

Abstract
Memory T cells, including the well-known CD4+ and CD8+ T cells, are central components of the acquired immune system and are the basis for successful vaccination. After infection, CD4+ and CD8+ T cells expand into effector cells, and then differentiate into long-lived memory cells. We show that a rare population of CD4−CD8−CD3+ αβ + γδ −NK1.1− T cells has similar functions. These cells potently and specifically inhibit the growth of the intracellular bacteria Mycobacterium tuberculosis (M. tb.) or Francisella tularensis Live Vaccine Strain (LVS) in macrophages in vitro, promote survival of mice infected with these organisms in vivo, and adoptively transfer immunity to F. tularensis LVS. Furthermore, these cells expand in the spleens of mice infected with M. tb. or F. tularensis LVS, and then acquire a memory cell phenotype. Thus, CD4−CD8− T cells have a role in the control of intracellular infection and may contribute to successful vaccination.
Mycobacterium tuberculosis (M. tb.) is one of the leading causes of death by infectious disease worldwide (1). In contrast, Francisella tularensis is not a major public health problem, but virulent strains cause an acute febrile illness with a high mortality rate, and F. tularensis is classified as a category A bioterrorism agent (2). Vaccine options for both tularemia and tuberculosis are limited, and further studies of the immune responses to these intracellular organisms are essential to facilitate the development of new vaccines.
CD4+, CD8+, and TCR γδ+ T cells are well-characterized T cell populations that respond to intracellular pathogens (3). Several features of T cells define their response to an invading pathogen. Upon an initial antigen encounter, naive T cells expand into a large effector cell population, of which some possess cytotoxic activities and produce essential macrophage-activating cytokines, such as IFN-γ and TNF. These effector T cell activities work to limit the growth of the pathogen within its host cell. After the peak of the T cell response, a small subset of effector cells survives and develops into a long-lived memory population. These memory cells can react rapidly to a second antigen encounter and are essential for successful vaccination.
Studies of these three major T cell subpopulations in both M. tb. and F. tularensis Live Vaccine Strain (LVS) infections have established roles for each in both infections (1, 4, 5). Aerosol infection of mice with M. tb. results in a lung disease accompanied by bacterial dissemination to other organs of the reticuloendothelial system, i.e., lymph nodes, spleen, and liver. In all tissues, development of M. tb.–specific CD4+ T cells appears critical in limiting further bacterial growth; a chronic infection ensues that is rapidly fatal if CD4+ T cells are removed. CD8+ T cells are also important, particularly in the later stages of infection, whereas the contributions of TCR γδ+ T cells are subtle (1, 6, 7). The relative importance of each T cell subpopulation in murine infection has clear parallels in human infection (1, 8). In contrast to chronic M. tb. infection, infection of mice with LVS results in either clearance and survival, or death, depending on the route of introduction: the i.p. LD50 is essentially 1 bacterium, but when given subcutaneously or intradermally (i.d.), the LD50 is much higher, on the order of 106 bacteria (4, 9, 10). Survival of a sublethal primary i.d. infection consistently leads to very strong and easily measurable specific protective immunity against a secondary lethal i.p. challenge. Similar to M. tb. infection, CD4+, but also CD8+, T cells contribute to resolution of primary i.d. infection, as well as to protection against secondary challenge (5, 11).
In contrast to the well-known CD4+ and CD8+ T cell subpopulations, CD4−CD8− double negative (DN) T cells comprise a mixed population of cells that are found in small numbers in WT mice. A subset of this DN T cell population is TCRαβ+CD3+Thy1+, and some cells lack expression of NK1.1 and TCR γδ markers (12). Previous studies in both rodents and humans have described alternative non-CD4, non-CD8 α/β+ T cells, particularly in autoimmune syndromes (13, 14) or KO mice (15, 16), but the biological role of such DN T cells is unclear. We (5) and others (11) observed the presence of a CD4−CD8−Thy1+ population during in vivo F. tularensis and Listeria monocytogenes infections (17), prompting our further interest. Our previous in vitro studies, using a culture system designed to measure the impact of LVS-immune T cells on intracellular bacterial growth, suggested activity for CD4−CD8−Thy1+ cells that were TCR αβ+ (12). Here we further investigate in detail the role of this DN T cell subset in immune responses to infections with LVS, as well as M. tb. We show that DN T cells are potent effector cells that expand after infection with F. tularensis and M. tb., can control the intracellular growth of these organisms, and acquire a memory cell phenotype after the contraction phase of the T cell response. These findings demonstrate a previously unappreciated role for this DN T cell subset in in vivo protection against intracellular infections.
Results
Pathogen-specific DN T cells control intramacrophage growth of LVS and M. tb.
Our previous studies demonstrated that antigen-specific CD4−CD8−Thy1+ cells control F. tularensis LVS growth in murine BM-derived macrophages (BMMØs) in vitro, suggesting T cell-related functions rather than NKT cell–like functions (12). To further characterize the role of DN cells during LVS infection, as well as other intracellular infections such as tuberculosis, we isolated these cells from the spleens of mice infected with either M. tb. or LVS and compared their ability to inhibit intramacrophage growth of bacteria using an in vitro culture system. The culture system used for these experiments was designed to detect the ability of immune cells to limit the growth of intracellular bacteria in their preferred host cells, i.e., primary resting macrophages. Of note, only immune T cells have activity in both the LVS and M. tb. culture systems, and most if not all bacterial growth occurred inside cells (18, 19). Mice immune to either LVS or M. tb. were depleted in vivo of CD4+, CD8+, NK1.1+, and TCR γδ+ cells before harvest of splenocytes, and the remaining cells were further purified to yield a Thy1+CD4−CD8−NK1.1−B220−TCRγδ− T cell population (DN T cells; see Materials and methods). These Thy1+ cells were predominantly (>80–90%) TCRαβ+CD3+ (see Fig. 3), and also lacked expression of the NKT cell marker DX5 (not depicted). The small number of contaminating Thy1− cells consisted of macrophages, neutrophils, and granulocytes; none of these cells exhibit anti-LVS or anti–M. tb. activities in these in vitro systems (18, 19). As seen previously (12), co-culture of highly enriched LVS-immune DN T cells with LVS-infected BMMØs resulted in a significant (P < 0.001) inhibition of LVS growth as compared with naive splenocytes (Fig. 1 a). Similarly, M. tb.–immune DN T cells significantly inhibited M. tb. intramacrophage growth (P < 0.001 as compared with naive splenocytes) with a magnitude similar to the control provided by whole M. tb.–immune splenocytes (Fig. 1 b). Investigation of the specificity of DN T cells isolated from M. tb.–immune, LVS-immune, and L. monocytogenes–immune mice in the LVS and M.tb BMMØ co-culture systems revealed that DN T cells only inhibit intramacrophage growth of the organism for which they were primed (Fig. 2, a and b). DN T cells were obtained from L. monocytogenes–immune mice as a specificity control; i.e, an immune population derived from an intracellular bacterial infection but with no known antigens in common with M. tb. or LVS. Thus, DN T cells contributed to in vitro control of both murine tuberculosis and LVS infection in a pathogen-specific manner.
Figure 3.

