

Abstract
Engagement of the Fas receptor (CD95) initiates multiple signaling pathways that lead to apoptosis, such as the formation of death-inducing signaling complex (DISC), activation of caspase cascades, and the generation of the lipid messenger, ceramide. Sphingomyelin (SM) is a major component of lipid rafts, which are specialized structures that enhance the efficiency of membrane receptor signaling and are a main source of ceramide. However, the functions of SM in Fas-mediated apoptosis have yet to be clearly defined, as the responsible genes have not been identified. After cloning a gene responsible for SM synthesis, SMS1, we established SM synthase–defective WR19L cells transfected with the human Fas gene (WR/Fas-SM(−)), and cells that have been functionally restored by transfection with SMS1 (WR/Fas-SMS1). We show that expression of membrane SM enhances Fas-mediated apoptosis through increasing DISC formation, activation of caspases, efficient translocation of Fas into lipid rafts, and subsequent Fas clustering. Furthermore, WR/Fas-SMS1 cells, but not WR/Fas-SM(−) cells, showed a considerable increase in ceramide generation within lipid rafts upon Fas stimulation. These data suggest that a membrane SM is important for Fas clustering through aggregation of lipid rafts, leading to Fas-mediated apoptosis.
Fas, also known as CD95, is a death domain–containing member of the TNFR super family (1). Currently, two distinct pathways that require different initiator caspases (caspase-8, -9, and -10) but converge at activation of executor caspases (caspase-3, -6, and -7) are proposed in Fas-mediated apoptosis signaling (2, 3). In type I cell apoptosis (mitochondrion independent), engagement by Fas ligand (FasL), or anti-Fas antibody (CH11), leads to receptor trimerization and recruitment of the cytoplasmic adaptor protein, Fas-associated death domain (FADD), and procaspase-8 and -10, thus forming a death-inducing signaling complex (DISC). On the other hand, in type II cell apoptosis (mitochondrion dependent), minimal caspase-8 can cleave the Bcl-2 family member Bid. The truncated Bid translocates to mitochondria, where it perturbs the mitochondrial membrane potential (ΔΨm) and facilitates the formation of mitochondrial permeability transition pores, resulting in the release of cytochrome c (4).
Aggregation and clustering of cell surface receptors on binding to their specific ligands has been reported for a variety of receptors, including the TCR–CD3 complex, B cell receptor, TNFR, epithelial-derived growth factor receptor, CD2, CD44, CD11a/CD18, and Fas, and is facilitated by localization of receptor and proximal signaling components within cholesterol, glycosphingolipids, and sphingomyelin (SM)-rich membrane microdomains, known as lipid rafts (5–9). Recently, there is accumulating evidence that rafts are involved in Fas-induced apoptosis through the translocation and clustering of Fas into rafts on stimulation (7, 10, 11). Studies have reported that Fas ligation triggers translocation of the acid sphingomyelinase (aSMase) from an intracellular compartment onto the cell surface, where it hydrolyzes SM to ceramide, and that accumulation of ceramide contributes to transforming small rafts into larger signaling platforms that trap and cluster Fas (7, 10). Rafts have been detected using choleratoxin B (CTx), which binds to ganglioside GM1 colocalized in rafts, and the role of rafts has been evaluated by the disruption of rafts using the cholesterol-chelating reagent methyl-β-cyclodextrin. However, there has been no direct evidence that membrane SM, a major raft component, is involved in raft functions or Fas-mediated apoptosis. Very recently, we and others have succeeded in cloning the human cDNA for SM synthase (SMS1; references 12 and 13). Using SM synthesis–deficient cells, and cells in which function has been restored by transfection with the novel SMS1 gene, we demonstrated that expression of membrane SM enhanced Fas-mediated apoptosis through efficient clustering of Fas itself with a concomitant increase in DISC formation, the activation of caspases, and the loss of ΔΨm.
Results
Characterization of SM-deficient and functionally restored cells by transfection with the SM synthase gene
Lysenin is an SM-directed cytolysin purified from the earthworm (14), which binds to membrane SM and induces pore formation in the plasma membrane and subsequent cell death (14). During investigation of the sphingolipid metabolism in SM synthase–defective WR19L mouse lymphoid cells transfected with the human Fas gene (WR19L/Fas; reference 15), we established membrane sphigomyelin-deficient cells, WR/Fas-SM(−), which were resistant to lysenin-mediated cell lysis. Recently, we established a functional revertant cell line, designated WR/Fas-SMS1 cells, by transfection of SMS1 (12). WR/Fas-SMS1 cells exhibited restored SM synthesis assayed by radiolabeling of cellular lipids with [14C]serine (Fig. 1 A) and recovered sensitivity against lysenin-mediated cytolysis (not depicted; reference 12).
Figure 1.

Characterization of WR/Fas-SM(−) and WR/Fas-SMS1 cells. (A) SM synthase activity of WR/Fas-SM(−) (lane 1) and WR/Fas-SMS1 (lane 2) cells. The cellular lipids were labeled with [14C]serine, extracted by the Bligh and Dyer method (reference 53), and assessed by TLC. PE, phosphatidylethanolamine; PS, phosphatidylserine. (B) Analysis of membrane SM expression by confocal microscopy. Cells were stained with lysenin-MBP, and FITC-conjugated anti–mouse IgG mAb, then examined by laser scan confocal microscopy. (C) FACS analysis of membrane sphingolipids. To detect membrane SM, cells were stained with lysenin-MBP (Lysenin), CH11, and FITC-conjugated anti–mouse IgG mAb. Surface expression of ganglioside GM1, cholesterol, and human Fas were analyzed using FITC-conjugated CTx, cholesterol-PEG (cholesterol), or anti-Fas mAB (h-Fas), respectively.
Recently, Yamaji-Hasegawa et al. produced a mutant lysenin, which specifically binds to SM without induction of cell death (16). Using the mutant lysenin conjugated with maltose-binding protein (MBP), we examined SM expression on the plasma membrane of WR/Fas-SM(−) and WR/Fas-SMS1 cells by confocal microscopy. Expression of SM detected by lysenin–MBP plus FITC-labeled anti-MBP antibody was positive in WR/Fas-SMS1 but not in WR/Fas-SM(−) cells (Fig. 1 B). Membrane expression of ganglioside GM1 detected by FITC-labeled CTx was observed on both cells (not depicted; reference 12).
We confirmed the accumulation of SM on the surface of WR/Fas-SM(−) and WR/Fas-SMS1 cells by FACS analysis. Binding of lysenin–MBP was positive in WR/Fas-SMS1 cells but not in WR/Fas-SM(−) cells. We also examined the expression of other lipid components of the plasma membrane, such as ganglioside GM1 and cholesterol, as well as the expression of human Fas. Binding of CTx, which binds the oligosaccharide portion of ganglioside GM1, considered to be a maker of lipid rafts, and binding of fluorescein ester of polyethylene glycol-derivatized cholesterol (fPEG-cholesterol; reference 17), which specifically binds membrane cholesterol, as well as expression of Fas, was detected equally on both cells (Fig. 1 C).
Fas-mediated apoptosis and loss of ΔΨm in WR/Fas-SM(−) and WR/Fas-SMS1 cells
Although Itoh et al. have reported that cross-linking of human Fas on WR19L/Fas cells promotes type I cell apoptosis (15), we found that WR/Fas-SM(−) cells are resistant to Fas-mediated apoptosis. Therefore, we used WR/Fas-SM(−) and WR/Fas-SMS1 cells to determine whether membrane SM plays an important role in Fas-mediated apoptosis. Because high antibody concentrations might cause Fas clustering (7), we determined the optimal conditions for Fas-mediated apoptosis by analyzing the effect of concentration of agonistic CH11 (mouse IgM) and the time course of response of these cells. WR/Fas-SMS1 cells underwent stronger apoptosis at 50 ng/ml CH11 than WR/Fas-SM(−) cells, exhibiting 31 and 9.9% apoptosis, respectively (Fig. 2 A). Although apoptosis was increased in a dose-dependent manner in both cells, high concentrations of CH11 (up to 500 ng/ml) induced only 24.3% apoptosis in WR/SMS(−) cells (Fig. 2 B). In subsequent experiments, cells were treated with 50 ng/ml of CH11 as the optimum condition for apoptosis. Next, cells were stimulated with 50 ng/ml CH11 for the indicated time, and WR/Fas-SMS1 cells showed stronger apoptosis compared with WR/Fas-SM(−) cells at both 3 and 6 h (Fig. 2 C).
