

Abstract
The lineage relationships of central–memory T cells (TCM) cells and effector–memory T cells (TEM), as well as their homeostasis and recall capacities, are still controversial. We investigated these issues in a murine model using two complementary approaches: T cell receptor repertoire analysis and adoptive transfer experiments of purified H-Y–specific TCM and TEM populations. Repertoire studies showed that approximately two thirds of TCM and TEM clones derived from a common naive precursor, whereas the other third was distinct. Both approaches highlighted that TCM and TEM had drastically distinct behaviors in vivo, both in the absence of antigen or upon restimulation. TCM clones were stable in the absence of restimulation and mounted a potent and sustained recall response upon secondary challenge, giving rise to both TCM and TEM, although only a fraction of TCM generated TEM. In contrast, TEM persisted for only a short time in the absence of antigen and, although a fraction of them were able to express CD62L, they were unable to mount a proliferative response upon secondary challenge in this model.
Due to the persistence of memory T and B lymphocytes and long-lived plasma cells, the immune system is able to keep track of previously encountered pathogens and to give rise to a rapid and efficient response in case of reencounter with the same pathogen (1, 2). Memory T cells persist with an elevated frequency and display enhanced functional capacities (3). Unfortunately, our knowledge of the memory differentiation pathway is still limited, which might explain why we fail to generate efficient immune memory against some pathogens or other antigens such as tumor cells.
This difficulty may stem in part from the heterogeneity of the memory T cell pool in terms of phenotype, migration, and functional capacities (3, 4). One recently recognized heterogeneity is the existence of memory cells residing within peripheral tissues beside those recirculating between secondary lymphoid organs (5). In humans, two subsets can be defined according to their expression levels of CCR7 and CD62L (6), both markers being necessary for entry in peripheral lymph nodes through HEV (7). The CCR7+CD62L+CD45RO+ central–memory T cells (TCM) recirculate through lymphoid organs and do not display immediate effector functions, whereas the CCR7−CD62L− cells are effector–memory T cells (TEM) that reside within, or recirculate through, peripheral tissues and have immediate effector functions. All subsets can be found in blood and in the spleen (6, 8, 9). TCM and TEM subsets can also be distinguished in mice depending on CD62L or CCR7 expression levels (9, 10).
The distinction between the central– and peripheral memory subsets raises two fundamental issues: (a) what is the relationship between these subsets? and (b) are both subsets equally potent to generate a recall response? Three models have recently been proposed regarding their relationship. The first one is the “progressive differentiation model” (11) characterized by three hallmarks. Depending on the strength and duration of the stimulation received by a naive T cell in the course of the immune response, this cell will either follow the differentiation pathway to acquire effector functions to its end, or stop in an intermediate differentiation state. Next, at the end of the immune response, intermediates of differentiation give rise to TCM, whereas TEM stem from fully differentiated cells. Finally, in the absence of restimulation, a proportion of TCM differentiates into TEM to replenish the effector–memory pool. This model is supported by several studies, both in humans and mice; only nonpolarized in vitro–generated effectors recirculate in lymph nodes (8, 12), and TEM have a lower proliferative potential (9, 13) and enhanced effector functions (14–16), suggesting that TEM are more differentiated. Moreover, in the presence of IL-7 and IL-15, both CD4 and CD8 TCM were shown to generate TEM in vitro (17, 18).
In the second model, the two subsets also belong to one lineage continuum, but it is the TCM that derive from the TEM, the latter being viewed as a transitory state (9, 19). The rate at which these cells convert to TCM depends on the magnitude of the stimulation. Consistent with this model is the observed patterns of gene expression during memory T cell differentiation (20).
No mandatory lineage relationship between the two subsets is assumed in the third model. In a previous study, we analyzed the TCR repertoires of sorted TCM and TEM PBMCs in healthy donors and found that they were mainly distinct at a given time point. No conversion from one subset to the other was observed over a 9-mo period (21). Therefore, we suggested that both subsets could arise at least partly independently, appearing in distinct secondary lymphoid organs and/or under distinct primary stimulation conditions. Alternatively, if most effector cells were to give rise to both TCM and TEM, at least one of these memory cell population had to be transient or could not recirculate in the blood.
As a result, the differentiation pathway of TCM and TEM subsets remains controversial, and additional complexity arises concerning the self-renewal capacities of both subsets or the conversion of cells from one subset to the other in long-term steady-state conditions or upon restimulation (5).
The other key issue deals with the capacity of either subset to mount a recall response. Several studies showed that TCM and TEM may have the same effector capacities, in both human or murine CD4 and CD8 T cells (22, 23), and that TCM may include fully differentiated effectors (24, 25). However, others have suggested that TCM and TEM might have differential protective capacities upon reencounter with their cognate antigen. Indeed, although several papers suggested that the presence of effector cells may be necessary for protection against a peripheral challenge (26, 27), other studies highlighted the crucial role of TCM in host defense (9, 14, 28). The comprehension of the respective roles of TCM and TEM subsets in host protection in case of secondary challenge is thus still limited and needs further investigation.
The study of lineage relationships requires the in vivo production of measurable numbers of memory cells, which is best achieved by the adoptive transfer of antigen-specific naive T cells (29). Past studies have used TCRαβ transgenic T cells as a homogeneous source of naive T cells. However, with this experimental design, it is impossible to determine whether two memory cells would derive from a single or two distinct naive precursors. To analyze the lineage relationships between TCM and TEM and their respective ability to mount a recall response, we used a murine model enabling us to generate CD8 TCM and TEM subsets specific for the H-Y male antigen with heterogeneous TCR. Mice transgenic for the β chain of a Db-restricted Smcy3 peptide-specific TCR were used as a source of CD8+ H-Y–specific naive T cell precursors. These cells were transferred into C57BL/6 naive female mice that were subsequently immunized intravenously with male syngeneic bone marrow cells. We have chosen this model for two reasons. First, the H-Y antigen has been described as a nonpersistent and noncross-reactive antigen (30). And second, fixing only the TCRβ chain combines the advantages of making the TCRα chain a signature of each T cell clone and of restricting the repertoire of Db-Smcy3–specific CD8 T cells (31), which permits a global study of the memory TCRα repertoire.
We report here on the analysis of the TCR repertoires of both subsets in lymphoid organs as well as peripheral tissues, their evolution in steady-state conditions or after a secondary challenge, and the fate of the transferred purified TCM or TEM populations after this challenge.
