

Abstract
Schnurri (Shn) is a large zinc finger protein implicated in cell growth, signal transduction, and lymphocyte development. Vertebrates possess at least three Shn orthologues (Shn-1, Shn-2, and Shn-3), which appear to act within the bone morphogenetic protein, transforming growth factor β, and activin signaling pathways. However, the physiological functions of the Shn proteins remain largely unknown. In Shn-2–deficient mice, mature peripheral T cells exhibited normal anti–T cell receptor–induced proliferation, although there was dramatic enhancement in the differentiation into T helper type (Th)2 cells and a marginal effect on Th1 cell differentiation. Shn-2–deficient developing Th2 cells showed constitutive activation of nuclear factor κB (NF-κB) and enhanced GATA3 induction. Shn-2 was able to compete with p50 NF-κB for binding to a consensus NF-κB motif and inhibit NF-κB–driven promoter activity. Thus, Shn-2 plays a crucial role in the control of Th2 cell differentiation by regulating NF-κB function.
CD4 T cell–dependent immune responses are controlled by the balance of antigen-specific Th1 and Th2 cells (1–3). IL-12–induced activation of STAT4 is crucial for Th1 cell differentiation, whereas IL-4–induced STAT6 activation is crucial for Th2 cell differentiation (4–8). In addition to cytokine-induced signals, the activation of the TCR-mediated signaling is indispensable for both Th1 and Th2 cell differentiation. In particular, Th2 cell differentiation is largely dependent on the activation of p56lck, calcineurin, and the Ras-ERK MAPK signaling cascade (9–11). A negative regulator of the above signaling pathways, SHP-1, also controls the efficiency of Th2 cell differentiation and Th2 cell–dependent immune responses (12). Master transcription factors for Th1 and Th2 cell differentiation have been revealed, i.e., GATA3 for Th2 cells and T-bet for Th1 cells (13–17).
In Drosophila, Mad-Medea and Schnurri (Shn), a large zinc finger protein, are reported to interact with each other and act as nuclear targets in the Drosophila decapentaplegic signaling pathway (18–20). In vertebrates, the Drosophila decapentaplegic signaling pathway may equate to the bone morphogenetic protein/TGF-β/activin signaling pathways that play diverse roles in developmental processes (21). Vertebrates have at least three orthologues of Shn: Shn-1 (also known as HIV-EP1, MBP-1, PRDII-BF1, and αA-CRYBP1), Shn-2 (also known as HIV-EP2, MBP-2, AGIE-BP1, and MIBP1), and Shn-3 (also known as HIV-EP3, KRC, and ZAS3). The mRNA expression of Shn-2 was detected mostly in the brain, heart, and spleen (22–24). Recently, the requirement of Shn-2 in positive selection of thymocytes was reported (25) and Shn-3–deficient CD4+ CD8+ thymocytes were shown to exhibit a defect in cell survival (26). However, the precise physiological roles of these Shn family members remain largely unknown.
NF-κB is a critical transcription factor that regulates Th2 cell differentiation and Th2 cell–dependent airway inflammation (27–29). NF-κB–deficient (p50 subunit–deficient) mice were unable to mount OVA-induced airway inflammation (28). The lack of inflammation was not due to defects in T cell priming, T cell proliferation, or expression of important cell adhesion molecules, but rather to the impaired induction of GATA3 (30). Recently, one of the Shn family members, Shn-3 (KRC), was reported to associate with TNF receptor–associated factor (TRAF)2 to repress nuclear translocation of NF-κB (31). In addition, Shn-3 (ZAS3) was shown to bind to the NF-κB motif directly, and the competition with NF-κB binding resulted in the repression of transactivation of the NF-κB target gene (32, 33). A direct repressive activity of Shn-3 was also demonstrated (33). These emerging findings suggest that the Shn protein is involved in NF-κB activation and/or NF-κB–mediated gene transactivation.
Here, we investigate the role of Shn-2 in Th1/Th2 cell differentiation by using Shn-2–deficient (Shn-2−/−) mice. Our results suggest that Shn-2 plays crucial roles in the control of Th2 cell differentiation by regulating NF-κB activation and GATA3 expression.
Results
Phenotypic and functional characterization of peripheral CD4 T cells in Shn-2–deficient (Shn-2−/−) mice
Shn-2−/−mice were previously shown to have a defect in T cell generation due to the impairment of positive thymic selection (25). However, we found that moderate numbers of CD4 and CD8 T cells were present in Shn-2−/−mice of a BALB/c background (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20040733/DC1). The cell surface expression of TCR-β, CD3ɛ, common γ (γC), IL-4Rα, CD25, CD69, CD44, and CD62L on splenic CD4 T cells was found to be comparable to those of controls (Fig. S1 B). The TCR Vβ chain repertoire in the peripheral Shn-2−/− T cells was similar to that of Shn-2+/+T cells (Fig. S1 C). Anti–TCR-β– or anti-CD3ɛ–induced proliferative responses were the same in Shn-2+/+ and Shn-2−/− CD4 T cells (Fig. S2 A). Furthermore, the phosphorylation status of MAPKs (Erk1 and Erk2) after TCR cross-linking in Shn-2−/−CD4 T cells was comparable to that of controls (Fig. S2 B). Thus, no obvious defect in the phenotype or the activation of Shn-2−/−splenic CD4 T cells was noted. The expression of Shn-2 mRNA was detected in freshly prepared CD4 T cells, and it was decreased after anti-TCR stimulation in Th1 and Th2 cell differentiation cultures (Fig. S3).
Enhanced Th2 cell differentiation in Shn-2−/−× DO11.10 transgenic (Tg) naive CD4 T cells
Next, we assessed the capability of Shn-2−/−CD4 T cells to differentiate into Th1/Th2 cells. Naive splenic CD4 T cells (CD4+ CD44low) were purified by cell sorting (purity >98%) and then subjected to in vitro Th1/Th2 cell differentiation culture and analyzed by intracellular cytokine staining and ELISA. Naive CD4 T cells from Shn-2−/−× OVA–specific TCR-αβ Tg (DO11.10 Tg) mice were stimulated with 0.1 μM of antigenic OVA peptide and irradiated BALB/c splenocytes in the presence of appropriate cytokines and anti-cytokine mAbs under three different conditions: Th2-skewed (IL-4 with anti–IL-12 mAb and anti–IFN-γ mAb), Th1-skewed (IL-12 with anti–IL-4 mAb), and nonskewed (IL-2 with anti–IL-4 mAb, anti–IL-12 mAb, and anti–IFN-γ mAb) conditions. As shown in Fig. 1 A, Th2 cell differentiation was enhanced substantially in Shn-2−/−T cells (17.3 vs. 37.1%), whereas Th1 cell differentiation was unaffected (83.0 vs. 80.5%). Under nonskewed conditions, the baseline levels of IL-4–producing and IFN-γ–producing cells were increased in Shn-2−/−T cell cultures (Fig. 1 A, left). Portions of the same cell cultures that were used for intracellular cytokine staining were restimulated, and cytokine production was measured by ELISA. Basically, a similar pattern among cultures was observed (Fig. 1 B). The production of IL-4 and IL-5 was increased about twofold in Shn-2−/−Th2 cells, whereas similar levels of IFN-γ production in Th1 cells were detected. The levels of IL-4 and IFN-γ production in nonskewed cultures were very low, but they were slightly increased in the Shn-2−/−cultures.
Figure 1.
