

Abstract
The exquisite ability of the liver to regenerate is finite. Identification of mechanisms that limit regeneration after massive injury holds the key to expanding the limits of liver transplantation and salvaging livers and hosts overwhelmed by carcinoma and toxic insults. Receptor for advanced glycation endproducts (RAGE) is up-regulated in liver remnants selectively after massive (85%) versus partial (70%) hepatectomy, principally in mononuclear phagocyte-derived dendritic cells (MPDDCs). Blockade of RAGE, using pharmacological antagonists or transgenic mice in which a signaling-deficient RAGE mutant is expressed in cells of mononuclear phagocyte lineage, significantly increases survival after massive liver resection. In the first hours after massive resection, remnants retrieved from RAGE-blocked mice displayed increased activated NF-κB, principally in hepatocytes, and enhanced expression of regeneration-promoting cytokines, TNF-α and IL-6, and the antiinflammatory cytokine, IL-10. Hepatocyte proliferation was increased by RAGE blockade, in parallel with significantly reduced apoptosis. These data highlight central roles for RAGE and MPDDCs in modulation of cell death–promoting mechanisms in massive hepatectomy and suggest that RAGE blockade is a novel strategy to promote regeneration in the massively injured liver.
The explosive growth of liver surgery and transplantation as a therapeutic modality over the last two decades mandates the development of novel protective strategies to optimize outcomes. The ability of the liver to regenerate upon extensive resection or toxic insult, and to restore its mass to homeostasis by programmed regeneration or limited activation of apoptotic pathways, belies an emerging challenge in the management of liver disease (1, 2). Despite widespread interest in the biology of liver regeneration, the clinically relevant limits of the ability of the liver to regenerate are not well understood, and there has been little progress in the development of therapy to enhance regeneration and optimize survival when the liver is excessively small.
Our group has had a long-standing interest in the use of subtotal hepatectomy as a highly relevant preclinical model with which to probe the limits of regeneration in vivo and to dissect underlying mechanisms and test potential therapeutic modalities (3). In rodent models, 70% hepatectomy is well tolerated and regeneration is permitted to nearly full recovery in all animals. However, beyond 70% resection, progressive increments in the extent of resected liver evoke a step-wise acceleration of morbidity and mortality (3). These observations established that the state of being “too small” is inherently toxic to the liver. Therefore, protective strategies might modify the extent of tolerable resection.
The development of partial liver transplantation from living human donors made it essential to determine prospectively the lower limits of graft size compatible with a successful outcome (4, 5). Clinically, this is manifested by the development of a cholestatic syndrome of hepatic injury in recipients of “small for size” livers. The state of the excessively small liver is highly correlated with mortality in clinical and experimental settings (3, 5). Although the direct cause of death in liver failure remains elusive, it is nevertheless established that damage to the small liver occurs in an environment characterized by up-regulation of proinflammatory pathways; the host experiences failure of restoration of liver function as well as local and systemic inflammatory injury (3–5). Thus, dissection of the molecular mechanisms that thwart the small liver's attempts to restore homeostatic mass and function represents a critical step in exploiting the regenerative capacity of the organ. We hypothesized that inflammatory mediators routinely cleared and detoxified by the liver rapidly overwhelm the hepatic remnant after massive resection, thereby diverting pathways normally signaled to restore liver mass and function to those leading to cell death. Alternatively, the liver remnant, suddenly overwhelmed by the increased metabolic burden, faces intense metabolic stress, which might indirectly initiate inflammatory mechanisms.
As the receptor for advanced glycation endproducts (RAGE) has been linked to escalation of cellular stress subsequent to engagement by its ligands (6–8), we tested the premise that RAGE mediates injurious stress responses in massive liver resection and initiates events that critically curtail the limits of regeneration.
Results
Blockade of RAGE improves survival after massive hepatectomy
To define the limits of liver regeneration, we used a murine model of incremental liver injury, partial and well-tolerated hepatectomy (70% resection; references 9, 10), and established a model of massive hepatectomy (85% resection). Sham-treated animals underwent anesthesia and laparotomy, without liver resection. C57BL/6 mice subjected to 70% hepatectomy displayed an ∼90% probability of survival at 7 d (Fig. 1 A). In contrast, mice subjected to 85% hepatectomy displayed significantly reduced probability of survival, ∼30% of the animals were alive by day 7; P < 0.0035 versus 70% resection (Fig. 1 A). These findings demonstrated that beyond resection of 70% of the liver, mechanisms are engaged that significantly increase mortality. To explore the potential role of RAGE in massive (85%) hepatectomy, we first assessed levels of the receptor. An ∼3.2-fold increase in RAGE transcripts by quantitative PCR was observed in the hepatic remnant 8 h after massive (85%) hepatectomy versus 70% hepatectomy or sham procedure; P < 0.05 (Fig. 1 B). Importantly, RAGE transcripts in the remnant did not differ between sham-treated mice and those animals subjected to 70% hepatectomy, thereby suggesting that perturbation triggered by partial (70%) resection did not trigger up-regulation of this receptor. Immunohistochemistry revealed that remnant tissue expressed increased RAGE epitopes compared with sham-treated livers at 8 h after massive hepatectomy (Fig. 1, C and D). RAGE expression localized to cells expressing both CD68 and Cd11c, thereby indicating that RAGE was principally expressed in dendritic cells, likely mononuclear phagocyte (MP)-derived DCs (MPDDCs), in the injured remnant (Fig. 1, E and F). Based on the merged images in Fig. 1 (E and F), RAGE antigen was not detected to appreciable degrees in CD68+/CD11c− cells, thereby suggesting that RAGE was not significantly expressed in Kupffer cells. Furthermore, RAGE-expressing cells did not express α-smooth muscle actin, thereby indicating that they were not stellate or smooth muscle cells (unpublished data). In addition, there was no evidence of colocalization with anti-CD31 IgG, indicating that RAGE-expressing cells were not endothelial cells (unpublished data).
Figure 1.

Up-regulation of RAGE in hepatic remnants after massive (85%) hepatectomy: cellular localization and impact of ligand/RAGE blockade. Male C57BL/6 mice were subjected to partial (70%) or massive (85%) hepatectomy or sham surgery. Mice subjected to 85% resection were treated with murine sRAGE, 100 μg per day, or vehicle, PBS daily until death. (A) Kaplan-Meier Product Limit Estimate. The times of death were recorded for mice undergoing hepatectomy, and survival was plotted. (B) Quantitative PCR. Quantitative PCR was performed on liver remnants retrieved from mice undergoing sham, 70% or 85% liver resection, or 85% resection in the presence of sRAGE. After normalization to internal controls, levels of murine RAGE transcripts in sham-treated mice livers were arbitrarily defined as “1.” In this experiment, n = 5 mice per group. *, P < 0.05 versus 70% hepatectomy. **, P < 0.05 versus PBS treatment/85% hepatectomy. (C–F) Immunofluorescence microscopy. Immunofluorescence for detection of RAGE antigen was performed in sections prepared 8 h after sham surgery (C) or massive resection (D, 85%). In tissues prepared from remnants after massive resection, double immunofluorescence staining was performed with anti-RAGE IgG (green) and an anti-CD68 IgG (E, red), or with anti-RAGE IgG (green) and anti-CD11c IgG (F, red) as described before. Single and merged images are shown. (C, E, and F) Bars, 50 μm. (G) Kaplan-Meier Product Limit Estimate. Male C57BL/6 mice were subjected to massive hepatic resection and treated with the indicated F(ab′)2 fragments of rabbit anti-RAGE, anti-S100/calgranulin, antiamphoterin, or nonimmune IgG. The times of death were recorded for mice undergoing hepatectomy, and survival was plotted.