Results
In vivo generation of T CD8 memory subsets after intravenous immunization
First, we verified that we could generate measurable numbers of memory T cells of both subsets in our model. Naive C57BL/6 female mice received 105 CD8 naive T cells from a female mouse transgenic for the β chain of TCR H-Y, corresponding to ∼3 × 104 CD8 T cells specific for the H2-Db–restricted Smcy3738-746 peptide (H-Y antigen; reference 31). The transferred mice were immunized intravenously with 5 × 106 male syngenic bone marrow cells (32), and we used H2-Db-Smcy3 MHC tetramers to measure the immune response in vivo. In peripheral blood, we observed a classical T CD8 response beginning with an expansion of specific CD8 cells, followed by a contraction phase prolonged by a plateau that was maintained throughout the memory phase (Fig. 1 A). Using CD45.1+ C57BL/6 mice as hosts, we showed that the vast majority of Db-Smcy3–specific cells generated derived from the transferred β-tg CD45.2+ CD8 cells (see Fig. 4 C). CD62L expression by Db-Smcy3–specific PBLs cells was first down-regulated during the expansion phase, almost all specific CD8 effector cells being CD62L− at the peak of the response. Next, the proportion of CD62L+ cells steadily increased to reach approximately half of the CD8 Db-Smcy3–specific population after 6 wk and slowly continued to increase later on (Fig. 1 B). At 6 wk, we analyzed the presence of Db-Smcy3–specific cells in lymphoid (blood, spleen, PLN, MLN) and peripheral (liver, gut lamina propria) organs and their expression of the CD62L molecule. Db-Smcy3–specific cells were present in all organs analyzed (Fig. 1 C), and their percentage was higher in peripheral organs such as liver or lamina propria. We observed two subsets defined by their expression level of CD62L (low/high) in all organs except lamina propria in which virtually all cells displayed a CD62L− phenotype. Thus, our murine model was able to generate two subsets of CD44hi Db-Smcy3–specific CD8 memory cells defined by their expression level of the CD62L molecule corresponding to TCM (CD62L+) and TEM (CD62L−), allowing us to study their TCR repertoires and their recall response capacities.
Figure 1.

In vivo generation of T CD8 memory subsets after intravenous immunization with male bone marrow cells of a C57BL/6 female host transferred with naive β-tg CD8 cells. (A) Analysis of the percentage of Db-Smcy3+ cells among CD8 PBLs for a naive C57BL/6 mouse (open circles) or immunized hosts (closed squares). Data from 12 mice were pooled. (B) Evolution of the percentage of CD62L+ cells among CD8+ Db-Smcy3+ PBLs. (C) Analysis of the percentages and absolute numbers (in brackets) of Db-Smcy3–specific cells among CD8+ cells in several organs 6 wk after immunization. (D) Expression of CD62L by CD8+ Db-Smcy3+ cells in several organs at least 6 wk after immunization. FACS profiles are representative of all mice analyzed.
Figure 4.

TCM but not TEM can mount a recall response after transfer and challenge. CD62L+ and CD62L− cells were purified from the spleen and PLN of hosts transferred with naive β-tg CD8 cells and immunized at least 6 wk earlier (see Materials and methods) and equal numbers of Db-Smcy3–specific TCM (closed symbols) or TEM (open symbols) were transferred into naive CD45.1+ female hosts that were immediately immunized with syngeneic CD45.1+ male bone marrow cells. (A) Evolution of the percentage of CD45.2+ transferred cells among host blood CD8 cells. The percentage observed at day 2 is normalized as 100%, and all subsequent percentages are expressed as a percentage of this standard. (B) Analysis of the percentage of Db-Smcy3+ cells among blood CD8 cells. (C) Expression of the CD45.2 molecule by CD8+ Db-Smcy3+ blood cells at day 15 or >1 mo after challenge, after transfer of naive, TCM, or TEM. (D) CD62L expression profiles and absolute numbers (in parentheses) of CD8+ Db-Smcy3+ cells in organs of a CD45.1+ female host transferred with TCM and challenged at least 6 wk before analysis. All results shown are representative of at least three experiments.
Repertoires of CD8 TCM and TEM in lymphoid and nonlymphoid organs are partly distinct
We sorted the Db-Smcy3–specific TCM and TEM subsets from lymphoid (spleen, PLN, MLN) and peripheral (LP) organs from C57BL/6 mice transferred with naive β-tg CD8 cells and immunized at least 6 wk before, and analyzed their TCRα repertoires. Having previously shown that most Db-Smcy3–specific cells express a TCRα chain using the TCRAV9 segment (31), we focused our analysis on TCRAV9-TCRAC rearrangements, which were sequenced extensively for splenic TCM and TEM subsets sorted from several individual mice. Both Db-Smcy3–specific subsets displayed a restricted repertoire comprised of ≤200 different clones. Moreover, the different clones had a restricted usage of TCRAJ segments (Fig. S1 B, available at http://www.jem.org/cgi/content/full/jem.20040876/DC1) and displayed heterogeneous sizes, clone frequencies ranging from 10−3 to 10−1 (Fig. S1 C).
Although it was difficult to identify every clone represented within a particular sample, we could estimate the repertoire diversity and overlap of both subsets using statistical tools (see Materials and methods). Altogether, the TCRα repertoires of TCM subsets were more diverse than those of TEM, but only by a 1.5 factor (115 and 175 clones on average, respectively; Fig. S1 D). The low diversity observed implied at least two consequences. First, because the number of specific sorted cells analyzed by sequencing ranged between 6,000 and 27,000 cells, this sampling was large enough to study a >30-fold lower clonal diversity. Second, because we transferred ∼105 transgenic cells per mouse, ∼30% of which were specific for the Smcy3 peptide, the presence of CD8+ memory T cells with the same TCR in both subsets might occur even if these cells were not derived from the same stimulated naive T cell.
To measure this probability of artifactually shared clones between the two subsets, we compared the repertoire overlap of TCM and TEM subsets from the same mouse (intraindividual overlap) or from two distinct mice (interindividual overlap), using the observed percentages of shared clones and the statistical overlap index (Table I and Fig. S1 E). The interindividual percentages of shared clones were important for both subsets (34.0 ± 7.9% for TEM and 24.5 ± 3.6% for TCM), as well as the overlap index (0.38 ± 0.03), showing that a substantial sharing of clones between TCM and TEM subsets could be attributed to the restricted diversity of the Db-Smcy3–specific memory repertoire. However, intraindividual percentages of shared clones were significantly higher (55.4 ± 7.4% for TEM and 38.6 ± 4.7% for TCM), as was the mean intraindividual overlap index (0.56 ± 0.03). This indicates that a significant proportion of the shared clones derived from common naive precursor T cells. Taking into account the baseline percentage of sharing due to the restricted diversity of the memory repertoire and the maximal percentage of sharing observable with our technology, we estimated that two out of three clones were shared between the TCM and TEM subsets (Fig. S1 E). Interestingly, most of the well-represented clones were shared between the TCM and TEM subsets (Fig. S1 C).
Table I.
TCM and TEM clone sharing
| Interindividual mean | Intraindividual mean | |
|---|---|---|
| Overlap index | 0.38 ± 0.03 | 0.56 ± 0.03 |
| % shared clones | % shared clones | |
| CD62L+ | 24.5 ± 3.6 | 38.6 ± 4.7 |
| CD62L− | 34.0 ± 7.9 | 55.4 ± 7.4 |
| Total | 16.5 ± 3.1 | 29.3 ± 3.9 |
Mean interindividual and intraindividual overlap indexes and percentages of shared clones for TCM, TEM, and total clones are displayed.
To confirm that the presence of unshared clones in the subset repertoire was not due to an insufficient accumulation of sequences, nor to sequestering of some clones within particular lymphoid or peripheral organs, and to have a broader idea of the repartition of clones in the body, we measured by quantitative clonotypic PCR the frequency of individual clones in TCM and TEM subsets sorted from the spleen, PLN, MLN, and LP from two individual mice transferred and immunized as described before (Fig. 2 and Fig. S2 B, available at http://www.jem.org/cgi/content/full/jem.20040876/DC1). Out of eight clones detected by sequencing, only in the TCM or TEM subset, we confirmed that six of them were readily detected in one subset only. In addition, this observation could be extended to other organs; when a clone was present in one subset of the spleen only, it was either undetected in other lymphoid organs, or remained within the same subset in these organs.
