patients infected with the human immunodeficiency virus. N. Engl. J. Med. 335:1350–1356. [DOI] [PubMed] [Google Scholar]
- 18.Bruner, R.J., and S.S. Farag. 2003. Monoclonal antibodies for the prevention and treatment of graft-versus-host disease. Semin. Oncol. 30:509–519. [DOI] [PubMed] [Google Scholar]
- 19.Jacobsohn, D.A., and G.B. Vogelsang. 2004. Anti-cytokine therapy for the treatment of graft-versus-host disease. Curr. Pharm. Des. 10:1195–1205. [DOI] [PubMed] [Google Scholar]
- 20.Gillis, S., and K.A. Smith. 1977. Long term culture of tumour-specific cytotoxic T cells. Nature. 268:154–156. [DOI] [PubMed] [Google Scholar]
- 21.Cousens, L.P., J.S. Orange, and C.A. Biron. 1995. Endogenous IL-2 contributes to T cell expansion and IFN-gamma production during lymphocytic choriomeningitis virus infection. J. Immunol. 155:5690–5699. [PubMed] [Google Scholar]
- 22.Malek, T.R., A. Yu, V. Vincek, P. Scibelli, and L. Kong. 2002. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 17:167–178. [DOI] [PubMed] [Google Scholar]
- 23.Lenardo, M.J. 1991. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 353:858–861. [DOI] [PubMed] [Google Scholar]
- 24.Refaeli, Y., L. Van Parijs, C.A. London, J. Tschopp, and A.K. Abbas. 1998. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 8:615–623. [DOI] [PubMed] [Google Scholar]
- 25.Sadlack, B., H. Merz, H. Schorle, A. Schimpl, A.C. Feller, and I. Horak. 1993. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 75:253–261. [DOI] [PubMed] [Google Scholar]
- 26.Kundig, T.M., H. Schorle, M.F. Bachmann, H. Hengartner, R.M. Zinkernagel, and I. Horak. 1993. Immune responses in interleukin-2-deficient mice. Science. 262:1059–1061. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki, H., T.M. Kundig, C. Furlonger, A. Wakeham, E. Timms, T. Matsuyama, R. Schmits, J.J. Simard, P.S. Ohashi, H. Griesser, et al. 1995. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 268:1472–1476. [DOI] [PubMed] [Google Scholar]
- 28.Willerford, D.M., J. Chen, J.A. Ferry, L. Davidson, A. Ma, and F.W. Alt. 1995. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 3:521–530. [DOI] [PubMed] [Google Scholar]
- 29.Lohr, J., B. Knoechel, E.C. Kahn, and A.K. Abbas. 2004. Role of B7 in T cell tolerance. J. Immunol. 173:5028–5035. [DOI] [PubMed] [Google Scholar]
- 30.Ferrara, J.L., and H.J. Deeg. 1991. Graft-versus-host disease. N. Engl. J. Med. 324:667–674. [DOI] [PubMed] [Google Scholar]
- 31.Reddy, P. 2003. Pathophysiology of acute graft-versus-host disease. Hematol. Oncol. 21:149–161. [DOI] [PubMed] [Google Scholar]
- 32.Prlic, M., B.R. Blazar, A. Khoruts, T. Zell, and S.C. Jameson. 2001. Homeostatic expansion occurs independently of costimulatory signals. J. Immunol. 167:5664–5668. [DOI] [PubMed] [Google Scholar]
- 33.Gudmundsdottir, H., and L.A. Turka. 2001. A closer look at homeostatic proliferation of CD4+ T cells: costimulatory requirements and role in memory formation. J. Immunol. 167:3699–3707. [DOI] [PubMed] [Google Scholar]
- 34.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 35.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 36.Walker, L.S., A. Chodos, M. Eggena, H. Dooms, and A.K. Abbas. 2003. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J. Exp. Med. 198:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dooms, H., E. Kahn, B. Knoechel, and A.K. Abbas. 2004. IL-2 induces a competitive survival advantage in T lymphocytes. J. Immunol. 172:5973–5979. [DOI] [PubMed] [Google Scholar]
- 38.Malek, T.R., and A.L. Bayer. 2004. Tolerance, not immunity, crucially depends on IL-2. Nat. Rev. Immunol. 4:665–674. [DOI] [PubMed] [Google Scholar]
- 39.Granucci, F., C. Vizzardelli, N. Pavelka, S. Feau, M. Persico, E. Virzi, M. Rescigno, G. Moro, and P. Ricciardi-Castagnoli. 2001. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat. Immunol. 2:882–888. [DOI] [PubMed] [Google Scholar]
- 40.Anderson, B.E., J.M. McNiff, C. Matte, I. Athanasiadis, W.D. Shlomchik, and M.J. Shlomchik. 2004. Recipient CD4+ T cells that survive irradiation regulate chronic graft-versus-host disease. Blood. 104:1565–1573. [DOI] [PubMed] [Google Scholar]