Figure 2.

Fas-mediated apoptosis in WR/Fas-SM(−) and WR/Fas-SMS1 cells. (A and B) Dose dependency of Fas-mediated apoptosis. Cells were incubated for 3 h with the indicated concentration of CH11. After incubation, cells were harvested and analyzed by flow cytometry for DNA fragmentation using nuclear staining with PI. The numbers in each box represent the percentages of apoptotic cells. (C) Time kinetics of Fas-mediated apoptosis. Cells were incubated with 50 ng/ml of CH11 for the indicated time, and apoptosis was analyzed by flow cytometry using PI. Error bars represent SEM. (D) Time–kinetics of loss of ΔΨm in apoptotic cells. Cells were incubated with 50 ng/ml of CH11 for the indicated time. ΔΨm was determined by intracellular staining with DiOC6(3) and flow cytometry. (E) Inhibition of Fas-mediated apoptosis by caspase inhibitors. Cells were stimulated with 50 ng/ml of CH11 for 6 h in the presence of the indicated amounts of Ac-DEVD-CHO or Ac-IETD-CHO. After incubation, apoptosis was analyzed by flow cytometry using PI.
ΔΨm during apoptosis is likely to contribute to the death of the cell through the loss of mitochondrial function before DNA fragmentation in both type I and II cell apoptosis. As shown in Fig. 2 D, the treatment of WR/Fas-SMS1 cells with CH11 dramatically induced a loss of ΔΨm as determined by staining with DiOC6(3), a dye taken up by mitochondria, with similar time kinetics as Fas-mediated apoptosis. However, A majority of WR/Fas-SM(−) cells exhibited a normal ΔΨm through the assay (Fig. 2 D). The time kinetics of loss of ΔΨm were comparable to those of apoptosis.
Next, we examined whether Fas-mediated apoptosis of WR/Fas-SM(−) and WR/Fas-SMS1 cells depends on caspase activation. Cells were stimulated with 50 ng/ml CH11 for 6 h in the presence of the specific caspase-3 inhibitor (Ac-DEVD-CHO) or the specific caspase-8 inhibitor (Ac-IETD-CHO). As shown in Fig. 2 E, 100 μM of each inhibitor completely suppressed Fas-mediated apoptosis of WR/Fas-SMS1 cells.
Fas-mediated caspase-3 activation of WR/Fas-SM(−) and WR/Fas-SMS1 cells
Cells were stimulated with 50 ng/ml CH11 for the indicated time (Fig. 3 A) and at the indicated concentration for 30 min (Fig. 3 B). Western blot analysis using a caspase-3–specific antibody revealed that the active fragments (p17) of caspase-3 cleaved from 32-kD pro–caspase-3 was expressed at a higher level in WR/Fas-SMS1 cells compared with WR/Fas-SM(−) cells in time-dependent (Fig. 3 A) and concentration of Fas antibody–dependent manners (Fig. 3 B). As shown in Fig. 3 C, colorimetric assay revealed that cytoplasmic caspase-3 activity of WR/Fas-SMS1 cells increased more than twofold over baseline by Fas cross-linking. However, caspase-3 activity of WR/Fas-SM(−) cells remained at baseline for the entire 6 h.
Figure 3.
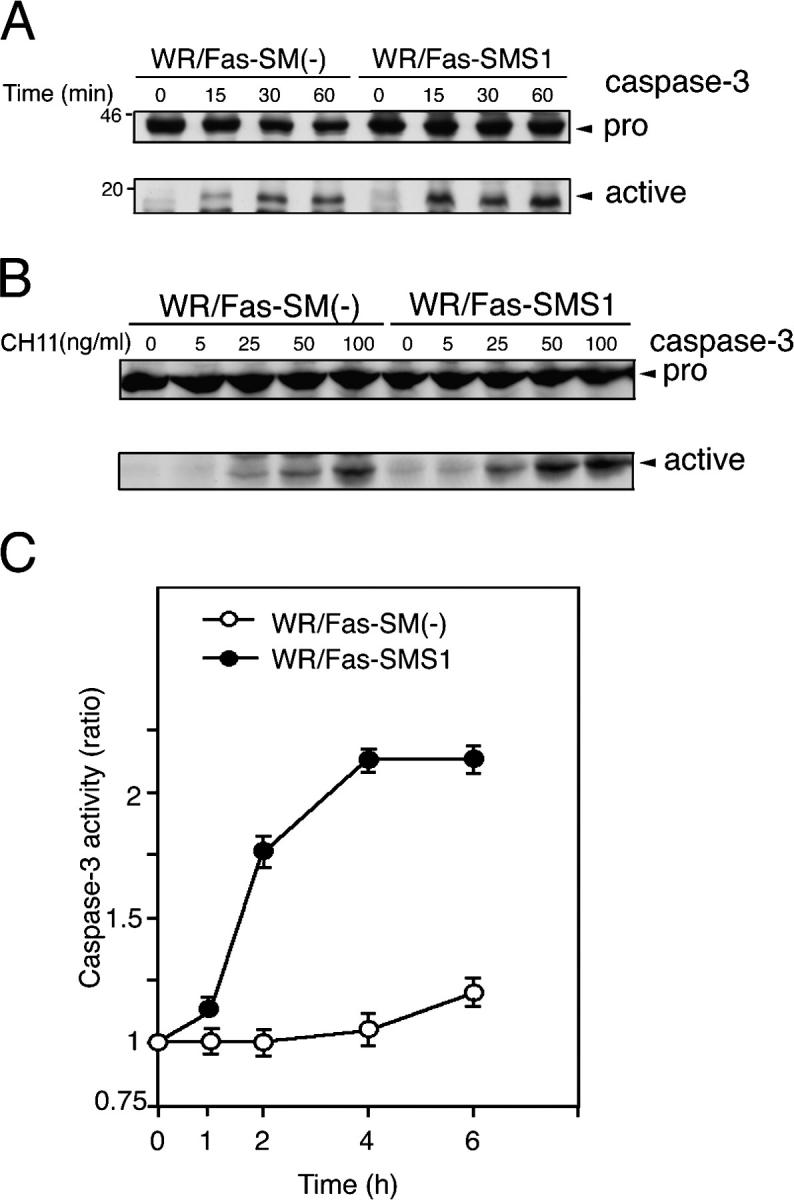
Fas-mediated caspase-3 activation in WR/Fas-SM(−) and WR/Fas-SMS1 cells. Time kinetics (A) and dose dependency (B) of caspase-3 activation by Western blot analysis. Cells were stimulated with 50 ng/ml CH11 for the indicated time (A), or stimulated with the indicated concentration of CH11 for 15 min (B). Total cell lysates were analyzed by Western blot using mouse mAb to caspase-3. The arrows indicate the bands corresponding to 32 kD for procaspase-3 (pro) and 17 kD for the active caspase (active). (C) Cells were stimulated with 50 ng/ml CH11 for the indicated time, and caspase-3 activities were measured in extracts of cell lysates using colorimetric assay kits. Each experiment was done in triplicate. Data are representative of five independent experiments. Error bars represent SEM.