Figure 2.
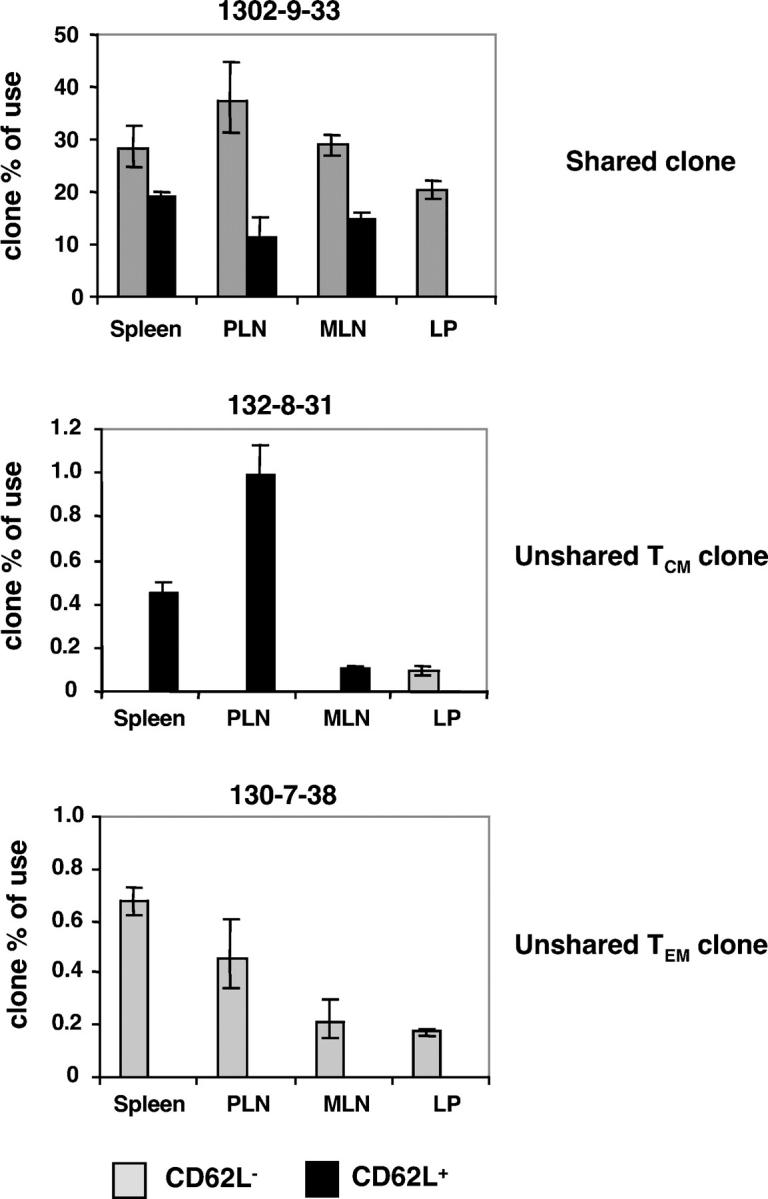
Frequency of individual clones in lymphoid and peripheral organs of an immune mouse. The frequency of each clone among TCRAV9+ cells was measured by quantitative clonotypic PCR within each subset. An example of each type of clone (shared, unshared TCM, unshared TEM) is displayed. The frequency observed in the LP CD62L− subset for clone 132–8-31 corresponds to several cells <10 and, therefore, is considered as nonspecific amplification.
Altogether, 6 wk after immunization, approximately two thirds of total memory T cell clones were shared between both subsets, so that TCM and TEM repertoires could be considered as only partly distinct. Clonotypic analyses confirmed the presence of unshared clones and showed no sequestration of clones in particular organs, suggesting that the situation observed in the spleen was representative of the whole body.
Evolution of TCM and TEM TCRα repertoires in physiological conditions or after restimulation
This repertoire analysis was performed at a given time point. It has been proposed that TEM can generate TCM (9) and vice versa (17, 18). Therefore, we analyzed the stability of both populations in the same mouse, with or without challenge. For this purpose, we immunized mice after transfer of transgenic naive T cells as described before, and performed hemisplenectomy 6 wk after immunization (T1), the other spleen half being recovered 10 wk later (T2), with or without a secondary intravenous immunization 4 wk after hemisplenectomy (Fig. 3 A). Thus, we could perform repertoire analyses of both subsets on sorted specific splenocytes at two time points distant of 10 wk. We extensively sequenced TCRAV9-TCRAC rearrangements and analyzed the percentages of shared clones and the overlap indexes between each mouse (four samples) with the aim of assessing possible passages of clones between T1 and T2 from one subset to the other. The only significant modification was observed in the nonreimmunized mouse (Fig. 3 B). In that case, the percentage of shared clones between the TEM (T1) and TCM (T2) subsets was significantly higher than that between TEM (T1) and TCM (T1) subsets (78.3 vs. 53.0%). Accordingly, the overlap index also increased (from 0.51 to 0.60). This evolution strongly suggested that a sizable fraction of TEM (T1) clones had reexpressed the CD62L molecule between T1 and T2, also consistent with the observed increase of the estimated diversity in the TCM subset from T1 to T2 (from 150 to 201 estimated clones), and with an increase of the percentage of CD62L+ cells among Db-Smcy3–specific sorted cells (37% at T1 vs. 78% at T2). Interestingly, not all cells of these TEM clones had apparently converted to a CD62L+ phenotype because the estimated diversity of the TEM (T1) and TEM (T2) repertoires remained close (111 and 121, respectively).
Figure 3.

TCRα repertoire analysis of splenic Db-Smcy3–specific TCM and TEM subsets at two time points (T1 and T2), with or without a challenge in between. TCRAV9-TCRAC rearrangements were sequenced and analyzed. (A) Illustration of the experimental protocol. (B) In the nonchallenged mouse, evolution of the overlap index and of the percentage of shared clones between TEM (T1) and TCM at T1 and T2. (C) Evolution of TEM (T1) unshared clones at T2 in both conditions. (D and E) Evolution of the frequency of T1 unshared clones (D) in the absence of restimulation or (E) in the presence of an intravenous challenge. Frequencies were determined as in Fig. 2.
To further analyze the possibility of TEM converting to a CD62L+ phenotype, we studied the fate of the clones found at T1 in the TEM subset only (Fig. 3 C). In the absence of a second challenge, most TEM (T1) unshared clones (82%) were readily detected at T2, and 59% of these TEM (T1) unshared clones appeared in the TCM subset, confirming that cells from a substantial fraction of TEM (T1) clones had converted to a CD62L+ phenotype in the 10-wk interval, although most of them were still present in the TEM (T2) subset. These observations were further confirmed by quantitative PCR clonotypic analyses. We measured the frequency of five clones present in the TEM subset at T1 (Fig. S2 C, top). Two of them were only detected in the TEM subset at T1 and both converted, at least partly, to a CD62L+ phenotype in the absence of restimulation (Fig. 3 D, TEM→TCM and TEM). The other three clones were already shared by both subsets at T1, and two of them displayed a decrease of their frequency in the TEM subset at T2, though without a corresponding increase of their TCM frequency.