Highly purified LVS-immune DN T cells control the intracellular growth of F. tularensis LVS. Enriched DN T cells from LVS-immune mice were prepared as in Fig. 1. The resulting cells were labeled with a panel of fluorescently labeled antibodies to cell surface markers and further selected via flow sorting on the basis of their expression of both TCR β and Thy 1.2, as well as their lack of expression of CD4, CD8, TCR δ, NK1.1, B220, and CD11b. In this experiment, 81–83% of the enriched cells expressed Thy1.2 before sorting (a and b); after sorting, the purified cells were 99.6% positive for both TCR β and Thy1.2 (c). To compare the function of the enriched and purified DN T cells, these cells were co-cultured with LVS-infected BMMØs (d). BMMØ co-cultures contained splenocytes from either uninfected mice (naive spleen); highly enriched LVS-immune DN T cells (presort); DN T cells purified by flow sorting (TCRβ+Thy1+); or no spleen cells (BMMØ + LVS). Bacterial growth was determined as described in Fig. 1. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Bacterial uptake by the BMMØs was determined to be log10 1.52 ± 0.187 CFU/ml at time 0. *, P < 0.01 compared with co-cultures containing naive spleen cells.
Figure 1.
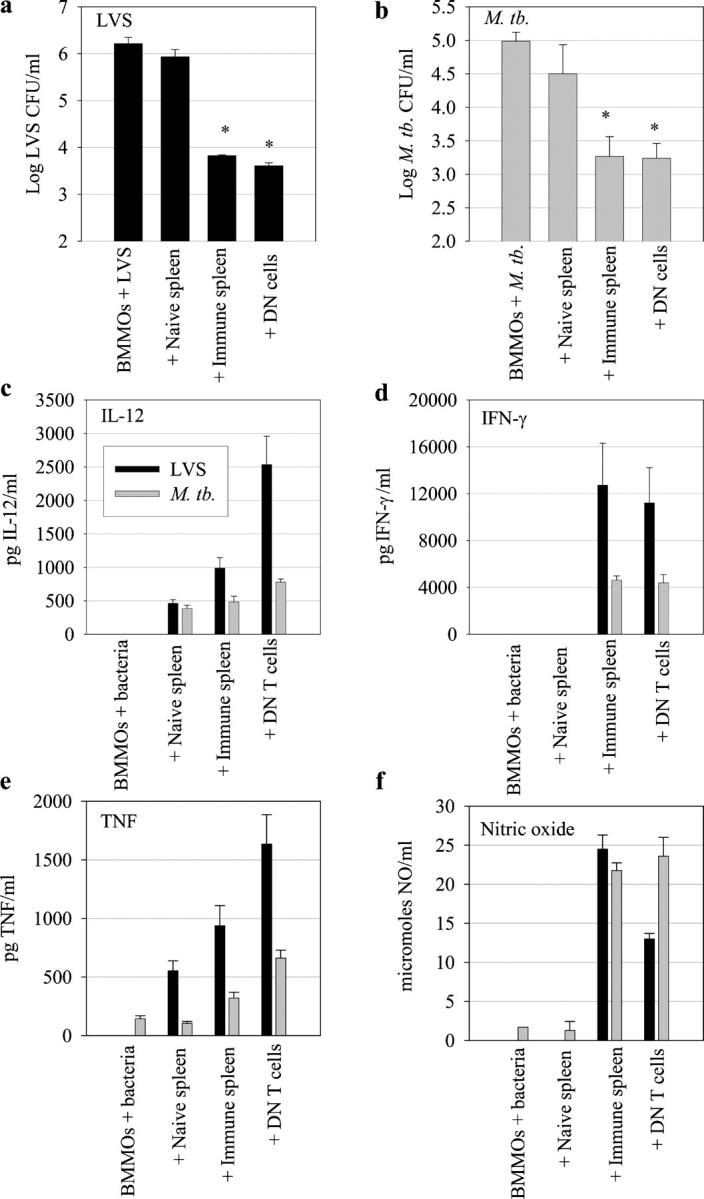
Control of F. tularensis LVS and M. tb. macrophage intracellular growth by DN T cells in vitro. BMMØs from WT mice were infected with LVS at an MOI of 1:20 (bacterium/macrophage) or with M. tb. at an MOI of 1:100. Infected BMMØs were co-cultured with splenocytes from uninfected mice (naive spleen); splenocytes from C57BL/6J mice infected intradermally with LVS 4 wk earlier (a) or with M. tb. by aerosol 4 wk earlier (b, immune spleen); highly enriched DN T cells (prepared as described in Materials and methods) from LVS-immune or M. tb.–immune spleens; or no spleen cells (BMMØ + LVS or M. tb.). Immediately after infection of the BMMØs, splenocytes were added to the indicated wells at a ratio of 1:2 (splenocyte/BMMØ). 3 d (LVS) or 8 d (M. tb.) after infection, BMMØs were washed, lysed, and plated to determine the levels of intracellular bacteria. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Bacterial uptake by the BMMØs was determined to be log10 2.040 ± 0.154 CFU/ml (LVS) and 3.441 ± 0.200 CFU/ml (M. tb.) at time 0. These data are representative of eight (a) or five (b) experiments of similar design. *, P < 0.001 compared with co-cultures containing naive spleen cells. (c–f) Supernatants from triplicate samples obtained from LVS co-cultures (black bars) or M. tb. co-cultures (gray bars) immediately before macrophage lysis from the indicated cultures were assessed by ELISA for IL-12, IFN-γ, or TNF-α, or by Griess reaction as an indirect measure of NO. Values shown in panels c–e are mean pg/ml ± SEM of cytokine. Values shown in panel f are mean μmol/ml ± SEM of NO.
Figure 2.

DN T cell specificity: assessment of the ability of DN T cells isolated from immune and nonimmune sources to control bacterial intracellular growth. Highly enriched DN T cells were prepared from LVS-immune, M. tb.–immune, L. monocytogenes–immune, or naive mice. The enriched DN T cell populations were co-cultured with BMMØs infected with either F. tularensis LVS (a) or M. tb. (b). Bacterial growth was determined as described in Fig. 1. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). Bacterial uptake by the BMMØs was determined to be log10 1.318 ± 0.275 CFU/ml (LVS) and 2.448 ± 0.114 CFU/ml (M. tb.) at time 0. These data are representative of two experiments of similar design. *, P < 0.01 (a) and P < 0.05 compared with co-cultures containing naive spleen cells.
To investigate the cytokine response generated by DN T cells, cytokine and nitric oxide (NO) levels in the co-culture supernatants were examined (Fig. 1, c–f). In both the LVS and M. tb. co-cultures, supernatants from infected macrophages alone, or cultures containing naive splenocytes, had low to undetectable levels of IFN-γ, TNF-α, IL-12, and NO. In contrast, cultures containing whole primed splenocytes or purified DN T cells contained high levels of these factors; in repeated experiments, the levels of these factors were roughly comparable between cultures containing whole primed splenocytes, enriched T cells, and purified DN T cells. Levels of IL-10 and IL-4 were below the limit of detection. Thus, DN T cells in both the LVS and M. tb. co-cultures are Th1-like.
As the DN T cells commonly copurified with a small TCRαβ−Thy1+ population, we used flow cytometry to sort these two populations from LVS-immune splenocytes (Fig. 3, a–c) and examined their ability to control LVS growth in BMMØs in vitro (Fig. 3 d). These highly purified TCR αβ+ DN T cells (>99% CD3+Thy1+) also significantly controlled (P < 0.01) LVS growth, with a magnitude similar to the starting DN T cell population (Fig. 3 d).