To test the concept that engagement of RAGE mediated deleterious stress responses after massive hepatectomy, we used several means to block the receptor. First, we administered murine soluble RAGE (sRAGE), the extracellular ligand binding domain of RAGE that serves as a decoy, thereby suppressing ligand-induced stimulation of cell surface receptor (8, 11). Mice were treated with murine sRAGE (100 μg per day by intraperitoneal route, beginning 1 d before massive hepatectomy and then daily until sacrifice or death), or with equal volumes of vehicle, PBS. Kaplan Meier product limit estimate studies revealed that compared with mice undergoing 85% hepatectomy receiving PBS, in whom an ∼30% probability of survival was observed at 7 d, mice receiving sRAGE displayed significantly increased survival, ∼90%; P < 0.0001 (Fig. 1 A). The probability of survival in sRAGE-treated mice undergoing 85% hepatectomy was not significantly different than that observed in mice undergoing less extensive resection (70%); P > 0.05 (Fig. 1 A).
In addition to treatment with sRAGE, we administered blocking F(ab′)2 fragments of either RAGE or its ligands to wild-type mice undergoing massive resection. Administration of blocking F(ab′)2 fragments prepared from anti-RAGE IgG significantly increased survival of mice undergoing massive hepatectomy compared with animals treated with nonimmune F(ab′)2 fragment; P < 0.04 (Fig. 1 G). These findings underscored the key role of the cell surface receptor RAGE in mediating enhanced injury after massive resection. Furthermore, supportive of a key role of RAGE's proinflammatory ligands, S100/calgranulin and amphoterin, in mediating liver injury upon massive hepatectomy, administration of blocking F(ab′)2 fragments prepared from anti-S100/calgranulin or antiamphoterin IgG resulted in significantly increased probability of survival compared with mice receiving nonimmune F(ab′)2 fragments; P < 0.04 (Fig. 1 G). Consistent with the concept that one consequence of ligand–RAGE engagement is up-regulation of RAGE itself; compared with PBS-treated mice, remnants retrieved from sRAGE-treated animals displayed decreased transcripts for RAGE (P < 0.05), which were not different than those observed in sham-treated or 70% hepatectomized mice (Fig. 1 B).
The effects of sRAGE were dose dependent; although mice undergoing 85% hepatectomy in the presence of sRAGE, 100 μg per day, displayed significantly increased survival compared with mice receiving PBS (P < 0.0001), mice receiving sRAGE at 50, 10, or 2 μg per day did not display significantly improved survival compared with vehicle treatment (Fig. 2 A). Therefore, in further studies to delineate the mechanisms by which RAGE blockade enhanced survival after massive hepatectomy, sRAGE was administered to the mice at 100 μg/d. Importantly, when we extended these experiments to 14 d, we found no further mortality in either sRAGE- or PBS-treated groups that had survived to 7 d (unpublished data). These findings indicated that in this murine model, the key mechanisms linked to survival and regeneration was operative within the first 7 d after massive resection.
Figure 2.

Administration of sRAGE improves survival and enhances regeneration after massive hepatectomy. Male C57BL/6 mice were subjected to 85% hepatectomy in the presence of the indicated once daily dose of sRAGE or vehicle, PBS. (A) Kaplan-Meier Product Limit Estimate. The times of death were recorded for mice undergoing hepatectomy in the presence of sRAGE versus vehicle, and survival was plotted. Survival curves for sRAGE, 100 μg/d- and PBS-treated mice, reflect the same animals as in Fig. 1 A. (B–D) Indices of injury and regeneration. At the indicated times, plasma or hepatic remnant was retrieved and analyzed for ALT (B) and Prothrombin Time (C). (D) Liver weight/body weight ratios are shown. (B and C) n = 3–8 mice per condition. (D) n = 10 (PBS) or n = 27 (sRAGE) mice per condition. (B–D) *, P < 0.05. (E and F) Histology. Hepatic remnants were retrieved and grading was performed based on the percentage of necrosis as described before; mean ± standard error is shown. (E) n = 5 mice per condition. *, P < 0.05. (F) Representative sections from PBS- and sRAGE-treated mice at 24 h are illustrated. Bar, 80 μm (inset) 160 μm. (G) Immunoprecipitation. 16 h after 85% hepatectomy, plasma from sRAGE- or PBS-treated mice was retrieved and subjected to immunoprecipitation using rabbit anti-RAGE IgG. Immunoprecipitated material was subjected to blotting using anti-S100/calgranulin IgG. Plasma was pooled from n = 3 sRAGE- or n = 3 PBS-treated plasma for immunoprecipitation studies.
These data led us to consider if inherent differences in RAGE expression in the liver accounted for the ability of the relatively few PBS-treated animals to survive massive resection. Thus, we tested levels of RAGE transcripts in the resected tissue (85% of the liver) to determine if RAGE expression differed in this tissue retrieved from mice that survived or succumbed (at 7 d). We found that there were no significant differences in RAGE mRNA transcripts in the resected tissue of mice that survived or died by 7 d, irrespective of sRAGE or PBS treatment in the 85% resection group (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20040934/DC1). These findings suggest that basal differences in RAGE expression in the liver did not account for the likelihood of survival or death after 85% resection.
Indices of liver necrosis (release of alanine transferase), function (prothrombin time), and restoration of liver mass (% liver weight/body weight), were significantly improved in mice treated with sRAGE, 100 μg per day, versus vehicle, PBS (Fig. 2, B–D). The differences in survival between sRAGE-treated mice and those receiving vehicle were greatest early after hepatectomy. Inevitably, only innately resistant survivors populated the vehicle-treated group beyond 40 h. Therefore, survival in the relatively fewer vehicle- versus sRAGE-treated mice both converged toward full restoration of mass and liver function over time.
We examined the remnants from vehicle- and sRAGE-treated mice to determine the impact of massive hepatectomy on the remnant and the potential influence of RAGE blockade. Sections were prepared from liver remnants and stained with hematoxylin and eosin. Frank necrosis was visible in centrilobular regions (graded 0–3; reference 12). Destruction of the liver parenchyma, in the form of necrosis, was observed in mice treated with PBS at 24 h (Fig. 2, E and F). However, preservation of liver parenchyma with an absence of necrosis was observed in the remnant tissue of mice treated with sRAGE (Fig. 2, E and F).