We also analyzed the fate of TEM (T1) unshared clones in the reimmunized mouse. In contrast with what was observed in the absence of restimulation, 50% of the unshared TEM (T1) clones were no longer detected at T2, neither in the TEM nor in the TCM subset, suggesting that a sizable fraction of TEM (T1) clones had either migrated into peripheral tissues or died.
Likewise, we investigated the possibility that some TCM clones could acquire a TEM phenotype over time. We analyzed the variation of the percentage of shared clones between TCM (T1) and TEM clones. No significant difference could be observed in both conditions (unpublished data), which could receive two explanations: either TCM clones were very stable and did not convert to TEM, or the TCM clones that converted to TEM were already shared at T1. To further analyze this point, we measured in both conditions the frequency of several unshared TCM (T1) clones in both subsets at T1 and T2 (Fig. S2 C). In the absence of restimulation, out of six analyzed clones, five were stable and only found in the TCM subset at T2 (the last one was undetectable at T2 in both TCM and TEM subsets), which shows that TCM clones were quite stable in the absence of restimulation (Fig. 3 D, TCM→TCM). In the reimmunized mouse, out of five TCM (T1) unshared clones, four were stable in the TCM subset at T2 (Fig. 3 E, TCM→TCM), whereas the last one gave rise to TEM at T2 (Fig. 3 E, TCM→TCM and TEM), indicating that the antigenic stimulation mainly drove already shared TCM clones to differentiate into TEM clones, and that few new clones appeared in the TEM subset upon restimulation.
This protocol using hemisplenectomies to analyze antigen-specific TCM and TEM repertoires at two time points in the same mouse in the presence or absence of a secondary challenge thus enabled us to point out several features of the fate of TCM and TEM in vivo. First, in the absence of antigen, whereas TCM clones were quite stable, cells of ∼25% of the observed TEM clones converted to a CD62L+ phenotype. Second, in the case of restimulation, TCM and TEM subsets had a drastically different behavior; whereas TCM clones either remained stable or gave rise to TEM, a majority of TEM unshared clones at T1 disappeared.
TCM, but not TEM, can mount a recall immune response after an i.v. challenge
This last observation raised the question of the memory capacity of TEM. To refine our analysis of the properties of TCM and TEM subsets, we examined their behavior after an adoptive transfer in a new host, immediately followed by a secondary intravenous immunization. At least 6 wk after the primary immunization, we purified CD62L+ and CD62L− cells from the spleen and PLN of C57BL/6 hosts (Fig. S1 A). Each of these populations containing 3 × 104 Db-Smcy3–specific TCM or TEM was transferred into a new female C57BL/6 host expressing the CD45.1 allele. These hosts were immunized with 5 × 106 bone marrow cells from male C57BL/6 CD45.1 mice and the Db-Smcy3–specific response was followed by flow cytometry in blood.
When TCM were transferred, a rapid increase of the percentage of the CD45.2+ transferred cells was observed in blood (Fig. 4 A), correlated with the increase of the percentage of CD8+ Db-Smcy3–specific cells (Fig. 4 B). This Db-Smcy3–specific response was more rapid than that observed in hosts after primary immunization of naive β-tg CD8 cells, and was characteristic of a recall response. On the contrary, when TEM were transferred, after a possible initial increase, the percentage of CD45.2+ transferred cells among host CD8+ cells dramatically decreased until these cells became barely detectable (Fig. 4 A), in agreement with our conclusion on the spleen repertoire study. However, a Db-Smcy3 response did occur in blood (Fig. 4 B), but it was due to the CD45.1+ naive cells of the host (Fig. 4 C). These results were further extended to all organs analyzed after 6 wk. In particular, the CD45.2+ CD8+ cells were barely detectable in all organs analyzed including liver and gut lamina propria when CD62L− cells had been transferred (unpublished data), indicating that the decrease in the percentage of CD45.2+ cells observed in the blood was not due to their migration to peripheral tissues, but most likely to their death.
The sequencing and clonotypic PCR studies performed in the spleen at two time points with a secondary immunization in between indicated that only a fraction of the TCM clones could give rise to TEM clones after a secondary response. However, this fraction was difficult to determine from the hemisplenectomy experiments. Therefore, we analyzed the CD62L phenotype of the Db-Smcy3–specific memory cells generated upon transfer with, and subsequent challenge of, TCM. As shown in Fig. 4 D, the TCM transferred cells expanded and gave rise to both TCM and TEM in the different organs analyzed. To determine the actual proportion of the transferred clones that generated the TEM, we sorted the Db-Smcy3–specific TCM and TEM splenic subsets of two individual C57BL/6 CD45.1 hosts after transfer and challenge, and analyzed their TCRAV9 repertoire. Once again, the TCM subset repertoire was more diverse. Moreover, a majority of TEM clones (56 ± 3%) had a counterpart in the TCM subset, suggesting that most TCM clones that gave rise to TEM also remained within the TCM subset. However, only 47 ± 7% of the TCM clones were also detected within the TEM subset, demonstrating that less than half of TCM clones gave rise to TEM clones in case of restimulation (unpublished data).
These transfer experiments of purified CD62L+ or CD62L− memory cells showed that TCM and TEM had drastically different behaviors in response to a secondary intravenous immunization; whereas TCM mounted a secondary response giving rise to an heterogeneous memory pool comprised of both TCM and TEM, TEM did not expand significantly and were even less efficient than naive β-tg CD8 cells to mount a proliferative Db-Smcy3–specific response.
In the absence of antigen, a proportion of TEM apparently convert to TCM but remain generally unable to mount a secondary response
The results of our splenocyte repertoire studies showed that, in the absence of antigenic restimulation, a fraction of TEM converted to a TCM phenotype. Here, we wanted to determine whether this conversion would be restricted to the mere expression of CD62L, or would permit the converted cells to regain the full capacity to respond as true TCM. We used the same transfer model as for the analysis of TCM and TEM recall capacities and monitored the evolution of the percentage of the transferred population among CD8+ cells and its CD62L phenotype in blood in the absence of restimulation. CD62L+ cells persisted after transfer, with only a slow decrease in the percentage of CD45.2+ cells among CD8 PBL cells with time (Fig. 5 A). Transferred CD8+ CD44+ cells, as well as Db-Smcy3–specific cells, remained in great majority CD62L+ in all organs analyzed (Fig. 5 B and not depicted).
Figure 5.

Adoptive transfer into naive hosts of CD62L+/− subsets purified from immune mice: overall TCM stability versus TEM death and partial CD62L reexpression. (A, B, and D) TCM and TEM were purified from spleen and PLN of immune hosts and transferred into naive female CD45.1 hosts. (A) Analysis of the persistence of CD8+CD62L+/− transferred cells 1 wk or >1 mo after transfer. The percentage of the CD45.2+ transferred cells among CD8 compared with day 2 is displayed. Each circle represents an individual transferred host. The mean value is shown for each transferred population. (B) CD62L expression and absolute numbers (in parentheses) of CD8+ Db-Smcy3+CD45.2+ cells 6 wk after transfer of purified CD62L+ cells. (C) Analysis of the propensity of TCM and TEM to undergo apoptosis. CD62L+/− subsets were purified from the spleen and PLN cells from an old C57BL/6 mouse, and the expression of annexin V by CD8+CD44+ cells was analyzed either directly ex vivo or after a 4-h stimulation of both subsets with PMA and ionomycin. (D) Kinetic analysis of the percentage of CD62L+ cells among CD8+CD44+CD45.2+ cells in blood after transfer of CD45.2+CD62L+ (closed circles) or CD45.2+CD62L− (open circles) cells in a naive CD45.1 host. (E) CD62L expression profiles of CD8+CD45.2+ remaining cells in various organs of a CD45.1 female host 6 wk after transfer of CD62L− spleen and PLN cells sorted from an immune donor. (F) Naive CD45.1 female mice were transferred with CD45.2+CD62L− cells, allowed to rest for various durations, and challenged with 5 × 106 CD45.1+ male bone marrow cells. Indicated for each duration are the number of mice analyzed, the percentage of transferred CD8 cells expressing CD62L, and the number of individual mice in which a host or a donor response was observed, respectively.
