

Abstract
Systemic autoimmune disease in humans and mice is characterized by loss of immunologic tolerance to a restricted set of self-nuclear antigens. Autoantigens, such as double-stranded (ds) DNA and the RNA-containing Smith antigen (Sm), may be selectively targeted in systemic lupus erythematosus because of their ability to activate a putative common receptor. Toll-like receptor 9 (TLR9), a receptor for CpG DNA, has been implicated in the activation of autoreactive B cells in vitro, but its role in promoting autoantibody production and disease in vivo has not been determined. We show that in TLR9-deficient lupus-prone mice, the generation of anti-dsDNA and antichromatin autoantibodies is specifically inhibited. Other autoantibodies, such as anti-Sm, are maintained and even increased in TLR9-deficient mice. In contrast, ablation of TLR3, a receptor for dsRNA, did not inhibit the formation of autoantibodies to either RNA- or DNA-containing antigens. Surprisingly, we found that despite the lack of anti-dsDNA autoantibodies in TLR9-deficient mice, there was no effect on the development of clinical autoimmune disease or nephritis. These results demonstrate a specific requirement for TLR9 in autoantibody formation in vivo and indicate a critical role for innate immune activation in autoimmunity.
Systemic lupus erythematosus (SLE) is the prototypical human autoimmune disease. Although the underlying causes of SLE remain unknown, the characteristic loss of immunologic tolerance to a restricted set of self-nuclear antigens is a common and defining feature of disease (1–3). Autoantibodies to macromolecular complexes of protein and nucleic acid, such as chromatin and small nuclear ribonucleoproteins (snRNPs), are predominant in SLE patients; antibodies directed against the individual components of double-stranded (ds) DNA, histones, and the several Smith antigen (Sm) polypeptides can also be found in many lupus patients (4–6). Because the ubiquitous autoantigens targeted in SLE all contain some form of nucleic acid, it is possible that the canonical antinuclear antibodies of lupus arise because these autoantigens can stimulate invariant receptors that recognize conserved nucleic acid determinants (3).
Toll-like receptors (TLRs) are a class of germline-encoded receptors that can be activated by pathogen-associated molecular patterns. They are essential for the generation of adaptive immune responses against a wide variety of microbial components (7, 8). Recent evidence, however, indicates that certain TLRs may also be activated by nonmicrobial endogenous ligands. TLR9, a receptor for hypomethylated CpG DNA motifs (9), is expressed by B cells in humans (10) and mice (9) and has been implicated in the breakdown of immunologic tolerance to self-nucleic acids in SLE. In vitro, rheumatoid factor B cells proliferate in the presence of chromatin-containing IgG immune complexes; anti-dsDNA B cells similarly respond to free chromatin. Both of these responses require signaling through TLR9 (11–13). TLR3 is a receptor for dsRNA and is thought to play a role in the immune response to RNA-containing viruses (14). Although TLR3 has not been directly linked to autoimmunity, there is evidence that mRNA released from necrotic cells can activate signaling pathways downstream of TLR3 (15). Moreover, the RNA-containing Sm and U1-snRNP autoantigens contain stemloop and double-stranded structures and may therefore represent endogenous TLR3 ligands (16). Such activation of TLR9 and TLR3 by the nuclear remnants of dying cells may focus the autoimmune response on DNA and RNA antigens in lupus.
Despite several lines of intriguing in vitro data, there is as of yet no direct in vivo evidence for a TLR-mediated mechanism of autoreactive B cell activation in SLE. Furthermore, the relevance of TLR activation to autoimmune pathogenesis and end organ disease has not been determined. In this report, we have used the MRL/Mplpr/lpr murine lupus model to investigate the requirements for TLR9 and TLR3 in autoantibody production and clinical autoimmune disease. We find that TLR9, but not TLR3, plays a critical role in determining autoantibody specificity, as TLR9-deficient mice failed to generate anti-DNA antibodies. However, the lack of TLR9 and resultant block of anti-DNA antibodies did not inhibit clinical disease, indicating that multiple mechanisms, perhaps mediated by other TLRs, contribute to SLE pathogenesis.
Results
ANA profiles in TLR9- and TLR3-deficient mice
The inbred MRL/Mp mouse strain develops a lupus-like syndrome marked by characteristic autoantibodies, dermatitis, nephritis, and early mortality, which are all accelerated in the presence of the Faslpr/lpr mutation (17). To investigate the role of TLR9 in autoimmune disease, we generated lupus-prone TLR9-deficient (TLR9−/−) mice by making F2 crosses of TLR9−/− mice and Fas-deficient MRL/Mplpr/lpr mice. We selected those TLR9−/− × MRL/Mplpr/lpr F2 littermates that were homozygous for Fas deficiency and either wild-type TLR9 (TLR9+/+; n = 19) or TLR9−/− (n = 16) mice. The heterogeneous genetic composition of the F2 generation was controlled with the use of large cohorts of littermate controls such that background genes were evenly divided between the two groups. Furthermore, the development of autoimmunity in Fas-deficient mice has been documented on multiple genetic backgrounds (18). Similar breeding strategies have been used previously to study the effects of single genes on autoimmune disease (19, 20).
Because the fluorescent antinuclear antibody (ANA) assay is the most sensitive detection method for antibodies to a variety of nuclear components in their native antigenic form (21, 22), we used it as an initial measure of autoantibody production in the serum of F2 mice. By classification of the ANA staining pattern, we were able to determine the specificity of the autoimmune response and identify the dominant autoantigens targeted by individual mice. Homogenous nuclear staining is known to correlate with anti-dsDNA antibodies, whereas a coarsely speckled nuclear staining pattern corresponds to antibodies directed against RNA-containing antigens such as snRNPs or Sm (6, 23). Serum from 7 out of 19 TLR9+/+ mice exhibited homogenous nuclear staining, whereas 0 out of 16 TLR9−/− sera showed this pattern (Fig. 1, A and C; P = 0.009, Fisher's exact test), suggesting an impairment in the generation of anti-dsDNA autoantibodies. Moreover, equatorial staining of chromosomes in metaphase cells, indicative of antichromatin antibodies, was observed in 17 out of 19 TLR9+/+ sera and 0 out of 16 TLR9−/− sera (Fig. 1, A, B, and D; P < 0.0001). The high prevalence of metaphase chromatin staining in TLR9+/+ sera presumably reflects the presence of multiple different anti-DNA and antinucleosome antibody specificities. TLR9−/− mice were incapable of producing any of these autoantibodies.
Figure 1.

TLR9-deficient sera lack anti-DNA and antichromatin staining patterns. (A) ANAs were determined in sera (1:200) from 20-wk-old F2 mice. TLR9+/+ sera are shown at the top (left, homogenous pattern; middle and right, speckled pattern); TLR9−/− sera are shown at the bottom (left and middle, speckled pattern; right, cytoplasmic pattern). Some TLR9+/+ sera had predominantly speckled patterns superimposed on faint homogenous staining (middle). White arrows indicate cells in metaphase that demonstrate positive (top, TLR9+/+) or negative (bottom, TLR9−/−) chromatin staining. Original magnification, 400. (B) Digitally enlarged images of metaphase cells with positive (top, TLR9+/+) or negative (bottom, TLR9−/−) chromatin staining. (C) Serum ANAs were classified as nuclear homogenous, nuclear speckled, or cytoplasmic staining patterns. Black bars indicate TLR9+/+ sera (n = 19), and white bars indicate TLR9−/− sera (n = 16). (D) As in C, but serum ANAs were classified as either positive or negative for mitotic chromatin staining. *, P < 0.02; **, P < 0.01; and ***, P < 0.0001 by Fisher's exact test.
In contrast to the lack of staining for anti-dsDNA and antichromatin autoantibodies in TLR9−/− sera, there was no decrease in the proportion of mice with predominantly speckled ANA patterns, though TLR9−/− sera had a qualitatively different speckled pattern compared with TLR9+/+ sera. Although wild-type sera demonstrated mixed speckled and homogenous staining, TLR9−/− sera had exclusively speckled patterns in the absence of any superimposed homogenous staining (Fig. 1 A and not depicted). In addition, TLR9−/− sera had an increased incidence of primarily cytoplasmic staining compared with wild-type littermates (Fig. 1, A and C; P = 0.013), although the sera with cytoplasmic patterns also stained nuclei in a faint speckled pattern. These findings indicate that TLR9−/− mice were capable of targeting the typical RNA-containing autoantigens of lupus and account for the equivalence of quantitative ANA titers in the two groups of mice (median titer >1:2,000; unpublished data). Furthermore, the distribution of ANA patterns in TLR9−/− mice suggests that in the absence of anti-DNA antibodies, other autoantibody specificities are maintained and may even become more prominent.