Consistent with the premise that administration of sRAGE trapped proinflammatory ligands of RAGE, immunoprecipitation of plasma retrieved from mice receiving sRAGE, but not PBS, with anti-RAGE IgG revealed S100/calgranulin antigen by blotting (Fig. 2 G). In contrast, immunoprecipitation of sRAGE-treated plasma with nonimmune rabbit IgG failed to reveal bands immunoreactive with S100/calgranulin (unpublished data).
Blockade of RAGE and cytokine generation after massive hepatectomy
To explore the mechanisms underlying diminished regeneration associated with massive hepatectomy and the protective effects of sRAGE, we assessed levels of proinflammatory mediators linked to these processes. We examined transcripts for TNF-α, a multifactorial cytokine that displays pleiotropic effects; in partial (70%) hepatectomy, TNF-α promotes regeneration and suppresses apoptosis; however, in a highly inflamed milieu, TNF-α has been linked to tissue injury (13–15). At 8 h after massive hepatectomy, TNF-α transcripts by quantitative PCR were significantly increased in the hepatic remnant in sRAGE-treated mice versus those receiving PBS; P < 0.05. In addition, levels of TNF-α transcripts in PBS-treated remnants were not different than those observed in sham-treated livers (Fig. 3 A). Examination of transcripts encoding TNF-α in remnants retrieved from mice undergoing 70% resection revealed significantly lower levels compared with sRAGE-treated mice undergoing 85% resection; P < 0.05 (Fig. 3 A). These findings underscore the concept that distinct mechanisms are recruited upon 85% versus less extensive 70% resection.
Figure 3.

Blockade of RAGE modulates expression of inflammatory and regenerative mediators after massive hepatectomy. 8 h after sham surgery, 70 or 85% resection in the presence of vehicle or sRAGE treatment, hepatic remnants were retrieved and subjected to quantitative PCR for detection of transcripts for the following: A, TNF-α; B, IL-6; C, IL-10; D, VCAM-1; and E, IFN-γ. After normalization to internal controls, fold changes relative to sham surgery remnant transcript levels (“1.0”) were reported. n = 5 mice per condition. *, P < 0.05 versus sham control. **, P < 0.05 versus PBS treatment.
Next, we examined levels of IL-6, as this cytokine, regulated largely by TNF-α, has been importantly linked to regenerative processes after liver injury (16–18). Previous studies demonstrated that infusion of IL-6 in mice subjected to partial hepatectomy in genetically modified mice resistant to TNF-α secondary to deletion of type 1 TNF-α receptor restored regeneration (14). Analogous to the pattern of expression of TNF-α after massive hepatectomy, IL-6 transcripts in the hepatic remnant were significantly higher in sRAGE-treated mice versus PBS at 8 h; P < 0.05, and versus PBS-treated mice undergoing 70% resection; P < 0.05 (Fig. 3 B). These findings underscore the premise that both TNF-α and IL-6 expression are associated with regeneration, particularly after massive hepatectomy, and that RAGE blunts expression of these protective mediators after largely lethal resection of the liver.
The remnants of sRAGE-treated mice also displayed significantly increased transcripts for an antiinflammatory cytokine IL-10 (19) at 8 h after resection compared with PBS-treated mice; P < 0.05 (Fig. 3 C). Levels of vascular cell adhesion molecule-1 (VCAM-1) transcripts were not significantly different at 8 h in sRAGE- versus PBS-treated mice after massive hepatectomy. Although levels of IFN-γ transcripts were significantly higher in remnants after 85% resection versus sham surgery (P < 0.05), there was no significant impact of sRAGE treatment in 85% resection. Together, these findings suggest that VCAM-1 and IFN-γ likely did not critically contribute to failure of regeneration after massive resection (Fig. 3, D and E).
Blockade of RAGE and modulation of NF-κB activation after massive liver resection
To determine the impact of massive resection on key pathways regulating transcription and cell fate in the remnant, we examined activation of NF-κB, a pleiotropic transcription factor that mediates proinflammatory pathways and proliferative/antiapoptotic programs in developing and injured liver (20–22). Multiple studies report that NF-κB is significantly activated by 70% hepatectomy (23). At 2 h after massive hepatectomy (85%), nuclear translocation of NF-κB in the PBS-treated hepatic remnants was not significantly increased compared with 70% partial hepatectomy (Fig. 4 A). However, in the presence of sRAGE/85% hepatectomy, an approximately fivefold increase in activation of NF-κB was noted compared with vehicle treatment or 70% hepatectomy at 2 h; P < 0.05 (Fig. 4 A). Supershift assays revealed that the NF-κB complex after massive hepatectomy consisted of both p50 and p65 antigens, but especially p50, as demonstrated by incubation of the nuclear extract with anti-p50 and/or anti-p65 IgG (Fig. 4 B). In competition studies, addition of a 10-fold excess of unlabeled NF-κB probe resulted in disappearance of the band.
Figure 4.
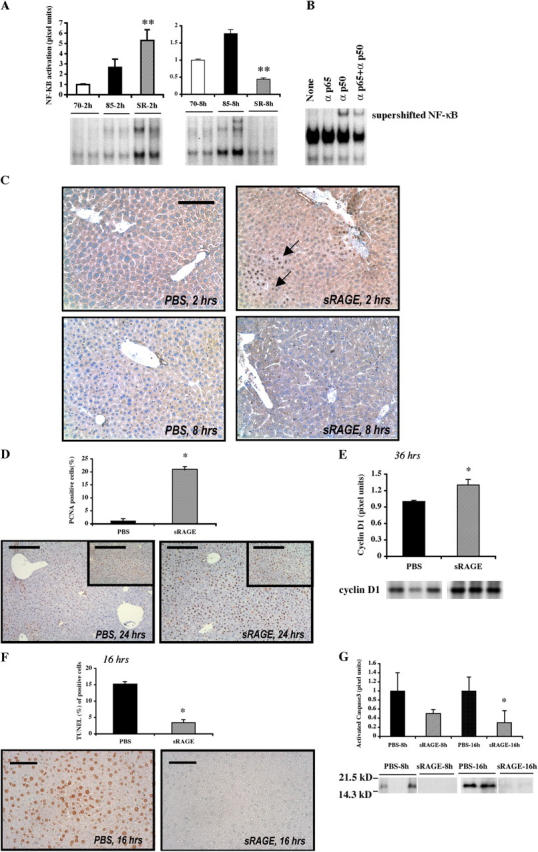
Blockade of RAGE modulates activation of NF-κB after massive hepatectomy. (A) EMSA. Nuclear extracts were prepared from the remnants of the indicated mice and the EMSA was performed. The illustrated bands are representative of n = 4–6 mice per condition. **, P < 0.05 versus PBS treatment/85% resection. (B) Supershift assay. Remnants retrieved from mice undergoing 85% hepatectomy in the presence of sRAGE at 2 h were retrieved and subjected to incubation with anti-p50, anti-p65, or both anti-p50 and anti-p65 IgG before EMSA. (C) Immunohistochemistry. Hepatic remnants at 2 and 8 h were retrieved and subjected to immunohistochemistry with anti-p65 NF-κB subunit antibodies. Bar, 50 μm. (D) Proliferation. Hepatic remnants were retrieved and subjected to immunohistochemistry with anti-PCNA IgG and mean numbers of PCNA+ cells were determined from n = 10 fields per section/mouse. The indicated results are representative of n = 5 mice per condition. Bars, 80 μm. (inset) 160 μm. (E) Western blotting of hepatic remnant lysates was performed at the indicated times using anti-cyclin D1 IgG. The illustrated bands are representative of n = 3–4 mice per condition. (F) Apoptosis. Hepatic remnants were retrieved and subjected to TUNEL assay and mean numbers of TUNEL+ cells determined from n = 10 fields per section/mouse. The indicated results are representative of n = 5 mice per condition. Bar, 40 μm. (G) Western blotting on remnants was performed using anti-activated caspase-3 IgG. The illustrated bands are representative of n = 5 mice per condition. In all cases, bands were scanned into a densitometer and relative pixel units of band density reported. *, P < 0.05 versus PBS/85% resection.