Mechanisms used by DN T cells to control M. tb. and LVS intracellular growth
To characterize the mechanisms used by DN T cells to control LVS and M. tb. intracellular growth, we co-cultured LVS-immune or M. tb.–immune WT DN T cells with infected BMMØs obtained from WT IFN-γR KO, TNFR 1/2 KO, iNOS KO, and P2X7R KO mice (Fig. 4, a and b). The uptake of either LVS or M. tb. bacteria was comparable in all types of macrophages (see legend to Fig. 4). In the LVS-immune DN T cell co-cultures, partial but significant loss of LVS growth control was observed in co-cultures containing IFN-γR KO, TNFR 1/2 KO, and iNOS KO BMMØs as compared with WT BMMØs (P < 0.05), but not P2X7R KO BMMØs (P >0.05). Similarly, in the M. tb.–immune DN T cell co-cultures, significant loss of control of M. tb. growth was observed in the IFN-γR KO and TNFR 1/2 KO BMMØs as compared with WT BMMØs (P < 0.05). In repeated experiments, the magnitude of M. tb. and LVS growth inhibition in the TNFR 1/2 KO and iNOS KO BMMØ cultures was variable; for both pathogens, loss of growth control was significant (P < 0.05) in some experiments and not significant in others. In contrast, loss of M. tb. and LVS growth inhibition was significant in all experiments for the IFN-γR KO BMMØ cultures.
Figure 4.
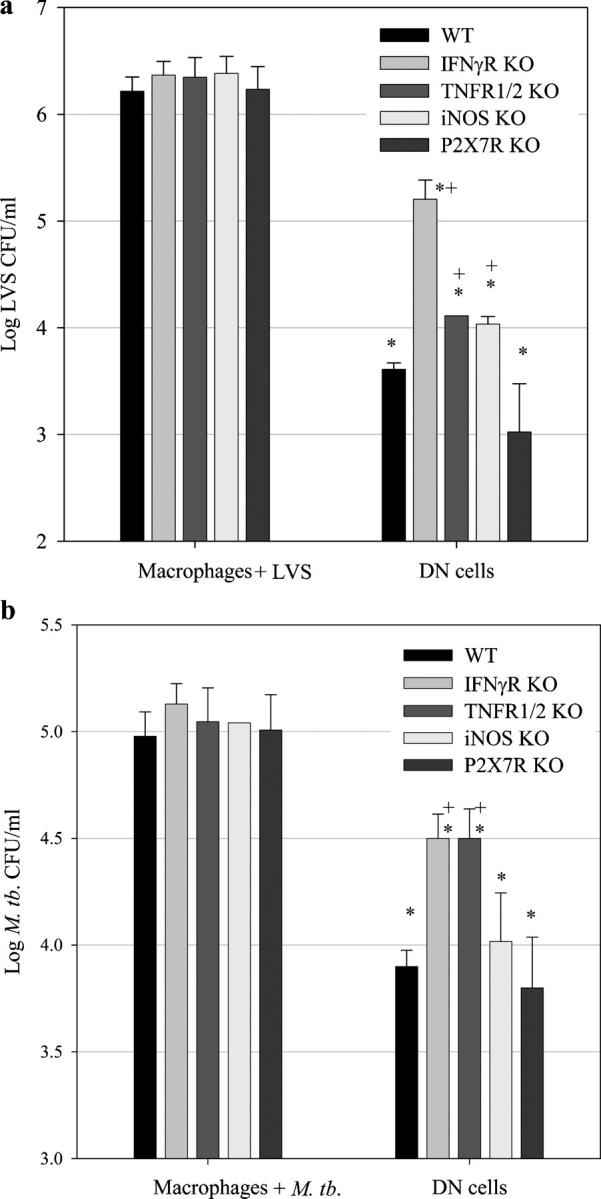
LVS or M. tb.–immune DN T cells control the intracellular growth of F. tularensis LVS and M. tb. in part by production of IFN-γ and TNF-α. BMMØs from WT mice, IFN-γR KO mice, TNFR 1/2 KO mice, iNOS KO mice, or P2X7R KO mice (as indicated by the respective shaded bars) were infected with LVS at an MOI of 1:20 (bacterium/macrophage) or with M. tb. at an MOI of 1:100. The indicated infected BMMØs were co-cultured with highly enriched WT DN T cells (prepared as described in Materials and methods) from LVS-immune spleens (a), M. tb.–immune spleens (b), or no spleen cells (a, BMMØ + LVS and b, BMMØ + M. tb.). 3 d (LVS) or 8 d (M. tb.) after infection, BMMØs were washed, lysed, and plated to determine the levels of intracellular bacteria. Values shown are the mean numbers of CFU/ml ± the SEM of viable bacteria (triplicate samples). LVS uptake by the different BMMØ monolayers at time 0 was as follows: log10 1.401 ± 0.174 CFU/ml (WT), log10 1.492 ± 0.199 CFU/ml (IFN-γR KO), log10 1.46 ± 0.151 CFU/ml (TNFR 1/2 KO), log10 1.360 ± 0.102 CFU/ml (iNOS KO), and log10 1.360 ± 0.318 CFU/ml (P2X7R KO). M. tb. uptake for the different BMMØ monolayers at time 0 was as follows: log10 3.367 ± 0.096 CFU/ml (WT), log10 3.390 ± 0.154 CFU/ml (IFN-γR KO), log10 3.284 ± 0.255 CFU/ml (TNFR 1/2 KO), log10 3.340 ± 0.297 CFU/ml (iNOS KO), and log10 3.465 ± 0.061 CFU/ml (P2X7R KO). In all cases, bacterial uptake in the KO BMMØs was not significantly different from their WT counterparts (P > 0.20). These data are representative of three (a and b) experiments of similar design. *, P < 0.05 compared with co-cultures containing macrophages + LVS; +, P < 0.05 compared with co-cultures containing DN T cells and WT BMMØs.
DN T cells contribute to control of intracellular infections in vivo
We next investigated the ability of DN T cells to control primary in vivo LVS infection in the absence of all other T cell types. Before and throughout a primary sublethal LVS infection, groups of mice were depleted in vivo of either all Thy1+ T cells or CD4+, CD8+, NK1.1+, and TCR γδ+ cells (antibody combination); thus, DN T cells are the only known T cell subpopulation remaining after the latter treatment. Of note, previous studies indicate that T cells are absolutely required for the clearance of sublethal LVS infection, and B cells, macrophages, DCs, and neutrophils are insufficient (4, 20–23). Survival of mice and growth of LVS in the organs of the depleted mice were compared with that of PBS-treated mice (Fig. 5 a). By day 7, a significant (P < 0.01) difference in spleen bacterial burdens was evident between the Thy1+-depleted mice and the other two treatment groups (antibody combination and PBS mice). The Thy1+-depleted mice succumbed to the infection, with a mean survival time of 11.8 ± 1.2 d. In contrast, both the PBS-treated and antibody combination–treated mice survived for the duration of the experiment (68 d). Furthermore, although the PBS-treated mice had no detectable bacterial burden in their organs by day 21 after infection, the antibody combination–depleted mice exhibited a chronic infection for the entire treatment period (68 d; Fig. 5 a). Thus, during a primary sublethal LVS infection, DN T cells controlled bacterial growth and promoted survival, but were unable to completely eliminate infection from tissues.
Figure 5.

DN T cells contribute to control of bacterial infection in vivo and expand during the course of in vivo bacterial infection. To assess the contribution of DN T cells to primary infection, mice were treated with either PBS, a combination of antibodies designed to remove all T cells except DN T cells (anti-CD4, anti-CD8, anti-NK1.1, and anti–TCR γδ), or with anti-Thy1.2 to remove all T cells, and then infected with a sublethal i.d. dose of LVS (a) or by aerosol with M. tb. (b) Numbers of CFU in organs (n = 4) were assessed for bacterial burdens at the indicated time points (a), or 30 d after aerosol infection (b). In panel a, mice treated with anti-Thy1.2 died by day 11, precluding further analysis of that group. Values shown are mean numbers of CFU/ml ± the SEM of viable bacteria. *, P < 0.01 compared with CFUs in organs obtained from PBS-treated mice. To assess the status of responding DN T cells during primary infection, mice were infected with a sublethal i.d. dose of LVS (c) or with a low aerosol dose of M. tb. (d), and total numbers of the indicated T cell populations in spleens (±SEM) of four infected mice were assessed by flow cytometry for each time point. In panel c, the numbers of DN T cells in normal mice were also compared with the numbers in mice depleted of all other T cells before infection (Ab tx, antibody combination). These data are representative of three (a and c) or two (b and d) experiments of similar design. Black circles, numbers of CD4+ T cells; white circles, numbers of CD8+ T cells; black triangles, numbers of DN T cells in normal mice; white triangles, numbers of DN T cells in antibody combination–treated mice.