Fas-mediated DISC formation and caspase-8 activation in WR/Fas-SM(−) and WR/Fas-SMS1 cells
According to the current model of type I apoptosis, binding of either the FasL or an agonistic antibody induces aggregation of Fas followed by a conformational change in its cytoplasmic domain that results in formation of the DISC (2, 3, 18). Therefore, we examined DISC formation in WR/Fas-SM(−) and WR/Fas-SMS1 cells. Cells were stimulated with 50 ng/ml CH11 for the indicated time (Fig. 4 A) and at the indicated concentration (Fig. 4 B) for 30 min. After lysis, Fas was immunoprecipitated with anti–mouse IgM antibody, and precipitated proteins were examined by immunoblotting with anti-FADD or anti–caspase-8 antibody. The results of the time course study revealed that FADD associated with Fas within 5 min in both cells. However, the association of FADD and Fas in WR/Fas-SMS1 cells was stronger and sooner (with the maximum association at 5 min) than in WR/Fas-SM(−) cells, in which the association gradually increased over the 30-min period. Caspase-8 appeared within the DISC in WR/Fas-SMS1 cells after 5 min of stimulation and reached maximum levels at 15 min. However, caspase-8 within DISC in WR/Fas-SM(−) cells was barely detectable throughout the assay (Fig. 4 A). The results of a dose-dependent experiment revealed that association of FADD and caspase-8 with Fas in WR/Fas-SMS1 cells appeared much stronger than in WR/Fas-SM(−) cells at any concentration of antibody (Fig. 4 B). Although FADD was detected in the DISC after stimulation with 5 ng/ml CH11 in both cell types, caspase-8 appeared in the DISC at a relatively high concentration of CH11; i.e., 25 ng/ml for WR/Fas-SMS1 and 50 ng/ml for WR/Fas-SM(−) cells, respectively (Fig. 4 B).
Figure 4.
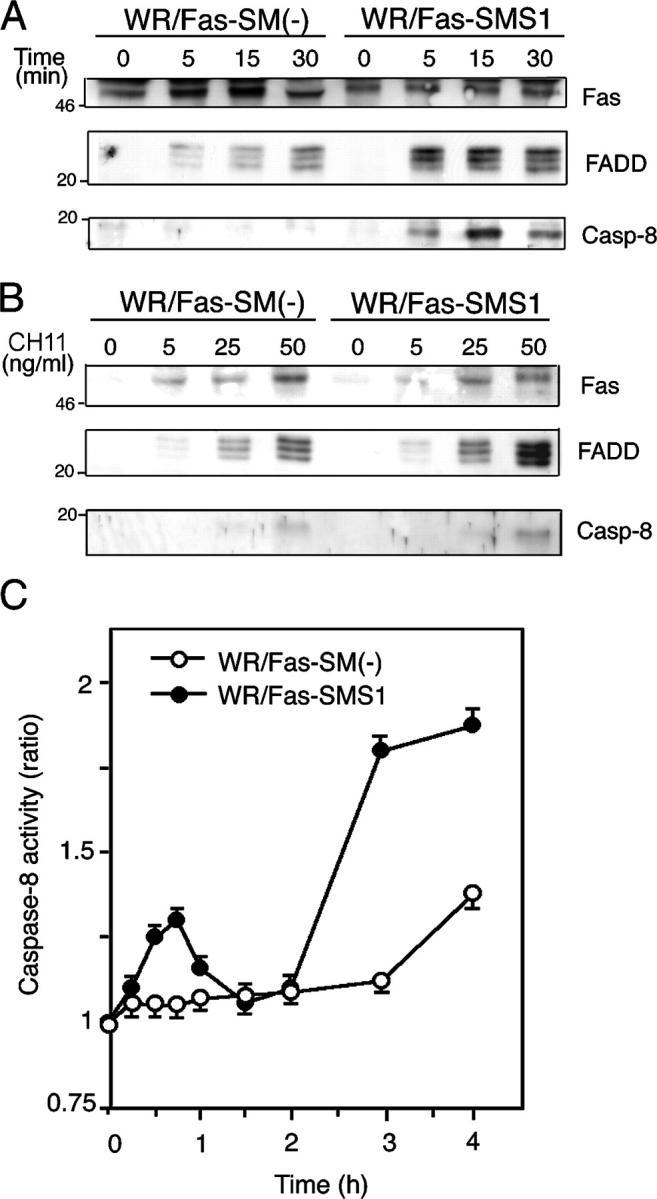
Fas-mediated DISC formation and caspase-8 activation in WR/Fas-SM(−) and WR/Fas-SMS1 cells. Time kinetics (A) and dose dependency (B) of Fas-mediated DISC formation. 2 × 107 cells were stimulated with 50 ng/ml CH11 for the indicated time (A) or stimulated for 15 min with the indicated concentration of CH11 (B). After stimulation, Fas was immunoprecipitated with anti–mouse IgM antibody from Brij 97 lysates. Immunoprecipitates were subjected to 12% SDS-PAGE and immunoblotted with anti-Fas death domain (3D5), anti-FADD, and anti–caspase-8 mAb. Data are representative of five independent experiments. (C) Fas-mediated activation of caspase-8. Cells were stimulated with 50 ng/ml CH11 for the indicated time, and caspase-8 activities were measured in extracts of cell lysates using colorimetric assay kits. Each experiment was done in triplicate. Data are representative of five independent experiments. Error bars represent SEM.
We also examined the cytoplasmic caspase-8 activity in WR/Fas-SM(−) and WR/Fas-SMS1 cells after Fas cross-linking by colorimetric assay. Interestingly, caspase-8 activities in WR/Fas-SMS1 cells exhibited a biphasic peak after stimulation: the first small peak appeared within 1 h; and the second high peak, after 2 h. However, caspase-8 activity in WR/Fas-SM(−) cells remained at baseline for 3 h, with slight increase at 4 h (Fig. 4 C).
Fas aggregation and capping in WR/Fas-SM(−) and WR/Fas-SMS1 cells
It has been reported that Fas, as well as TNFR, assemble into trimers in the absence of ligands (19, 20). Although Fas trimers do not trigger apoptosis in the resting condition, clustering of these trimers is crucial for Fas-mediated apoptosis (18, 21). CH11 causes the formation of Fas multimers, thereby inducing complete cellular activation leading to capping. Kamitani et al. reported that cross-linking of Fas on the cell surface by CH11 resulted in the formation of high molecular mass Fas aggregates, which were stable in 2% SDS and 5% β-mercaptoethanol (22). As shown in Fig. 5, stimulation with CH11 induced the formation of SDS- and 2-ME–stable, high molecular aggregates (>200 kD) of Fas in both cells, as reported by others (22, 23). Fas aggregates appeared more strongly in WR/Fas-SMS1 cells compared with WR/Fas-SM(−) cells in time-dependent (Fig. 5 A) and CH11 dose–dependent (Fig. 5 B) fashions.
Figure 5.

Fas clustering and capping in WR/Fas-SM(−) and WR/Fas-SMS1 cells. Time kinetics (A) and dose dependency (B) of Fas multimer formation. 4 × 107 cells were stimulated with 50 ng/ml CH11 for the indicated time (A), or stimulated for 10 min with the indicated concentration of CH11 (B). After stimulation, cell pellets were snap frozen and treated at 45°C for 1 h in 300 μl of a 1% NP-40–treating solution. DNA in the samples was sheared with a 25-gauge needle, and solubilized samples were loaded on 12% SDS-PAGE. Fas multimers were detected with antibody to the intracellular death domain of human Fas (3D5). Data are representative of more than three independent experiments. (C) Activation-induced capping of Fas. Cells were stained for 20 min with 50 ng/ml CH11 at 4°C. Afterwards, capping was induced by warming cells to 37°C for 30 min in a water bath with mild agitation. Cells were harvested at the indicated times and fixed with 4% paraformaldehyde for 20 min at 22°C. Fixed cells were washed twice and mounted in FITC-conjugated secondary antibody. Fluorescence was detected using a confocal microscope equipped with a SPOT digital camera. The data are representative of more than five experiments. Arrowheads indicate Fas capping. (D) Time kinetics of activation-induced Fas capping. Capping was induced for the indicated time, and cells with Fas clustering were counted by two independent observers. Percentages of capping were calculated in 150–200 total cells. These results are the mean of three independent experiments. Error bars represent SEM. *, P < 0.01.