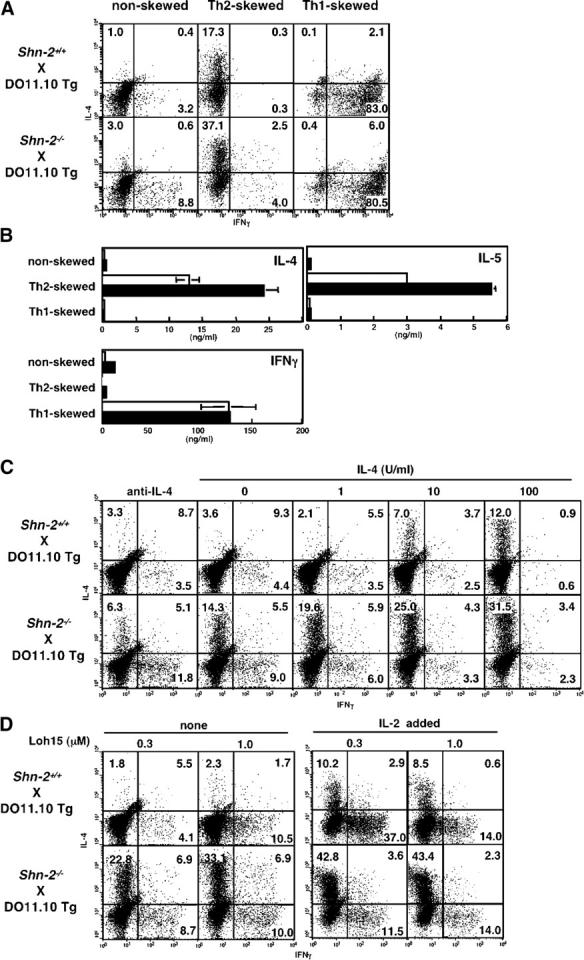
Enhanced Th2 cell differentiation in Shn-2−/−× DO11.10 TCR Tg T cells. (A) Naive (CD44low) CD4 T cells from Shn-2−/−× DO11.10 Tg mice were purified by cell sorting and stimulated with antigenic OVA peptide (Loh15: 0.1 μM) and irradiated BALB/c APCs for 5 d. Th2 cell–skewed (IL-4 with anti–IL-12 mAb and anti–IFN-γ mAb), Th1 cell–skewed (IL-12 with anti–IL-4 mAb), and nonskewed (IL-2 with anti–IL-4 mAb, anti–IL-12 mAb, and anti–IFN-γ mAb) conditions were used. Intracellular staining was performed with FITC-conjugated anti–IFN-γ mAb and PE-conjugated anti–IL-4 mAb. Under the typical Th2 cell–skewed conditions, the levels of Th2 cell differentiation in five experiments were 17.5 ± 2.9% in Shn-2 + / + cultures and 38.0 ± 11.8% in Shn-2−/−cultures. P < 0.006. (B) A portion of the same differentiated cell cultures used in A were restimulated with antigenic peptide and APCs for 24 h, and the concentrations of cytokines (IL-4, IL-5, and IFN-γ) in the culture supernatant were determined by ELISA. (C) Naive (CD44low) CD4 T cells from Shn-2−/−× DO11.10 Tg mice were stimulated with a specific OVA peptide (Loh15: 0.1 μM) and irradiated normal BALB/c APCs in the presence of indicated doses of IL-4. Intracellular staining profiles of IFN-γ and IL-4 are shown with percentages of cells in each area. (D) Purified naive (CD44low) CD4 T cells were stimulated with indicated doses of specific OVA peptides (Loh15: 0.3 and 1.0 μM) and irradiated BALB/c APCs in the presence or absence of 30 U/ml of exogenous IL-2. Two independent experiments were performed with similar results.
To examine further the effect on Th2 cell differentiation, naive CD4 T cells from Shn-2−/−× DO11.10 Tg mice were stimulated with a minimum dose of antigenic peptide (0.1 μM) and APCs in the presence of graded doses of exogenous IL-4. As shown in Fig. 1 C, IL-4–producing Th2 cells were generated in an exogenous IL-4 dose-dependent manner in wild-type Shn-2+/+T cell cultures. As expected, the generation of Th2 cells in Shn-2−/−mice was dramatically enhanced at all doses of exogenous IL-4. The number of IL-5–producing cells was also enhanced in Shn-2−/−T cell cultures (not depicted). Also, we detected increased numbers of IFN-γ–producing cells at all groups. Next, we examined the effect of the concentration of antigenic peptide and IL-2 in the culture. Naive CD4 T cells purified by cell sorting were stimulated with two different doses of antigenic peptide (0.3 or 1.0 μM) and irradiated APCs from normal BALB/c mice in the absence of exogenous IL-4. A dramatic increase in the numbers of IL-4–producing Th2 cells was observed in Shn-2−/−cultures at either dose of antigenic peptide (Fig. 1 D, left). In the presence of exogenous IL-2, Th2 cell generation was enhanced in both Shn-2+/+and Shn-2−/−T cell cultures, although the levels were substantially higher in Shn-2−/−groups as compared with Shn-2+/+.
Enhanced Th2 cell differentiation in Shn-2−/−splenic CD4 T cells after anti-TCR mAb stimulation
To further assess the efficiency of Th2 cell differentiation in the splenic Shn-2−/−T cells, purified splenic naive CD4 T cells (purity >98%) were stimulated with immobilized anti–TCR-β mAb in the presence of graded doses of exogenous IL-4. No APCs were added. 30 U/ml of exogenous IL-2 was added to the differentiation cultures. As shown in Fig. 2 A, IL-4–producing Th2 cells were generated in an exogenous IL-4 dose-dependent manner in the Shn-2+/+wild-type T cell cultures, and as expected, the generation of Th2 cells in Shn-2−/−mice was substantially enhanced in the cultures at all doses of exogenous IL-4 (Fig. 2 A). These results suggest that the ability to differentiate into Th2 cells was enhanced in Shn-2−/−splenic CD4 T cells. The number of Th2 cells generated in Shn-2−/−× STAT6−/−T cell cultures was insignificant, suggesting that the enhanced Th2 cell differentiation in Shn-2−/−T cell cultures was STAT6 dependent (not depicted).
Figure 2.
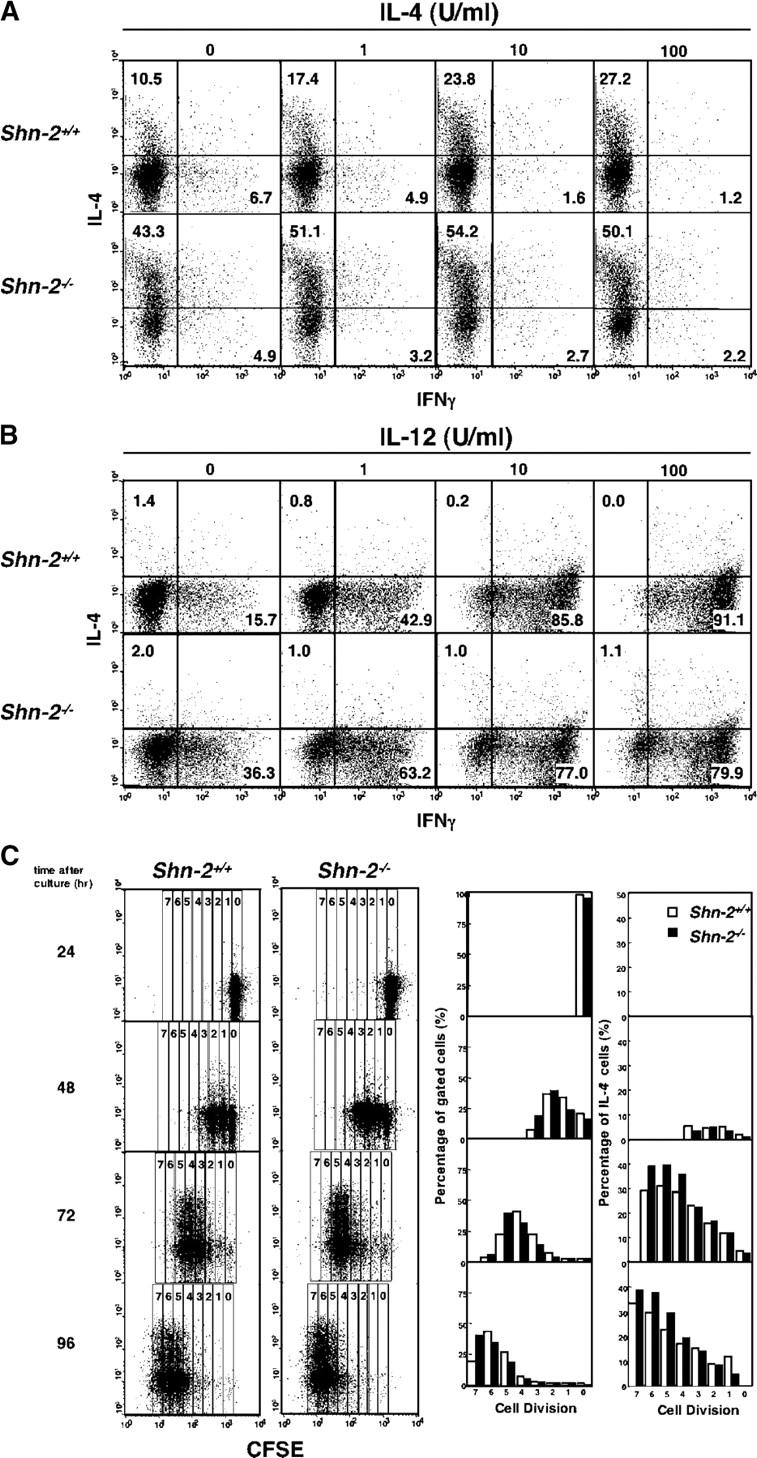
Enhanced Th2 cell differentiation in Shn-2−/−T cells. (A) Splenic naive CD4 T cells from Shn-2−/−mice were stimulated with immobilized anti–TCR-β mAb, 30 U/ml of exogenous IL-2, and indicated concentrations of IL-4 for 5 d. Intracellular staining profiles of IFN-γ and IL-4 are shown with percentages of cells in each area. The absolute numbers of cells harvested in these cultures were similar. Three independent experiments were performed with similar results. (B) Splenic naive CD4 T cells from Shn-2−/−mice were stimulated with immobilized anti–TCR-β mAb in the presence of indicated doses of IL-12 and anti–IL-4 mAb. Intracellular staining profiles of IFN-γ and IL-4 are shown with percentages of cells in each area. The absolute numbers of cells harvested in these cultures were similar. Three independent experiments were performed with similar results. (C) Splenic naive CD4 T cells were labeled with CFSE and stimulated with immobilized anti–TCR-β mAb in the presence of IL-4 and IL-2. After culturing for the indicated times, cells were restimulated and subjected to intracellular staining with APC-conjugated anti–IL-4 mAb. Percentages of the cells in the gates representing numbers of cell division (0–7) and percentages of IL-4–producing Th2 cells in each gate are shown in the right panels. Percentages of IL-4–producing cells in total (without gating) are 0.7% (24 h), 5.4% (48 h), 28.6% (72 h), and 29.3% (96 h) in Shn-2+/+ cultures and 0.6% (24 h), 5.4% (48 h), 36.9% (72 h), and 37.9% (96 h) in Shn-2−/− cells. Two independent experiments were performed with similar results.