Immunohistochemistry using an antibody selective for activated NF-κB (p65) showed strong nuclear positivity within clusters of hepatocytes most prominently in a perivascular distribution around central and portal venules at 2 h after massive resection in sRAGE-treated mice (Fig. 4 C). Appreciable NF-κB immunoreactivity was not detectable in the hepatic remnants after massive resection in the PBS-treated group at 2 h.
In contrast, by 8 h after massive hepatectomy, nuclear translocation of NF-κB was only marginally increased in the PBS-treated group versus 70% hepatectomy, ∼1.7-fold (Fig. 4 A). Electrophoretic mobility shift assays (EMSAs) revealed that in sRAGE-treated remnants at 8 h, levels of activated NF-κB were not different than those observed in 70% resection, but were significantly less than levels seen in PBS-treated remnants after 85% resection; P < 0.05 (Fig. 4 A). Immunohistochemistry revealed that at 8 h after massive resection in PBS-treated mice, nuclear staining for NF-κB did not localize to hepatocytes; rather, it scattered nonparenchymal liver cells, such as Kupffer cells, endothelial cells, and occasional inflammatory cells, largely expressing the activated form of this transcription factor (Fig. 4 C).
Blockade of RAGE enhances proliferation and reduces apoptosis in hepatocytes after massive hepatectomy
Next, we addressed the potential consequences of activated NF-κB, particularly in the sRAGE-treated remnants. At 24 h after massive hepatectomy, sRAGE-treated remnants displayed increased immunoreactivity for proliferating cell nuclear antigen (PCNA) in hepatocytes; P < 0.05 (Fig. 4 D). Consistent with increased proliferation in the sRAGE-treated remnant, levels of cyclin D1 antigen (24), a target gene of NF-κB (25), were higher in sRAGE- versus PBS-treated remnants by Western blotting; P < 0.05 (Fig. 4 E). TUNEL+ cells were markedly reduced in sRAGE- versus PBS-treated remnants at 16 h after massive resection (Fig. 4 F), in parallel with decreased activated caspase-3 antigen at 8 and 16 h; P < 0.05 (Fig. 4 G).
Furthermore, although a central role has been ascribed to hepatocyte growth factor-1 in hepatocyte replication and regeneration (26, 27), serial analysis after massive hepatectomy revealed approximately 5-, 10- and 4-fold higher antigen levels of hepatocyte growth factor-1 in hepatic remnants retrieved from PBS- versus sRAGE-treated mice at 8, 16, and 24 h, respectively (unpublished data). Thus, in massive resection, despite up-regulation of this factor, regenerative programs appear to be ineffective.
RAGE-expressing cells of MP lineage mediate failure of regeneration after massive liver resection
Cellular localization studies suggested that CD68+/CD11c+ MPDDCs were the principal RAGE-expressing cells in the liver remnant after massive hepatectomy. To ascribe a role for RAGE in cells of MP lineage to the tissue-injurious pathways activated upon massive hepatectomy, we used transgenic mice bearing selective introduction of cytosolic tail–deleted human RAGE into MP lineage cells, driven by the type A scavenger receptor promoter (SR; references 28, 29). Introduction of cytosolic tail–deleted RAGE into endogenously RAGE-bearing cells causes a “dominant negative” (DN) effect both in vitro and in vivo upon ligand engagement (30–32). Tissue localization studies indicated that RAGE was expressed to enhanced degrees in cells expressing CD68 and CD11c, suggesting that DN RAGE was expressed in both Kupffer cells and MPDDCs in these transgenic mice (Fig. 5, A–D).
Figure 5.


Transgenic mice expressing DN RAGE in cells of MP lineage display enhanced survival and liver regeneration after massive hepatectomy. Transgenic mice expressing human DN RAGE selectively in cells of MP lineage using the scavenger receptor type A promoter were used for these studies. (A–D) Immunofluorescence microscopy. Immunofluorescence for detection of RAGE antigen was performed in sections prepared from the remnants of transgenic mice 8 h after sham surgery (A) or massive resection (B, 85%). Double immunofluorescence staining was performed on sections prepared from transgenic mice 8 h after massive resection using the following antibodies: anti-RAGE IgG (C and D, green) and anti-CD68 IgG (C, red) or anti-CD11c IgG (D, red). Single and merged images are shown. (A and B) Bars, 100 μm; (C and D) Bars, 50 μm. (E) Kaplan-Meier Product Limit Estimate. The times of death were recorded for transgenic versus littermate control mice undergoing massive hepatectomy and survival was plotted. (F and G) Quantitative PCR. Massive hepatectomy was performed in transgenic (DN) and wild-type littermate mice (WT). Hepatic remnants were retrieved at 8 h after resection and subjected to quantitative PCR for detection of transcripts for TNF-α (F) and IL-6 (G). After normalization to internal controls, fold changes relative to WT mice remnant transcript levels (“1.0”) are reported. n = 5 mice per condition. *, P < 0.05 versus WT mice. (H) EMSA. Massive hepatectomy was performed in transgenic (DN) and wild-type littermate mice (WT). Remnants were retrieved at 2 and 8 h, and nuclear extracts were prepared for EMSA. Bands were scanned into a densitometer and relative pixel units of band density reported. *, P < 0.05 versus WT mice/85% resection. (I) Western blotting. At the indicated times after massive resection, hepatic remnants were retrieved and subjected to Western blotting using anti-cyclin D1 IgG. (H and I) The illustrated bands are representative of n = 3–4 mice per condition.