When CD62L− spleen and PLN cells were transferred, we observed a progressive decrease in the percentage of CD45.2+ transferred cells among CD8 PBLs with time, which often became undetectable after 3 or 4 wk (Fig. 5 A). This result and the fact that the transferred TEM were unable to mount a recall response and disappeared within a few days from reimmunized hosts suggested that they could have an increased susceptibility to apoptosis. Thus, we analyzed the propensity of purified CD62L+/− cells to undergo apoptosis after in vitro stimulation. As shown in Fig. 5 C, the percentage of annexin V+ cells among CD8+CD44+ cells was already much higher in the CD62L− than in the CD62L+ population ex vivo. Moreover, contrary to CD62L+CD8+ CD44+ cells, the percentage of apoptotic annexin V+ cells in the CD62L− population increased significantly after a 4-h stimulation, suggesting that the TEM subset comprised a subpopulation of memory cells that had an increased susceptibility to apoptosis in comparison to TCM.
The expression of the CD62L molecule by the transferred CD8 cells was altered with time. In blood, we observed a progressive reexpression of the CD62L molecule (Fig. 5 D). In some cases, when the transferred CD45.2+ cells persisted at detectable frequencies, we could analyze the evolution of the CD62L phenotype in organs 6 wk after transfer. Due to the already low percentage of transferred cells, we performed our analysis on the total CD8+ transferred TEM population. The CD8+CD45.2+CD62L profiles obtained in various organs showed that a substantial fraction of the persisting transferred CD8 cells were reexpressing CD62L (Fig. 5 E). Moreover, although the percentage of transferred CD8 cells in PLN was 1.61 ± 0.7 and 2.11 ± 0.7 times lower than in spleen and liver, respectively, some converted CD62L+ transferred cells even gained access to these lymphoid organs. This suggested that, in the absence of antigen, although a majority of TEM had died after a few weeks, a fraction of them was able to progressively convert to TCM.
Next, we analyzed the capacity of the CD62L+ converted cells to mount an anamnestic response. We transferred the purified spleen and PLN CD45.2+CD62L+/− populations into CD45.1+ female hosts and intravenously reimmunized these mice 2, 7, 15, or 30 d after transfer with male CD45.1+ bone marrow cells. We monitored the anti–Db-Smcy3–specific response in PBLs (Fig. 5 F). For mice transferred with CD62L− cells 2, 7, or 15 d before immunization, although respectively 26 ± 7%, 19 ± 7%, and 26 ± 7% of the transferred cells had converted to a CD62L+ phenotype, the specific response observed after immunization was always due to host naive cells. In some cases, we did see a short and abortive proliferation of the transferred CD8+CD45.2+ cells. In five out of seven hosts transferred 30 d before immunization, the transferred cells had reached an undetectable frequency on day 30 and a host-specific response was observed after immunization. However, in the two others, the transferred cells were still fully detectable at day 30, although the percentage of CD62L+ converted cells among the CD8+CD45.2+ cells was drastically different in the two mice (35 vs. 91%). In both cases, a CD45.2+ Db-Smcy3–specific memory response was observed without a substantial host response. These two mice were from distinct experiments in which, although several other mice had been transferred with the same batch of purified TEM, no other donor response was observed.
Altogether, in contrast with TCM that can persist in the absence of antigenic restimulation, most of the TEM were susceptible to apoptosis, and the small fraction of cells that could reexpress CD62L+ were generally unable to mount a secondary response.
Discussion
TCM and TEM lineage relationships and homeostasis
The recent recognition that the CD8+ T lymphocyte memory pool is heterogeneous raises the fundamental issue of lineage relationship of TCM and TEM subsets, for which several models have been proposed. In the model of independent differentiation of TCM and TEM subsets, no relationship is assumed between the two subsets; TCM and TEM could arise independently, on a stochastic basis, based on precommitment of the individual naive T cell, or depending on its stimulation conditions. In the present work, the TCM and TEM repertoires analyzed 6 wk after immunization were partly distinct, some clones being found in one subset only in all organs studied. This observation could be interpreted as the result of an independent differentiation occurring in particular locations or in response to some stimulation conditions. The TCR affinity is probably not a discriminating factor for this matter, as we observed no difference in TCR affinity between TCM and TEM clones based on their TCRα sequences (31). All stimulated cells appeared to display TCRα sequences considered as high affinity (unpublished data), and identical clones could be unshared TCM clones in one host and unshared TEM clones in another one. Among other factors, particular cytokine environments have been shown to favor the differentiation of TCM or TEM in vitro (8, 12) and in vivo (33), and the stimulation of naive CD8+ T cells in the absence of CD4 help resulted in memory cells with a TEM phenotype (34), indicating that particular stimulation conditions could preferentially induce the differentiation of TCM or TEM.
Our results clearly demonstrate that, during a systemic immunization, approximately two thirds of the stimulated naive clones can give rise to both TCM and TEM that are able to coexist, at least transiently, in peripheral and lymphoid organs, which argues against an exclusive model of independent differentiation pathways for TCM and TEM. This sharing percentage is much higher than that we had reported in a previous paper on total PBL CD8+ TCM and TEM human subsets, although it was computed in a different way (21). This variation can now be explained by the fact that we had mainly analyzed whole human repertoires, irrespective of the antigenic specificity, therefore a long time after memory has probably been established for most antigens, and we know from this and another paper that many TEM are able to regain the CD62L expression with time (9). Also, subsequent immunizations with other antigens could differentially alter TCM and TEM repertoires (35).
In the frequent case where a naive T cell generates both TEM and TCM, three models can be proposed: the two cell types could arise in parallel or sequentially, TEM giving rise to TCM (9) or vice versa (17, 18). We were unable to detect any significant production of TEM from TCM clones in the absence of antigenic challenge, neither after hemisplenectomy nor using adoptive transfer. Indeed, in both approaches, the TCM subset was very stable, consistent with the known persistence of memory T cells in lymphoid organs (3). In the hemisplenectomy experiment, although we cannot rule out that the shared TCM clones could produce TEM, the presence of unshared TCM clones indicates that at least some TCM clones did not produce TEM. In addition, when we transferred purified TCM into naive recipients, we were unable to detect any significant number of TEM in the absence of restimulation. This result challenges in vitro studies showing conversions of some TCM clones to TEM in the presence of homeostatic IL-15 and IL-7 cytokines (17, 18). There are two main possible explanations: first, in these studies, inflammatory DC-derived cytokines that could mimic inflammation mediated by a heterologous infection were added in most cases. Heterologous immunity has been shown to affect memory homeostasis, with consequences such as attrition and/or expansion of cross-reactive T cells (35). Therefore, these conditions might drive the differentiation of TCM clones into TEM, as was the case here when TCM were challenged with male cells. Similarly, we also observed some phenotype conversion of non–Db-Smcy3–specific CD8 cells in mice transferred with CD62L+ cells and reimmunized with male cells, but it might also be accounted for by other H-Y epitopes' ongoing responses (32). Alternatively, this phenomenon of TCM conversion to TEM in the absence of antigen could be a much slower process in vivo compared with in vitro and we might have observed it at later time point.