We used confocal microscopy to examine Fas capping on the plasma membrane of WR/Fas-SM(−) and WR/Fas-SMS1 cells after treatment with 50 ng/ml CH11 as described in Materials and methods. In unstimulated cells, Fas was diffusely distributed across the surface on both WR/Fas-SM(−) and WR/Fas-SMS1 cells (Fig. 5 C, 0 min). Cross-linking of Fas with antibody induced small patches of Fas along the plasma membrane, which appeared to fuse to large clusters/capping in WR/Fas-SMS1 cells (Fig. 5 C, 30 min). The frequency of Fas capping on WR/Fas-SMS1 cells was significantly greater than that of WR/Fas-SM(−) cells at each time point (P < 0.01; Fig. 5 D).
Fas distribution in lipid rafts of WR/Fas-SM(−) and WR/Fas-SMS1 cells
Recent studies have shown that rafts play an important role in signal transduction pathways, including apoptosis and, in particular, through the organization of surface receptors, signaling enzymes, and adaptor molecules into rafts (5, 24). Muppidi et al. have reported that translocation of Fas into lipid rafts increased sensitivity to Fas-mediated apoptosis (25). To determine Fas distribution of WR/Fas-SM(−) and WR/Fas-SMS1 cells, rafts were isolated using equilibrium sucrose density gradients. The position of the membrane rafts in the sucrose gradient was determined by the presence of lck, a well-established raft-associated molecule. As shown in Fig. 6, lck was enriched in the upper part of the sucrose gradient (fractions 4 and 5), with a secondary localization at the bottom of the gradient in which a cytoskeletal protein, tublin, was localized (fractions 10–12), indicating a separation of the lipid rafts (fractions 4 and 5) from the Triton X-100–soluble membrane. Fas was detected in raft fractions of WR/Fas-SM(−) and WR/Fas-SMS1 cells before stimulation (Fig. 6 A, a and b). Although a substantial shift of Fas into raft fractions was observed in both cells after stimulation, amounts of Fas in raft fractions were greater in WR/Fas-SMS1 than in WR/Fas-SM(−) cells (Fig. 6 A, a and c vs. Fig. 6 A, b and d). The mean of three independent experiments revealed that Fas redistribution in raft fractions was significantly greater in WR/Fas-SMS1 cells than in those of WR/Fas-SM(−) cells after stimulation (P < 0.01; Fig. 6, B and C).
Figure 6.

Distribution of Fas into lipid rafts of WR/Fas-SM(−) and WR/Fas-SMS1 cells. (A) 108 cells were left unstimulated (a and b) or were stimulated with CH11 for 30 min (c and d), and Triton X-100 lysates were subjected to sucrose density gradient fractionation. Fractions were run on 15% SDS-PAGE and immunoblotted with antibodies against lck (a marker for the raft fractions), tublin (a marker for the nonraft fractions), and Fas (3D5). The blots shown are representative of four independent experiments. (B) Redistribution of Fas into lipid rafts on stimulation. Raft fractions (fraction 4) were run on 15% SDS-PAGE and immunoblotted with antibodies against lck and Fas. (C) Quantification of Fas contents in lipid rafts was performed densitometrically and normalized to the amount of lck. Data are expressed as the mean ± SEM for relative increase of three independent experiments. *, P < 0.01.
Ceramide generation in lipid rafts of WR/Fas-SM(−) and WR/Fas-SMS1 cells
Ceramide modulates the activity of a large number of proteins, and a role in apoptosis induction has been proposed (26–28). However, the signals that activate the enzyme responsible for ceramide production are not well defined, and the actual contribution of ceramide to the apoptotic response remains poorly understood. It has been reported that ceramide formation is associated with the execution phase of apoptosis as a consequence of processes downstream of the activation of caspases (29, 30). Consistent with this, we observed delayed generation of cytosolic ceramide in WR/Fas-SMS1 compared with WR/Fas-SM(−) cells after a 2-h Fas stimulation (unpublished data). However, stress-induced SM hydrolysis occurs rapidly, suggesting that the early event is located in close proximity to the plasma membrane. Recently, it has been reported that aSMase translocates into rafts within minutes after cell stimulation and catalyzes the formation of ceramide from SM (11, 31, 32). Therefore, we analyzed membrane ceramide within lipid rafts in WR/Fas-SM(−) and WR/Fas-SMS1 cells. Although WR/Fas-SM(−) cells, because of a lack of SM synthase, contain more ceramide than WR/Fas-SMS1 cells, both cells showed a similar distribution of ceramide among fractions (Fig. 7 A). Thus, in both cell types, 70% of ceramide is located in raft fractions with ∼40% in fraction 4 (Fig. 7 A, bottom). Because membrane SM is one of the major sources of ceramide generation, we next examined Fas-stimulated ceramide generation in raft fractions of WR/Fas-SM(−) and WR/Fas-SMS1 cells. After 5 min of Fas stimulation, cells were lysed, and rafts were isolated using equilibrium sucrose density gradients. Analysis of ceramide in the raft fraction revealed that Fas cross-linking increased ceramide content by 50% in WR/Fas-SMS1 cells (from 2,955 to 4,386 pmol/106 cells), but marginally decreased ceramide in WR/Fas-SM(−) cells (from 5,960 to 5,651 pmol/106 cells; Fig. 7 B). The mean of three independent experiments revealed that ceramide contents of raft fractions in WR/Fas-SMS1 cells were significantly greater than those of WR/Fas-SM(−) cells (P < 0.01; Fig. 7 C).
Figure 7.

Ceramide generation in lipid rafts of WR/Fas-SM(−) and WR/Fas-SMS1 cells. (A) Ceramide contents in membrane fractions of sucrose density gradient fractionation. 108 cells were lysed in Triton X-100 buffer and subjected to sucrose density gradient fractionation. Lipids of each fraction were extracted by the method of Bligh and Dyer (reference 53), and ceramide content was measured by the diacylglycerol kinase assay. After separation of ceramide-1-phosphates by TLC, radioactivity was visualized and estimated. The results are representative of three independent experiments and expressed as the percentage of total PSL arbitrary units. (B) Fas-mediated ceramide generation in lipid rafts. 108 cells were left unstimulated or were stimulated with CH11 for 5 min and Triton X-100 lysates were subjected to sucrose density gradient fractionation. Lipids of raft fraction (fraction 4) were extracted by the method of Bligh and Dyer, and ceramide generation was measured by the diacylglycerol kinase assay. Radioactivity was visualized and estimated. The results are representative of three independent experiments. (C) The mean of three independent experiments revealed that ceramide contents of lipid rafts in WR/Fas-SMS1 cells were significantly greater than those of WR/Fas-SM(−) cells. The results were the mean of three independent experiments and expressed as the percentage increase of PSL arbitrary units. Error bars represent SEM. *, P < 0.01.
Effects of exogenous ceramide on Fas-mediated apoptosis in WR/Fas-SM(−) and WR/Fas-SMS1 cells
We and others have shown that ceramide is a proapoptotic lipid mediator because diverse mechanisms of cell stress, including CH11 cross-linking, TNF-α treatment, irradiation, heat shock, and anticancer drugs, increases intracellular ceramide during the execution phase of apoptosis (27, 28, 31, 33–37). It has been reported that the addition of natural or C16-ceramide, which by themselves did not induce apoptosis, enabled soluble FasL to cap and completely restore the apoptosis in aSMase−/− hepatocytes (10, 11, 38). Therefore, we examined the effects of exogenous natural or C16-ceramide on Fas-mediated apoptosis and the formation of Fas multimers in WR/Fas-SM(−) and WR/Fas-SMS1 cells. Cells were pretreated with the indicated concentration of natural or C16-ceramide for 1 h. After washing, cells were stimulated with 50 ng/ml of agonist CH11 for 6 h before assaying for apoptosis, or 30 min before assessing Fas multimer formation. Although natural and C16-ceramide did not induce apoptosis by themselves, both ceramides enhanced Fas-induced apoptosis. However, high concentrations of ceramides, even at 5 μM, could not restore apoptosis of WR/Fas-SM(−) cells to the levels of WR/Fas-SMS1 cells (Fig. 8, A and B). We also examined the effects of natural or C16-ceramide on Fas multimer formation and found that pretreatment of cells with 5 μM natural or C16-ceramides did not enhance Fas multimer formation in WR/Fas-SM(−) and WR/Fas-SMS1 cells (unpublished data).