To determine the impact of introduction of DN RAGE in cells of MP lineage in massive hepatectomy, we assessed survival. Kaplan-Meier product-limit estimates revealed that upon massive hepatectomy, transgenic SR DN-RAGE mice displayed significantly increased survival compared with wild-type littermates (∼90% vs. 20%, respectively; P = 0.004; Fig. 5 E). At 8 h, TNF-α and IL-6 transcripts were significantly increased in the remnants of transgenic SR DN-RAGE mice compared with littermate controls; P < 0.05 (Fig. 5, F and G). EMSAs performed on nuclear extracts from transgenic SR DN RAGE mouse remnants at 2 h after massive hepatectomy revealed a significant increase in activated NF-κB versus wild-type littermates subjected to the same degree of injury; P < 0.05 (Fig. 5 H). Addition of excess unlabeled NF-κB probe resulted in disappearance of the band (Fig. 5 H). In contrast, at 8 h, although no differences were observed in activation of NF-κB in transgenic SR DN RAGE mice versus littermates after 70% hepatectomy, a significant attenuation of NF-κB activation was noted in transgenic SR DN RAGE versus littermate mice at 8 h; P < 0.05 (Fig. 5 H). Supershift studies confirmed that the NF-κB complex in both sets of mice consisted of both p50 and p65 (unpublished data). Levels of cyclin D1 antigen in the hepatic remnants of transgenic SR DN RAGE mice were increased ∼7.5- and ∼5-fold higher at 8 and 36 h compared with littermate controls, respectively; P < 0.05 (Fig. 5 I). By 7 d after massive hepatectomy, liver/body weight ratios were fully restored in the transgenic mice (unpublished data).
Discussion
In homeostasis, the portal vein continually bathes the liver with toxic products released from microorganisms or delivered from the intestinal tract. In addition, a wide array of antigens is continually delivered to and processed by the liver, without evoking untoward inflammatory or immune responses. Even upon removal of 70% of the liver, the small remnant facilitates regeneration-provoking programs to restore mass and function. A key element of liver regeneration in 70% hepatectomy is up-regulation of hepatocyte NF-κB, beginning as early as 30 min after resection and peaking at 1–2 h (23), in parallel with enhanced expression of regeneration-promoting cytokines TNF-α and IL-6.
Our findings provide experimental evidence that supports the clinical observation that the regenerative capacity of the liver is finite. These data demonstrate for the first time that RAGE-dependent mechanisms contribute to remnant hepatocyte necrosis/apoptosis and failure of proliferation after massive liver resection. In contrast, in sRAGE-treated mice or transgenic mice in which RAGE signaling in cells of MP lineage is disrupted, the remnants displayed significant up-regulation of NF-κB, particularly in hepatocytes, beyond that observed in 70% resection or vehicle-treated 85% hepatectomy. In parallel, increased transcripts for TNF-α and IL-6 were observed. The failure to observe significant activation of hepatocyte NF-κB after 85% hepatectomy in vehicle-treated animals at 2 h suggests that RAGE prevents its activation, thereby leading to preferential activation of proapoptotic mechanisms and suppression of proliferation. In sRAGE-treated remnants, early activation of NF-κB, within the first 2 h, is likely critically linked to the transcriptional up-regulation of one of its key targets, the cell cycle regulatory molecule, cyclin D1 (24, 25, 33). In parallel, the proregenerative effects of TNF-α are directly linked to its ability to facilitate the priming phase (G0–G1) of the cell cycle (34). These experiments provide yet an additional contrast with the biology of partial (70%) hepatectomy, as it has recently been shown that blockade of hepatocyte NF-κB causes apoptosis after injection of TNF, but not after partial hepatectomy (35). In 85% resection, suppressed activation of NF-κB was associated with increased apoptosis. Because the principal RAGE-expressing cells in the remnants were CD68+/CD11c+ inflammatory cells, these findings suggest that the products of activated inflammatory cells in the remnant generate an environment in which rapid up-regulation of hepatocyte NF-κB is subsequently thwarted.
What are the RAGE-expressing cells that mediate injury consequent to massive hepatectomy? An intriguing finding in our studies is that the hepatocytes themselves do not express appreciable RAGE epitopes, either in the basal state or after massive resection. Rather, MPDDCs are the principal RAGE-expressing cells in the remnant after massive resection (36, 37). In contrast, Kupffer cells, distributed widely throughout the liver, did not express RAGE in the basal or perturbed state. Although our studies using sRAGE or anti-RAGE F(ab′)2 fragments do not indicate the specific cellular target, experiments using transgenic mice expressing signaling-deficient mutant RAGE in cells of MP lineage provide direct insights into the role of RAGE signaling in the target cells of this promoter.
Based on the use of the type A SR to drive expression of DN RAGE, at least three distinct classes of cells may express DN RAGE in the transgenic mice. Although Kupffer cells prominently expressed the DN RAGE transgene, we posit that their impact in RAGE-modulated injury after massive hepatectomy is not a key determining factor linking massive resection to failure of regeneration. Little to no Kupffer cell RAGE expression was noted in the remnant in wild-type mice after massive resection; yet, pharmacological blockade of RAGE in wild-type animals significantly modulated survival and regeneration. The presence of RAGE staining localized largely to CD11c+/CD68+ cells (thus, MPDDCs, not Kupffer cells) in wild-type mice suggests that MPDDCs are the principal RAGE-expressing cells in the remnant. In the transgenic mice, as well, DN RAGE was expressed in MPDDCs under the control of this promoter. It is important to note that peripheral blood–derived monocytes may also express the DN RAGE transgene, and that these cells might contribute to RAGE-mediated injury after massive resection. Although our pathologic analysis did not demonstrate detectable inflammatory infiltrates at early or late times within the remnant after massive resection, it is not possible to fully exclude the potential impact of low numbers of infiltrating peripheral blood–derived MP after massive resection on the response to injury in the present studies.
In this context, novel roles for MPDDCs in multiple facets of the immune/inflammatory response in the liver are emerging (36, 37). Indeed, studies suggest that MPDDCs are an heterogeneous population of cells, whose distinct functions may range from antigen recognition and processing to generation of cytokines with varied effects on the adaptive immune response, such as IL-12, IFN-γ, TNF-α, and IL-6 (38). Discrete subsets have been identified; each of which likely plays distinct roles in diverse settings, from allogeneic transplantation to infection. Furthermore, these cells display migratory properties, and may traverse the liver into peripheral lymphoid compartments; processes potentially linked to induction of tolerance (38).
Our studies point to deleterious functions of MPDDCs, at least in part via RAGE, in massive resection. Future studies must address two central questions; first, what is the impact of massive resection on MPDDCs properties and, second, what classes of MPDDCs express RAGE in both the unperturbed state and postmassive resection? Importantly, our findings by quantitative PCR revealed that transcripts for IFN-γ were increased in PBS-treated mice after massive resection, but that treatment with sRAGE did not significantly attenuate levels of this cytokine. However, levels of IL-10 transcripts were significantly higher in sRAGE-treated remnants. These data suggest that proinflammatory effects of MPDDCs, at least those expressing RAGE, may dominate the remnant upon massive resection, as treatment with sRAGE enhanced expression of a key antiinflammatory cytokine, IL-10. Indeed, studies in an autoimmune model of type 1 diabetes in NOD mice revealed that blockade of RAGE attenuated the time to development of hyperglycemia in NOD/SCID mice injected with diabetogenic splenocytes from diabetic NOD animals (39). In parallel with decreased islet expression of proinflammatory cytokines, increased islet expression of antiinflammatory cytokines such as IL-10 and TGF-β was shown (39). These studies, together with the present work, suggest that RAGE activates proinflammatory pathways that, either directly or indirectly, attenuate the expression and biologic impact of antiinflammatory mediators. In these present studies, potential roles for subsets of MPDDCs expressing RAGE in pro- versus antiinflammatory mechanisms must be dissected. Such concepts form the basis of ongoing investigation.