TLR3-deficient lupus-prone mice were generated by F2 crosses with MRL/Mplpr/lpr mice in an analogous fashion to TLR9 mice. Unlike the TLR9 cohort, there was no difference in the distribution of either homogenous or speckled nuclear ANA patterns between TLR3+/+ (n = 15) and TLR3−/− (n = 17) littermates (Fig. 2, A and C). In addition, nearly all mice in both groups produced antibodies to chromatin, as evidenced by bright staining of metaphase chromosomes in mitotic cells (Fig. 2, A, B, and D). Because TLR3 is a receptor for dsRNA and may be involved in the generation of autoantibodies to RNA-containing antigens, we examined speckled ANA staining patterns in TLR3−/− sera but could detect no difference in either the quality or intensity of staining for RNA-containing antigens. Both TLR3−/− and TLR3+/+ mice produced autoantibodies that stained nuclear substrates with fine speckled, discrete speckled, and mixed speckled/homogenous patterns (Fig. 2 A and not depicted). Based on the ANA, the autoantibody profile of TLR3−/− mice was thus indistinguishable from wild-type littermates.
Figure 2.

Unaltered ANA patterns in TLR3-deficient sera. (A) Serum ANAs were determined as in Fig. 1. TLR3+/+ sera are shown at the top (left, homogenous pattern; middle and right, speckled pattern); TLR3−/− sera are shown at the bottom (left, homogenous pattern; middle and right, speckled pattern). White arrows indicate cells in metaphase that demonstrate positive chromatin staining. Original magnification, 400. (B) Digitally enlarged images of metaphase cells with positive chromatin staining. (C) Serum ANAs were classified as nuclear homogenous, nuclear speckled, or cytoplasmic staining patterns. Black bars indicate TLR3+/+ sera (n = 15), and white bars indicate TLR3−/− sera (n = 17). (D) As in C, but serum ANAs were classified as either positive or negative for mitotic chromatin staining.
Lack of anti-dsDNA autoantibodies in TLR9-deficient mice
The loss of homogenous nuclear and mitotic chromatin staining in TLR9−/− sera implied a block in the generation of autoantibodies to DNA-containing antigens; thus, we sought to confirm this with a specific assay for anti-dsDNA antibodies. Indirect immunofluorescence on Crithidia luciliae substrates is an established assay for the detection of anti-dsDNA autoantibodies in SLE (24). Because the kinetoplast organelle of C. luciliae is composed exclusively of circular dsDNA in the absence of single-stranded (ss) DNA, RNA, or histones, the C. luciliae assay is clinically the most specific test for antibodies to native dsDNA (25, 26). To further enhance the detection of anti-DNA antibodies, we costained the C. luciliae DNA with DAPI, thus permitting the differentiation of irrelevant cytoplasmic structures from true staining of kinetoplast and nuclear dsDNA (Fig. 2 A).
Among the TLR9 F2 mice, a subset of TLR9+/+ sera contained specific anti-dsDNA antibodies as detected by strong staining of the C. luciliae kinetoplast, but these antibodies were absent in sera from TLR9−/− mice (Fig. 3, A and B; P = 0.0121, Mann-Whitney U test). 7 out of 19 wild-type and 3 out of 16 TLR9−/− sera were also found to have borderline staining of dsDNA, as evidenced by a faint ring around the kinetoplast (intensity score, 1; Fig. 3, A and B). It was unclear whether these staining patterns represented low-affinity anti-dsDNA or nonspecific antibodies. We therefore analyzed the incidence of anti-dsDNA using intensity scores of either >0 or >1 as positive. With both methods of analysis, we observed a significant reduction in anti-dsDNA antibodies in TLR9−/− sera (P < 0.05, Fisher's exact test). In contrast, TLR3−/− mice appeared to have no impairment in the generation of anti-dsDNA autoantibodies, as there was no difference in the incidence or intensity of kinetoplast staining between TLR3+/+ and TLR3−/− sera (Fig. 3 C). Thus, TLR9, but not TLR3, was required for the generation of specific autoantibodies to dsDNA in the context of murine lupus.
Figure 3.
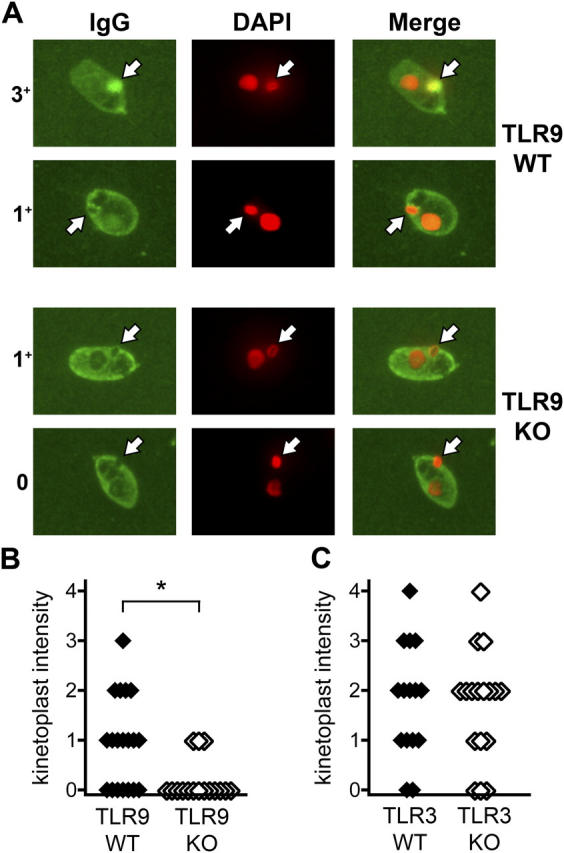
Reduced anti-dsDNA autoantibodies in TLR9−/− but not TLR3−/− mice. (A) Anti-dsDNA antibodies were detected by C. luciliae immunofluorescence. IgG antibodies to C. luciliae DNA are shown in green (left), and DAPI staining of DNA is shown in red (middle). White arrows indicate the kinetoplast. Specific anti-dsDNA antibodies are identified by colocalization of IgG and DAPI staining in the kinetoplast and appear in yellow (right). Representative TLR9+/+ sera are shown in the top two rows (intensity scores of 3+ and 1+), and TLR9−/− sera are shown in the bottom two rows (intensity scores of 1+ and 0). Original magnification, 1,000. (B and C) Specific anti-dsDNA staining of C. luciliae kinetoplasts was scored from 0 to 4 as in A for either TLR9+/+ (n = 19) and TLR9−/− (n = 16) sera (B), or TLR3+/+ (n = 15) and TLR3−/− (n = 17) sera (C). *, P < 0.02 by the Mann-Whitney U test.
Anti-Sm and anticardiolipin autoantibodies in TLR-deficient mice
Autoantibodies to Sm are exclusive to SLE and occur in ∼25% of diseased humans and mice (27, 28). We performed Western blots to detect antibodies against the protein components of the Sm complex. Mice in both the TLR9 and TLR3 F2 cohorts generated antibodies reacting with a cluster of bands in the 29–34-kD region of the gel, corresponding to the complex of B and B′ proteins (Fig. 4, A and B). As expected, TLR9−/− mice produced anti-Sm autoantibodies. In fact, there was a significant increase in the proportion of TLR9−/− mice with anti-Sm reactivity compared with TLR9+/+ littermates (Fig. 4, A and C; P = 0.0049, Fisher's exact test). We confirmed these findings with a solid phase ELISA using purified whole Sm antigen as the target and again found higher levels of anti-Sm autoantibodies in TLR9−/− mice (Fig. 4 D; P = 0.0014, Mann-Whitney U test). Consistent with the presence of speckled ANA patterns, TLR3−/− mice also generated anti-Sm autoantibodies at frequencies equivalent to their wild-type counterparts (Fig. 4, B, C, and E). Thus, neither TLR9 nor TLR3 was required for the production of autoantibodies to the RNA-containing Sm antigen, even though, unexpectedly, there was a relative increase in anti-Sm autoantibodies in TLR9−/− mice.