To assess the effect on Th1 cell differentiation in a non-TCR Tg system, Shn-2−/−splenic CD4 T cells were stimulated with anti–TCR-β mAb in the presence of various doses of exogenous IL-12 and anti–IL-4 mAb (Fig. 2 B). Moderate increases in the generation of Th1 cells were observed in groups with no exogenous IL-12 or a low dose of IL-12 (1 U/ml). However, no further increases but rather a slight decrease in the number of Th1 cells generated was observed at higher doses of IL-12 (10 or 100 U/ml). A similar pattern was obtained in STAT6-deficient Th1 cell cultures (not depicted). We observed similar enhancement in the generation of Th2 cells in Shn-2−/−mice with either a B6 background or a B6 × BALB/c background (not depicted).
Anti-TCR–induced cell division of Shn-2−/−CD4 T cells
Because some cycles of cell divisions are reported to be required for the generation of IL-4–producing Th2 cells (34), we examined the anti-TCR–induced cell division of Shn-2−/−CD4 T cells. Carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled naive CD4 T cells were stimulated with anti–TCR-β mAb in the presence of IL-2 and IL-4 (Th2 cell–skewed condition), and 24, 48, 72, and 96 h later, IL-4 production in the developing Th2 cells was assessed (Fig. 2 C). 24 h after the stimulation, most cells had not undergone cell division (cell division no. 0), and IL-4–producing Th2 cells were not detected. At 48 h, cells had undergone up to three cell divisions, and there was a slight increase in the numbers of cells with three cell divisions detected in Shn-2−/−CD4 T cells. The percentages of IL-4–producing T cells generated were not apparently different between Shn-2−/−and Shn-2+/+CD4 T cell cultures at this time point. After 72 and 96 h in culture, the rate of cell division appeared to be slightly increased in Shn-2−/−CD4 T cells, and the generation of IL-4–producing T cells was higher at all numbers of cell division. These results suggest that although Shn-2−/−CD4 T cells show a slightly increased rate in cell division, the enhanced Th2 cell generation in Shn-2−/−CD4 T cells could not be simply explained by the difference in the number of cell divisions that the developing Th2 cells had undergone.
We reported previously that CD28 costimulation induced increased Th2 cell differentiation (35) and induced NF-κB activation and enhanced GATA3 expression in developing Th2 cells to facilitate chromatin remodeling of the IL-5 gene loci (36). Shn-2−/−T cells were stimulated with anti-TCR plus anti-CD28 mAb under Th2 cell–skewed conditions (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20040733/DC1). Shn-2−/−T cells showed increased levels of Th2 cell differentiation without anti-CD28 mAb as compared with wild-type (46.2 vs. 32.4%). No obvious additional enhancement of Th2 cell differentiation was detected in the presence of CD28 costimulation under the conditions where the CD28-mediated enhancing effect was observed in Shn-2+/+T cell cultures.
No functional defect was observed in DCs from Shn-2−/−mice
The results thus far suggested that the observed enhancement of Th2 cell differentiation is due to an alteration in T cells because we used purified T cells (Figs. 1 and 2) and APCs from normal mice (Fig. 1). However, Shn-2 mRNA is expressed in both CD4 T cells and APCs (not depicted), and thus, we wished to evaluate the effect of Shn-2 deficiency on APC function using bone marrow–derived DCs (BMDCs). The expression levels of CD11c, I-Ad, H-2Kd, CD40, B7-1, B7-2, and Fas were comparable between Shn-2+/+and Shn-2−/−BMDCs (not depicted). OVA-pulsed BMDCs from Shn-2+/+or Shn-2−/−mice were cocultured with DO11.10 Tg CD4 T cells, and proliferative responses of T cells were assessed by [3H]thymidine uptake. Under these conditions, the APC function of both immature and mature BMDCs was found to be comparable between Shn-2−/−or Shn-2+/+mice (Fig. 3 A). We then examined the ability of OVA-pulsed BMDCs as APCs to induce Th1/Th2 cell differentiation. Stimulation with either immature or mature BMDCs resulted in similar levels of Th1/Th2 cell differentiation in the cultures with Shn-2+/+and Shn-2−/−BMDCs (Fig. 3 B). To further evaluate the APC function of Shn-2−/−mice, spleen cells were irradiated and tested for their ability to induce Th1/Th2 cells. The numbers of Th2 cells generated were found to be comparable between cultures with Shn-2−/−and Shn-2+/+APCs (Fig. 3 C). Taken together, the enhanced Th2 cell generation in Shn-2−/−T cell cultures appears to be due to an alteration in CD4 T cells themselves and not to any change in APC function.
Figure 3.
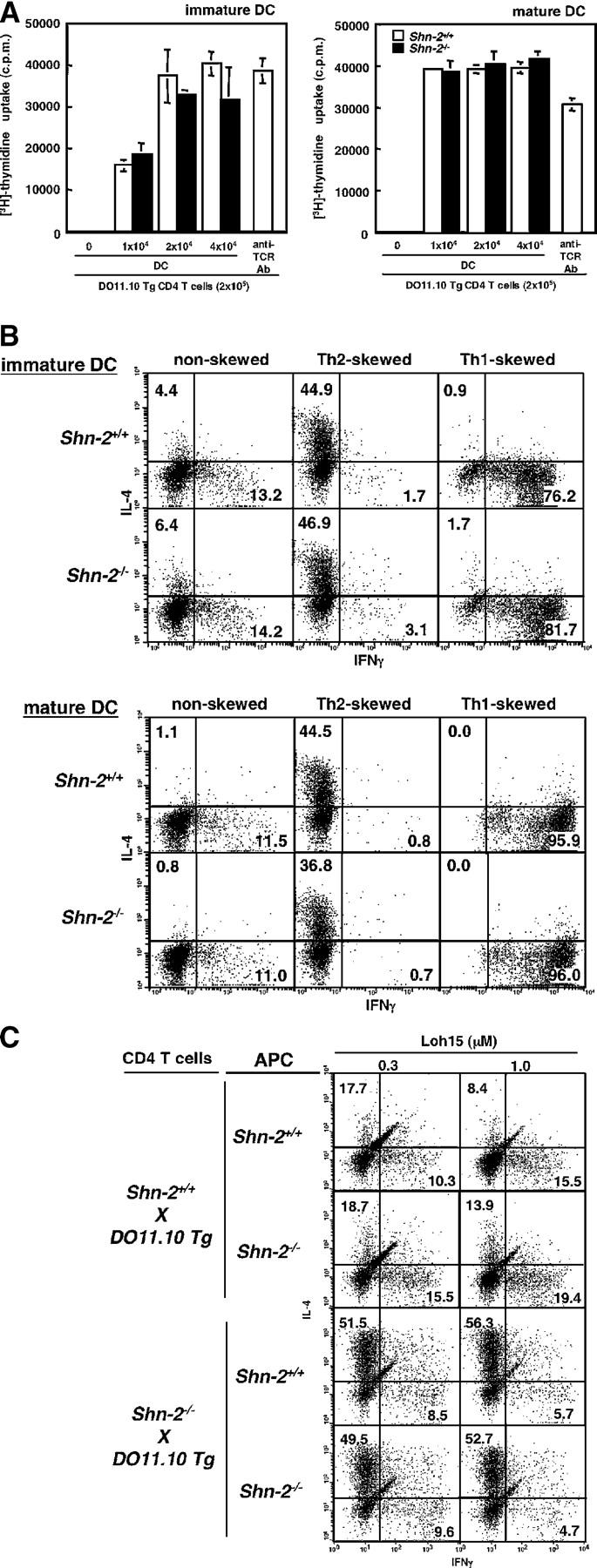
The function of Shn-2−/− APCs. (A) OVA-pulsed immature or mature BMDCs were cocultured with DO11.10 Tg CD4 T cells for 72 h. [3H]thymidine (37 kBq/well) was added to the stimulation culture for the last 16 h. The results (mean and standard deviation) of [3H]thymidine incorporation are shown. Three independent experiments were performed with similar results. (B) OVA-pulsed immature or mature BMDCs (2 × 104 cells) were cocultured with DO11.10 Tg naive (CD44low) CD4 T cells (2 × 105 cells) under nonskewed, Th2 cell–skewed, and Th1 cell–skewed conditions for 5 d. Intracellular staining profiles (IFN-γ/IL-4) of the cultured cells are shown with percentages of the cells in each quadrant. Three independent experiments were performed with similar results. (C) Naive (CD44low) CD4 T cells (1.5 × 104 cells) were purified from Shn-2−/−× DO11.10 Tg mice by cell sorting and stimulated with indicated doses of specific OVA peptides (Loh15: 0.1 μM) and irradiated CD4− spleen cells (105 cells) from non-DO11.10 Tg Shn-2+/+or Shn-2−/−mice. Two independent experiments were performed with similar results.