Strikingly, in the absence of challenge, cells from TEM clones were shown to reexpress CD62L over a 10-wk period, consistent with a recent paper (9). Although a high proportion of TEM clones were implicated in this process, all cells of these clones did not reexpress CD62L in a synchronized manner, and most TEM clones found 6 wk after immunization were still present in the TEM subset 10 wk later. This is consistent with other papers showing an overall stability of the TEM subset in humans (6, 13, 15, 21), although most of these studies were performed either on the total memory population, independently of antigen specificity, or in chronically infected patients. Moreover, despite the persistence of TEM clonotypes over a 10-wk period in the nonreimmunized mouse, the percentage of CD62L+ cells among total Db-Smcy3–specific sorted cells significantly increased from T1 to T2 in this mouse, showing that the number of TEM globally decreased in the absence of restimulation. This may in part stem from the reexpression of CD62L by TEM but certainly also results from the general poor survival capacity of TEM that was observed after their adoptive transfer into naive recipients.
Indeed, although our findings concerning TEM phenotypic conversion to TCM are in line with the results obtained by Wherry et al., we found that only a fraction of the transferred TEM converted to a TCM CD62L+ phenotype, whereas many others were disappearing, probably because of apoptosis. These results are in agreement with several studies showing a progressive decrease of nondividing TEM in peripheral tissues (16, 26, 36) and others showing that the intrinsic ability of an effector T cell to differentiate into a memory T cell is inversely correlated with its effector functions (37).
Overall, our results show that the differentiation process of TCM and TEM is highly complex and heterogeneous and that several features of the different proposed models have to be taken into account. Indeed, although approximately two thirds of TCM and TEM clones derive from a common naive precursor, it is still possible that some TCM clones arise without passing through a TEM stage, as suggested by the presence of unshared TCM clones and the higher repertoire diversity of the TCM subset as early as 6 wk after immunization. This would be in agreement with many studies suggesting that TCM contain fewer differentiated cells (6, 13, 15). Concerning the stability of both subsets in vivo in the absence of antigen, our results show that, whereas TCM are quite stable, both in terms of persistence and phenotype, TEM can follow three different fates: death, persistence as TEM, or conversion to a TCM phenotype (Fig. 6). Only a fraction of these cells were prone to apoptosis, whereas the remaining cells were able to survive for several weeks, even upon transfer into a new host, therefore in the absence of antigen. This threefold fate of TEM could be stochastic, only a certain percentage of the TEM progeny of a naive precursor being able to differentiate further into TCM. This could explain why a potent recall response was observed only in 2 out of 15 recipients transferred with purified TEM and immunized at least 7 d later, whereas in none of the other mice transferred with the same batches of TEM was a donor response observed. Alternatively, some particular exogenous signals might be needed for a TEM to turn into a potent TCM. In any case, for the TEM capable of converting to TCM, the differentiation process appears to be progressive and the reexpression of CD62L is probably only a first step. Indeed, although a significant proportion of TEM transferred cells had already converted to a CD62L+ phenotype 2 d only after transfer, no recall response of the transferred cells was observed in any of the 14 animals that were transferred <30 d before the challenge. This aspect is consistent with the fact that the transcriptome of activated antigen-specific cells starts stabilizing only 6 wk after immunization (20).
Figure 6.

Proposed model for the differentiation pathways of TCM and TEM after a primary response, in the absence of antigen, or in the case of antigenic restimulation. TCM are depicted as white cells and TEM are depicted as dark gray cells. The three possible fates of the latter are displayed. Light gray cells represent transitory TEM→TCM reexpressing CD62L but with various recall capacities depending on their position in the TEM→TCM differentiation pathway.
Respective roles of TCM and TEM
Our results show that most TEM have a short persistence in vivo, which prompts the question of the real memory capacity of these cells in vivo. In this work, TEM were less efficient than TCM to eliminate the recall antigen, as reported recently in another model (9). In particular, they were unable to mount a sustained recall proliferative response. If they did participate in the elimination of the male cells, their action was certainly not potent enough so as to prevent any endogenous primary host response to occur. However, despite their inability to proliferate upon restimulation, TEM probably mediated some kind of memory (Fig. 6) because the host response was somewhat attenuated in recipients transferred with TEM compared with nontransferred ones or those transferred with naive T cells (Fig. 4 and not depicted), suggesting that TEM provided a first effector wave to eliminate male cells. It should be emphasized here that we only tested splenocytes as a source of TEM, in combination with the intravenous route for injection of a nonpathogenic antigen. Therefore, we cannot exclude that the transfer of TEM from different peripheral tissues, such as LP TEM and/or the choice of a pathogenic antigen, and/or its administration locally, would lead to a different conclusion. Our model is more akin to conditions that prevail for a systemic immunological response against autologous virally infected or malignant cells.
The short-term memory role of the TEM could be a general phenomenon, as several studies suggest that peripheral TEM, and maybe recruited circulating TEM, do not divide in peripheral infected tissue, but rather provide a first line of defense against replication of the pathogen, the resolution of the recall infection being mediated by new effector cells derived from memory cells proliferating in the draining lymph node (38). Moreover, although several studies have implicated remaining effector or effector–memory cells in protection against some short-term secondary challenges, particularly for local peripheral restimulations (26, 27), most data point out the crucial role of TCM in secondary responses. Similarly, we found that transferred TCM mounted a rapid and sustained proliferative response upon secondary challenge, generating a large pool of secondary effectors that, after clearance of male cells, gave rise to a heterogeneous memory population, comprised of both TCM and TEM. However, this paper is the first to demonstrate that not all TCM clones give rise to TEM after a secondary response in vivo (Fig. 6) and that the TCM clones already shared after the primary response may preferentially differentiate into TEM upon secondary challenge.
Altogether, this paper provides new insights into the drastically different behaviors of TCM and TEM CD8 subsets in vivo and shows that vaccinologists should be careful not to generate the maximum quantity of memory T cells, but rather the right quality of memory cells, as was suggested recently by Seaman et al. (33).
Materials and Methods
Mice and immunizations
CD45.1+ and CD45.2+ C57BL/6 mice were purchased from CDTA and Charles River Laboratories, respectively. Female mice transgenic for the β chain of the H-Y TCR (β-tg mice) were raised in our animal facility. In these animals, ∼30% of the CD8 naive cells are specific for the Db–Smcy3 complex (31). All animal experiments were performed according to institutional guidelines for animal care and use.
For the generation of T CD8 memory cells, 105 female β-tg CD8 naive cells containing ∼3 × 104 Db-Smcy3–specific CD8 cells were adoptively transferred into C57BL/6 female hosts, which were intravenously injected with 5 × 106 bone marrow cells from male syngeneic mice. All studies were performed at least 6 wk after immunization.