Figure 4.
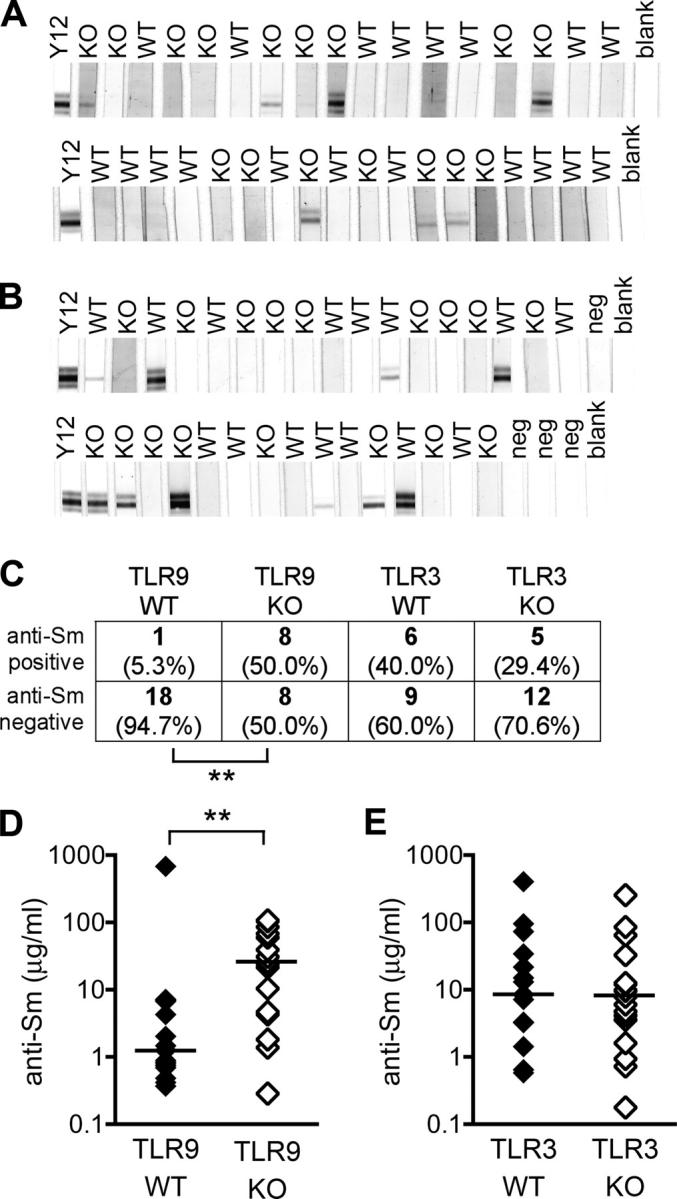
Presence of anti-Sm autoantibodies in TLR-deficient mice. (A) Anti-Sm antibodies were detected by Western blot in TLR9+/+ (WT; n = 19) and TLR9−/− (KO; n = 16) sera. The monoclonal anti-Sm antibody Y12 was used as a positive control. Depicted portion of the gel is from 29–34 kD, corresponding to the B and B′ cluster of Sm proteins. (B) Anti-Sm antibodies were detected in TLR3+/+ (WT; n = 15) and TLR3−/− (KO; n = 17) sera as in A. Nonautoimmune control sera are also depicted (neg; n = 4). (C) Sera from TLR9 and TLR3 cohorts was scored as either positive or negative for anti-Sm antibodies by Western blot as in A and B. (D and E) Anti-Sm antibodies were confirmed by ELISA in either TLR9+/+ (n = 19) and TLR9−/− (n = 16) sera (D), or TLR3+/+ (n = 15) and TLR3−/− (n = 17) sera (E). Bars represent median values. *, P < 0.005 by Fisher's exact test (C) or the Mann-Whitney U test (D).
Antiphospholipid antibodies are also commonly found in autoimmune disease and are associated with coagulation abnormalities and thrombosis (6). Because of the polyanionic nature of the lipid antigen, antiphospholipid antibodies can cross-react with DNA, and anti-DNA antibodies can likewise have dual specificity for lipids such as phosphatidylserine (29). To determine if TLR9 deficiency or the loss of anti-dsDNA antibodies in TLR9−/− mice affected the generation of antiphospholipid antibodies, we measured autoantibodies to cardiolipin, a prototypical phospholipid antigen. We found that TLR9−/− mice developed serum titers of anticardiolipin antibodies that were comparable to TLR9+/+ littermates (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050338/DC1), indicating that anti-dsDNA and antiphospholipid antibodies are controlled by separate mechanisms in these mice.
Global immune activation in TLR-deficient lupus-prone mice
Antichromatin and antiribonucleoprotein antibodies are among the earliest detectable autoantibodies in SLE and are known to precede the appearance of overt clinical disease in both humans and mice (30, 31). The breakdown of tolerance to DNA-containing antigens may thus be a critical first step in systemic autoimmunity, and the early appearance of antichromatin autoantibodies may facilitate disease progression and epitope spreading to other nuclear autoantigens (32). We therefore determined whether the lack of TLR9 or TLR3 had any effect on several parameters of global autoimmune disease. TLR-intact and TLR-deficient mice from both the TLR9 and TLR3 cohorts exhibited massive lymphadenopathy and splenomegaly (Fig. 5 A), characteristic of the accumulation of activated lymphocytes in Fas-deficient autoimmunity. Both groups of mice also had elevated levels of total IgG and IgG2a in the serum relative to nonautoimmune C57BL/6 mice (Fig. 5 B). To further define the autoimmune phenotype, we enumerated T cells, B cells, and CD4−/CD8− double-negative T cells, which accumulate with age in MRL/Mplpr/lprmice (17). These cell populations were not decreased in TLR-deficient mice in the spleen (Fig. 5 C) or lymph nodes (unpublished data), nor was there any block in the accumulation of activated and memory phenotype CD4+ T cells (Fig. 5 D). Thus, neither deficiency of TLR9 or TLR3 nor the specific absence of anti-DNA autoantibodies in TLR9−/− mice inhibited the disregulated immune activation and aberrant lymphocyte accumulation of systemic autoimmune disease.
Figure 5.

Lymphadenopathy, hypergammaglobulinemia, and lymphocyte accumulation in TLR-deficient mice. TLR9+/+ (n = 19), TLR9−/− (n = 16), TLR3+/+ (n = 15), and TLR3−/− (n = 17) mice were killed at 20 wk of age and assessed for evidence of aberrant immune activation; nonautoimmune C57BL/6 control mice (n = 4) were killed at 26 wk of age. (A) Spleens and the two largest axillary lymph nodes were removed and weighed. (B) Total serum IgG and IgG2a were determined. (C) Splenocyte subsets were enumerated by FACS analysis for T cells (Thy1.2+), DNTC (CD4−/CD8− double-negative T cells), and B cells (CD22+). (D) Splenic CD4+ T cells were classified as either naive (CD44− CD62L+), activated (CD44+ CD62L+), or memory (CD44+ CD62L−) phenotype. The analysis in D was performed on 12 TLR3+/+ and 12 TLR3−/− mice. Horizontal lines represent mean values.
There was, however, an unexpected increase in immune activation in TLR9−/− animals compared with wild-type littermates. This was not observed for animals in the TLR3 cohort. Lymph node weight (P = 0.0016), splenic T cell count (P = 0.0277), double-negative T cell count (P = 0.0150), and activated CD4+ T cell count (P = 0.0327) were all significantly greater in TLR9−/− mice compared with TLR9+/+ littermates. Because the vast majority of the lymph node in these animals is composed of activated and double-negative T cells (unpublished data), these findings all point toward an increased level of T cell activation in TLR9-deficient mice.
In addition to hypergammaglobulinemia and the accumulation of activated lymphocytes, increased production of type I IFNs is associated with SLE in humans and mice (33–35). Moreover, genetic ablation of the type I IFNR markedly reduced clinical and immunologic signs of lupus in New Zealand Black mice (36), although this was not observed in other autoimmune strains (37). Because activation of TLR9 and TLR3 is known to stimulate the secretion of type I IFNs (38, 39), we determined whether lack of either of these receptors influenced spontaneous IFN levels in autoimmune mice. Using an ELISA to detect serum IFN-α, we could not detect any impairment in IFN production by TLR9−/− or TLR3−/− F2 mice (unpublished data). Although the sensitivity of this assay was limited to IFN-α, it was clear that a subset of mice exhibited markedly elevated levels type I IFNs even in the absence of TLR9 or TLR3.