Hyperactivation of NF-κB in Shn-2−/−CD4 T cells
We examined the NF-κB activation in Shn-2−/−CD4 T cells by electrophoretic mobility shift assays (EMSAs). CD4 T cells from Shn-2+/+and Shn-2−/−mice were stimulated with anti-TCR and anti-CD28 for 3 h, and subsequently, nuclear extracts were prepared. To facilitate the stimulation of the TCR complex on naive T cells, we used both anti-TCR and anti-CD28 mAbs. The NF-κB DNA binding activity was found to be increased after TCR stimulation in Shn-2+/+CD4 T cells (Fig. 4 A, lanes 2 and 3, and B, lanes 2 and 14). In contrast, in Shn-2−/−CD4 T cells, substantial levels of binding activity could already be detected even in nonstimulated T cells (Fig. 4 A, lane 4, and B, lanes 7 and 17), and an only moderate elevation in activity was observed after TCR stimulation (Fig. 4 A, lanes 5 and 6, and B, lanes 8 and 18). We performed a supershift assay to determine the specificity for NF-κB subunits and observed a substantial supershift with the anti-p65 antibody (Fig. 4 A, lanes 8 and 9, and B, lanes 4 and 15). A small, but reproducible shift was detected in Shn-2−/−nonstimulated samples (Fig. 4 A, lane 10, and B, lane 9). Increased amounts of p65 supershift band were detected after anti-TCR stimulation in Shn-2−/−T cells. Furthermore, an EMSA with anti-p50 antibody revealed that most of the NF-κB EMSA band detected in nonstimulated Shn-2−/−T cells contained the p50 subunit (Fig. 4 B, lane 11). There was a substantial increase in the level of the p50 band after anti-TCR stimulation (Fig. 4 B, lane 12). These results suggest that NF-κB is constitutively activated in Shn-2−/−CD4 T cells and it is hyperactivated after anti-TCR stimulation. We did not observe any obvious disappearance of specific bands in Shn-2−/−groups. This could be due to an insufficient amount of Shn-2 for visualization in the EMSA assay.
Figure 4.

Hyperactivation of NF-κB in Shn-2−/−CD4 T cells. (A and B) Splenic CD4 T cells were incubated with medium alone overnight and then stimulated with immobilized anti–TCR-αβ mAb in the presence or absence of agonistic anti-CD28 antibody for 3 h. Nuclear extracts of the cultured cells were prepared and subjected to EMSAs with NF-κB probes. The supershift assays were performed with antibodies specific for NF-κB p50 and p65 subunit detection. Three independent experiments were performed with similar results. (C) Splenic CD4 T cells were incubated with medium alone overnight and then stimulated with immobilized anti–TCR-β mAb for 3 h. Subsequently, both cytosol and nuclear extracts were prepared and these were subjected to immunoblotting using anti-p50, anti-p65, anti–tubulin-α, and anti–histone H1 antibodies. Arbitrary densitometric units are shown under each band. (D) RNAs were prepared from fresh splenic CD4 T cells, resting CD4 T cells that were incubated with medium alone overnight, and anti-TCR–stimulated CD4 T cells, and then quantitative PCR assay was performed. The expression levels of p50 and p65 were normalized with 18S expression. Two independent experiments were performed with similar results.
An assessment of the protein expression levels of NF-κB p50 and p65 subunits in the cytosol and in nuclear fractions revealed that no obvious difference was detected between Shn-2+/+and Shn-2−/−T cells before or after anti-TCR stimulation, suggesting that the nuclear translocation of the NF-κB p50 and p65 subunits is not altered in the absence of Shn-2 (Fig. 4 C). Furthermore, transcriptional levels of p50 and p65 were compared by quantitative RT-PCR assay using freshly prepared CD4 T cells, resting CD4 T cells, and those after anti-TCR stimulation for 3 h (Fig. 4 D). In all cases, basically no difference was detected between Shn-2+/+and Shn-2−/−groups.
We also examined the activation of AP-1, CREB, and IL-4 NFAT in Shn-2−/−CD4 T cells by EMSA (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20040733/DC1). The DNA binding ability of AP-1 was slightly, but reproducibly, enhanced in Shn-2−/−T cells, whereas the DNA binding activity of CREB and IL-4 NFAT appeared to be similar in Shn-2+/+and Shn-2−/−T cells.
Enhanced expression of GATA3 in anti-TCR–activated Shn-2−/−CD4 T cells
Because the levels of GATA3 expression are critical for Th2 cell differentiation (14) and because GATA3 expression is regulated by NF-κB (30), we next assessed the protein expression of GATA3 in cultured Shn-2−/−T cells under Th2 cell–skewed condition. Splenic CD4 T cells from Shn-2−/−mice were stimulated with anti–TCR-β mAb and IL-4, and 16 h later, the expression of GATA3 was assessed by immunoblotting with specific antibodies. The levels of GATA3 protein were found to be reproducibly increased in Shn-2−/−T cells (Fig. 5 A). No detectable GATA3 protein was observed in freshly prepared Shn-2−/−CD4 T cells (not depicted). Concurrently, the mRNA levels were assessed by semiquantitative RT-PCR analysis, and increased GATA3 mRNA levels were detected (Fig. 5 B). These results suggest that enhanced induction of GATA3 takes place in early developing Shn-2−/−Th2 cells as compared with that of control Shn-2+/+Th2 cells.
Figure 5.

Increased GATA3 expression in Shn-2−/−developing Th2 cells. (A) Splenic CD4 T cells were cultured under Th2 cell–skewed conditions for 16 h. The cells were lysed, and cell lysates with threefold serial dilutions of cell lysates were subjected to immunoblotting using anti-GATA3 or anti–tubulin-α antibodies. Arbitrary densitometric units are shown under each band. (B) Splenic CD4 T cells were cultured under Th2 cell–skewed conditions for 6 h, and the expression levels of GATA3 mRNA were determined by RT-PCR. Arbitrary densitometric units are shown under each band. (C) Expression of NFAT1 and NFAT2 in Shn-2−/−Th2 cells. Splenic CD4 T cells were stimulated under Th2 cell–skewed conditions for 2 or 5 d, and then both cytosol and nuclear cell lysates were prepared. Immunoblotting was performed with anti-NFAT1 or anti-NFAT2 antibodies. Arbitrary densitometric units are shown under each band. (D) Splenic CD4 T cells were cultured under Th1 cell–skewed conditions for 6 or 16 h, and the expression levels of T-bet mRNA were determined by quantitative PCR assay. The expression was normalized with 18S expression. Two independent experiments were performed with similar results.