Together, these findings highlight novel insights into pathogenic roles for RAGE, at least in part in MPDDCs, in inhibition of regeneration after massive liver resection. We propose that RAGE antagonism may provide a new modality to expand the limits of tolerable resection and, thus, lead to novel strategies to salvage livers and hosts overwhelmed by massive hepatic insult.
MATERIALS AND METHODS
Animals and surgical procedures
Male C57BL/6 mice (The Jackson Laboratory), weighing 25–28 g, were used in all experiments and maintained in a temperature-controlled room with alternating 12-h light/dark cycles. All experiments were approved by the Institutional Animal Care and Use Committee of Columbia University.
In n = 18 mice, each lobe was weighed and compared with the total weight of the liver; results are reported in the percent of total liver weight. The lobes of the liver reflect the following percentage/total weight: middle and left lobes, ∼65.5 ± 2.6%; right superior lobe, ∼13.7 ± 1.7%; right inferior lobe, ∼13.8 ± 1.9%; and the lobus caudatus, ∼6.8 ± 1.8%. In the first model, ∼65.5 ± 2.6% of the liver was resected by removing the left and middle lobes of the liver; the caudate and right lobes remained intact. In the second model, ∼85% of the liver was resected. In addition to the left and middle lobes of the liver, the caudate and right inferior lobes of the liver were removed. Approximately 86.2 ± 2.1% of the liver was resected; the hepatic remnant was composed of the right superior lobe. Animals were examined every 6 h for the duration of the study to estimate time of death. Certain mice were treated with murine sRAGE (11) or vehicle, PBS. Mice were treated once daily by intraperitoneal administration beginning 1 d before surgery, and once daily until they were killed at the indicated time points. In other experiments, F(ab′)2 fragments of rabbit nonimmune, anti-RAGE, anti-S100/calgranulin, or antiamphoterin IgG were prepared according to the manufacturer's instructions using a kit from Pierce Chemical Co. The indicated F(ab′)2 fragments were administered to the mice 12 h before the surgery and once daily until they were killed.
Analyses of plasma and serum
Mice were killed and blood was withdrawn through cardiac puncture. Plasma was assayed for prothrombin time and serum was assayed for levels of bilirubin, albumin, and alanine aminotransferase by Ani Lytics, Inc. Levels of blood glucose were assayed using a glucometer, Accu Chek Advantage (Boehringer).
Immunohistochemistry and histology
Consecutive sections (5 μm) from paraffin-embedded liver were prepared for hematoxylin-eosin staining, TUNEL, and PCNA immunohistochemistry. Sections of liver remnant were subjected to hematoxylin and eosin staining and the degree of necrosis was graded based on the percentage of cells involved, similar to a grading system used for the determination of hepatic steatosis (12) as follows: grade 0, no necrosis; grade 1, necrosis of ≤30% of (centrilobular) hepatocytes; grade 2, necrosis of 30–60% (centrilobular) hepatocytes; and grade 3, ≥60% (centrilobular) hepatocytes. The TUNEL+ cells were identified using an apoptosis kit from Intergen. Staining for detection of PCNA was performed using mouse anti-PCNA IgG (DakoCytomation) as described previously (40). To detect activated NF-κB, formalin-fixed sections were prepared and subjected to immunostaining with a rabbit polyclonal antibody against NF-κB, p65 subunit (1:50 dilution; Santa Cruz Biotechnology, Inc.) as described previously (40).
Immunofluorescence
To detect RAGE epitopes, and to localize RAGE antigen to distinct cell types, single and double immunofluorescence staining was performed in frozen sections as described previously (40). In brief, after blocking with normal serum, sections were incubated with rabbit anti-RAGE IgG (1:50 dilution; reference 40) and incubated with a biotinylated goat anti–rabbit IgG (1:100; Vector Laboratories) followed by incubation with streptavidin-conjugated to Alexa Fluor 555 green (1 μg/ml; Molecular Probes, Inc.). After blocking with avidin/biotin solution (Vector Laboratories), the same sections were incubated with rat anti–mouse CD68 IgG (1:50 dilution; Serotec) and incubated with a biotinylated rabbit anti–rat IgG (1:100; Vector Laboratories) followed by streptavidin conjugated to Alexa Fluor 555 red (1 μg/ml; Molecular Probes, Inc.). In other double staining procedures, hamster anti–mouse CD11c (1:25 dilution; BD Biosciences) was used as another primary antibody and incubated with a secondary biotinylated goat anti–hamster IgG (1:100; BD Biosciences) and Alexa Fluor 555 red. The slides were photographed using fluorescence microscopy.
Immunoprecipitation and Western blotting
Plasma was retrieved from sRAGE- and PBS-treated mice 16 h after massive hepatectomy. 300 μl of plasma was diluted with buffer A (1% NP-40; 100 mM Tris, pH 8.0; 20% glycerol; 0.2 mM EDTA; 400 mM NaCl; 200 μM Na3VO4; 100 mM NaF; 2 mM DTT; and 0.8 mM PMSF). Nonspecific binding to rabbit IgG was performed by a preclearing step; the diluted plasma was incubated with 2 μg of rabbit IgG (Sigma-Aldrich) and protein A agarose beads for 1 h at 4°C. Samples were centrifuged, and the supernatants were further incubated with 3 μg/ml rabbit anti–human RAGE IgG or 3 μg/ml nonimmune rabbit IgG and protein A agarose beads for 2 h at 4°C. The supernatants were discarded, and beads were resuspended in nonreducing sample buffer and subjected to SDS-PAGE. The gels were transferred to nitrocellulose and blotted using rabbit anti-S100/calgranulin IgG (Sigma-Aldrich).
The whole protein was extracted from frozen liver tissues using lysis buffer (10 mM Tris HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% Glycerol, 2 mM sodium orthovanadate, and complete protease inhibitor cocktail; Roche Diagnostics). Western blotting was performed as described previously (40).
EMSAs
Nuclear proteins were prepared from the liver tissues using the isolation kit from Pierce Chemical Co. NF-κB DNA binding activity was measured using end-labeled double-stranded NF-κB oligonucleotide (Promega) as described previously (40). Supershift analysis was performed by incubating nuclear extracts with 0.5 μg of antibodies against p50 or p65 subunit of NF-κB (Santa Cruz Biotechnology, Inc.) before adding the γ-[32P] ATP-labeled oligonucleotide (40).