Glomerulonephritis and immune complex deposition in the absence of anti-DNA autoantibodies
To assess renal disease in TLR9- and TLR3-deficient autoimmune mice, we first measured spot proteinuria at the time of sacrifice. There was no difference between TLR9−/− or TLR3−/− mice and their wild-type counterparts, with most mice exhibiting mild to moderate levels of proteinuria (unpublished data). Histologically, interstitial and perivascular lymphocytic infiltrates were present in the kidneys of all mice. There was a moderate increase in glomerular size and cellularity, with no detectable difference in any parameter between TLR9+/+ and TLR9−/− (Fig. 6, A, C, and E) or TLR3+/+ and TLR3−/− mice (Fig. 6, G, I, and K). Periodic acid Schiff (PAS) staining of kidney sections further revealed that TLR9−/− (Fig. 6, B, D, and F) and TLR3−/− (Fig. 6, H, J, and L) mice developed substantial glomerular protein deposition that was comparable to wild-type littermates, with marked thickening of capillary loops and obliteration of vessel lumens. These findings indicate that neither TLR9 nor TLR3 was essential for the induction of end organ disease in murine lupus.
Figure 6.
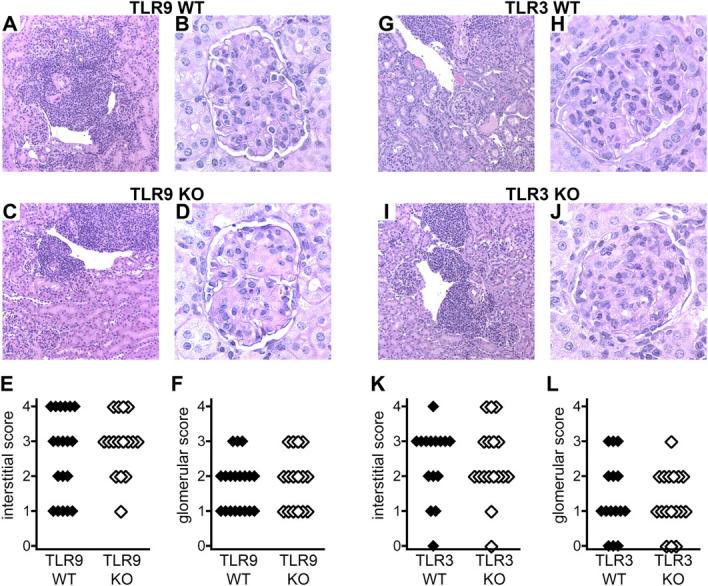
Glomerulonephritis in the absence of anti-DNA autoantibodies. Histological renal disease was assessed in TLR9+/+ (n = 19), TLR9−/− (n = 16), TLR3+/+ (n = 15), and TLR3−/− (n = 17) mice at 20 wk of age. (A–F) Paraffin kidney sections from TLR9+/+ (A and B) and TLR9−/− (C and D) mice were stained with H&E (A and C) or PAS (B and D). Interstitial infiltrates (E) and glomerular disease (F) were scored from 0 to 4 for all mice. (G–L) Paraffin kidney sections from TLR3+/+ (G and H) and TLR3−/− (I and J) mice were stained with H&E (G and I) or PAS (H and J). Interstitial infiltrates (K) and glomerular disease (L) were scored from 0 to 4 for all mice. Representative images are shown. Original magnification: 100 for H&E sections (A, C, G, and I) and 400 for PAS sections (B, D, H, and J).
Although interstitial renal disease and glomerulonephritis were not inhibited in TLR9-deficient mice, it was unclear whether this was caused by direct glomerular deposition of immunoglobulin and complement-containing immune complexes. We therefore assessed renal IgG and complement deposition in TLR9−/− mice lacking anti-dsDNA and antichromatin autoantibodies. Both TLR9+/+ and TLR9−/− mice had immune deposits throughout the glomerulus, in contrast to nonautoimmune C57BL/6 mice (Fig. 7 A). Although levels of IgG and C3 deposition varied among individuals, there was no difference in the mean glomerular fluorescence between the two groups of mice (Fig. 7 B). Thus, lupus nephritis and glomerular immune complex deposition were unaffected by the absence of anti-dsDNA and antichromatin autoantibodies in autoimmune TLR9−/− mice.
Figure 7.
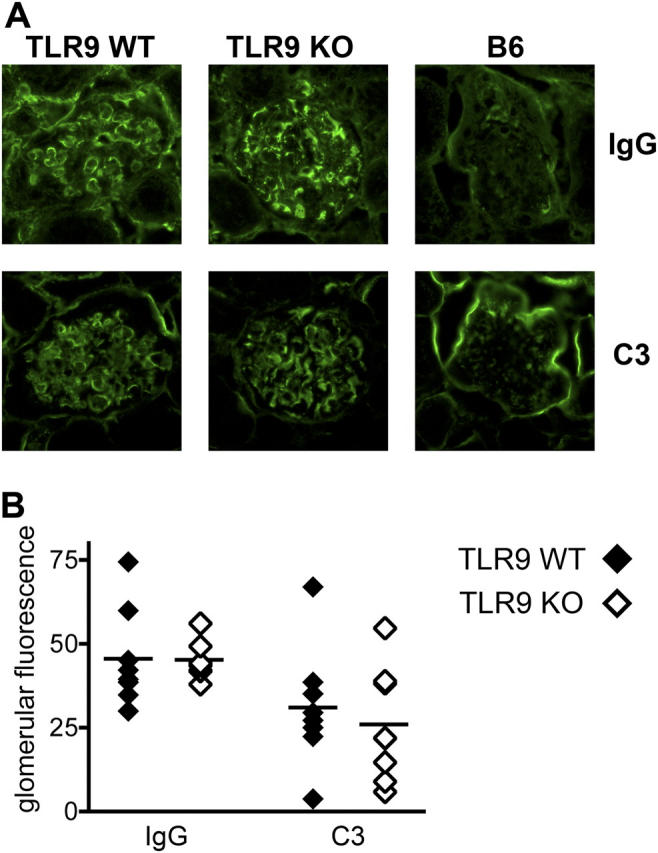
Glomerular immune deposits do not require anti-DNA antibodies. (A) Glomerular immune deposits were detected by direct immunofluorescence for IgG (top) and complement C3 (bottom) in frozen kidney sections from TLR9+/+ (left), TLR9−/− (middle), and nonautoimmune C57BL/6 control mice (right). Representative images are shown. Original magnification, 400. (B) Mean glomerular fluorescence intensity (arbitrary units) was determined for IgG and C3 in TLR9+/+ (n = 8) and TLR9−/− (n = 8) mice. Horizontal lines represent mean values.
Discussion
Since the initial report of a role for TLR9 in the activation of autoreactive B cells in vitro (11), there has been intense interest and speculation about the importance of TLRs for autoantibody production and clinical disease in SLE. Our findings represent the first demonstration that a TLR is required for specific autoantibody production in vivo. We found that TLR9, but not TLR3, was necessary for the generation of specific autoantibodies to chromatin and dsDNA in the context of murine lupus. The control of these autoantibodies by TLR9 indicates the central role of this receptor in systemic autoimmune diseases such as SLE, in which ANAs are a cardinal feature. Because of inherent limitations in the determination of autoantibody specificity, however, we could not completely exclude the presence of low-affinity or cross-reactive antibodies to DNA in TLR9−/− mice, which may arise by TLR9-independent mechanisms.
The requirement for TLR9 in anti-DNA antibody production was surprising because TLRs are thought to discriminate self from nonself by recognizing only pathogen-associated molecular patterns not present on mammalian cells. However, mounting evidence suggests that innate receptors such as TLR9 exhibit some degeneracy of ligand binding. Synthetic oligonucleotides containing methylated DNA or lacking canonical CpG activation motifs have both been shown to stimulate B cells in the presence of concurrent B cell receptor (BCR) stimulation (40, 41), and murine genomic DNA can induce APC activation both in vitro and in vivo (42). More relevant to autoimmunity, chromatin–antichromatin immune complexes provide a potent activation stimulus to dendritic cells and autoreactive rheumatoid factor B cells, both of which require TLR9 for complete stimulation (11, 13, 43).