We examined the cytosolic or nuclear protein expression levels of NFAT1 and NFAT2 in developing Th2 cells (Fig. 5 C). In either cytosolic or nuclear lysate, there was no clear difference in the expression of NFAT1 detected between Shn-2+/+and Shn-2−/−T cells, suggesting normal nuclear translocation of NFAT1 occurs. The nuclear translocation of NFAT2 was, however, slightly impaired in Shn-2−/−T cells. An assessment of the expression of other molecules that may regulate Th2 cell differentiation directly or indirectly, such as JunB, cMaf, Bcl-6, and TRAF2, revealed no clear difference in mRNA expression levels in Shn-2−/−developing Th2 cells (not depicted). In addition, mRNA levels of several NF-κB target genes (IL-6, IL-1β, IL-15, and IL-17) are increased in Shn-2–deficient T cells (not depicted). The levels of T-bet in the Shn-2+/+and Shn-2−/−cells cultured under Th1 cell–skewed conditions are shown in Fig. 5 D. No significant difference was observed in the levels of T-bet mRNA.
Shn-2 inhibits an NF-κB–dependent transcriptional activity
The ability of Shn-2 protein to bind to the consensus NF-κB binding motif was assessed. 293 T cells were transfected with a pAct-Flag-hShn-2 vector, and 2 d later, nuclear extracts were prepared. Biotinylated NF-κB and control AP-1 oligonucleotides were absorbed on to streptavidin-agarose beads and incubated with the nuclear extracts. The amount of Shn-2 protein in the precipitates of agarose beads was assessed by immunoblotting with anti-Flag mAb. Total nuclear extracts were also run in parallel as loading controls. As can be seen in Fig. 6 A, a substantial binding of Flag–Shn-2 protein to NF-κB oligo was observed. This result suggests that Shn-2 protein is able to bind to a consensus NF-κB binding motif.
Figure 6.

Inhibition of NF-κB–dependent transcriptional activity by Shn-2. (A) 293 T cells were transfected with a control pAct or a pAct-Flag-hShn-2 vector, and 2 d later, their cell lysates were prepared. Biotinylated NF-κB and control AP-1 oligonucleotides were absorbed by streptavidin-agarose beads, and then the beads were incubated with cell lysates. The amount of Shn-2 protein in the precipitates was assessed by immunoblotting with anti-Flag mAb. Total cell lysates (106 equivalent/lane) were also run as controls (input). Three independent experiments were performed with similar results. (B) Nuclear extracts were prepared from 293 T cells transfected with pAct-Flag-hShn2 or pCMX-p50 vectors. The extracts were mixed in a certain ratio and then incubated with NF-κB oligonucleotides absorbed with streptavidin-agarose beads. The numbers represent the volume of cell lysates (μl; 1 μl = 5 × 105 cell equivalent). none, nontransfected 293 T lysates. The bound protein was detected by immunoblotting with an anti-Flag mAb for Shn-2 and an anti-p50 mAb for p50 protein. Total cell lysates were also run as controls (input). The position of Shn-2 and p50 are indicated. Three experiments were performed with similar results. (C) 293 T cells were transfected with 25 ng of the 5× NF-κB reporter constructs with the indicated doses of a pAct control vector (Mock) or a pAct-Flag-hShn-2 vector (Shn-2). Additionally, 1 ng pRL-TK vector was added into each transfection as an internal control. 24 h after the transfection, cells were stimulated with 50 ng/ml PMA and 500 nM ionomycin or 10 ng/ml TNF-α for12 h and then assayed for luciferase activity.
Next, we wished to examine whether Shn2 competes effectively with p50 NF-κB for binding to a consensus NF-κB binding motif. Nuclear extracts were prepared from 293 T cells that were transfected with pAct-Flag-hShn2 or pCMX-p50 vectors. The extracts were mixed in specific ratios and then incubated with NF-κB oligonucleotides absorbed with streptavidin-agarose beads. The bound protein was detected by immunoblotting with an anti-Flag mAb for Shn-2 and an anti-p50 mAb for p50 protein. As shown in Fig. 6 B, left, the binding of p50 to NF-κB oligonucleotides was inhibited by Shn-2–containing extracts in a dose-dependent manner. In addition, the binding of Shn-2 to NF-κB oligonucleotides was inhibited by the presence of p50 (Fig. 6 B, right). These results clearly suggest that Shn-2 competes with p50 for binding to a consensus NF-κB motif.
Consequently, to determine whether Shn-2 protein plays a functional role in NF-κB–dependent transcription, NF-κB–induced luciferase assay was performed with 293 T cells transfected with the 5× NF-κB reporter constructs along with graded doses of pAct-Flag-hShn-2 vector after stimulation with PMA plus ionomycin or TNF-α (Fig. 6 C). Some level of luciferase activity was detected without stimulation, and a significant inhibition was observed in the presence of Shn-2. After PMA plus ionomycin stimulation, the luciferase activity was increased and this was efficiently inhibited by Shn-2 (Fig. 6 C, middle). The luciferase activity was increased dramatically by stimulation with TNF-α, and this was moderately inhibited in the presence of Shn-2 protein (Fig. 6 C, right). These results suggest that Shn-2 protein is able to inhibit the NF-κB–dependent transcriptional activity.
Discussion
In this report, we demonstrated a crucial role for Shn-2 in Th2 cell differentiation. Shn-2 appears to control NF-κB DNA binding activity and subsequent GATA3 induction in early developing Th2 cells.
The DNA binding of NF-κB was enhanced in Shn-2–deficient CD4 T cells (Fig. 4, A and B). The enhancement was already apparent in nonstimulated Shn-2–deficient CD4 T cells. However, we found the NF-κB protein equally expressed in the nucleus and cytoplasm of nonstimulated and stimulated Shn-2–deficient CD4 T cells (Fig. 4 C). Thus, the enhanced DNA binding of NF-κB might be regulated by nuclear events such as those involved in the DNA binding process. A previous report suggested that Shn-3 binds to a NF-κB motif and represses transactivation of NF-κB–dependent genes (33). We show here that Shn-2 protein is able to bind to a consensus NF-κB motif (Fig. 6 A) and compete with p50 for DNA binding in a dose-dependent manner (Fig. 6 B). In addition, we detected significant inhibition of the NF-κB–dependent transcriptional activity in 293 T cells. Thus, it is conceivable that Shn-2 is able to bind to NF-κB DNA binding motifs and thus compete with the binding of NF-κB complexes or to repress directly the transactivation of the NF-κB–dependent genes in early developing Th2 cells. This might be a mechanism to account for the elevated NF-κB DNA binding activity in nonstimulated Shn-2–deficient CD4 T cells (Fig. 4, A and B).
In addition, we detected a further enhancement of DNA binding of NF-κB after TCR stimulation (Fig. 4, A and B). This could explain the enhanced induction of GATA3 in early developing Shn-2−/−Th2 cells (Fig. 5, A and B). A similar regulation was demonstrated in an experimental system using p50−/− mice (30). In our preliminary results, the generation of IL-4–producing Th2 cells in wild-type BALB/c CD4 T cell cultures was increased by the ectopic expression of both p50 and p65 (unpublished data). Thus, it appears probable that the enhanced induction of GATA3 by NF-κB hyperactivation is a mechanism that is responsible for the enhanced Th2 cell differentiation observed in Shn-2–deficient T cells.
Another interesting possibility for the molecular targets of Shn-2 is the involvement of TRAF2-mediated NF-κB activation. A previous report suggested that there is a physical association of Shn-3 with TRAF2 (31). Our preliminary results suggest that a physical association of Shn-2 with TRAF2 in 293 T cells can be demonstrated (unpublished data). Thus, Shn proteins may bind to TRAF2 and inhibit the TNFR-induced NF-κB activation and nuclear translocation in T cells. However, this appeared to be unlikely because no obvious difference in the protein expression of p50 and p65 was observed in the cytosol or nucleus before or after anti-TCR stimulation between Shn-2+/+ and Shn-2−/− T cells (Fig. 4 C). Lieberson et al. (37) have reported that Th2 cell responses are enhanced in dominant-negative TRAF2 Tg CD4 T cells. This result would appear to be contradictory to the above hypothesis. However, the dominant-negative TRAF2 used in the study by Lieberson et al. inhibits JNK activity efficiently, but it does not affect NF-κB activity very significantly (38). Thus, it is still possible that the Shn-2–TRAF complex is formed in T cells and that it is involved in the control of Th2 cell differentiation by regulating NF-κB activation. A more precise investigation is needed to clarify this issue.