Figure 8.

Effects of exogenous ceramides on Fas-mediated apoptosis in WR/Fas-SM(−) and WR/Fas-SMS1 cells. Cells were pretreated with the indicated concentration of C16- (A) or natural (B) ceramide for 1 h. After washing, cells were stimulated with 50 ng/ml CH11 for 6 h and apoptosis was analyzed by flow cytometry using PI. Error bars represent SEM.
Discussion
Currently, two distinct pathways are proposed in Fas-mediated apoptosis signaling; i.e., type I and type II apoptosis. The consensus in type I apoptosis is that FasL, itself a homotrimer, engages three Fas monomers, leading to the assembly of a trimeric Fas receptor. This FasL-induced trimerization has been suggested to bring together death domains present in the cytoplasmic region of each Fas monomer, leading to a DISC formation. However, it has been reported that soluble FasL (sFasL) reduced apoptosis by <1,000-fold compared with membrane-bound FasL (39), and that sFasL or antibodies to Fas cause cell death more efficiently if polymerized by adsorption on plastic or by molecular cross-linking (40), suggesting that clustering of Fas is important in Fas-mediated apoptosis. Recently, Fas, as well as TNFR, has been reported to assemble into trimers in the absence of ligands through a preligand assembly domain in the extracellular, amino-terminal region of receptors (19, 41). Moreover, Siegel et al. showed that Fas trimers in the absence of FasL do not recruit the DISC, implying a lack of apoptotic signaling (19). Thus, it has been believed that the oligomerization of preassembled Fas trimers is essential for optimal acute signaling and direct activation of the caspase cascade through the DISC (18, 21). However, the molecular basis of Fas clustering is not clear.
It has been reported that Fas ligation triggers translocation of aSMase from an intracellular compartment onto the cell surface, where it hydrolyzes SM to ceramide, and that accumulation of ceramide contributes to transforming small rafts into larger aggregates for signaling platforms, which trap and cluster Fas (7, 10, 11, 31, 42). In contrast to the extensive studies of the acid or neutral SMase in cell death, the biological function of SM synthase has not been elucidated because of a lack of molecular cloning of its responsible genes. Recently, we have succeeded in cloning SMS1 (12) and established WR/Fas-SM(−) and WR/Fas-SMS1 cells (Fig. 1). Using this system to investigate the mechanisms of Fas-mediated apoptosis, we report here that restoration of membrane SM by transfection of SM synthase gene into SM-deficient cells restored Fas-mediated apoptosis (Fig. 2) through DISC formation (Fig. 4, A and B), activation of caspase-3 (Fig. 3) and caspase-8 (Fig. 4 C), and Fas clustering (Fig. 5). The equivalent expression of other lipid components in the plasma membrane, such as ganglioside GM1 and cholesterol (Fig. 1 C), and equivalent localization of raft fractions in sucrose density gradients (Fig. 6) argue against other lipid abnormalities in SM-deficient cells.
Because lipid rafts promote efficient formation of receptor-associated signaling complexes to produce the biological outcomes dictated by these complexes (24, 43–45), redistribution of Fas in lipid rafts is one possible mechanism for regulating the efficiency of Fas signaling. After titration of Triton X-100 for the isolation of lipid rafts (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20041685/DC1), we examined whether the redistribution of Fas in lipid rafts is different between WR/Fas-SM(−) and WR/Fas-SMS1 cells. Although Fas cross-linking enhanced redistribution of Fas into raft fractions in both cells, Fas contents in raft fractions were greater in WR/Fas-SMS1 than WR/Fas-SM(−) cells (Fig. 6). These results suggest that membrane SM plays a crucial role for raft partitioning of Fas, which is one of the important mechanisms in Fas-mediated apoptosis of these cells.
It has been believed that the trapping and clustering of Fas may promote the formation of multimers, which ultimately transmit a strong signal into the cell that induces apoptosis (18, 21). We examined the efficiency of Fas clustering in WR/Fas-SM(−) and WR/Fas-SMS1 cells and found that the aggregation and clustering of Fas in WR/Fas-SMS1 cells were markedly enhanced after Fas cross-linking compared with that in WR/Fas-SM(−) cells (Fig. 5), suggesting that oligomerization of Fas is crucial for optimal apoptosis signaling. Although the exact mechanisms of platform formation for Fas clustering on the membrane are not clear, it has been reported that aSMase translocates from an intracellular compartment to the extracellular surface of the cell membrane on Fas stimulation, and that translocation of aSMase to SM-rich rafts generates ceramide (11, 42). Therefore, using a diacylglycerol kinase assay, we measured ceramide content in raft fractions of WR/Fas-SM(−) and WR/Fas-SMS1 cells before and after Fas stimulation. Although ceramide generation in WR/Fas-SM(−) cells is not increased by Fas cross-linking, WR/Fas-SMS1 cells showed a 50% increase in ceramide contents in a raft fraction within 5 min after Fas stimulation (Fig. 7). Because it has been reported that ceramide displaces cholesterol from lipid rafts (46) and self-associates within lipid rafts through hydrogen bonding (47), ceramide may provide the driving force that results in the coalescence of microscopic rafts into large-membrane macrodomains (48), indicating that ceramide in rafts serves to form a signaling platform for Fas clustering (7, 49).
Ceramide has been recognized as an important intracellular lipid mediator related to a variety of cell functions, including cell differentiation and apoptosis (27, 28, 31, 33–37, 50). It has also been reported that the addition of exogenous natural or C16-ceramide enabled soluble FasL to cap and completely restored the apoptosis in aSMase−/− hepatocytes (10, 11, 38). Although exogenous ceramides partially enhanced Fas-mediated apoptosis, they could neither restore apoptosis nor enhance Fas multimer formation in WR/Fas-SM(−) cells to the levels in WR/Fas-SMS1 cells (Fig. 8). In this regard, Liu et al. have reported that IL-1β induced the loss of a resident population of SMs from the SM-rich plasma membrane (i.e., lipid rafts) and the concomitant appearance of ceramide (51). Therefore, we hypothesize that intact SM-enriched membrane domains may be essential for local ceramide production that compartmentalize lipid rafts to ceramide-enriched membrane platforms, leading to Fas capping.
Concerning the molecular ordering of the initial signaling events of CD95, Algeciras-Schimnich et al. have reported four stages: (a) ligand-induced formation of CD95 microaggregates at the cell surface; (b) recruitment of FADD to form a DISC; (c) formation of large CD95 surface clusters, which is positively regulated by DISC-generated caspase-8; and (d) internalization of activated CD95 through an endosomal pathway (52). We examined caspase-8 activation, ceramide generation within lipid rafts, redistribution of Fas within lipid rafts, and Fas multimer formation at very early time points (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20041685/DC1). Caspase-8 activation, ceramide generation, and Fas redistribution within lipid rafts were observed within 1 min in WR/Fas-SMS1 but not WR/Fas-SM(−) cells. In contrast, Fas multimer formation was observed in WR/Fas-SMS1 but not WR/Fas-SM(−) cells at later time points (after 2 min; Fig. S2). Thus, our findings support a crucial role for membrane SM in the extrinsic pathway of Fas-mediated apoptosis through the generation of ceramide in lipid rafts, which facilitates efficient Fas clustering, DISC formation, and the early caspase-8 activation leading to the downstream cascade of caspase-3 activation.
Materials and Methods
Cells and cell transfection
Mouse T cell lymphoma WR19L cells transfected with the cDNA for the human Fas gene (WR19L/Fas; reference 15) were a gift of S. Yonehara (Kyoto University, Kyoto, Japan). We isolated the SM-defective WR/Fas-SM(−) cells and the SM-containing WR/Fas-SM(+) cells from the original WR19L/Fas cells by limiting dilution. SMS1, subcloned into the pLIB expression vector, was transfected into the WR/Fas-SM(−) cells in VSV-G retroviral particles. These cells were designated WR/Fas-SMS1 cells (12).