Why are TLRs that can recognize self-antigens not constitutively activated? One important control mechanism appears to derive from intracellular compartmentalization. Normally, activation of TLR9 by endogenous nucleic acids may be inhibited because the receptor is sequestered in the endosomal pathway (44), where host-derived nuclear antigens are largely excluded. Other TLRs that bind nucleic acid ligands are similarly segregated in endosomal compartments (8). However, because anti-DNA B cells are uniquely poised to endocytose and deliver DNA-containing antigens (via the BCR) to TLR9 in restricted intracellular compartments, the spatial separation of TLR9 from inappropriate self-ligands may break down in these cells. This phenomenon could explain the selective tolerance breakdown of antinuclear B cells in autoimmunity and the highly restricted autoantibody repertoire of SLE. Such a mechanism of BCR-mediated endocytosis also suggests a B cell–intrinsic requirement for TLR9 costimulation. In vitro studies using rheumatoid factor and anti-DNA B cells have confirmed this two signal model of autoreactive B cell activation through both BCR and TLR9 signaling pathways (12).
Introduction of TLR9 ligands to restricted compartments may also occur when mechanisms for clearance of apoptotic cell debris are overwhelmed. Apoptotic cells are repositories of lupus autoantigens (45), and there is evidence that residual fragments of apoptotic DNA have a greater stimulatory capacity than native genomic DNA (12). Further support for innate immune activation by the products of cell death comes from studies of genetic ablation of DNaseII, a lysosomal endonuclease required for digestion of apoptotic DNA fragments by macrophages. In Drosophila, DNaseII deficiency results in dysregulated expression of antibacterial gene products (46), whereas in mice, the accumulation of residual DNA induces IFN-β secretion by fetal liver macrophages, inhibition of hematopoiesis, and resultant embryonic lethality (47). The development of autoimmunity in mice with impaired phagocytosis of apoptotic cells (48) and in humans and mice lacking the early components of complement (49) provides additional evidence for a model of tolerance breakdown initiated by inappropriate innate recognition of apoptotic or necrotic debris.
In contrast to the absence of anti-dsDNA and antichromatin, we found an increased incidence of anti-Sm autoantibodies in TLR9-deficient mice relative to wild-type littermates. This could be the result of several factors. For instance, TLR9−/− mice may have developed a compensatory increase in levels of autoantibodies to RNA-containing antigens as a direct result of the loss of anti-DNA antibodies. If the “immunizing” antigens in SLE derive from apoptotic debris and antinuclear antibodies can aid in the clearance of these particles, then the lack of anti-DNA antibodies in TLR9−/− mice could lead to a marked increase in circulating levels of autoantigens. In this situation, when the presumably dominant epitopes of DNA and chromatin are rendered nonimmunogenic, alternate epitopes such as Sm, snRNPs, and cytoplasmic antigens may become more prominent, as observed in TLR9−/− sera. Alternatively, the increase in anti-Sm antibodies may reflect competition and reciprocal suppression among different TLRs. Activation of various TLRs is known to inhibit subsequent responses by TLRs that share downstream signaling pathways (50), and stimulation of TLR9 by endogenous DNA may therefore prevent responses to other TLR ligands. Because it is probable that other TLRs control the autoantibody response to RNA antigens (as will be discussed), the removal of TLR9-mediated inhibition may allow these alternate mechanisms of autoantibody production to proceed unchecked. Finally, the increase in anti-Sm titers among TLR9−/− mice may reflect an increased severity of autoimmune disease, as suggested by the trend toward increased accumulation of activated T cells in these mice.
Unlike TLR9, TLR3 had no role in the generation of autoantibodies to DNA or the prototypical RNA-containing Sm antigen. One potential explanation for this is the highly restricted cellular expression of TLR3. TLR3 expression has only been detected in mature dendritic cells of the myeloid lineage (51), and was not observed in B cells by quantitative PCR (52). It is also possible that the specificity of TLR3 for dsRNA is not appropriate for the prototypical RNA-containing autoantigens of lupus, which are composed primarily of ssRNA alternating with regions of intrastrand base pairing. Another candidate for the regulation of autoantibodies to RNA antigens is TLR7, which has recently been identified as an innate receptor for ssRNA (53, 54). TLR7 is closely related to TLR9 in protein sequence and downstream signaling pathways (7), and, like TLR9, is expressed on B cells in endosomal compartments (8, 52). Other innate receptors capable of recognizing microbial RNA may also promote anti-RNA antibodies in SLE. These include the protein kinase regulated by RNA (55), TLR8 in humans (54), and, potentially, additional RNA receptors not yet identified (56, 57).
Although TLR9 can shape the autoantibody repertoire in SLE, lack of either TLR9 or TLR3 did not ameliorate clinical disease in lupus-prone mice. This was particularly surprising given the prevailing view that anti-dsDNA and antichromatin antibodies are principal mediators of renal pathology in SLE (2, 58). Although it is clear that anti-DNA antibodies are associated with nephritis (5), are found deposited in diseased kidneys (59), and can mediate nephritis in adoptive transfer experiments (60), there is an accumulating body of evidence to suggest that other autoantibody specificities can play equally prominent roles. Anti-Sm antibodies, for example, are also correlated with renal disease in clinical studies (61), and the elevated titers of anti-Sm antibodies in TLR9−/− mice may account for the persistence of nephritis and immune deposits in the absence of anti-dsDNA. A definitive experiment using NZM congenic mice further demonstrated that severe, chronic glomerulonephritis with early mortality could occur in the absence of any detectable ANA, including anti-DNA antibodies (62). Our data thus provide a novel demonstration of a previously observed finding: although anti-dsDNA autoantibodies may predict nephritis in SLE, they are not the exclusive pathogenic specificity. Finally, it remains possible that disease occurs in the absence of anti-dsDNA because autoantibodies by themselves are not required for target organ disease, and autoreactive B cells exert pathogenic effects independent of antibody secretion (63).
Given the potential of TLR9 to recognize endogenous DNA and the inherent predisposition toward anti-DNA reactivity in the germline V gene repertoire of the BCR (64), it is unclear why this dual propensity for self-reactivity should coexist in the same cell. It is possible that self-reactivity and the rare occurrence of anti-DNA antibodies are the unavoidable byproduct of an essential defense against pathogenic microorganisms. It is also possible, however, that TLR stimulation could lead to regulatory or even antiinflammatory effects in certain contexts. For example, the ability of TLR stimulation to promote homeostatic repair processes without the induction of an inflammatory or adaptive immune response was recently demonstrated in a model of induced intestinal injury (65). Similarly, the regulated production of low-affinity anti-DNA IgM antibodies in the absence of infection may facilitate clearance of dying cells, limit inappropriate inflammation, and suppress autoimmune disease. Thus, the increased lymphadenopathy and T cell activation observed in lupus-prone TLR9−/− mice may be caused by the lack of a protective subset of anti-DNA antibodies. Consistent with this idea, the absence of secreted IgM in lupus-prone mice exacerbates autoantibody formation, disease activity, and mortality (66). An analogous role for physiologic rheumatoid factor autoantibodies has been postulated, as these “natural” anti-IgG antibodies promote clearance of immune complexes in conditions of high antigenic burden (67, 68). The intriguing possibility that TLR-mediated cellular activation and autoantibody production could enhance the clearance of apoptotic cell debris, promote tissue repair or limit autoimmune pathology remains to be investigated.
Regardless of the exact roles of anti-dsDNA and antichromatin antibodies in promoting or regulating disease, the demonstration that TLR9 controls these canonical autoantibodies of SLE represents an important advance in our understanding of the mechanisms of systemic autoimmunity. The pathologic immune response of autoimmune disease, like the adaptive immune response to foreign antigens, may fundamentally require instruction by the innate immune system. Although prevention of anti-dsDNA and antichromatin autoantibody production did not ameliorate clinical disease, inhibition of TLRs or other innate signaling pathways may yet provide new therapeutic targets in the treatment of human lupus. This will require a more complete understanding of the complete complement of innate immune receptors and their individual roles in the pathogenesis of autoimmune disease.