As for the signaling pathways in which Shn-2 is involved, Shn-2 may regulate outcomes of both the TCR- and TNF-dependent pathways if the major function of Shn-2 is mediated by a mechanism proposed above, namely, competition with NF-κB for DNA binding in the nucleus. In this paper, we focused our analysis mostly on T cells where the TNF-dependent signaling pathway is not activated efficiently, and thus, further investigation will be required to address this issue. In addition, we did not observe CD28 costimulation-mediated enhancement of Th2 cell differentiation in Shn-2−/−cells (Fig. S4). CD28 costimulation induced NF-κB activation and enhanced GATA3 expression in developing Th2 cells (36). Thus, it is possible that the effect of CD28 costimulation was masked by hyperactivation of NF-κB in Shn-2−/−T cells.
It has been established that the expression of GATA3 is crucial for the efficient Th2 cell differentiation (16). GATA3 plays an essential role in chromatin-remodeling processes, such as acetylation of histone H3 and H4 of the Th2 cell cytokine gene loci (36). The activation of STAT6 is most critical for the induction of GATA3 transcription (16, 39). More recently, several regulatory molecules for GATA3 transcription have emerged. One is the NF-κB activation discussed above (30). We reported previously that a polycomb group gene product, mel-18, controls Th2 cell differentiation by regulating GATA3 transcription (40). Shn-2 is another example of a protein that regulates GATA3 transcription and expression in developing Th2 cells. Further investigation of the regulation of GATA3 expression is likely to provide a clearer molecular view of the initiation of Th2 cell differentiation.
Recombinant TGF-β inhibited Th2 cell differentiation of normal and Shn-2–deficient T cells very efficiently (unpublished data). In addition, Smad7 expression was not changed in Shn-2–deficient T cells (unpublished data). Thus, Shn-2 may not be involved in the TGF-β–mediated signaling pathway in T cells, at least not in the TGF-β–mediated inhibition of Th2 cell differentiation. However, because mammalian Shn has three orthologues (Shn-1, Shn-2, and Shn-3), it is still possible that Shn-1 and/or Shn-3 could substitute for the function of Shn-2 in Shn-2–deficient T cells.
As for the effects of Shn-2 deficiency on Th1 cell differentiation, Shn-2 may not play critical roles in Th1 cell differentiation. No effect was observed in typical Th1 cell–skewed cultures containing excess amounts of IL-12 (Fig. 1 A). However, we observed some enhancement in Th1 cell differentiation when IL-12 is limiting (Fig. 2 B). Also, IFN-γ–producing cells generated under Th2 cell–skewed conditions were increased consistently (Fig. 1 C). Thus, it is still possible that Shn-2 may act as a negative regulator of the generation of IFN-γ–producing cells.
In summary, the results of this study indicate that Shn-2 plays crucial roles in the control of Th2 cell differentiation by regulating the activation of NF-κB and subsequent GATA3 induction in early developing Th2 cells.
Materials and Methods
Mice
Shn-2–deficient (Shn-2−/−) mice were described previously (25). Animals used in this study were backcrossed to BALB/c more than 12 times and were 6–8 wk old. Anti-OVA–specific TCR-αβ (DO11.10) Tg mice were provided by D. Loh (Washington University School of Medicine, St. Louis, MO; reference 41). Shn-2−/−× DO11.10 Tg mice were used at 10–12 wk of age. STAT6-deficient (STAT6−/−) mice were provided by S. Akira (Osaka University, Osaka, Japan; reference 39). All mice used in this study were maintained under specific pathogen-free conditions. Animal care was in accordance with the guidelines of Chiba University.
Immunofluorescent staining and flow cytometry analysis
In general, 106 cells were incubated on ice for 30 min with the appropriate staining reagents according to a standard method (42). The reagents used in this study were as follows: anti-CD4–PE (RM4-1–PE), anti-CD4–FITC (RM4-1–FITC), anti-CD44–FITC (IM7-FITC), anti-CD44–PE (IM7-PE), anti-CD69–FITC (H1.2F3), anti-CD62L–FITC (MEL-14), anti-CD25–FITC (7D4), anti–IL-4Rα antibody, and anti-γC antibody-PE were purchased from BD Biosciences. Anti–rat Ig-FITC was purchased from CAPPEL. Anti–TCR-β–FITC (H57-FITC), anti-CD3–FITC (2C11-FITC), and anti-CD8–Cy5 were prepared in our laboratory. Flow cytometry analysis was performed on a FACSCalibur (Becton Dickinson), and results were analyzed with CELLQuest software (Becton Dickinson). Intracellular staining of IL-4 and IFN-γ was performed as described previously (11). FITC- conjugated anti–IFN-γ antibody (XMG1.2), APC-conjugated anti–IL-5 antibody, and PE-conjugated anti–IL-4 antibody (11B11; all from BD Biosciences) were used for detection.
Cell purification
Splenic CD4 T cells were purified by using magnetic beads and an Auto-MACS Sorter (Miltenyi Biotec) yielding a purity of >98%. Where indicated, CD4 T cells with naive phenotype (CD44low) were isolated from spleens on a FACSVantage cell sorter (Becton Dickinson) yielding a purity of >98% as described previously (11).
Bone marrow DC cultures
A modified method of Inaba et al. (43) and Chen-Woan et al. (44) was used for bone marrow DC cultures. In brief, bone marrow cells depleted of FcR+ cells (0.5–1.0 × 106 cells) were cultured in 1 ml medium in the presence of 10 ng/ml GM-CSF. The culture medium was changed every other day by gently swirling the plates, aspirating ∼75% of the medium, and adding back fresh medium with cytokines. On day 6, the cells were harvested and then CD11c+ cells (immature DCs) were purified using anti-CD11c mAb-conjugated micro beads and a MACS LS column (Miltenyi Biotec). The purified immature CD11c+ DCs were cultured for an additional 2 d with 10 ng/ml GM-CSF and 1 μg/ml LPS to allow further maturation and were used as mature DCs.
Proliferation assay
2 × 105 splenic CD4 T cells were stimulated in 200-μl cultures for 40 h with immobilized anti–TCR-β mAb (H57-597) and anti-CD3ɛ mAb (2C11). Immature or mature DCs were pulsed with 1 μM OVA peptide for 2 h at 37°C and then cocultured with CD4 T cells from DO11.10 Tg mice for 72 h. [3H]thymidine (37 kBq/well) was added to the stimulation culture for the last 16 h, and the incorporated radioactivity was measured by using a β plate (40).
ELISA for the measurement of cytokine concentration
The production of IL-2, IL-4, IL-5, IL-13, and IFN-γ was measured by ELISA as described previously (40).
In vitro Th1/Th2 cell differentiation cultures.
Naive splenic CD4 T cells were stimulated with 3 μg/ml of immobilized anti–TCR-β mAb (H57-597) in the presence of 30 U/ml IL-2 and 1–100 U/ml IL-4 as described previously (10). Where indicated, naive splenic CD4 T cells were labeled with CFSE (Molecular Probes) using the same procedure as described previously (40). For Th1 cell differentiation, naive splenic CD4 T cells were stimulated with 3 μg/ml of immobilized anti–TCR-β mAb (H57-597) in the presence of 30 U/ml IL-2, IL-12, and anti–IL-4 mAb. 1.5 × 104 sorted DO11.10 Tg CD44low CD4 T cells were stimulated with antigenic OVA peptide (Loh15, OVA; 323–339) and 105 irradiated (3,000 rad) normal BALB/c splenocytes. For stimulation with purified DCs, 2 × 105 sorted DO11.10 Tg CD44low CD4 T cells were cocultured with 2 × 104 OVA-pulsed immature or mature DCs. Appropriate cytokines and anti-cytokine antibodies were added in Th1/Th2 cell differentiation cultures as described previously (11). In typical DO11.10 Tg T cell cultures, Th2 cell–skewed (IL-4 with anti–IL-12 mAb and anti–IFN-γ mAb), Th1 cell–skewed (IL-12 with anti–IL-4 mAb), and nonskewed (IL-2 with anti–IL-4 mAb, anti–IL-12 mAb, and anti–IFN-γ mAb) conditions were used.
Immunoblotting
The Immunoblotting for the detection of GATA3, tubulin-α, NFAT1, and NFAT2 was performed as described previously (45). For NF-κB detection, anti-p65 (F-6), anti-p50 (E-10), and anti–histone H1 (AE-4, all from Santa Cruz Biotechnology, Inc.) antibodies were used.