Unlike LVS infection of WT mice, which is cleared within 3–4 wk, murine tuberculosis is chronic (1). Mice deficient in either CD4+ or CD8+ T cells exhibit greatly increased susceptibility to M. tb. as compared with WT mice, indicating that both cell types are essential for resistance to primary infection (1). To examine the ability of DN T cells to control an M. tb. infection in the absence of other T cell types, mice were given a 102-CFU aerosol M. tb. Erdman challenge. 2 wk after infection, mice were depleted in vivo of either all Thy1+ T cells or CD4+, CD8+, NK1.1+ and TCR γδ+ cells. Survival of mice and growth of M. tb. in the organs of depleted mice were compared with that of PBS-treated mice. By day 28, both the Thy1+-depleted mice and the antibody combination–depleted mice exhibited significantly (P < 0.01) increased lung, liver, and spleen organ burdens as compared with the PBS-treated mice (Fig. 5 b). Multicolor immunohistochemistry analyses of sections of lungs from long-term infected and antibody-depleted mice, stained with various antibodies to cell surface markers, further supported the efficiency of depletion, indicating the absence of CD4+ cells and the presence of the remaining CD3+ cells infiltrating lung tissue (unpublished data). The PBS-treated mice all survived for >6 mo, when observation was terminated. Both the Thy1+-depleted and antibody combination–depleted treatment groups succumbed to the infection; however, the mean survival time of the Thy1+-depleted mice was 40 ± 1 d, whereas the antibody combination–depleted mice had a significantly increased survival time of 51 ± 3 d (P < 0.001). Thus, DN T cells alone were sufficient to significantly prolong survival.
DN T cells expand and undergo effector to memory cell transition during infection
Naive CD4+ and CD8+ T cells encountering specific antigens expand into effector cell populations. This expansion is followed by a contraction phase, resulting in a population of memory T cells (24–26). We therefore wished to determine whether DN T cells also followed this program of expansion, contraction, and differentiation into memory cells. To this end, we followed the absolute numbers of CD4+, CD8+, and DN T cells in the spleens of PBS-treated mice during a sublethal LVS infection; the numbers of DN T cells in the spleens of antibody combination–depleted mice were also measured (Fig. 5 c). CD4+, CD8+, and DN T cell populations in PBS-treated mice expanded by day 7 after infection; numbers peaked at day 14, shortly after LVS growth began to diminish (Fig. 5 c). Flow cytometric analysis revealed that 3.5% of naive DN T cells express CD25 (IL-2Rα), which increased to 35% by day 7 after infection, during the expansion phase of the T cell response. This was followed by an apparent contraction phase for these cells and concomitant loss of CD25 expression. In this experiment, CD4+, CD8+, and DN T cell numbers increased in spleens by approximately six-, three-, and eightfold, respectively, before returning to their initial levels. In contrast, DN T cell numbers in antibody combination–depleted mice reached a peak at day 14 that remained high for the duration of the experiment, which was consistent with the chronic infection observed in these mice (Fig. 5 a). Interestingly, the numbers of DN T cells expand in spleens after depletion but before infection (Fig. 5 c), possibly suggesting homeostatic proliferation. Expansion of CD4+, CD8+, and DN T cells in the spleens of normal mice after a low-dose M. tb. aerosol infection was also evident (Fig. 5 d). Consistent with the slow growth of M. tb. in the tissues and the chronic nature of the infection, DN T cell numbers peaked at day 28 after infection and remained high for the duration of the experiment. Thus, DN T cells followed a time course of expansion and contraction similar to CD4+ and CD8+ T cells in the spleens of mice infected with either LVS or M. tb.
Changes in expression of other surface markers on CD4+ and CD8+ T cells can be used to describe the progression of cells from naive (CD44lo CD45RBhi CD62Lhi IL-7Rαhi), to effector (CD44hi CD45RBhi CD62Llo IL-7Rαlo), to a memory cell phenotype (CD44hi CD45RBlo CD62Lhi and lo IL-7Rαhi; references 24–26). In particular, recent experiments indicate an important role for IL-7 in T cell survival, promoting long-term persistence of both naive and memory T cells through the regulation of apoptosis (27). Here we examined the expression of these markers on DN T cells during a sublethal LVS infection. Consistent with CD4 and CD8 T cell experiments, 42% of naive DN T cells expressed IL-7Rα, but this number diminished to only 12% at the peak of the DN T cell response (day 14 after infection), followed by the emergence of a larger IL-7Rα–positive population (32%) by day 28 (Fig. 6 a). Similarly, CD62L, CD44, and CD45RB expression on DN T cells followed a pattern consistent with their development from naive, to effector, and finally to memory T cells (Fig. 6 a). Thus, DN T cells acquired typical memory cell surface markers after LVS infection.
Figure 6.


DN T cells acquire memory cell markers during primary sublethal LVS infection and transfer immunity to LVS in vivo. Mice infected with a sublethal i.d. dose of LVS were treated with a combination of antibodies designed to remove all T cells except DN T cells (anti-CD4, anti-CD8, anti-NK1.1, and anti–TCR γδ). Spleens were removed from naive mice (naive) or at the indicated time points after LVS infection. Splenocytes were labeled with a panel of fluorescent antibodies to cell surface markers and gated on the basis of their expression of Thy 1.2, as well as their lack of expression of CD4, CD8, TCR δ, NK1.1, B220, and CD11b. This gated DN T cell population was then further assessed by flow cytometry for expression of the memory cell markers IL-7Rα, CD62L, CD44, and CD45RB (a). The numbers represent the percentage of cells in each population that falls within the designated gate. Red lines, isotype control; black lines, antibody-specific staining. Similar results were observed using this flow cytometry analysis on mice not depleted before analyses. In panel b, enriched CD4+, CD8+, and DN T cells were prepared from LVS-immune mice (described in Materials and methods). Whole naive and LVS-immune splenocytes were also prepared, and 107 of each of these cell preparations were adoptively transferred i.p. to naive C57/BL6 mice. 2 h later, all mice were given a lethal 102 LVS i.p. challenge. Bacterial liver and spleen burdens were determined 3 d after infection and are represented as the mean numbers of CFU/organ ± the SEM of viable bacteria. The number of survivors/total mice for each group is also shown. These data are representative of three (a) or two (b) experiments of similar design. *, P < 0.01 compared with CFUs in organs obtained from mice given naive spleen.
We next sought to determine whether DN memory T cells adoptively transferred immunity in vivo. DN T cells were isolated from the spleens of LVS-immune mice and were transferred to naive mice before a lethal LVS i.p. challenge. Bacterial burdens in spleens were assessed 3 d after challenge, and survival of the recipient mice was monitored. The ability of DN memory T cells to transfer immunity was compared with that of naive splenocytes, whole LVS-immune splenocytes, and LVS-immune CD4+ and CD8+ T cells (Fig. 6 b). Bacterial burdens in spleens and livers of mice receiving whole LVS-immune splenocytes, CD4+, CD8+, and DN T cells were significantly diminished as compared with mice receiving naive splenocytes (P < 0.01). Consistent with these observations, all mice that received CD4+, CD8+, or DN LVS-immune T cells survived lethal LVS challenge, whereas all mice receiving naive cells succumbed to the infection (Fig. 6 b). These results indicate that DN T cells functioned as true memory cells in transferring protective immunity to naive mice.