Antibodies, peptides, and apoptosis detection
All antibodies used in this study were purchased from BD Biosciences. Smcy3738-746 peptide and Db-Smcy3 tetramers were described previously (31). For apoptosis experiment, purified splenic CD62L+ or CD62L− cells from C57BL/6 mice were triple stained with anti-CD8 allophycocyanin, anti-CD44 Cychrome antibodies, and annexin V solution (Boehringer) as recommended by the manufacturer, either directly ex vivo or after 4-h culture with RPMI 1640 medium supplemented with 10% FCS, 50 ng/ml PMA, and 0.5 μg/ml ionomycin.
Purification of TCM and TEM subsets
Lamina propria cells were prepared as described previously (39). For repertoire analysis, splenic cells from immunized mice were enriched in CD8 cells using MACS CD8 purification kit (Miltenyi Biotec). Cells from all organs were stained with Db-Smcy3 tetramers, with anti-CD8–allophycocyanin and anti-CD44–Cy, and anti-CD62L–FITC antibodies and CD8+Db-Smcy3+CD44+CD62L+/− (TCM/TEM) cells were sorted on Moflo sorter (DakoCytomation). For functional analyses and transfer experiments, splenic CD62L+ and CD62L− subsets were purified using the automacs (Miltenyi Biotec), either by staining cells with anti-CD62L–allophycocyanin antibodies and anti-allophycocyanin microbeads, or directly with anti-CD62L microbeads. Purity data and representative CD62L expression profiles of sorted samples are provided in Fig. S1 A.
Transfer experiments
To analyze the behavior of TCM and TEM subsets in the absence of antigen or in case of restimulation, purified spleen and PLN CD45.2+ CD62L+/− cells containing 3 × 104 CD8+ Db-Smcy3–specific memory cells were transferred into CD45.1+ C57BL/6 female mice. For recall response experiments, these mice were injected intravenously with 5 × 106 bone marrow cells from CD45.1+ C57BL/6 male mice on day 0, 2, 7, 15 or 30 after transfer of the purified CD62L+/− populations.
TCRα repertoire analysis
For sequencing analysis, TCRAV9-TCRAC transcripts were amplified using TCRAV9.1-specific, 5′-GACACCGTTGTTAAAGGCACC-3′, TCRAV9.2-specific, 5′-GACGCCGTTGTTAAAGGCAC-3′, and TCRAC-specific, 5′-GGCACATTGATTTGGGAGTCA-3′ primers; cloned; and sequenced as described previously (40). For clonotypic quantitative analysis, real-time PCR was performed as described previously (40) and in Fig. S2 A using the TCRAV9-specific primers, the TCRAV9-specific nested Taqman probe TM_mTCRAV9 6-FAM-ACTCGGCCTCAAAGC-MGB, and either a nested TCRAC-specific primer, 5′–CTGAGACCGAGGATCTTTTAACTGG-3′ or one of the clonotypic primers described in Fig. S2 (B and C).
Statistical analyses
To assess the repertoire diversity of a T cell subset, we computed the abundance-based coverage estimator (41) with EstimateS software (http://purl.oclc.org/estimates). We accumulated sequences until the estimator reached a plateau. Representative curves for TCM and TEM sorted populations are displayed in Fig. S1 D and show that, for most samples, the estimator plateaued after 300–400 sequences. To estimate the repertoire overlap between two populations, we computed the nonparametric maximum likelihood estimator (42). This index gives an estimation of the percentage of shared clones among the pooled TCM and TEM sequences, taking into account their frequencies. Two controls were performed to normalize these values (Fig. S1 E).
Online supplemental material
Fig. S1 illustrates the purification of CD62L+/− subsets and the analysis of TCRAV9-TCRAC rearrangements of sorted Db-Smcy3–specific TCM and TEM spleen cells. Fig. S2 illustrates the analysis of individual clonotypes by quantitative PCR (Q-PCR). Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040876/DC1.
Acknowledgments
We thank P. Bousso and J. Di Santo for critical reading of the text, A. Louise and H. Kiefer-Biasizo for cell sorting, F. Lemaître for his help with tetramer preparation, and S. Celli for helping with hemisplenectomy.
C. Bouneaud was supported by the French Délégation Générale pour l'Armement and the Ligue contre le cancer.
The authors have no conflicting financial interests.
Abbreviations used: TCM, central–memory T cells; TEM, effector–memory T cells.
References
- 1.Ahmed, R., and D. Gray. 1996. Immunological memory and protective immunity: understanding their relation. Science. 272:54–60. [DOI] [PubMed] [Google Scholar]
- 2.McHeyzer-Williams, M.G., and R. Ahmed. 1999. B cell memory and the long-lived plasma cell. Curr. Opin. Immunol. 11:172–179. [DOI] [PubMed] [Google Scholar]
- 3.Sprent, J., and C.D. Surh. 2002. T cell memory. Annu. Rev. Immunol. 20:551–579. [DOI] [PubMed] [Google Scholar]
- 4.Woodland, D.L., and R.W. Dutton. 2003. Heterogeneity of CD4(+) and CD8(+) T cells. Curr. Opin. Immunol. 15:336–342. [DOI] [PubMed] [Google Scholar]
- 5.Lefrancois, L., and D. Masopust. 2002. T cell immunity in lymphoid and non-lymphoid tissues. Curr. Opin. Immunol. 14:503–508. [DOI] [PubMed] [Google Scholar]
- 6.Sallusto, F., D. Lenig, R. Forster, M. Lipp, and A. Lanzavecchia. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 401:708–712. [DOI] [PubMed] [Google Scholar]
- 7.Cyster, J.G. 2000. Leukocyte migration: scent of the T zone. Curr. Biol. 10:R30–R33. [DOI] [PubMed] [Google Scholar]
- 8.Iezzi, G., D. Scheidegger, and A. Lanzavecchia. 2001. Migration and function of antigen-primed nonpolarized T lymphocytes in vivo. J. Exp. Med. 193:987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry, E.J., V. Teichgraber, T.C. Becker, D. Masopust, S.M. Kaech, R. Antia, U.H. Von Andrian, and R. Ahmed. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4:225–234. [DOI] [PubMed] [Google Scholar]
- 10.Oehen, S., and K. Brduscha-Riem. 1998. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J. Immunol. 161:5338–5346. [PubMed] [Google Scholar]
- 11.Lanzavecchia, A., and F. Sallusto. 2002. Progressive differentiation and selection of the fittest in the immune response. Nat. Rev. Immunol. 2:982–987. [DOI] [PubMed] [Google Scholar]
- 12.Weninger, W., M.A. Crowley, N. Manjunath, and U.H. von Andrian. 2001. Migratory properties of naive, effector, and memory CD8+ T cells. J. Exp. Med. 194:953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Champagne, P., G.S. Ogg, A.S. King, C. Knabenhans, K. Ellefsen, M. Nobile, V. Appay, G.P. Rizzardi, S. Fleury, M. Lipp, et al. 2001. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 410:106–111. [DOI] [PubMed] [Google Scholar]