Antibodies and reagents
Anti-Fas (CH11, mouse IgM) and anti-FADD (1F7, mouse IgG1) were purchased from MBL International Corporation. FITC-conjugated anti–mouse IgM, anti–mouse IgG2a, anti–human Fas (DX2), and anti–caspase-3/CPP32 pAb were purchased from BD Biosciences. Anti-Fas death domain (3D5) and anti–caspase-8 (1G12) antibodies were purchased from Qbiogene. Lysenin, FITC-conjugated CTx, peroxidase-conjugated CTx, and methyl-β-cyclodextrin were purchased from Sigma-Aldrich. Rabbit anti–mouse IgG mAb was purchased from Cappel. Ac-DEVD-CHO and Ac-IETD-CHO were purchased from Peptide Institute. RNase and saponin were purchased from Nacalai Tesque. The cell viability assay kit using WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium) was purchased from Wako Co. Ltd. l-[14C]serine and γ-[32P]ATP were purchased from GE Healthcare. Rabbit anti–mouse IgM (μ chain specific) antibody was purchased from Zymed Laboratories. The ECL immunodetection system and horseradish peroxidase–conjugated goat anti–mouse or anti–rabbit IgG mAb were obtained from GE Healthcare.
FACS analysis
DNA fragmentation was quantified by analyzing cell cycle and total DNA content to measure cell death by apoptosis. In brief, cells were treated with CH11 at the indicated concentration and incubated for the indicated time shown in the figures. After harvesting, cells were resuspended in permeabilization solution (0.5% paraformaldehyde and 0.5% saponin) and treated with 50 μg/ml RNase A for 30 min at room temperature, and propidium iodide (PI; Molecular Probes) was added to a final concentration of 20 μg/ml. After 20 min, the fluorescence of the PI-stained DNA was quantitated on a per cell basis using a FACSCalibur (BD Biosciences), and cells with subdiploid content were considered to be apoptotic cells.
To detect SM localized at the outer leaflet of the plasma membrane, cells were stained on ice for 30 min with nontoxic lysenin–MBP (16), incubated for 30 min with FITC-conjugated anti–mouse IgG (Sigma-Aldrich), and analyzed with a FACSCalibur. Surface expression of ganglioside GM1 and cholesterol was analyzed using FITC-conjugated CTx) or fPEG-cholesterol (17), respectively. Data were analyzed using CellQuest software (Becton Dickinson).
To assess ΔΨm, healthy or dying cells were incubated for 15 min at 37°C in buffer containing 40 nM 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3); Molecular Probes) before the addition of 5 μg/ml PI. After compensation to exclude nonviable cells, fluorescence was recorded at 525 nM (FL-1) for DiOC6(3) and 600 nM (FL-3) for PI on a FACScan (26).
Confocal microscopy
To assess Fas capping, cells were stained for 20 min with 50 ng/ml of CH11 at 4°C, and capping was induced by warming cells to 37°C in a water bath with mild agitation. Cells were harvested at the indicated times shown in the figures and fixed with 4% paraformaldehyde for 10 min at 22°C. Fixed cells were washed twice and mounted in FITC-conjugated secondary antibody. Fluorescence was detected with a confocal microscope (LSM-5 Pascal; Carl Zeiss MicroImaging, Inc.) equipped with a SPOT digital camera. Large clusters of Fas were defined as cells in which the fluorescence condenses onto >25% of the cell surface, whereas fluorescence was homogeneously distributed on the membrane of resting cells.
For visualization of SM localized at the outer leaflet of the plasma membrane, cells were allowed to settle onto slides coated with poly–l-lysine, fixed in 4% formaldehyde, stained with lysenin–MBP at 4°C for 45 min, and treated with anti-MBP.
Cell labeling and lipid separation
The method for detection of SM synthesis is described elsewhere (28). In brief, cells were reseeded at 5 × 105 cells/ml in RPMI 1640 with 2% FBS and l-[14C]serine (specific activity, 155 mCi/mmol) and incubated at 37°C in 5% CO2 for 36 h. The cell lipids were extracted by the method of Bligh and Dyer (53), applied on silica gel TLC plate (Whatman), and developed with solvent containing methyl acetate/propanol/chloroform/methanol/0.25% KCl (25:25:25:10:9). The radioactive spots were visualized and quantified using an image analyzer (BAS 2000; Fuji Photo Film).
Immunoprecipitation, Western blotting, and immunoblotting
Cells were solubilized with lysis buffer containing 50 mM Tris-HCl, pH 7.6, 1% Brij 97, 300 mM NaCl, 5 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF, and 1 mM sodium orthovanadate with gentle rocking for 30 min at 4°C. Immunoprecipitated proteins were eluted by boiling in SDS-containing sample buffer and fractionated by SDS-PAGE (8–12% polyacrylamide gels; reference 54). Proteins were electrophoretically transferred to polyvinylidene difluoride (Immobilon-P) membranes (Sigma-Aldrich). Peroxidase-conjugated secondary antibodies (GE Healthcare) were used at a 1:1,000 dilution, and immunoreactive bands were visualized using ECL. Densitometry of the protein bands was performed using National Institutes of Health image software (55). Quantitation of Fas in raft fractions was corrected to the amount of lck.
The aggregated form of Fas was detected by the method of Kamitani et al. (22). In brief, cell pellets were snap frozen to prevent protein degradation. Frozen pellets were treated at 45°C for 1 h in 300 ml of 2% treating solution containing 5% β-mercaptoethanol. DNA in the samples were sheared with a 25-gauge needle, and solubilized samples were loaded on 12% SDS-PAGE. Aggregated Fas was detected by 3D5, a mouse mAb (IgG1) specific for the intracellular death domain of human Fas (Qbiogene).
Caspase activity assay
Activities of caspase-3 and -8 were determined using a colorimetric assay kit (MBL International Corporation) according to the manufacturer's protocol. Apoptosis inhibition was assayed in cells stimulated for 6 h with 50 ng/ml CH11 in the presence of the indicated doses of Ac-DEVD-CHO or Ac-IETD-CHO shown in the figures.
Isolation of a raft fraction in equilibrium density gradients
Raft fractions were prepared as described by Rodgers and Rose (56) with minor modifications (8). In brief, 108 cells were lysed with 1 ml MS-buffered saline (MBS; 25 mM MES, pH 6.5, and 150 mM NaCl) containing 1% Triton X-100, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM PMSF, 1 mM sodium orthovanadate, and 5 mM EDTA. The lysate was homogenized with 20 strokes of a Dounce homogenizer (Iwaki Glass Co.), gently mixed with an equal volume of 80% sucrose (wt/vol) in MBS, and placed in the bottom of a 14 × 95 mm clear centrifuge tube (model 344060; Beckman Coulter). The sample was then overlaid with 6.5 ml of 30% sucrose and 3.5 ml of 5% sucrose in MBS and centrifuged at 200,000 g in a rotor (model SW40Ti; Beckman Coulter) at 4°C for 16 h. After centrifugation, 12 1-ml fractions (excluding the pellet) were collected from the top of the gradient.
Ceramide measurement
Lipids were extracted from cell lysates by the method of Bligh and Dyer (53), and ceramide mass measurement using Escherichia coli diacylglycerol kinase, which phosphorylates ceramide to ceramide-1-phosphate, was performed as described previously (50). The solvent system to separate ceramide-1-phosphate and phosphatidic acid on TLC plates consists of chloroform/acetone/methanol/acetic acid/H2O (10:4:3:2:1). Radioactivity within spots of ceramide-1-phosphate was estimated with an image analyzer system (BASE III; Fuiji) and expressed as PSL arbitrary units (54), and ceramide levels were corrected for phospholipid phosphate as described elsewhere (57).
Statistical analysis
Statistical significance was evaluated by unpaired t tests and by analysis of variance as applicable; P < 0.01 was considered statistically significant.
Online supplemental material
Fig. S1 shows concentration of Triton X-100 for lipid raft separation. Fig. S2 shows early timecourse of caspase-8 activity, ceramide generation in lipid rafts, Fas redistribution into lipid rafts, and Fas multimer formation. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20041685/DC1.