Discussion
Improvements in vaccine development rely on advances in our knowledge of the cell types and cytokines that contribute to protective immunity. For intracellular pathogens, CD4+ and CD8+ T cells are considered the primary mediators of long-lived protective memory. We show, for the first time, that another T cell type contributes considerably to in vivo control of infections with two intracellular pathogens, M. tb. and F. tularensis LVS. These DN T cells, derived from LVS and M. tb.–immune mice, are pathogen-specific, Th1-like T cells expressing TCR αβ and CD3; importantly, these cells acquire a memory phenotype after infection. DN T cells potently control the intracellular growth of M. tb. and LVS in vitro, expand in vivo, and contribute to control of in vivo infection by several mechanisms, including cytokine production and possibly cytotoxicity.
In both mice and humans, protective immune responses to M. tb. and to Francisella depend on cell-mediated immunity provided by T cells and appear to rely heavily on appropriate CD4+ T cell activity (1, 4). This is clearly illustrated by the impact of HIV infection and subsequent reduction in CD4+ T cell numbers on susceptibility to tuberculosis (1). CD8+ cells also contribute notably to both infections (1, 4). The effector mechanisms used by CD4+ and CD8+ T cells remain the subject of intense study, but clearly include production of IFN-γ and TNF-α for the activation of macrophages, leading to production of toxic reactive oxygen and nitrogen species, as well as classical CD8+ T cell cytotoxic activities (1, 3). We have taken advantage of an in vitro culture system to further understand the various effector mechanisms used by DN T cells. We find that TNF, iNOS, and especially IFN-γ contribute to M. tb. and LVS growth control by DN T cells (Fig. 4), but other mechanisms that remain to be defined are also important. However, stimulation of macrophage P2X7R by DN T cell products is not a likely alternative mechanism. Others have presented evidence that control of latent M. tb. infection in mice is dependent on IFN-γ but independent of iNOS (28). One iNOS-independent candidate is LRG-47, a 47-kD guanosine triphosphatase family member (29), and another is the recently identified product of the Ipr1 gene, a putative IFN-γ–regulated transcriptional cofactor (30). The role of these molecules in the control of LVS and M. tb. intracellular growth by CD4+, CD8+, and DN T cells is currently under investigation. Preliminary studies indicate LVS-immune DN T cells derived from perforin KO mice retain anti-LVS activity, and abrogation of DN T cell FasL function has no impact on anti-LVS activity (unpublished data). Interestingly, however, ∼20% and 5% of LVS-immune DN T cells express intracellular granzyme B on stimulation through CD3 or via LVS-infected BMMØs, respectively (unpublished data), indicating that these cells possess unconventional cytotoxic activity that merits further detailed study.
Current studies are also seeking to determine the developmental origin and MHC restriction profiles of these DN T cells, which appear to be complex. The lack of expression of the NK1.1 and DX5 markers on these cells indicate that they are very unlikely to be NKT cells. To date, we have obtained functional DN T cells from LVS-immune class II KO mice, β2microglobulin KO mice, and CD8 KO mice (unpublished data), indicating that DN T cell development and function does not rely exclusively on any one of these elements. For several reasons, we find it unlikely that these DN cells arise from normal CD4+ or CD8+ T cells that have lost expression of their respective costimulatory molecules after activation, such as been observed after HIV infection (31). First, DN T cells were readily detected in and obtained from normal uninfected mice (see Figs. 2, 5, and 6). Second, in this study, DN cells expanded in mice that were depleted of all CD4 and CD8 cells before infection. Third, retroviruses, in particular, commonly down-regulate their own receptors, whereas LVS and M. tb. are not known to interact directly with either CD4 or CD8. However, whether these cells naturally lost expression of CD4 and CD8 because of high-avidity TCR signals during thymic development (32, 33) remains to be determined.
Other studies have described DN T cells in a variety of genetically deficient mice, including TCR transgenic mice (34), CD4 KO mice (15, 16, 35), and autoimmune syndromes including lpr and FasL-deficient mice (14, 33, 36). These animals may have expanded or abnormal accumulations of DN T cells as a result of disregulated immune systems; cells lacking expression of CD4 and/or CD8 in these mouse models arise as a consequence of the mutation, and do not reflect normal physiology. There have only been sporadic descriptions of DN T cells in normal mice. DN T cells that possess regulatory T cell activities in tissue allograft rejection models have been described (37, 38); in one case, these cells acquired and presented alloantigen to syngeneic CD8+ T cells, suppressing their function (38). In addition to these artificial models, isolated reports previously suggested that DN T cells in normal mice might contribute to immune responses. A population of DN T cells proliferates and produces IFN-γ and TNF-α in response to L. monocytogenes (39) and mouse CMV (40) infections. DN T cells are 70–90% of lymphocytes in the genital tract of Chlamydia trachomatis–infected mice (41). However, the few murine descriptions of the presence of DN T cells have not addressed their biological importance or determined whether they contribute to memory immune responses. In humans, DN T cell numbers increase during staphylococcal toxic shock syndrome (42) and HIV infection (43, 44) and respond to lipid M. tb. antigens (45, 46). Thus, it is possible that humans have a similar population with an important role in immune defense.
We demonstrate that a small DN T cell population is found in the spleens of normal WT mice before, during, and after intracellular bacterial infections, and that these cells clearly participate in control of infection. These experiments unambiguously assign a normal physiological function to cells that were previously of uncertain utility and consequence. Most importantly, we show that these cells acquire a memory T cell phenotype and, thus, may be a new target for vaccination.
Materials and Methods
Bacteria.
M. tb. Erdman (originally obtained from the collection of F. Collins, Center for Biologics Evaluation and Research [CBER]/Food and Drug Administration [FDA], Rockville, MD) was grown and frozen as previously described (18), as was F. tularensis LVS (29684; American Type Culture Collection; references 4, 12). Viable bacteria were quantified by plating serial dilutions on Mueller-Hinton agar or 7H11 plates, respectively.
Animals, infections, and determination of bacterial organ burdens.
Male-specific, pathogen-free C57BL/6J mice, as well as all C57 KO mice (except P2X7), were purchased from the Jackson Laboratory. P2X7 KO mice (47) were obtained from C. Gabel (Pfizer, Groton, CT) and bred at CBER/FDA. All animals were housed in a barrier environment at CBER/FDA, and procedures were performed according to approved protocols under Animal Care and Use Committee guidelines. Throughout, “LVS immune” refers to splenoctyes obtained from mice given a sublethal priming dose of 104 LVS i.d. 1–3 mo before death. “M. tb. immune” refers to splenocytes obtained from mice given a low-dose aerosol infection of M. tb. Erdman using an inhalation exposure system (model A4212; Glas-Col), as previously described (18), 1–3 mo before death. “L. monocytogenes immune” refers to splenoctyes obtained from mice given a sublethal priming dose of 103 L. monocytogenes, strain EGD, i.d. or i.p. 1–3 mo before death. Lethal LVS infections for adoptive transfer studies were given i.p. All materials, including bacteria, were diluted in PBS (Cambrex) containing <0.01 ng/ml endotoxin, including 0.05% Tween 80 for M. tb. The numbers of CFUs were determined in emulsified organs from infected mice as previously described (4, 12, 18), using groups of three to five mice.