Materials and Methods
Mice.
Heterozygous TLR9+/− mice (9) of a mixed genetic background (B6 and 129Sv) were bred to MRL/Mplpr/lpr mice in our colony under specific pathogen-free conditions. Intercrossing of F1 offspring in five matings produced 22 Fas-deficient (Faslpr/lpr) F2 mice, 5 of which were TLR9+/+ and 4 of which were TLR9−/−. Intercrossing of heterozygous TLR9+/− F2 mice in four additional matings produced 56 Faslpr/lpr mice, 15 of which were TLR9+/+ and 13 of which were TLR9−/−. The genetic composition of both generations of mice was ∼50% MRL/Mp. One mouse in each group died before planned analysis at 19–21 wk of age. In a separate cohort, homozygous TLR3−/− mice (14) of a mixed genetic background (B6 and 129Sv) were bred to MRL/Mplpr/lpr mice as above. Intercrossing of F1 offspring in nine matings produced 33 Faslpr/lpr mice, 9 of which were TLR3+/+ and 11 of which were TLR3−/−. Intercrossing of heterozygous TLR3+/− F2 mice in three additional matings produced 26 Faslpr/lpr mice, 8 of which were TLR3+/+ and 6 of which were TLR3−/−. Two TLR3+/+ mice died before planned analysis at 19–21 wk of age. C57BL/6 mice (Charles River Laboratories) were maintained in our mouse colony to an age of 26 wk. All animal studies were conducted in accordance with protocols approved by the Yale University Institutional Animal Care and Welfare Committee.
ANA and anti-dsDNA immunofluorescence.
Serum was obtained via retroorbital puncture just before death. For ANA, serum was diluted 1:200 and used for indirect immunofluorescence on fixed Hep-2 ANA slides (Antibodies Inc.) with fluorescein-conjugated goat anti–mouse IgG (Southern Biotechnology Associates, Inc.) as the detection reagent. Slides were read on a fluorescent microscope (BX-40; Olympus) at 400× and scored as either nuclear homogenous, nuclear speckled, or cytoplasmic staining patterns by a reader blinded to the genotype of the mice. In addition, each serum sample was scored for staining of mitotic chromosomes. Images were captured with a cooled CCD camera (Spot-RT Slider; Diagnostic Instruments) with a constant exposure time of 4 s for TLR9 sera and 2.5 s for TLR3 sera. For TLR9 F2 mice, endpoint titration of serum was performed at reciprocal dilutions of 50, 200, 800, 2,000, 104, 4 × 104, and 105, and samples were scored for the presence of nuclear staining, regardless of pattern or cytoplasmic staining. Serum samples from 26-wk-old C57BL/6 mice were negative for ANA at 1:50 dilution (unpublished data).
For anti-dsDNA, 1:10 diluted serum was applied to fixed C. luciliae slides (Antibodies Inc.), with biotin-conjugated goat anti–mouse IgG (Southern Biotech) and Alexa Fluor 555–conjugated streptavidin (Molecular Probes) as detection reagents. C. luciliae DNA was colocalized with DAPI (Molecular Probes). Slides were read at 1,000× and scored 0–4 for intensity of kinetoplast staining by a reader blinded to the genotype of the mice. Images were captured with a constant exposure time of 0.5 s in red (IgG) and blue (DAPI) channels, and then transferred into green (IgG) and red (DAPI) channels.
Anti-Sm Western blot and ELISA.
For Western blot, purified bovine Sm antigen (Immunovision) was separated by SDS-PAGE, transferred to nitrocellulose membrane, and probed with 1:200 diluted serum, using alkaline phosphatase-conjugated goat anti–mouse IgG (Southern Biotech) and BCIP/NBT substrate (KPL, Inc.) as detection reagents. The monoclonal mouse anti-Sm antibody Y12 (28) was used as a standard. Blots were scored for the presence of visible anti-Sm bands at 29–34 kD. For ELISA, polystyrene plates were coated with Sm antigen, blocked with 1% BSA in PBS, and serial dilutions of serum from 1:50 to 1:36,450 were added. Anti-Sm antibodies were detected with alkaline phosphatase-conjugated goat anti–mouse IgG (Southern Biotech), and absorbance at 405/630nm was compared with the Y12 standard to quantitate.
Anticardiolipin ELISA.
Polystyrene plates were coated with cardiolipin by incubation with 100 μg/ml bovine cardiolipin (Avanti Polar Lipids, Inc.) in 100% ethanol for 12–18 h at 4°C until completely evaporated. Plates were blocked with 1% BSA in PBS, and serial dilutions of serum from 1:100 to 1:2,700 were added. Anticardiolipin antibodies were detected with alkaline phosphatase–conjugated goat anti–mouse IgG (Southern Biotech), and absorbance at 405/630 nm was compared with the FD1 standard to quantitate. Anticardiolipin standards were provided by M. Monestier (Temple University School of Medicine, Philadelphia, PA).
Analysis of lymphocyte subsets and serum IgG.
Spleen and lymph node cells were isolated, counted, and stained with the following antibodies for fluorescence-activated cell sorting analysis: anti-Thy1.2 (30H12), anti-CD4 (GK1.5), anti-CD8 (TIB105), anti-CD44 (IM7), anti-CD62L (Mel-14), and anti-CD22.2 (Cy34.1). All antibodies except anti-CD22.2 (BD Biosciences) were made in our laboratory. Total serum IgG and IgG2a were determined by ELISA. Polystyrene plates were coated with goat anti–mouse IgG (Southern Biotech), blocked with 1% BSA in PBS, and serial dilutions of serum from 1:104 to 1:7.3 × 106 were added. IgG or IgG2a was detected with alkaline phosphatase–conjugated goat anti–mouse IgG or goat anti–mouse IgG2a (Southern Biotech), and absorbance at 405/630nm was compared with known mouse IgG or IgG2a standards to quantitate.
Serum IFN-α ELISA.
Serum was collected at the age of 17–18 wk for TLR9, or at the time of death for TLR3 mice, and IFN-α was measured by ELISA. Polystyrene plates were coated with monoclonal rat anti–mouse IFN-α (PBL Biomedical Laboratories), blocked with 1% BSA in PBS, and 1:10 diluted serum was added. IFN-α was detected with rabbit anti–mouse IFN-α (PBL Biomedical Laboratories) and alkaline phosphatase–conjugated goat anti–rabbit IgG (Southern Biotech). Absorbance at 405/630nm was compared with recombinant mouse IFN-α standard (PBL Biomedical Laboratories) to quantitate. Limit of detection of the assay was 1.0 ng/ml in undiluted serum.
Analysis of kidney disease.
Kidneys were bisected, and one half was fixed in formalin and the other in paraformaldehyde (0.7% in PBS with 1.37% l-lysine and 0.2% sodium periodate). Paraformaldehyde-fixed kidneys were then dehydrated in 30% sucrose and flash-frozen in OCT. Formalin-fixed kidneys were paraffin-embedded, sectioned, and hematoxylin and eosin (H&E) or PAS stained (Histo-Scientific Research Laboratories). Stained sections were scored for glomerular and interstitial disease by a renal pathologist (M. Kashgarian) who was blinded to the genotype of the mice. For IgG deposition, frozen kidneys from eight TLR9+/+ and eight TLR9−/− mice were sectioned and stained with biotin-conjugated goat anti–mouse IgG (γ chain specific; Southern Biotechnology Associates, Inc.) and Alexa Fluor 647–conjugated streptavidin (Molecular Probes). For C3 deposition, frozen kidney sections were stained with monoclonal rat anti–mouse C3 (Connex) and Alexa Fluor 647–conjugated goat anti–rat IgG (cross-adsorbed against mouse IgG; Molecular Probes). Stained sections were read on a fluorescent microscope at 400×, and images were captured with a constant exposure time of 0.5 s. To quantitate IgG and C3 deposition, mean fluorescence was calculated from captured images; two representative glomeruli per mouse were outlined, and mean pixel intensity was calculated with Photoshop (Adobe).
Online supplemental material
Fig. S1 shows the results of determinations of anticardiolipin levels in sera from TLR9-deficient and intact mice, using the assay described in Anticardiolipin ELISA section. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050338/DC1.