Acknowledgments
We thank Drs. H. Inoue and S. Goda of Osaka Dental University for technical assistance.
This work was supported by grants 13557160, 15024236, and 15390313 (to H. Umehara) from the Japanese Ministry of Education and Science and Culture, by the Uehara Memorial Foundation, and by grant P04244 (to Z.-X. Jin) from the Japan Society for the Promotion of Science.
The authors have no conflicting financial interests.
Abbreviations used: aSMase; acid sphingomyelinase, CTx; choleratoxin B; ΔΨm, mitochondrial membrane potential; DISC, death-inducing signaling complex; FADD, Fas-associated death domain; FasL, Fas ligand; MBP, maltose-binding protein; PI, propidium iodide; SM; sphingomyelin.
References
- 1.Marsden, V.S., and A. Strasser. 2003. Control of apoptosis in the immune system: Bcl-2, BH-3 only proteins and more. Annu. Rev. Immunol. 21:71–105. [DOI] [PubMed] [Google Scholar]
- 2.Scaffidi, C., S. Fulda, A. Srinivasan, C. Friesen, F. Li, K.J. Tomaselli, K.M. Debatin, P.H. Krammer, and M.E. Peter. 1998. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wajant, H. 2002. The Fas signaling pathway: more than a paradigm. Science. 296:1635–1636. [DOI] [PubMed] [Google Scholar]
- 4.Hengartner, M.O. 2000. The biochemistry of apoptosis. Nature. 407:770–776. [DOI] [PubMed] [Google Scholar]
- 5.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 6.Brown, D.A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- 7.Grassme, H., A. Jekle, A. Riehle, H. Schwarz, J. Berger, K. Sandhoff, R. Kolesnick, and E. Gulbins. 2001. CD95 signaling via ceramide-rich membrane rafts. J. Biol. Chem. 276:20589–20596. [DOI] [PubMed] [Google Scholar]
- 8.Inoue, H., O. Yoneda, Y. Minami, Y. Tanaka, T. Okazaki, H. Imai, E. Bloom, N. Domae, and H. Umehara. 2002. Lipid rafts as the signaling scaffold for NK cell activation: tyrosine phosphorylation and association of LAT with PI 3-kinase and PLC-γ following CD2 stimulation. Eur. J. Immunol. 32:2188–2198. [DOI] [PubMed] [Google Scholar]
- 9.Delmas, D., C. Rebe, S. Lacour, R. Filomenko, A. Athias, P. Gambert, M. Cherkaoui-Malki, B. Jannin, L. Dubrez-Daloz, N. Latruffe, and E. Solary. 2003. Resveratrol-induced apoptosis is associated with Fas redistribution in the rafts and the formation of a death-inducing signaling complex in colon cancer cells. J. Biol. Chem. 278:41482–41490. [DOI] [PubMed] [Google Scholar]
- 10.Cremesti, A., F. Paris, H. Grassme, N. Holler, J. Tschopp, Z. Fuks, E. Gulbins, and R. Kolesnick. 2001. Ceramide enables Fas to cap and kill. J. Biol. Chem. 276:23954–23961. [DOI] [PubMed] [Google Scholar]
- 11.Grassme, H., A. Cremesti, R. Kolesnick, and E. Gulbins. 2003. Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene. 22:5457–5470. [DOI] [PubMed] [Google Scholar]
- 12.Yamaoka, S., M. Miyaji, T. Kitano, H. Umehara, and T. Okazaki. 2004. Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growth in sphingomyelin synthase-deficient cells. J. Biol. Chem. 279:18688–18693. [DOI] [PubMed] [Google Scholar]
- 13.Huitema, K., J.V.D. Dikkenberg, J.F. Brouwers, and J.C. Holhuis. 2004. Identification of a family of animal sphingomyelin synthases. EMBO J. 23:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaji, A., Y. Sekizawa, K. Emoto, H. Sakuraba, K. Inoue, H. Kobayashi, and M. Umeda. 1998. Lysenin, a novel sphingomyelin-specific binding protein. J. Biol. Chem. 273:5300–5306. [DOI] [PubMed] [Google Scholar]
- 15.Itoh, N., S. Yonehara, A. Ishii, M. Yonehara, S. Mizushima, M. Sameshima, A. Hase, Y. Seto, and S. Nagata. 1991. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 66:233–243. [DOI] [PubMed] [Google Scholar]
- 16.Yamaji-Hasegawa, A., A. Makino, T. Baba, Y. Senoh, H. Kimura-Suda, S.B. Sato, N. Terada, S. Ohno, E. Kiyokawa, M. Umeda, and T. Kobayashi. 2003. Oligomerization and pore formation of a sphingomyelin-specific toxin, lysenin. J. Biol. Chem. 278:22762–22770. [DOI] [PubMed] [Google Scholar]
- 17.Sato, S.B., K. Ishii, A. Makino, K. Iwabuchi, A. Yamaji-Hasegawa, Y. Senoh, I. Nagaosa, H. Sakuraba, and T. Kobayashi. 2004. Distribution and transport of cholesterol-rich membrane domains monitored by a membrane-impermeant fluorescent polyethylene glycol-derivatized cholesterol. J. Biol. Chem. 279:23790–23796. [DOI] [PubMed] [Google Scholar]
- 18.Mundle, S.D., and A. Raza. 2002. Defining the dynamics of self-assembled Fas-receptor activation. Trends Immunol. 23:187–194. [DOI] [PubMed] [Google Scholar]
- 19.Siegel, R.M., J.K. Frederiksen, D.A. Zacharias, F.K.M. Chan, M. Johnson, D. Lynch, R.Y. Tsien, and M. Lenardo. 2000. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 288:2354–2357. [DOI] [PubMed] [Google Scholar]
- 20.Chan, F.K.M., H.J. Chun, L. Zheng, R.M. Siegel, K.L. Bui, and M.J. Lenardo. 2000. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 288:2351–2354. [DOI] [PubMed] [Google Scholar]
- 21.Golstein, P. 2000. FasL binds preassembled Fas. Science. 288:2328–2329. [DOI] [PubMed] [Google Scholar]
- 22.Kamitani, T., H.P. Nguyen, and E.T.H. Yeh. 1997. Activation-induced aggregation and processing of the human Fas antigen. J. Biol. Chem. 272:22307–22314. [DOI] [PubMed] [Google Scholar]
- 23.Lee, Y.-J., and E. Shacter. 2001. Fas aggregation does not correlate with Fas-mediated apoptosis. J. Immunol. 167:82–89. [DOI] [PubMed] [Google Scholar]
- 24.Dykstra, M., A. Cherukuri, H.W. Sohn, S.J. Tzeng, and S.K. Pierce. 2003. Location is everything: lipid rafts and immune cell signaling. Annu. Rev. Immunol. 21:457–481. [DOI] [PubMed] [Google Scholar]
- 25.Muppidi, J., and R.M. Siegel. 2004. Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic death. Nat. Immunol. 5:182–189. [DOI] [PubMed] [Google Scholar]
- 26.Kondo, T., K. Iwai, T. Kitano, M. Watanabe, Y. Taguchi, T. Yabu, H. Umehara, N. Domae, T. Uchiyama, and T. Okazaki. 2002. Control of ceramide-induced apoptosis by IGF-1: involvement of PI-3 kinase, caspase-3 and catalase. Cell Death Differ. 9:682–692. [DOI] [PubMed] [Google Scholar]
- 27.Iwai, K., T. Kondo, M. Watanabe, T. Yabu, Y. Taguchi, H. Umehara, A. Takahashi, T. Uchiyama, and T. Okazaki. 2003. Ceramide increases oxidative damage due to inhibition of catalase by caspase-3-dependent proteolysis in HL-60 cell apoptosis. J. Biol. Chem. 278:9813–9822. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe, I., T. Kitano, T. Kondo, T. Yabu, Y. Taguchi, M. Tashima, H. Umehara, N. Domae, T. Uchiyama, and T. Okazaki. 2004. Increase of nuclear ceramide through caspase-3-dependent regulation of the “sphingomyelin (SM) cycle” in Fas-induced apoptosis. Cancer Res. 64:1000–1007. [DOI] [PubMed] [Google Scholar]