In vitro assessment of control of intracellular bacterial growth in BMMØs.
The in vitro culture systems used, and the validation of the culture systems' abilities to reflect known parameters of T cell activities during in vivo control of bacterial growth, have been described in detail elsewhere (18, 19). In brief, BMMØs were used as the target cells. BM was flushed from femurs of healthy mice with DMEM (Life Technologies) supplemented with 10% heat-inactivated FCS (HyClone), 10% L-929–conditioned medium, 0.2 mM l-glutamine (Life Technologies), 1 mM Hepes buffer (Life Technologies), and 0.1 mM nonessential amino acids (Life Technologies); the resulting mixture was termed complete DMEM (cDMEM). Cells were washed, and a single-cell suspension was prepared, plated at 106 viable cells per well in 48-well plates in cDMEM supplemented with 50 μg/ml gentamycin (Life Technologies) and incubated at 37°C in 5% CO2. After 1 d, the medium was replaced with antibiotic-free cDMEM and incubated for an additional 6 d at 37°C in 5% CO2. The medium was replaced with fresh, gentamicin-free cDMEM every 2 d during the 7-d incubation.
After the 7-d culture period, the BMMØs formed a confluent monolayer, and the concentration of BMMØs was estimated to be 5 × 106 cells/well in 48-well plates. BMMØs were then infected with F. tularensis LVS or M. tb. Erdman according to the following protocol: bacteria were diluted from frozen stocks in cDMEM and added at a multiplicity of infection (MOI) of 1:20 (LVS) or 1:100 (M. tb.) bacterium/BMMØ. A low MOI was chosen to mimic initial in vivo exposure to relatively small numbers of organisms and permit controlled infection of the macrophage monolayer over 3 d (LVS), or 8 d (M. tb.). These different time points were selected to accommodate the different growth rates of the two organisms and were based on previous data indicating the respective peaks of bacterial growth that coincided with the maximal impact of co-cultured immune cells (18, 19). LVS or M. tb. was coincubated with BMMØs at 37°C in 5% CO2 for 2 h and washed three to five times with PBS (Cambrex). The LVS-infected monolayers were then incubated for 45 min to 1 h in cDMEM supplemented with 50 μg/ml gentamicin to eliminate extracellular bacteria, and infected BMMØs were washed extensively. M. tb.–infected monolayers were not treated with antibiotics, but were washed extensively. After the last wash, PBS was replaced with 1 ml/well cDMEM, and spleen cells or separated subpopulations were added to the indicated wells for co-culture. Cultures were incubated at 37°C in 5% CO2 for the remainder of the experiment. To determine bacterial uptake, some BMMØs were lysed with sterile distilled water (LVS) or sterile distilled water containing 0.01% saponin (M. tb.) for 2–3 min immediately after infection and washing with PBS. Culture lysates were serially diluted in PBS (LVS) or PBS + 0.01% Tween 80 (M. tb.) and plated on Mueller-Hinton agar (LVS) or 7H11 agar (M. tb.) plates. Plates were incubated for 2–3 d (LVS) or 3 wk (M. tb.) at 37°C in 5% CO2, and colonies were counted. The growth of bacteria in BMMØs was further monitored by lysing cultures at the indicated time points after co-culture, plating lysates, and counting colonies, as described above.
Viability of the macrophage monolayer was routinely assessed during this time using both trypan blue exclusion and light microscopy. Macrophage monolayers at the end of the 3-d (LVS) or 8-d (M. tb.) incubation periods exhibited morphological changes consistent with infection; however, the monolayers were still intact, viable, and adherent (18, 19).
Spleens used for co-culture were aseptically removed from selected mice and disrupted with a 3-ml syringe plunger. A single-cell suspension was prepared, and erythrocytes were lysed with ammonium chloride. Cells were washed, their viability was assessed by exclusion of trypan blue, and cells were resuspended in Dulbecco PBS–2% FCS to the appropriate concentration and added to BMMØ cultures. Unless otherwise stated, 2.5 × 106 splenocytes (or their separated subpopulations) were added to the 48-well cultures (∼1 splenocyte to 2 BMMØs).
Enrichment of T cell subpopulations (for in vitro co-culture and in vivo adoptive transfer).
DN T cells were enriched using a combination of in vivo depletion by antibody treatment, in vitro depletion using MACS beads, and positive selection via MACS beads using a magnetic cell sorting system (Miltenyi Biotech). This approach is a further refinement of previously described methods (12). In brief, LVS- or M. tb.–immune mice were depleted in vivo of CD4+, CD8+, NK1.1+, and TCR γδ+ cells before harvest of spleens (see In vivo depletion…subpopulations section); splenocytes were further depleted in vitro using B220+ MACS cell depletion columns, and the remaining DN T cells were obtained using a Thy1.2+ MACS cell enrichment column. Separated and purified cell populations of CD4+ or CD8+ T cells were similarly prepared from the respective depleted mice, followed by enrichment with the appropriate MACS columns. All separated subpopulations were analyzed by flow cytometry to determine the purity of the cells. The purified cells were routinely >80–90% of the intended cell type; the identity of the other contaminating cells were always accounted for and included macrophages, neutrophils, or granulocytes that do not have antibacterial activity on the BMMØ monolayer in the in vitro system (18, 19).
Quantitation of cytokines and NO in BMMØ culture supernatants.
Culture supernatants were assayed for IFN-γ, IL-12, TNF-α, and IL-4 by standard sandwich ELISAs as previously described (12, 18, 19), using reagents obtained from BD Biosciences and quantified by comparison to recombinant standards. NO was detected in culture supernatants by the Griess reaction as previously described (48), using commercial Griess reagent (Sigma-Aldrich).
In vivo depletion of cell subpopulations.
Antibodies for in vivo depletion of CD4+ T cells (clone GK1.5), CD8+ T cells (clone 2.43), TCR γδ+ T cells (clone GL3), NK1.1+ cells (clone PK136), and Thy1.2+ cells (clone 30-H12) were produced as ascites in BALB/c nu/nu mice, precipitated with 50% ammonium sulfate, quantified using a capture ELISA specific for rat IgG or mouse IgM, and tested for endotoxin levels (Cambrex); all mAb preparations used in vivo contained <10 EU/ml endotoxin. Antibodies were administered to mice i.p. as previously described (5, 12, 18). For CD4+, CD8+, and TCR γδ+ T cell subpopulations, cell depletions routinely reduced the depleted cell type to <0.5% detectable remaining cells in spleens of treated mice. However, the effectiveness of NK1.1+ cell depletion approached 80% and was more variable.
Flow cytometry analysis and sorting.
Single cell suspensions were prepared and stained for a panel of murine cell surface markers and analyzed using a flow cytometer (LSR II; Becton Dickinson) and FACSDiVa BD Biosciences) or FlowJo (Tree Star, Inc.) software, essentially as previously described (5, 12, 18, 19). Clones used included RM4-5 (anti-CD4), 53-6.7 (anti-CD8a), GL3 (anti–TCR γδ), NK1.1 (anti-NK1.1), 53-2.1 (anti-Thy1.2), H57-597 (anti–TCR β), 17A2 (anti-CD3), M1/70 (anti-CD11b), RA3-6B2 (anti–CD45/B220), M1/70 (anti-CD11b), NK1.1 (anti-NK1.1), DX5 (anti-CD49b), and GL3 (anti–TCR γδ). All preceding antibodies, as well as FcBlock, were obtained from BD Biosciences, and optimal concentrations were determined in separate experiments for use in three- to five-color staining protocols as required using appropriate fluorochrome-labeled isotype control antibodies.