Quantification of gene expression using real-time PCR
Total RNA was isolated using TRIzol reagent (Life Technologies) and was processed directly to cDNA synthesis using the TaqMan Reverse Transcription Reagents Kit (Applied Biosystems). Primers and TaqMan probes were designed by Primer Express 2.0 software (Applied Biosystems), except for GAPDH, which was commercially available. The primers and probes were as follows: mouse RAGE forward primer, 5′-GGACCCTTAGCTGGCACTTAGA-3′, reverse primer, 5′-GAGTCCCGTCTCA-GGGTGTCT-3′, and probe, 6FAM-ATTCCCGATGGCAA-AGAAACACTCGTG-TAMRA. Mouse TNF-α forward primer, 5′-AATGGCCTCCCTCTCAT-CAGT-3′, reverse primer, 5′-GCTACAGGCTTGTCACTCGAATT-3′, and probe, 6FAM-ATGGCCCAGACCCTCACA-CTCAGATC-TAMRA. Mouse IL-6 forward primer, 5′-TATGAAGTTCCTCTCTGCAAGAGA-3′, reverse primer, 5′-TAGGGAAGGCCGTGGTT-3′, and probe, 6FAM-CCAGCATCAGTCCCAAGAAGGCAACT-TAMRA. VCAM-1 forward primer, 5′-CTGAATACAAAACGATCGCTCAA-3′, reverse primer, 5′-GCGTTTAGTGGGC-TGTCTATCTG-3′, and probe, 6FAM-CGGTGACTCCATGGCCCTCACTTG-TAMRA. IL-10 and IFN-γ probe and primers were purchased from Applied Biosystems. Quantitative real-time PCR was performed using the ABI Prism 7900 Sequence Detection System (Applied Biosystems). Data are calculated by 2-ΔΔCt method as described by the manufacturer and are expressed as the fold increases over the indicated controls (“1.0”) in each figure.
Transgenic SR-DN RAGE mice
Transgenic SR DN RAGE mice were prepared using the type A SR provided to us by C. Glass (University of California, San Diego, La Jolla, CA; reference 28). The generation and characterization of these mice has been described previously (29). Mice were generated and hemizygous mice, from two different founders, were backcrossed eight generations into C57BL/6 before study. Littermates not expressing the transgene were used as controls in these studies.
Statistical analysis
All data are reported as mean ± SD. Data were analyzed by analysis of variance and, as indicated, subject to post hoc comparisons using two-tailed Student's t test. For survival studies, the Kaplan-Meier method was used to analyze survival. The log-rank test was used to compare survival curves. Differences were considered significant when P < 0.05.
Online supplemental material.
To determine if RAGE expression differed in the resected tissue retrieved from mice that survived or succumbed after 70% or 85% hepatectomy by 7 d, real-time PCR for detection of RAGE transcript was performed for Fig. S1. Similarly, the resected tissues at the time of surgery or remnants from mice that survived by 14 d were subjected to PCR for detection of TNF-α transcript. Liver weight/body weight was measured. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040934/DC1.
Acknowledgments
This work was supported by the Surgical Research Fund of the College of Physicians and Surgeons, Columbia University, and by grants from the United States Public Health Service. A.M. Schmidt is a recipient of a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research.
A.M. Schmidt receives research support from TransTech Pharma, Inc. The authors have no other conflicting financial interests.
Abbreviations used: DN, dominant negative; EMSA, electrophoretic mobility shift assay; MP, mononuclear phagocyte; MPDDC, MP-derived DC; PCNA, proliferating cell nuclear antigen; RAGE, receptor for advanced glycation endproducts; SR, scavenger receptor promoter; sRAGE, soluble RAGE; VCAM-1, vascular cell adhesion molecule-1.
G. Cataldegirmen and S. Zeng contributed equally to this work.
References
- 1.Fausto, N. 2000. Liver regeneration. J. Hepatol. 32:19–31. [DOI] [PubMed] [Google Scholar]
- 2.Schulte-Hermann, R., B. Grasl-Kraupp, and W. Bursch. 1995. Apoptosis and hepatocarcinogenesis. In Liver Regeneration and Carcinogenesis. Molecular and Cellular Mechanisms. R.L. Jirtle, editor. Academic Press, San Diego, CA. 141–178.
- 3.Panis, Y., D.M. McMullan, and J.C. Emond. 1997. Progressive necrosis after hepatectomy and the pathophysiology of liver failure after massive resection. Surgery. 121:142–149. [DOI] [PubMed] [Google Scholar]
- 4.Francavilla, A., Q. Zeng, and L. Polimeno. 1994. Small-for-size liver transplanted into larger recipient: a model of hepatic regeneration. Hepatology. 19:210–216. [PMC free article] [PubMed] [Google Scholar]
- 5.Emond, J.C., J.F. Renz, R.C. Lim, J.P. Roberts, and N.L. Ascher. 1996. Functional characterization of liver grafts from living donors: implications for the treatment of older children and adults. Ann. Surg. 224:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang, H., O. Bloom, M. Zhang, J.M. Vishnubhakat, M. Ombrellino, J. Che, A. Frazier, H. Yang, S. Ivanova, L. Borovikova, et al. 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–251. [DOI] [PubMed] [Google Scholar]
- 7.Andersson, U., H. Wang, K. Palmblad, A.C. Aveberger, O. Bloom, H. Erlandsson-Harris, A. Janson, R. Kokkola, M. Zhang, H. Yang, and K.J. Tracey. 2000. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 192:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann, M.A., S. Drury, C. Fu, W. Qu, A. Taguchi, Y. Lu, C. Avila, N. Kambham, A. Bierhaus, P. Nawroth, et al. 1999. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 97:889–901. [DOI] [PubMed] [Google Scholar]
- 9.Bucher, N.L.R., and S.R. Farmer. 1998. Liver regeneration following partial hepatectomy: genes and metabolism. In Liver Growth and Repair. A.J. Strain and A.M. Diehl, editors. Chapman and Hall, London. 3–27.