EMSA
EMSAs were performed using Gel Shift Assay Systems (Promega) according to the manufacturer's instructions. In brief, nuclear extracts were incubated at 4°C with 32P-labeled double-stranded oligo (NF-κB oligo; AGTTGAGGGGACTTTCCCAGGC, AP-1 oligo; CGCTTGATGAGTCAGCCGGAA, CREB oligo; AGAGATTGCCTGACGTCAGAGAGCTAG, IL-4 NFAT; oligo; and GTAATAAAATTTTCCAATGTAAA) in DNA binding buffer (Promega). For supershift assays, nuclear extracts were incubated with anti-p50 (C-19; Santa Cruz Biotechnology, Inc.) or anti-p65 (F-6; Santa Cruz Biotechnology, Inc.) antibody at 4°C for 60 min and then incubated with 32P-labeled NF-κB double-stranded oligo at 4°C. Electrophoresis was performed on a 4% native polyacrylamide gel (0.5× TBE; acrylamide/bisacrylamide, 29:1), and the radioactivity was visualized by autoradiography.
Pull-down assay
A detailed protocol was described elsewhere (46). In brief, 293 T cells were transfected with 1.0 μg pAct or pAct-Flag-hShn-2, and 2 d later, the transfected cells were lysed for immunoblotting with anti-Flag mAb (M2; Sigma-Aldrich). Cell lysates were incubated with biotinylated oligonucleotides of NF-κB (AGTTGAGGGGACTTTCCCAGGC) and AP-1 (CGCTTGATGAGTCAGCCGGAA). The bound protein were eluted and separated on an SDS polyacrylamide gel and then subjected to immunoblot analysis using specific antibodies (anti-Flag Tag; M2; Sigma-Aldrich). For the competition assay shown in Fig. 6 B, 293 T cells were transfected with pAct-Flag-hShn-2 or pCMX-p50, and their cell extracts were mixed in certain ratios and then the mixed extracts were incubated with NF-κB oligonucleotides as described above. pCMX-p50 was provided by J. Inoue (University of Tokyo, Tokyo, Japan). Anti-Flag Tag (M2; Sigma-Aldrich) mAb and anti-p50 (E-10; Santa Cruz Biotechnology, Inc.) mAb were used for detection.
Transfection and luciferase assays
293 T cells were transfected with 5× NF-κB luciferase reporter plasmid together with pAct vector or pAct-Flag-hShn-2 vector and pRL-TK as normalization control. Transfected cells were stimulated with or without 50 ng/ml PMA and 500 nM ionomycin or 10 ng/ml TNF-α for 12 h before luciferase assays (Promega).
PCR analysis
Total RNA was isolated from cultured cells using the TRIzol reagent. Reverse transcription was performed with Superscript II RT (Invitrogen). Threefold serial dilutions of template cDNA were performed. The PCR reaction was performed as described previously (46). The primers used are as follows: β actin forward, GAGAGGGAAATCGTGCGTGA-3′; β actin reverse, 5′-ACATCTGCTGGAAGGTGGAC; GATA3 forward, gAAggCATCCAgACCCgAAAC-3′; GATA3 reverse, 5′-ACCCATggCggTgACCATgC; Shn-2 forward, ggAAAgAgggAAAggAgAgATTCACggAgAT-3′; and Shn-2 reverse, 5′-ATCTgAgTgTCATCACAAgAgTCACTgggT.
For quantitative PCR assay, first-strand cDNA was synthesized using random primers and Superscript II RT (Invitrogen). Samples were then subjected to real-time PCR analysis on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems) under standard conditions. The primers and TaqMan probes for the detection of p50, p65, T-bet, and 18S were purchased from Applied Biosystems. p50, p65, and T-bet expression were normalized using the 18S signal.
Online supplemental material
Fig. S1 shows the phenotypic feature of splenic CD4 T cells from Shn-2−/−mice. (A) The yields of thymocytes and splenocytes are shown in boxed numbers. (B) Each histogram depicts the expression of the indicated marker antigens on electronically gated splenic CD4 T cells. Background staining is shown as hatched areas. (C) The histogram shows the percentages of indicated Vβ cells among splenic CD4 T cells. Fig. S2 shows the proliferative responses and Erk phosphorylation status after TCR stimulation. Arbitrary densitometric units are shown under each band in B. Fig. S3 shows the expression of Shn-2 mRNA in developing Th1 and Th2 cells. The mRNA levels were determined by RT-PCR with threefold serial dilutions of template cDNA. Fig. S4 shows the effects of CD28 stimulation on Th2 cell differentiation. Gel shift assay for AP-1, CREB, or IL-4 NFAT are shown in Fig. S5. Figs. S1–S5 are available at http://www.jem.org/cgi/content/full/jem.20040733/DC1.
Acknowledgments
The authors are grateful to Dr. Ralph T. Kubo for helpful comments and constructive criticisms in the preparation of the manuscript. The authors also thank Ms. Kaoru Sugaya for excellent technical assistance.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (Japan; Grants-in-Aid for Scientific Research; Priority Areas Research nos. 13218016 and 16043211; Scientific Research B no. 14370107; Scientific Research C nos. 16616003 and 15790248; and Special Coordination Funds for Promoting Science and Technology), the Ministry of Health, Labor and Welfare (Japan), the Program for Promotion of Fundamental Studies in Health Science of the Organization for Pharmaceutical Safety and Research (Japan), The Japan Health Science Foundation, Uehara Memorial Foundation, Kanae Foundation, and the Mochida Memorial Foundation.
The authors have no conflicting financial interests.
Abbreviations used: BMDC, bone marrow–derived DC; CFSE, carboxyfluorescein diacetate succinimidyl ester; EMSA, electrophoretic mobility shift assay; Shn, Schnurri; Tg, transgenic; TRAF, TNF receptor–associated factor.
References
- 1.Mosmann, T.R., and R.L. Coffman. 1989. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7:145–173. [DOI] [PubMed] [Google Scholar]
- 2.Seder, R.A., and W.E. Paul. 1994. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12:635–673. [DOI] [PubMed] [Google Scholar]
- 3.Reiner, S.L., and R.M. Locksley. 1995. The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 13:151–177. [DOI] [PubMed] [Google Scholar]
- 4.Abbas, A.K., K.M. Murphy, and A. Sher. 1996. Functional diversity of helper T lymphocytes. Nature. 383:787–793. [DOI] [PubMed] [Google Scholar]
- 5.Constant, S.L., and K. Bottomly. 1997. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 15:297–322. [DOI] [PubMed] [Google Scholar]
- 6.Nelms, K., A.D. Keegan, J. Zamorano, J.J. Ryan, and W.E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 7.O'Garra, A. 1998. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 8:275–283. [DOI] [PubMed] [Google Scholar]
- 8.Murphy, K.M., W. Ouyang, J.D. Farrar, J. Yang, S. Ranganath, H. Asnagli, M. Afkarian, and T.L. Murphy. 2000. Signaling and transcription in T helper development. Annu. Rev. Immunol. 18:451–494. [DOI] [PubMed] [Google Scholar]
- 9.Yamashita, M., K. Hashimoto, M. Kimura, M. Kubo, T. Tada, and T. Nakayama. 1998. Requirement for p56lck tyrosine kinase activation in Th subset differentiation. Int. Immunol. 10:577–591. [DOI] [PubMed] [Google Scholar]
- 10.Yamashita, M., M. Katsumata, M. Iwashima, M. Kimura, C. Shimizu, T. Kamata, T. Shin, N. Seki, S. Suzuki, M. Taniguchi, and T. Nakayama. 2000. T cell receptor–induced calcineurin activation regulates T helper type 2 cell development by modifying the interleukin 4 receptor signaling complex. J. Exp. Med. 191:1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamashita, M., M. Kimura, M. Kubo, C. Shimizu, T. Tada, R.M. Perlmutter, and T. Nakayama. 1999. T cell antigen receptor-mediated activation of the Ras/mitogen-activated protein kinase pathway controls interleukin 4 receptor function and type-2 helper T cell differentiation. Proc. Natl. Acad. Sci. USA. 96:1024–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamata, T., M. Yamashita, M. Kimura, K. Murata, M. Inami, C. Shimizu, K. Sugaya, C.R. Wang, M. Taniguchi, and T. Nakayama. 2003. src homology 2 domain-containing tyrosine phosphatase SHP-1 controls the development of allergic airway inflammation. J. Clin. Invest. 111:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang, D.H., L. Cohn, P. Ray, K. Bottomly, and A. Ray. 1997. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J. Biol. Chem. 272:21597–21603. [DOI] [PubMed] [Google Scholar]