To assess expansion of CD4+, CD8+, and DN T cell populations, spleen cells were harvested at the time points after infection specified in the figures, total cell numbers were counted, and the percentages of CD4+ and CD8+ T cells in each spleen were determined. Spleens from LVS-infected mice were prepared using pressure disruption, whereas spleens from M. tb.–infected mice were prepared using Stomacher bags for greater biosafety containment. The latter technique yields slightly fewer total recovered cells, resulting in slightly lower T cell subpopulation numbers in M. tb. experiments (compare day 0 of Fig. 3 c with d). Spleen cells that were negative for CD4, CD8, NK1.1, TCR γδ, B220, and CD11b, but positive for Thy1, were designated DN T cells, and total cell numbers calculated.
To follow the expression of memory cell markers, LVS-infected mice were treated in vivo with antibodies specific for the depletion of CD4+ T cells, CD8+ T cells, NK1.1+ cells, and TCR γδ+ cells. All lymphocyte-gated cells that were negative for CD4, CD8, NK1.1, TCR γδ, B220, and CD11b, but positive for Thy1, were considered DN T cells, gated, and analyzed for expression of CD25 (clone PC61), CD44 (clone IM7), CD45RB (clone C363.16A), CD62L (clone MEL-14), and IL-7Rα (clone A7R34).
Purification of DN T cells via sorting was performed at the National Institute of Allergy and Infectious Diseases (NIAID)/National Institutes of Health (NIH) Flow Cytometry Facility on a FACStar Plus (Becton Dickinson). DN T cells were enriched from LVS-immune mouse spleens as described in Enrichment of T cell…transfer section. The enriched Thy1+ DN T cell population was further sorted, selecting for cells that were Thy1+TCRαβ+ or Thy1+TCRαβ−, as well as negative for CD4, CD8, NK1.1, TCR γδ, and B220.
Adoptive transfer of LVS-immune cell subpopulations.
Splenocytes from LVS-immune mice were prepared and enriched for various T cell subpopulations, as described in detail in Enrichment of T…transfer section, and transferred i.p. to naive C57BL/6J mice and mice challenged i.p. with a lethal 102 dose of LVS IP. Bacterial burdens were assessed 3 d after i.p. challenge at the peak of the secondary infection (9, 10, 21), and other recipient mice were observed for survival of challenge for 30 d.
Acknowledgments
The authors are grateful to their colleagues, Sheldon Morris, Suzanne Epstein, Jerry Weir, and Alan Sher, for helpful advice, discussions, and reviews of the manuscript; to Kevin Holmes and Thomas Moyer of the NIAID/NIH Flow Cytometry Facility for cell sorting; and to Susan Colombini and Michael Goldberg for excellent technical assistance.
The authors have no conflicting financial interests.
Abbreviations used: BMMØ, BM-derived macrophage; DN, double negative; i.d., intradermal(ly); LVS, Live Vaccine Strain; MOI, multiplicity of infection; M. tb., Mycobacterium tuberculosis.
References
- 1.Flynn, J.L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129. [DOI] [PubMed] [Google Scholar]
- 2.Dennis, D.T., T.V. Inglesby, D.A. Henderson, J.G. Bartlett, M.S. Ascher, E. Eitzen, A.D. Fine, A.M. Friedlander, J. Hauer, M. Layton, et al. 2001. Tularemia as a biological weapon: medical and public health management. JAMA. 285:2763–2773. [DOI] [PubMed] [Google Scholar]
- 3.Schaible, U.E., H.L. Collins, and S.H. Kaufmann. 1999. Confrontation between intracellular bacteria and the immune system. Adv. Immunol. 71:267–377. [DOI] [PubMed] [Google Scholar]
- 4.Elkins, K.L., S.C. Cowley, and C.M. Bosio. 2003. Innate and adaptive immune responses to an intracellular bacterium, Francisella tularensis live vaccine strain. Microbes Infect. 5:135–142. [DOI] [PubMed] [Google Scholar]
- 5.Yee, D., T.R. Rhinehart-Jones, and K.L. Elkins. 1996. Loss of either CD4+ or CD8+ T cells does not affect the magnitude of protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 157:5042–5048. [PubMed] [Google Scholar]
- 6.Ladel, C.H., C. Blum, A. Dreher, K. Reifenberg, and S.H.E. Kaufmann. 1995. Protective role of α/β T cells and γ/δ T cells in tuberculosis. Eur. J. Immunol. 25:2877–2881. [DOI] [PubMed] [Google Scholar]
- 7.D'Souza, C.D., A.M. Cooper, A.A. Frank, R.J. Mazzaccaro, B.R. Bloom, and I.M. Orme. 1997. An anti-inflammatory role for gamma delta T lymphocytes in acquired immunity to Mycobacterium tuberculosis. J. Immunol. 158:1217–1221. [PubMed] [Google Scholar]
- 8.Ellner, J.J., C.S. Hirsch, and C.C. Whalen. 2000. Correlates of protective immunity to Mycobacterium tuberculosis in humans. Clin. Infect. Dis. 30:S279–S282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elkins, K.L., R.K. Winegar, C.A. Nacy, and A.H. Fortier. 1992. Introduction of Francisella tularensis at skin sites induces resistance to infection and generation of protective immunity. Microb. Pathog. 13:417–421. [DOI] [PubMed] [Google Scholar]
- 10.Fortier, A.H., M.V. Slayter, R. Ziemba, M.S. Meltzer, and C.A. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 59:2922–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conlan, J.W., A. Sjöstedt, and R.J. North. 1994. CD4+ and CD8+ T-cell-dependent and -independent host defense mechanisms can operate to control and resolve primary and secondary Francisella tularensis LVS infection in mice. Infect. Immun. 62:5603–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cowley, S.C., and K.L. Elkins. 2003. Multiple T cell subsets control Francisella tularensis LVS intracellular growth without stimulation through macrophage interferon γ receptors. J. Exp. Med. 198:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Straus, S.E., M. Sneller, M.J. Lenardo, J.M. Puck, and W. Strober. 1999. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann. Intern. Med. 130:591–601. [DOI] [PubMed] [Google Scholar]
- 14.Cohen, P.L., and R.A. Eisenberg. 1991. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 9:243–269. [DOI] [PubMed] [Google Scholar]
- 15.Locksley, R.M., S.L. Reiner, F. Hatam, D.R. Littman, and N. Killeen. 1993. Helper T cells without CD4: control of leishmaniasis in CD4-deficient mice. Science. 261:1448–1451. [DOI] [PubMed] [Google Scholar]
- 16.Tyznik, A.J., J.C. Sun, and M.J. Bevan. 2004. The CD8 population in CD4-deficient mice is heavily contaminated with MHC class II–restricted T cells. J. Exp. Med. 199:559–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunn, P.L., and R.J. North. 1991. Resolution of primary murine listeriosis and acquired resistance to lethal secondary infection can be mediated predominantly by Thy-1+ CD4− CD8− cells. J. Infect. Dis. 164:869–877. [DOI] [PubMed] [Google Scholar]
- 18.Cowley, S.C., and K.L. Elkins. 2003. CD4+ T cells mediate IFN-gamma-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J. Immunol. 171:4689–4699. [DOI] [PubMed] [Google Scholar]