Acknowledgments
We thank Joe Craft, Ruslan Medzhitov, and Ann Marshak-Rothstein for helpful comments and discussions. We thank Marc Monestier for advice and positive control standards for the anticardiolipin assay. We thank Michelle Horniak, Kasie Stevens, Terrence Hunt, and the Yale Animal Resources Center staff for the expert animal care that made this work possible.
Sean Christensen was supported by National Institutes of Health (NIH)/National Institute of General Medical Sciences medical scientist training grant GM07205. These studies were supported by NIH grants R01 AI061090, P01 AR050256, and P01 AI36529.
The authors have no conflicting financial interests.
Abbreviations used: ANA, antinuclear antibody; BCR, B cell receptor; ds, double-stranded; PAS, periodic acid Schiff; SLE, systemic lupus erythematosus; Sm, Smith antigen; snRNP, small nuclear ribonucleoprotein; ss, single-stranded; Toll-like receptor, TLR.
References
- 1.Notman, D.D., N. Kurata, and E.M. Tan. 1975. Profiles of antinuclear antibodies in systemic rheumatic diseases. Ann. Intern. Med. 83:464–469. [DOI] [PubMed] [Google Scholar]
- 2.Kotzin, B. 1996. Systemic lupus erythematosus. Cell. 85:303–306. [DOI] [PubMed] [Google Scholar]
- 3.Plotz, P.H. 2003. The autoantibody repertoire: searching for order. Nat. Rev. Immunol. 3:73–78. [DOI] [PubMed] [Google Scholar]
- 4.Monestier, M., and B.L. Kotzin. 1992. Antibodies to histones in systemic lupus erythematosus and drug-induced lupus syndromes. Rheum. Dis. Clin. North Am. 18:415–436. [PubMed] [Google Scholar]
- 5.Hahn, B.H. 1998. Antibodies to DNA. N. Engl. J. Med. 338:1359–1368. [DOI] [PubMed] [Google Scholar]
- 6.Egner, W. 2000. The use of laboratory tests in the diagnosis of SLE. J. Clin. Pathol. 53:424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 8.Iwasaki, A., and R. Medzhitov. 2004. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5:987–995. [DOI] [PubMed] [Google Scholar]
- 9.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745. [DOI] [PubMed] [Google Scholar]
- 10.Bernasconi, N.L., N. Onai, and A. Lanzavecchia. 2003. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 101:4500–4504. [DOI] [PubMed] [Google Scholar]
- 11.Leadbetter, E.A., I.R. Rifkin, A.M. Hohlbaum, B.C. Beaudette, M.J. Shlomchik, and A. Marshak-Rothstein. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 416:603–607. [DOI] [PubMed] [Google Scholar]
- 12.Viglianti, G.A., C.M. Lau, T.M. Hanley, B.A. Miko, M.J. Shlomchik, and A. Marshak-Rothstein. 2003. Activation of autoreactive B cells by CpG dsDNA. Immunity. 19:837–847. [DOI] [PubMed] [Google Scholar]
- 13.Marshak-Rothstein, A., L. Busconi, C.M. Lau, A.S. Tabor, E.A. Leadbetter, S. Akira, A.M. Krieg, G.B. Lipford, G.A. Viglianti, and I.R. Rifkin. 2004. Comparison of CpG s-ODNs, chromatin immune complexes, and dsDNA fragment immune complexes in the TLR9-dependent activation of rheumatoid factor B cells. J. Endotoxin Res. 10:247–251. [DOI] [PubMed] [Google Scholar]
- 14.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 15.Kariko, K., H. Ni, J. Capodici, M. Lamphier, and D. Weissman. 2004. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 279:12542–12550. [DOI] [PubMed] [Google Scholar]
- 16.Will, C.L., and R. Luhrmann. 2001. Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 13:290–301. [DOI] [PubMed] [Google Scholar]
- 17.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 18.Izui, S., V.E. Kelley, K. Masuda, H. Yoshida, J.B. Roths, and E.D. Murphy. 1984. Induction of various autoantibodies by mutant gene lpr in several strains of mice. J. Immunol. 133:227–233. [PubMed] [Google Scholar]
- 19.Shlomchik, M.J., M.P. Madaio, D. Ni, M. Trounstine, and D. Huszar. 1994. The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 180:1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma, J., J. Xu, M.P. Madaio, Q. Peng, J. Zhang, I.S. Grewal, R.A. Flavell, and J. Craft. 1996. Autoimmune lpr/lpr mice deficient in CD40 ligand: spontaneous Ig class switching with dichotomy of autoantibody responses. J. Immunol. 157:417–426. [PubMed] [Google Scholar]
- 21.Tan, E.M., A.S. Cohen, J.F. Fries, A.T. Masi, D.J. McShane, N.F. Rothfield, J.G. Schaller, N. Talal, and R.J. Winchester. 1982. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 25:1271–1277. [DOI] [PubMed] [Google Scholar]
- 22.Emlen, W., and L. O'Neill. 1997. Clinical significance of antinuclear antibodies: comparison of detection with immunofluorescence and enzyme-linked immunosorbent assays. Arthritis Rheum. 40:1612–1618. [DOI] [PubMed] [Google Scholar]
- 23.Bradwell, A.R., R.P. Stokes, and G.D. Johnson, editors. 1995. Atlas of Hep-2 Patterns. The Binding Site Ltd., Birmingham, UK. 118 pp.
- 24.Aarden, L.A., E.R. de Groot, and T.E. Feltkamp. 1975. Immunology of DNA. III. Crithidia luciliae, a simple substrate for the determination of anti-dsDNA with the immunofluorescence technique. Ann. NY Acad. Sci. 254:505–515. [DOI] [PubMed] [Google Scholar]
- 25.Isenberg, D.A., C. Dudeney, W. Williams, I. Addison, S. Charles, J. Clarke, and A. Todd-Pokropek. 1987. Measurement of anti-DNA antibodies: a reappraisal using five different methods. Ann. Rheum. Dis. 46:448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kavanaugh, A., R. Tomar, J. Reveille, D.H. Solomon, and H.A. Homburger. 2000. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. American College of Pathologists. Arch. Pathol. Lab. Med. 124:71–81. [DOI] [PubMed] [Google Scholar]
- 27.Pettersson, I., M. Hinterberger, T. Mimori, E. Gottlieb, and J.A. Steitz. 1984. The structure of mammalian small nuclear ribonucleoproteins. Identification of multiple protein components reactive with anti-(U1)ribonucleoprotein and anti-Sm autoantibodies. J. Biol. Chem. 259:5907–5914. [PubMed] [Google Scholar]
- 28.Bloom, D.D., J.L. Davignon, M.W. Retter, M.J. Shlomchik, D.S. Pisetsky, P.L. Cohen, R.A. Eisenberg, and S.H. Clarke. 1993. V region gene analysis of anti-Sm hybridomas from MRL/Mp-lpr/lpr mice. J. Immunol. 150:1591–1610. [PubMed] [Google Scholar]
- 29.Cocca, B.A., S.N. Seal, P. D'Agnillo, Y.M. Mueller, P.D. Katsikis, J. Rauch, M. Weigert, and M.Z. Radic. 2001. Structural basis for autoantibody recognition of phosphatidylserine-beta 2 glycoprotein I and apoptotic cells. Proc. Natl. Acad. Sci. USA. 98:13826–13831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arbuckle, M.R., M.T. McClain, M.V. Rubertone, R.H. Scofield, G.J. Dennis, J.A. James, and J.B. Harley. 2003. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 349:1526–1533. [DOI] [PubMed] [Google Scholar]
- 31.Laderach, D., S. Koutouzov, J.F. Bach, and A.M. Yamamoto. 2003. Concomitant early appearance of anti-ribonucleoprotein and anti-nucleosome antibodies in lupus prone mice. J. Autoimmun. 20:161–170. [DOI] [PubMed] [Google Scholar]
- 32.Shlomchik, M.J., J. Craft, and M.J. Mamula. 2001. From T to B and back again: positive feedback in systemic autoimmune disease. Nat. Rev. Immunol. 1:147–153. [DOI] [PubMed] [Google Scholar]
- 33.Baechler, E.C., F.M. Batliwalla, G. Karypis, P.M. Gaffney, W.A. Ortmann, K.J. Espe, K.B. Shark, W.J. Grande, K.M. Hughes, V. Kapur, et al. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA. 100:2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett, L., A.K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, and V. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lian, Z.X., K. Kikuchi, G.X. Yang, A.A. Ansari, S. Ikehara, and M.E. Gershwin. 2004. Expansion of bone marrow IFN-alpha-producing dendritic cells in New Zealand Black (NZB) mice: high level expression of TLR9 and secretion of IFN-alpha in NZB bone marrow. J. Immunol. 173:5283–5289. [DOI] [PubMed] [Google Scholar]