- 14.Zheng, W., and R.A. Flavell. 1997. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 89:587–596. [DOI] [PubMed] [Google Scholar]
- 15.Ouyang, W., S.H. Ranganath, K. Weindel, D. Bhattacharya, T.L. Murphy, W.C. Sha, and K.M. Murphy. 1998. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 9:745–755. [DOI] [PubMed] [Google Scholar]
- 16.Lee, H.J., N. Takemoto, H. Kurata, Y. Kamogawa, S. Miyatake, A. O'Garra, and N. Arai. 2000. GATA-3 induces T helper cell type 2 (Th2) cytokine expression and chromatin remodeling in committed Th1 cells. J. Exp. Med. 192:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szabo, S.J., S.T. Kim, G.L. Costa, X. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 18.Affolter, M., T. Marty, M.A. Vigano, and A. Jazwinska. 2001. Nuclear interpretation of Dpp signaling in Drosophila. EMBO J. 20:3298–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arora, K., H. Dai, S.G. Kazuko, J. Jamal, M.B. O'Connor, A. Letsou, and R. Warrior. 1995. The Drosophila schnurri gene acts in the Dpp/TGFβ signaling pathway and encodes a transcription factor homologous to the human MBP family. Cell. 81:781–790. [DOI] [PubMed] [Google Scholar]
- 20.Staehling-Hampton, K., A.S. Laughon, and F.M. Hoffmann. 1995. A Drosophila protein related to the human zinc finger transcription factor PRDII/MBPI/HIV-EP1 is required for dpp signaling. Development. 121:3393–3403. [DOI] [PubMed] [Google Scholar]
- 21.Kingsley, D.M. 1994. The TGF-β superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 8:133–146. [DOI] [PubMed] [Google Scholar]
- 22.Makino, R., K. Akiyama, J. Yasuda, S. Mashiyama, S. Honda, T. Sekiya, and K. Hayashi. 1994. Cloning and characterization of a c-myc intron binding protein (MIBP1). Nucleic Acids Res. 22:5679–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ron, D., A.R. Brasier, and J.F. Habener. 1991. Angiotensinogen gene-inducible enhancer-binding protein 1, a member of a new family of large nuclear proteins that recognize nuclear factor kappa B-binding sites through a zinc finger motif. Mol. Cell. Biol. 11:2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell, D.B., and P. Levitt. 2003. Regionally restricted expression of the transcription factor c-myc intron 1 binding protein during brain development. J. Comp. Neurol. 467:581–592. [DOI] [PubMed] [Google Scholar]
- 25.Takagi, T., J. Harada, and S. Ishii. 2001. Murine Schnurri-2 is required for positive selection of thymocytes. Nat. Immunol. 2:1048–1053. [DOI] [PubMed] [Google Scholar]
- 26.Allen, C.E., N. Muthusamy, S.E. Weisbrode, J.W. Hong, and L.C. Wu. 2002. Developmental anomalies and neoplasia in animals and cells deficient in the large zinc finger protein KRC. Genes Chromosomes Cancer. 35:287–298. [DOI] [PubMed] [Google Scholar]
- 27.Barnes, P.J., and M. Karin. 1997. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336:1066–1071. [DOI] [PubMed] [Google Scholar]
- 28.Yang, L., L. Cohn, D.H. Zhang, R. Homer, A. Ray, and P. Ray. 1998. Essential role of nuclear factor κB in the induction of eosinophilia in allergic airway inflammation. J. Exp. Med. 188:1739–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donovan, C.E., D.A. Mark, H.Z. He, H.C. Liou, L. Kobzik, Y. Wang, G.T. De Sanctis, D.L. Perkins, and P.W. Finn. 1999. NF-κB/Rel transcription factors: c-Rel promotes airway hyperresponsiveness and allergic pulmonary inflammation. J. Immunol. 163:6827–6833. [PubMed] [Google Scholar]
- 30.Das, J., C.H. Chen, L. Yang, L. Cohn, P. Ray, and A. Ray. 2001. A critical role for NF-κB in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat. Immunol. 2:45–50. [DOI] [PubMed] [Google Scholar]
- 31.Oukka, M., S.T. Kim, G. Lugo, J. Sun, L.C. Wu, and L.H. Glimcher. 2002. A mammalian homolog of Drosophila schnurri, KRC, regulates TNF receptor-driven responses and interacts with TRAF2. Mol. Cell. 9:121–131. [DOI] [PubMed] [Google Scholar]
- 32.Bachmeyer, C., C.H. Mak, C.Y. Yu, and L.C. Wu. 1999. Regulation by phosphorylation of the zinc finger protein KRC that binds the κB motif and V(D)J recombination signal sequences. Nucleic Acids Res. 27:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong, J.W., C.E. Allen, and L.C. Wu. 2003. Inhibition of NF-κB by ZAS3, a zinc-finger protein that also binds to the κB motif. Proc. Natl. Acad. Sci. USA. 100:12301–12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bird, J.J., D.R. Brown, A.C. Mullen, N.H. Moskowitz, M.A. Mahowald, J.R. Sider, T.F. Gajewski, C.R. Wang, and S.L. Reiner. 1998. Helper T cell differentiation is controlled by the cell cycle. Immunity. 9:229–237. [DOI] [PubMed] [Google Scholar]
- 35.Kubo, M., M. Yamashita, R. Abe, T. Tada, K. Okumura, J.T. Ransom, and T. Nakayama. 1999. CD28 costimulation accelerates IL-4 receptor sensitivity and IL-4-mediated Th2 differentiation. J. Immunol. 163:2432–2442. [PubMed] [Google Scholar]
- 36.Inami, M., M. Yamashita, Y. Tenda, A. Hasegawa, M. Kimura, K. Hashimoto, N. Seki, M. Taniguchi, and T. Nakayama. 2004. CD28 costimulation controls histone hyperacetylation of the interleukin 5 gene locus in developing th2 cells. J. Biol. Chem. 279:23123–23133. [DOI] [PubMed] [Google Scholar]
- 37.Lieberson, R., K.A. Mowen, K.D. McBride, V. Leautaud, X. Zhang, W.K. Suh, L. Wu, and L.H. Glimcher. 2001. Tumor necrosis factor receptor–associated factor (TRAF)2 represses the T helper cell type 2 response through interaction with NFAT-interacting protein (NIP45). J. Exp. Med. 194:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, S.Y., A. Reichlin, A. Santana, K.A. Sokol, M.C. Nussenzweig, and Y. Choi. 1997. TRAF2 is essential for JNK but not NF-κB activation and regulates lymphocyte proliferation and survival. Immunity. 7:703–713. [DOI] [PubMed] [Google Scholar]
- 39.Takeda, K., T. Tanaka, W. Shi, M. Matsumoto, M. Minami, S. Kashiwamura, K. Nakanishi, N. Yoshida, T. Kishimoto, and S. Akira. 1996. Essential role of Stat6 in IL-4 signalling. Nature. 380:627–630. [DOI] [PubMed] [Google Scholar]
- 40.Kimura, M., Y. Koseki, M. Yamashita, N. Watanabe, C. Shimizu, T. Katsumoto, T. Kitamura, M. Taniguchi, H. Koseki, and T. Nakayama. 2001. Regulation of Th2 cell differentiation by mel-18, a mammalian polycomb group gene. Immunity. 15:275–287. [DOI] [PubMed] [Google Scholar]
- 41.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]
- 42.Nakayama, T., C.H. June, T.I. Munitz, M. Sheard, S.A. McCarthy, S.O. Sharrow, L.E. Samelson, and A. Singer. 1990. Inhibition of T cell receptor expression and function in immature CD4+CD8+ cells by CD4. Science. 249:1558–1561. [DOI] [PubMed] [Google Scholar]
- 43.Inaba, K., M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu, and R.M. Steinman. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176:1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen-Woan, M., C.P. Delaney, V. Fournier, Y. Wakizaka, N. Murase, J. Fung, T.E. Starzl, and A.J. Demetris. 1995. A new protocol for the propagation of dendritic cells from rat bone marrow using recombinant GM-CSF, and their quantification using the mAb OX-62. J. Immunol. Methods. 178:157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Omori, M., M. Yamashita, M. Inami, M. Ukai-Tadenuma, M. Kimura, Y. Nigo, H. Hosokawa, A. Hasegawa, M. Taniguchi, and T. Nakayama. 2003. CD8 T cell-specific downregulation of histone hyperacetylation and gene activation of the IL-4 gene locus by ROG, repressor of GATA. Immunity. 19:281–294. [DOI] [PubMed] [Google Scholar]
- 46.Yamashita, M., M. Ukai-Tadenuma, M. Kimura, M. Omori, M. Inami, M. Taniguchi, and T. Nakayama. 2002. Identification of a conserved GATA3 response element upstream proximal from the interleukin-13 gene locus. J. Biol. Chem. 277:42399–42408. [DOI] [PubMed] [Google Scholar]