- 19.Bosio, C.M., and K.L. Elkins. 2001. Susceptibility to secondary Francisella tularensis LVS infection in B cell deficient mice is associated with neutrophilia but not with defects in specific T cell mediated immunity. Infect. Immun. 69:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elkins, K.L., T. Rhinehart-Jones, C.A. Nacy, R.K. Winegar, and A.H. Fortier. 1993. T-cell-independent resistance to infection and generation of immunity to Francisella tularensis. Infect. Immun. 61:823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elkins, K.L., T.R. Rhinehart-Jones, S.J. Culkin, D. Yee, and R.K. Winegar. 1996. Minimal requirements for murine resistance to infection with Francisella tularensis LVS. Infect. Immun. 64:3288–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Culkin, S.J., T. Rhinehart-Jones, and K.L. Elkins. 1997. A novel role for B cells in early protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 158:3277–3284. [PubMed] [Google Scholar]
- 23.Elkins, K.L., C.M. Bosio, and T.R. Rhinehart-Jones. 1999. Importance of B cells, but not specific antibodies, in primary and secondary protective immunity to the model intracellular bacterium, Francisella tularensis live vaccine strain. Infect. Immun. 67:6002–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seder, R.A., and R. Ahmed. 2003. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat. Immunol. 4:835–842. [DOI] [PubMed] [Google Scholar]
- 25.Kaech, S.M., E.J. Wherry, and R. Ahmed. 2002. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2:251–262. [DOI] [PubMed] [Google Scholar]
- 26.Sallusto, F., J. Geginat, and A. Lanzavecchia. 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22:745–763. [DOI] [PubMed] [Google Scholar]
- 27.Kaech, S.M., J.T. Tan, E.J. Wherry, B.T. Konieczny, C.D. Surh, and R. Ahmed. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198. [DOI] [PubMed] [Google Scholar]
- 28.Flynn, J.L., C.A. Scanga, K.E. Tanaka, and J. Chan. 1998. Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 160:1796–1803. [PubMed] [Google Scholar]
- 29.MacMicking, J.D., G.A. Taylor, and J.D. McKinney. 2003. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 302:654–659. [DOI] [PubMed] [Google Scholar]
- 30.Pan, H., B.S. Yan, M. Rojas, Y.V. Shebzukhov, H. Zhou, L. Kobzik, D.E. Higgins, M.J. Daly, B.R. Bloom, and I. Kramnik. 2005. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 434:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marodon, G., D. Warren, M.C. Filomio, and D.N. Posnett. 1999. Productive infection of double-negative T cells with HIV in vivo. Proc. Natl. Acad. Sci. USA. 96:11958–11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budd, R.C., and P.F. Mixter. 1995. The origin of CD4−CD8−TCR alpha beta+ thymocytes: a model based on T-cell receptor avidity. Immunol. Today. 16:428–431. [DOI] [PubMed] [Google Scholar]
- 33.Mixter, P.F., J.Q. Russell, G.J. Morrissette, C. Charland, D. Aleman-Hoey, and R.C. Budd. 1999. A model for the origin of TCR-alphabeta+ CD4−CD8− B220+ cells based on high affinity TCR signals. J. Immunol. 162:5747–5756. [PubMed] [Google Scholar]
- 34.Blank, C., I. Brown, R. Marks, H. Nishimura, T. Honjo, and T.F. Gajewski. 2003. Absence of programmed death receptor 1 alters thymic development and enhances generation of CD4/CD8 double-negative TCR-transgenic T cells. J. Immunol. 171:4574–4581. [DOI] [PubMed] [Google Scholar]
- 35.Rahemtulla, A., W.P. Fung-Leung, M.W. Schilham, T.M. Kündig, S.R. Sambhara, A. Narendran, A. Arabian, A. Wakeham, C.J. Paige, R.M. Zinkernagel, R.G. Miller, and T.W. Mak. 1991. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 353:180-184. [DOI] [PubMed] [Google Scholar]
- 36.Ford, M.S., K.J. Young, Z. Zhang, P.S. Ohashi, and L. Zhang. 2002. The immune regulatory function of lymphoproliferative double negative T cells in vitro and in vivo. J. Exp. Med. 196:261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strober, S., L. Cheng, D. Zeng, R. Palathumpat, S. Dejbakhsh-Jones, P. Huie, and R. Sibley. 1996. Double negative (CD4−CD8− alpha beta+) T cells which promote tolerance induction and regulate autoimmunity. Immunol. Rev. 149:217–230. [DOI] [PubMed] [Google Scholar]
- 38.Zhang, Z.X., L. Yang, K.J. Young, B. DuTemple, and L. Zhang. 2000. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat. Med. 6:782–789. [DOI] [PubMed] [Google Scholar]
- 39.Kadena, T., G. Matsuzaki, S. Fujise, K. Kishihara, H. Takimoto, M. Sasaki, M. Beppu, S. Nakamura, and K. Nomoto. 1997. TCR alpha beta+ CD4− CD8− T cells differentiate extrathymically in an lck-independent manner and participate in early response against Listeria monocytogenes infection through interferon-gamma production. Immunology. 91:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hossain, M.S., H. Takimoto, T. Ninomiya, H. Yoshida, K. Kishihara, G. Matsuzaki, G. Kimura, and K. Nomoto. 2000. Characterization of CD4−CD8−CD3+ T-cell receptor-alphabeta+ T cells in murine cytomegalovirus infection. Immunology. 101:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johansson, M., and N. Lycke. 2003. A unique population of extrathymically derived alpha beta TCR+CD4−CD8− T cells with regulatory functions dominates the mouse female genital tract. J. Immunol. 170:1659–1666. [DOI] [PubMed] [Google Scholar]
- 42.Carulli, G., G. Lagomarsini, A. Azzara, R. Testi, R. Riccioni, and M. Petrini. 2004. Expansion of TcRalphabeta+CD3+CD4−CD8− (CD4/CD8 double-negative) T lymphocytes in a case of staphylococcal toxic shock syndrome. Acta Haematol. 111:163–167. [DOI] [PubMed] [Google Scholar]
- 43.Mathiot, N.D., R. Krueger, M.A. French, and P. Price. 2001. Percentage of CD3+CD4−CD8−gammadeltaTCR− T cells is increased HIV disease. AIDS Res. Hum. Retroviruses. 17:977–980. [DOI] [PubMed] [Google Scholar]
- 44.Moreau, J.F., J.L. Taupin, M. Dupon, J.C. Carron, J.M. Ragnaud, C. Marimoutou, N. Bernard, J. Constans, J. Texier-Maugein, P. Barbeau, et al. 1996. Increases in CD3+CD4−CD8− T lymphocytes in AIDS patients with disseminated Mycobacterium avium-intracellular complex infection. J. Infect. Dis. 174:969–976. [DOI] [PubMed] [Google Scholar]
- 45.Porcelli, S.A., and R.L. Modlin. 1999. The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu. Rev. Immunol. 17:297–329. [DOI] [PubMed] [Google Scholar]
- 46.Stenger, S., R.J. Mazzaccaro, K. Uyemura, S. Cho, P.F. Barnes, J.P. Rosat, A. Sette, M.B. Brenner, S.A. Porcelli, B.R. Bloom, and R.L. Modlin. 1997. Differential effects of cytolytic T cell subsets on intracellular infection. Science. 276:1684–1687. [DOI] [PubMed] [Google Scholar]
- 47.Labasi, J.M., N. Petrushova, C. Donovan, S. McCurdy, P. Lira, M.M. Payette, W. Brissette, J.R. Wicks, L. Audoly, and C.A. Gabel. 2002. Absence of the P2X7 receptor alters leukocyte function and attenuates an inflammatory response. J. Immunol. 168:6436–6445. [DOI] [PubMed] [Google Scholar]
- 48.Green, L.C., A. Wagner, J. Glogowski, P.L. Skipper, J.S. Wishnok, and S.R. Tannenbaum. 1982. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 126:131–138. [DOI] [PubMed] [Google Scholar]