- 41.Furtado, G.C., M.A. Curotto de Lafaille, N. Kutchukhidze, and J.J. Lafaille. 2002. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J. Exp. Med. 196:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McHugh, R.S., and E.M. Shevach. 2002. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J. Immunol. 168:5979–5983. [DOI] [PubMed] [Google Scholar]
- 43.Groux, H., A. O'Garra, M. Bigler, M. Rouleau, S. Antonenko, J.E. de Vries, and M.G. Roncarolo. 1997. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 389:737–742. [DOI] [PubMed] [Google Scholar]
- 44.Weiner, H.L. 2001. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol. Rev. 182:207–214. [DOI] [PubMed] [Google Scholar]
- 45.Bacchetta, R., C. Sartirana, M.K. Levings, C. Bordignon, S. Narula, and M.G. Roncarolo. 2002. Growth and expansion of human T regulatory type 1 cells are independent from TCR activation but require exogenous cytokines. Eur. J. Immunol. 32:2237–2245. [DOI] [PubMed] [Google Scholar]
- 46.Barrat, F.J., D.J. Cua, A. Boonstra, D.F. Richards, C. Crain, H.F. Savelkoul, R. de Waal-Malefyt, R.L. Coffman, C.M. Hawrylowicz, and A. O'Garra. 2002. In vitro generation of interleukin 10–producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)– and Th2-inducing cytokines. J. Exp. Med. 195:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen, W., W. Jin, N. Hardegen, K.J. Lei, L. Li, N. Marinos, G. McGrady, and S.M. Wahl. 2003. Conversion of peripheral CD4+ CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 198:1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blattman, J.N., J.M. Grayson, E.J. Wherry, S.M. Kaech, K.A. Smith, and R. Ahmed. 2003. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 9:540–547. [DOI] [PubMed] [Google Scholar]
- 49.Nelson, B.H. 2002. Interleukin-2 signaling and the maintenance of self-tolerance. Curr. Dir. Autoimmun. 5:92–112. [DOI] [PubMed] [Google Scholar]
- 50.Almeida, A.R., N. Legrand, M. Papiernik, and A.A. Freitas. 2002. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 169:4850–4860. [DOI] [PubMed] [Google Scholar]
- 51.Curotto de Lafaille, M.A., A.C. Lino, N. Kutchukhidze, and J.J. Lafaille. 2004. CD25− T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J. Immunol. 173:7259–7268. [DOI] [PubMed] [Google Scholar]
- 52.Klein, L., K. Khazaie, and H. von Boehmer. 2003. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc. Natl. Acad. Sci. USA. 100:8886–8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nussenblatt, R.B., D.J. Thompson, Z. Li, J.S. Peterson, R.R. Robinson, R.S. Shames, S. Nagarajan, M.T. Tang, M. Mailman, G. Velez, et al. 2003. Humanized anti-interleukin-2 (IL-2) receptor alpha therapy: long-term results in uveitis patients and preliminary safety and activity data for establishing parameters for subcutaneous administration. J. Autoimmun. 21:283–293. [DOI] [PubMed] [Google Scholar]
- 54.Belanger, C., H. Esperou-Bourdeau, P. Bordigoni, J.P. Jouet, G. Souillet, N. Milpied, X. Troussard, M. Kuentz, P. Herve, J. Reiffers, et al. 1993. Use of an anti-interleukin-2 receptor monoclonal antibody for GVHD prophylaxis in unrelated donor BMT. Bone Marrow Transplant. 11:293–297. [PubMed] [Google Scholar]
- 55.Anasetti, C., J.A. Hansen, T.A. Waldmann, F.R. Appelbaum, J. Davis, H.J. Deeg, K. Doney, P.J. Martin, R. Nash, R. Storb, et al. 1994. Treatment of acute graft-versus-host disease with humanized anti-Tac: an antibody that binds to the interleukin-2 receptor. Blood. 84:1320–1327. [PubMed] [Google Scholar]
- 56.Przepiorka, D., N.A. Kernan, C. Ippoliti, E.B. Papadopoulos, S. Giralt, I. Khouri, J.G. Lu, J. Gajewski, A. Durett, K. Cleary, et al. 2000. Daclizumab, a humanized anti-interleukin-2 receptor alpha chain antibody, for treatment of acute graft-versus-host disease. Blood. 95:83–89. [PubMed] [Google Scholar]
- 57.Lee, S.J., D. Zahrieh, E. Agura, M.L. MacMillan, R.T. Maziarz, P.L. McCarthy Jr., V.T. Ho, C. Cutler, E.P. Alyea, J.H. Antin, and R.J. Soiffer. 2004. Effect of up-front daclizumab when combined with steroids for the treatment of acute graft-versus-host disease: results of a randomized trial. Blood. 104:1559–1564. [DOI] [PubMed] [Google Scholar]
- 58.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 59.Grogan, J.L., M. Mohrs, B. Harmon, D.A. Lacy, J.W. Sedat, and R.M. Locksley. 2001. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity. 14:205–215. [DOI] [PubMed] [Google Scholar]