- 29.Tepper, A.D., P. Ruurs, T. Wiedmer, P.J. Sims, J. Borst, and W.J. van Blitterswijk. 2000. Sphingomyelin hydrolysis to ceramide during the execution phase of apoptosis results from phospholipid scrambling and alters cell-surface morphology. J. Cell Biol. 150:155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hetz, C.A., M. Hunn, P. Rojas, V. Torres, L. Leyton, and A.F.G. Quest. 2002. Caspase-dependent initiation of apoptosis and necrosis by the Fas receptor in lymphoid cells: onset of necrosis is associated with delayed ceramide increase. J. Cell Sci. 115:4671–4683. [DOI] [PubMed] [Google Scholar]
- 31.Blitterswijk, W.J.V., A.H. van der Luit, R.J. Veldman, and J. Borst. 2003. Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem. J. 369:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gulbins, E., and H. Grassme. 2002. Ceramide and cell death receptor clustering. Biochim. Biophys. Acta. 1585:139–145. [DOI] [PubMed] [Google Scholar]
- 33.Kolesnick, R., and Z. Fuks. 2003. Radiation and ceramide-induced apoptosis. Oncogene. 22:5897–5906. [DOI] [PubMed] [Google Scholar]
- 34.Pettus, B.J., C.E. Chalfant, and Y.A. Hannum. 2002. Ceramide in apoptosis: an overview and current perspective. Biochim. Biophys. Acta. 1585:114–125. [DOI] [PubMed] [Google Scholar]
- 35.Sawai, H., T. Okazaki, Y. Takeda, M. Tashima, H. Sawada, M. Okuma, S. Kishi, H. Umehara, and N. Domae. 1997. Ceramide-induced translocation of protein kinase C-delta and -epsilon to the cytosol. J. Biol. Chem. 272:2452–2458. [DOI] [PubMed] [Google Scholar]
- 36.Kondo, T., Y. Suzuki, T. Kitano, K. Iwai, M. Watanabe, H. Umehara, N. Daido, N. Domae, M. Tashima, T. Uchiyama, and T. Okazaki. 2002. Vesnarinone causes oxidative damage by inhibiting catalase function through ceramide action in myeloid cell apoptosis. Mol. Pharmacol. 61:620–627. [DOI] [PubMed] [Google Scholar]
- 37.Uchida, Y., M. Itoh, Y. Taguchi, S. Yamaoka, M. Umeda, S. Ichikawa, Y. Hirabayashi, W.M. Holleran, and T. Okazaki. 2004. Ceramide reduction and transcriptional up-regulation of glucosylceramide synthase through Doxorubicin-activated Sp1 in drug-resistant HL-60/ADR cells. Cancer Res. 64:6271–6279. [DOI] [PubMed] [Google Scholar]
- 38.Paris, F., H. Grassme, A. Cremesti, J. Zager, Y. Fong, A. Haimovitz-Friedman, Z. Fuks, E. Gulbins, and R. Kolesnick. 2001. Natural ceramide reverses Fas resistance of acid sphingomyelinase−/− hepatocytes. J. Biol. Chem. 276:8297–8305. [DOI] [PubMed] [Google Scholar]
- 39.Schneider, P., N. Holler, J.-L. Bodmer, M. Hahne, K. Frei, A. Fontana, and J. Tschopp. 1998. Conversion of membrane-bound Fas (CD95) ligand to its soluble form is associated with down-regulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 187:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang, D.C.S., M. Hahne, M. Schroeter, K. Frei, A. Fontana, A. Villunger, K. Newton, J. Tschopp, and A. Strasser. 1999. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-XL. Proc. Natl. Acad. Sci. USA. 96:14871–14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papoff, G., P. Hausler, A. Eramo, M.G. Pagano, G.D. Leve, A. Signore, and G. Ruberti. 1999. Identification and characterization of a ligand-independent oligomerization domain in the extracellular region of the CD95 death receptor. J. Biol. Chem. 274:38241–38250. [DOI] [PubMed] [Google Scholar]
- 42.Gulbins, E., and R. Kolesnick. 2003. Raft ceramide in molecular medicine. Oncogene. 22:7070–7077. [DOI] [PubMed] [Google Scholar]
- 43.Cherukuri, A., M. Dykstra, and S.K. Pierce. 2001. Floating the raft hypothesis: Lipid rafts play a role in immune cell activation. Immunity. 14:657–660. [DOI] [PubMed] [Google Scholar]
- 44.Bromley, S.K., W.R. Burack, K.G. Johnson, K. Somersalo, T.N. Sims, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 2001. The immunological synapse. Annu. Rev. Immunol. 19:375–396. [DOI] [PubMed] [Google Scholar]
- 45.Davis, D.M. 2002. Assembly of the immunological synapse for T cell and NK cells. Trends Immunol. 23:356–363. [DOI] [PubMed] [Google Scholar]
- 46.London, M., and L. Erwin. 2004. Ceramide selectively displaces cholesterol from ordered lipid domain (Rafts). J. Biol. Chem. 279:9997–10004. [DOI] [PubMed] [Google Scholar]
- 47.Veiga, M.P., J.L.R. Arrondo, F.M. Goni, and A. Alonso. 1999. Ceramides in phospholipid membranes: effects on bilayer stability and transition to nonlamellar phases. Biophys. J. 76:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holopainen, J.M., M. Subramanian, and P.K. Kinnunen. 1998. Sphingomyelinase induces lipid microdomain formation in a fluid phosphatidylcholine/sphingomyelin membrane. Biochemistry. 37:17562–17570. [DOI] [PubMed] [Google Scholar]
- 49.Grassme, H., V. Jendrossek, A. Riehle, G. von Kurthy, J. Berger, H. Schwarz, M. Weller, R. Kolesnick, and E. Gulbins. 2003. Host defence against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 9:322–330. [DOI] [PubMed] [Google Scholar]
- 50.Okazaki, T., R.M. Bell, and Y.A. Hannun. 1989. Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. J. Biol. Chem. 264:19076–19080. [PubMed] [Google Scholar]
- 51.Liu, P., and R.G. Anderson. 1995. Compartmentalized production of ceramide at the cell surface. J. Biol. Chem. 270:27179–27185. [DOI] [PubMed] [Google Scholar]
- 52.Algeciras-Schimnich, A., L. Shen, B.C. Barnhart, A.E. Murmann, J.K. Burkhardt, and M.E. Peter. 2002. Molecular ordering of the initial signaling events of CD95. Mol. Cell. Biol. 22:207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bligh, E., and W. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917. [DOI] [PubMed] [Google Scholar]
- 54.Umehara, H., J.-Y. Huang, T. Kono, F.H. Tabassam, T. Okazaki, E.T. Bloom, and N. Domae. 1997. Involvement of protein tyrosine kinase p72syk and phosphatidylinositol 3-kinase in CD2-mediated granular exocytosis in natural killer cell line. J. Immunol. 159:1200–1207. [PubMed] [Google Scholar]
- 55.Umehara, H., J.-Y. Huang, T. Kono, F.H. Tabassam, T. Okazaki, S. Gouda, Y. Nagano, E.T. Bloom, and N. Domae. 1998. Co-stimulation of T cells with CD2 augments TCR-CD3-mediated activation of protein tyrosine kinase p72syk, resulting in increased tyrosine phosphorylation of adapter proteins, Shc and Cbl. Int. Immunol. 10:833–845. [DOI] [PubMed] [Google Scholar]
- 56.Rodgers, W., and J.K. Rose. 1996. Exclusion of CD45 inhibits activity of p56lck associated with glycolipid-enriched membrane domains. J. Cell Biol. 135:1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okazaki, T., A. Bielawska, R.M. Bell, and Y.A. Hannun. 1990. Role of ceramide as a lipid mediator of 1 alpha, 25-dehydroxyvitamin D3-induced HL-60 cell differentiation. J. Biol. Chem. 265:15823–15831. [PubMed] [Google Scholar]