- 10.Michalopoulos, G.K., and M.C. DeFrances. 1997. Liver regeneration. Science. 276:60–66. [DOI] [PubMed] [Google Scholar]
- 11.Park, L., K.G. Raman, K.J. Lee, Y. Lu, L.J. Ferran Jr., W.S. Chow, D. Stern, and A.M. Schmidt. 1998. Suppression of accelerated diabetic atherosclerosis by soluble receptor for AGE (sRAGE). Nat. Med. 4:1025–1031. [DOI] [PubMed] [Google Scholar]
- 12.Brunt, E.M., C.G. Janney, A.M. Di Bisceglie, B.A. Neuschwander, and B.R.B.A. Bacon. 1999. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 94:2467–2474. [DOI] [PubMed] [Google Scholar]
- 13.Shinozuka, H., Y. Kubo, S.L. Katyal, P. Coni, G.M. Ledda-Columbano, A. Columbano, and T. Nakamura. 1994. Roles of hepatocyte growth factor and tumor necrosis factor-alpha on liver cell proliferation induced in rats by lead nitrate. Lab. Invest. 71:35–41. [PubMed] [Google Scholar]
- 14.Yamada, Y., I. Kirillova, J.J. Peschon, and N. Fausto. 1997. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type 1 tumor necrosis factor receptor. Proc. Natl. Acad. Sci. USA. 94:1441–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Webber, E.M., J. Bruix, R.H. Pierce, and N. Fausto. 1998. Tumor necrosis factor primes hepatocytes for DNA replication in the rat. Hepatology. 28:1226–1234. [DOI] [PubMed] [Google Scholar]
- 16.Cressman, D.E., L.E. Greenbaum, R.A. DeAngelis, G. Ciliberto, E.E. Furth, V. Poli, and R. Taub. 1996. Liver failure and defective hepatocyte regeneration in interleukin-6 deficient mice. Science. 274:1379–1383. [DOI] [PubMed] [Google Scholar]
- 17.Sakamoto, T., Z. Liu, N. Murase, T. Ezure, S. Yokomuro, V. Poli, and A.J. Demetris. 1999. Mitosis and apoptosis in the liver of interleukin-6 deficient mice after partial hepatectomy. Hepatology. 29:403–411. [DOI] [PubMed] [Google Scholar]
- 18.Wallenius, V., K. Wallenius, and J.O. Jansson. 2000. Normal pharmacologically-induced, but decreased regenerative liver growth in interleukin-6 deficient mice. J. Hepatol. 33:967–974. [DOI] [PubMed] [Google Scholar]
- 19.Li, L., J.F. Elliott, and T.R. Mosmann. 1994. IL-10 inhibits cytokine production, vascular leakage, and swelling during T helper 1 cell-induced delayed-type hypersensitivity. J. Immunol. 153:3967–3978. [PubMed] [Google Scholar]
- 20.Bergmann, M., L. Hart, M. Lindsay, P.J. Barnes, and R. Newton. 1998. IκBα degradation and nuclear factor κB DNA binding are insufficient for interleukin-1β and tumor necrosis factor-α-induced κB-dependent transcription. Requirement for an additional activation pathway. J. Biol. Chem. 273:6607–6610. [DOI] [PubMed] [Google Scholar]
- 21.Beg, A.A., W.C. Sha, R.T. Bronson, and D. Baltimore. 1995. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 376:167–170. [DOI] [PubMed] [Google Scholar]
- 22.Van Antwerp, D.J., S.J. Martin, T. Kafri, D.R. Green, and I.M. Verman. 1996. Suppression of TNF-α induced apoptosis by NF-κB. Science. 274:787–789. [DOI] [PubMed] [Google Scholar]
- 23.Cressman, D.E., L.E. Greenbaum, B.A. Haber, and R. Taub. 1994. Rapid activation of post-hepatectomy factor/nuclear factor kappa B in hepatocytes, a primary response in the regenerating liver. J. Biol. Chem. 269:30429–30435. [PubMed] [Google Scholar]
- 24.Albrecht, J.H., and L.K. Hanssen. 1999. Cyclin D1 promotes mitogen-independent cell cycle progression in hepatocytes. Cell Growth Differ. 10:397–404. [PubMed] [Google Scholar]
- 25.Guttridge, D.C., C. Albanese, J.Y. Reuther, R.G. Pestell, and A.S. Baldwin Jr. 1999. NF-kappa B controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 19:5785–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zarnegar, R., and G.K. Michalopoulos. 1995. The many faces of hepatocyte growth factor: from hepatopoeisis to hematopoiesis. J. Cell Biol. 129:1177–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Presnell, S.C., D.B. Stolz, W.M. Mars, M. Jo, G.K. Michalopoulos, and S.C. Strom. 1997. Modifications of the hepatocyte growth factor/c-met pathway by constitutive expression of TGF-α in rat liver epithelial cells. Mol. Carcinog. 18:244–255. [PubMed] [Google Scholar]
- 28.Wu, H., K. Moulton, A. Horvai, S. Parik, and C.K. Glass. 1994. Combinatorial interactions AP-1 and ets domain proteins contribute to the developmental regulation of the macrophage scavenger receptor gene. Mol. Cell. Biol. 14:2129–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rong, L.L., S.F. Yan, T. Wendt, D. Hans-Wagner, S. Pachydaki, L.G. Bucciarelli, A. Adebayo, W. Qu, Y. Lu, K. Kostov, et al. 2004. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 18:1818–1825. [DOI] [PubMed] [Google Scholar]
- 30.Kislinger, T., C. Fu, B. Huber, W. Qu, A. Taguchi, S.D. Yan, M. Hofmann, S.F. Yan, M. Pischetsrieder, D. Stern, and A.M. Schmidt. 1999. Ne (carboxymethyl)lysine modifications of proteins are ligands for RAGE that activate cell signalling pathways and modulate gene expression. J. Biol. Chem. 274:31740–31749. [DOI] [PubMed] [Google Scholar]
- 31.Huttunen, H.J., C. Fages, and H. Rauvala. 1999. RAGE-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signalling pathways. J. Biol. Chem. 274:19919–19924. [DOI] [PubMed] [Google Scholar]
- 32.Taguchi, A., D.C. Blood, G. del Toro, A. Canet, D.C. Lee, W. Qu, N. Tanji, Y. Lu, E. Lalla, C. Fu, et al. 2000. Blockade of amphoterin/RAGE signalling suppresses tumor growth and metastases. Nature. 405:354–360. [DOI] [PubMed] [Google Scholar]
- 33.Crossin, K.L. 2002. Cell adhesion molecules activate signaling networks that influence proliferation, gene expression, and differentiation. Ann. NY Acad. Sci. 961:59–160. [DOI] [PubMed] [Google Scholar]
- 34.Bruccoleri, A., R. Gallucci, D.R. Germolec, P. Blackshear, P. Simeonova, R.G. Thurman, and M.I. Luster. 1997. Induction of early-immediate genes by tumor necrosis factor alpha contribute to liver repair following chemical-induced hepatotoxicity. Hepatology. 25:133–141. [DOI] [PubMed] [Google Scholar]
- 35.Chaisson, M.L., J.T. Brooling, W. Ladiges, S. Tsai, and N. Fausto. 2002. Hepatocyte-specific inhibition of NF-κB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J. Clin. Invest. 110:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lau, A.H., and A.W. Thomson. 2003. Dendritic cells and immune regulation in the liver. Gut. 52:307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuno, K., H. Nomiyama, H. Yoneyama, and R. Uwatoku. 2002. Kupffer cell-mediated recruitment of dendritic cells to the liver crucial for a host defense. Dev. Immunol. 9:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lian, Z.X., T. Okada, X.S. He, H. Kita, Y.J. Liu, A.A. Ansari, K. Kikuchi, S. Ikehara, and M.E. Gershwin. 2003. Heterogeneity of dendritic cells in the mouse liver: identification and characterization of four distinct populations. J. Immunol. 170:2323–2330. [DOI] [PubMed] [Google Scholar]
- 39.Chen, Y., S.S. Yan, J. Colgan, H.P. Zhang, J. Luban, A.M. Schmidt, D. Stern, and K.C. Herold. 2004. Blockade of late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J. Immunol. 173:1399–1405. [DOI] [PubMed] [Google Scholar]
- 40.Zeng, S., N. Feirt, M. Goldstein, J. Guarrera, N. Ippagunta, U. Ekong, H. Dun, Y. Lu, W. Qu, A.M. Schmidt, and J.C. Emond. 2004. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology. 39:422–432. [DOI] [PubMed] [Google Scholar]