- 14.Harris, N.L., V. Watt, F. Ronchese, and G. Le Gros. 2002. Differential T cell function and fate in lymph node and nonlymphoid tissues. J. Exp. Med. 195:317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hislop, A.D., N.H. Gudgeon, M.F. Callan, C. Fazou, H. Hasegawa, M. Salmon, and A.B. Rickinson. 2001. EBV-specific CD8+ T cell memory: relationships between epitope specificity, cell phenotype, and immediate effector function. J. Immunol. 167:2019–2029. [DOI] [PubMed] [Google Scholar]
- 16.Masopust, D., V. Vezys, A.L. Marzo, and L. Lefrancois. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 291:2413-2417. [DOI] [PubMed] [Google Scholar]
- 17.Geginat, J., F. Sallusto, and A. Lanzavecchia. 2001. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4+ T cells. J. Exp. Med. 194:1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geginat, J., A. Lanzavecchia, and F. Sallusto. 2003. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 101:4260-4266. [DOI] [PubMed] [Google Scholar]
- 19.Kaech, S.M., E.J. Wherry, and R. Ahmed. 2002. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2:251–262. [DOI] [PubMed] [Google Scholar]
- 20.Kaech, S.M., S. Hemby, E. Kersh, and R. Ahmed. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 111:837–851. [DOI] [PubMed] [Google Scholar]
- 21.Baron, V., C. Bouneaud, A. Cumano, A. Lim, T.P. Arstila, P. Kourilsky, L. Ferradini, and C. Pannetier. 2003. The repertoires of circulating human CD8(+) central and effector memory T cell subsets are largely distinct. Immunity. 18:193–204. [DOI] [PubMed] [Google Scholar]
- 22.Ravkov, E.V., C.M. Myrick, and J.D. Altman. 2003. Immediate early effector functions of virus-specific CD8+CCR7+ memory cells in humans defined by HLA and CC chemokine ligand 19 tetramers. J. Immunol. 170:2461–2468. [DOI] [PubMed] [Google Scholar]
- 23.Unsoeld, H., S. Krautwald, D. Voehringer, U. Kunzendorf, and H. Pircher. 2002. Cutting edge: CCR7+ and CCR7− memory T cells do not differ in immediate effector cell function. J. Immunol. 169:638–641. [DOI] [PubMed] [Google Scholar]
- 24.Debes, G.F., U.E. Hopken, and A. Hamann. 2002. In vivo differentiated cytokine-producing CD4(+) T cells express functional CCR7. J. Immunol. 168:5441–5447. [DOI] [PubMed] [Google Scholar]
- 25.Kim, C.H., L. Rott, E.J. Kunkel, M.C. Genovese, D.P. Andrew, L. Wu, and E.C. Butcher. 2001. Rules of chemokine receptor association with T cell polarization in vivo. J. Clin. Invest. 108:1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hogan, R.J., E.J. Usherwood, W. Zhong, A.A. Roberts, R.W. Dutton, A.G. Harmsen, and D.L. Woodland. 2001. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J. Immunol. 166:1813–1822. [DOI] [PubMed] [Google Scholar]
- 27.Lauvau, G., S. Vijh, P. Kong, T. Horng, K. Kerksiek, N. Serbina, R.A. Tuma, and E.G. Pamer. 2001. Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science. 294:1735–1739. [DOI] [PubMed] [Google Scholar]
- 28.Ostler, T., T. Hussell, C.D. Surh, P. Openshaw, and S. Ehl. 2001. Long-term persistence and reactivation of T cell memory in the lung of mice infected with respiratory syncytial virus. Eur. J. Immunol. 31:2574–2582. [DOI] [PubMed] [Google Scholar]
- 29.Pape, K.A., E.R. Kearney, A. Khoruts, A. Mondino, R. Merica, Z.M. Chen, E. Ingulli, J. White, J.G. Johnson, and M.K. Jenkins. 1997. Use of adoptive transfer of T-cell-antigen-receptor-transgenic T cell for the study of T-cell activation in vivo. Immunol. Rev. 156:67–78. [DOI] [PubMed] [Google Scholar]
- 30.Tanchot, C., F.A. Lemonnier, B. Perarnau, A.A. Freitas, and B. Rocha. 1997. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 276:2057–2062. [DOI] [PubMed] [Google Scholar]
- 31.Bouneaud, C., P. Kourilsky, and P. Bousso. 2000. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity. 13:829–840. [DOI] [PubMed] [Google Scholar]
- 32.Millrain, M., P. Chandler, F. Dazzi, D. Scott, E. Simpson, and P.J. Dyson. 2001. Examination of HY response: T cell expansion, immunodominance, and cross-priming revealed by HY tetramer analysis. J. Immunol. 167:3756–3764. [DOI] [PubMed] [Google Scholar]
- 33.Seaman, M.S., F.W. Peyerl, S.S. Jackson, M.A. Lifton, D.A. Gorgone, J.E. Schmitz, and N.L. Letvin. 2004. Subsets of memory cytotoxic T lymphocytes elicited by vaccination influence the efficiency of secondary expansion in vivo. J. Virol. 78:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khanolkar, A., M.J. Fuller, and A.J. Zajac. 2004. CD4 T cell-dependent CD8 T cell maturation. J. Immunol. 172:2834–2844. [DOI] [PubMed] [Google Scholar]
- 35.Welsh, R.M., and L.K. Selin. 2002. No one is naive: the significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2:417–426. [DOI] [PubMed] [Google Scholar]
- 36.Hogan, R.J., L.S. Cauley, K.H. Ely, T. Cookenham, A.D. Roberts, J.W. Brennan, S. Monard, and D.L. Woodland. 2002. Long-term maintenance of virus-specific effector memory CD8+ T cells in the lung airways depends on proliferation. J. Immunol. 169:4976–4981. [DOI] [PubMed] [Google Scholar]
- 37.Wu, C.Y., J.R. Kirman, M.J. Rotte, D.F. Davey, S.P. Perfetto, E.G. Rhee, B.L. Freidag, B.J. Hill, D.C. Douek, and R.A. Seder. 2002. Distinct lineages of T(H)1 cells have differential capacities for memory cell generation in vivo. Nat. Immunol. 3:852–858. [DOI] [PubMed] [Google Scholar]
- 38.Ely, K.H., L.S. Cauley, A.D. Roberts, J.W. Brennan, T. Cookenham, and D.L. Woodland. 2003. Nonspecific recruitment of memory CD8+ T cells to the lung airways during respiratory virus infections. J. Immunol. 170:1423–1429. [DOI] [PubMed] [Google Scholar]
- 39.Arstila, T., T.P. Arstila, S. Calbo, F. Selz, M. Malassis-Seris, P. Vassalli, P. Kourilsky, and D. Guy-Grand. 2000. Identical T cell clones are located within the mouse gut epithelium and lamina propia and circulate in the thoracic duct lymph. J. Exp. Med. 191:823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim, A., V. Baron, L. Ferradini, M. Bonneville, P. Kourilsky, and C. Pannetier. 2002. Combination of MHC-peptide multimer-based T cell sorting with the Immunoscope permits sensitive ex vivo quantitation and follow-up of human CD8+ T cell immune responses. J. Immunol. Methods. 261:177–194. [DOI] [PubMed] [Google Scholar]
- 41.Chao, A., and S.M. Lee. 1992. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 87:210–217. [Google Scholar]
- 42.Yue, J.C., M.K. Clayton, and F.C. Lin. 2001. A nonparametric estimator of species overlap. Biometrics. 57:743–749. [DOI] [PubMed] [Google Scholar]