- 36.Santiago-Raber, M.L., R. Baccala, K.M. Haraldsson, D. Choubey, T.A. Stewart, D.H. Kono, and A.N. Theofilopoulos. 2003. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J. Exp. Med. 197:777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hron, J.D., and S.L. Peng. 2004. Type I IFN protects against murine lupus. J. Immunol. 173:2134–2142. [DOI] [PubMed] [Google Scholar]
- 38.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9–mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto, M., S. Sato, H. Hemmi, K. Hoshino, T. Kaisho, H. Sanjo, O. Takeuchi, M. Sugiyama, M. Okabe, K. Takeda, and S. Akira. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 301:640–643. [DOI] [PubMed] [Google Scholar]
- 40.Goeckeritz, B.E., M. Flora, K. Witherspoon, Q. Vos, A. Lees, G.J. Dennis, D.S. Pisetsky, D.M. Klinman, C.M. Snapper, and J.J. Mond. 1999. Multivalent cross-linking of membrane Ig sensitizes murine B cells to a broader spectrum of CpG-containing oligodeoxynucleotide motifs, including their methylated counterparts, for stimulation of proliferation and Ig secretion. Int. Immunol. 11:1693–1700. [DOI] [PubMed] [Google Scholar]
- 41.Wang, Y., and A.M. Krieg. 2003. Synergy between CpG- or non-CpG DNA and specific antigen for B cell activation. Int. Immunol. 15:223–231. [DOI] [PubMed] [Google Scholar]
- 42.Ishii, K.J., K. Suzuki, C. Coban, F. Takeshita, Y. Itoh, H. Matoba, L.D. Kohn, and D.M. Klinman. 2001. Genomic DNA released by dying cells induces the maturation of APCs. J. Immunol. 167:2602–2607. [DOI] [PubMed] [Google Scholar]
- 43.Boule, M.W., C. Broughton, F. Mackay, S. Akira, A. Marshak-Rothstein, and I.R. Rifkin. 2004. Toll-like receptor 9–dependent and –independent dendritic cell activation by chromatin–immunoglobulin G complexes. J. Exp. Med. 199:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Latz, E., A. Schoenemeyer, A. Visintin, K.A. Fitzgerald, B.G. Monks, C.F. Knetter, E. Lien, N.J. Nilsen, T. Espevik, and D.T. Golenbock. 2004. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 5:190–198. [DOI] [PubMed] [Google Scholar]
- 45.Casciola-Rosen, L.A., G. Anhalt, and A. Rosen. 1994. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 179:1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mukae, N., H. Yokoyama, T. Yokokura, Y. Sakoyama, and S. Nagata. 2002. Activation of the innate immunity in Drosophila by endogenous chromosomal DNA that escaped apoptotic degradation. Genes Dev. 16:2662–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida, H., Y. Okabe, K. Kawane, H. Fukuyama, and S. Nagata. 2005. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6:49–56. [DOI] [PubMed] [Google Scholar]
- 48.Cohen, P.L., R. Caricchio, V. Abraham, T.D. Camenisch, J.C. Jennette, R.A. Roubey, H.S. Earp, G. Matsushima, and E.A. Reap. 2002. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 196:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manderson, A.P., M. Botto, and M.J. Walport. 2004. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 22:431–456. [DOI] [PubMed] [Google Scholar]
- 50.Sato, S., O. Takeuchi, T. Fujita, H. Tomizawa, K. Takeda, and S. Akira. 2002. A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and -independent pathways. Int. Immunol. 14:783–791. [DOI] [PubMed] [Google Scholar]
- 51.Muzio, M., D. Bosisio, N. Polentarutti, G. D'Amico, A. Stoppacciaro, R. Mancinelli, C. van't Veer, G. Penton-Rol, L.P. Ruco, P. Allavena, and A. Mantovani. 2000. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 164:5998-6004. [DOI] [PubMed] [Google Scholar]
- 52.Hornung, V., S. Rothenfusser, S. Britsch, A. Krug, B. Jahrsdorfer, T. Giese, S. Endres, and G. Hartmann. 2002. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 168:4531–4537. [DOI] [PubMed] [Google Scholar]
- 53.Diebold, S.S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 54.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 55.Diebold, S.S., M. Montoya, H. Unger, L. Alexopoulou, P. Roy, L.E. Haswell, A. Al-Shamkhani, R. Flavell, P. Borrow, and C. Reis e Sousa. 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 424:324–328. [DOI] [PubMed] [Google Scholar]
- 56.Hoebe, K., E.M. Janssen, S.O. Kim, L. Alexopoulou, R.A. Flavell, J. Han, and B. Beutler. 2003. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat. Immunol. 4:1223–1229. [DOI] [PubMed] [Google Scholar]
- 57.Balachandran, S., E. Thomas, and G.N. Barber. 2004. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 432:401–405. [DOI] [PubMed] [Google Scholar]
- 58.Hardin, J.A. 2003. Directing autoimmunity to nucleoprotein particles: the impact of dendritic cells and interferon α in lupus. J. Exp. Med. 197:681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amoura, Z., H. Chabre, S. Koutouzov, C. Lotton, A. Cabrespines, J.F. Bach, and L. Jacob. 1994. Nucleosome-restricted antibodies are detected before anti-dsDNA and/or antihistone antibodies in serum of MRL-Mp lpr/lpr and +/+ mice, and are present in kidney eluates of lupus mice with proteinuria. Arthritis Rheum. 37:1684–1688. [DOI] [PubMed] [Google Scholar]
- 60.Vlahakos, D.V., M.H. Foster, S. Adams, M. Katz, A.A. Ucci, K.J. Barrett, S.K. Datta, and M.P. Madaio. 1992. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney Int. 41:1690–1700. [DOI] [PubMed] [Google Scholar]
- 61.Alba, P., L. Bento, M.J. Cuadrado, Y. Karim, M.F. Tungekar, I. Abbs, M.A. Khamashta, D. D'Cruz, and G.R. Hughes. 2003. Anti-dsDNA, anti-Sm antibodies, and the lupus anticoagulant: significant factors associated with lupus nephritis. Ann. Rheum. Dis. 62:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waters, S.T., M. McDuffie, H. Bagavant, U.S. Deshmukh, F. Gaskin, C. Jiang, K.S. Tung, and S.M. Fu. 2004. Breaking tolerance to double stranded DNA, nucleosome, and other nuclear antigens is not required for the pathogenesis of lupus glomerulonephritis. J. Exp. Med. 199:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chan, O.T., L.G. Hannum, A.M. Haberman, M.P. Madaio, and M.J. Shlomchik. 1999. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 189:1639–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li, H., Y. Jiang, E.L. Prak, M. Radic, and M. Weigert. 2001. Editors and editing of anti-DNA receptors. Immunity. 15:947–957. [DOI] [PubMed] [Google Scholar]
- 65.Rakoff-Nahoum, S., J. Paglino, F. Eslami-Varzaneh, S. Edberg, and R. Medzhitov. 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 118:229–241. [DOI] [PubMed] [Google Scholar]
- 66.Boes, M., T. Schmidt, K. Linkemann, B.C. Beaudette, A. Marshak-Rothstein, and J. Chen. 2000. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl. Acad. Sci. USA. 97:1184–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coulie, P., and J. Van Snick. 1983. Rheumatoid factors and secondary immune responses in the mouse. I. Frequent occurrence of hybridomas secreting IgM anti-IgG1 autoantibodies after immunization with protein antigens. Eur. J. Immunol. 13:895. [DOI] [PubMed] [Google Scholar]
- 68.Clarkson, A.B., Jr., and G.H. Mellow. 1981. Rheumatoid factor-like immunoglobulin M protects previously uninfected rat pups and dams from Trypanosoma lewisi. Science. 214:186–188. [DOI] [PubMed] [Google Scholar]