

Abstract
Elimination of apoptotic neurons without inflammation is crucial for brain tissue homeostasis, but the molecular mechanism has not been firmly established. Triggering receptor expressed on myeloid cells-2 (TREM2) is a recently identified innate immune receptor. Here, we show expression of TREM2 in microglia. TREM2 stimulation induced DAP12 phosphorylation, extracellular signal–regulated kinase phosphorylation, and cytoskeleton reorganization and increased phagocytosis. Knockdown of TREM2 in microglia inhibited phagocytosis of apoptotic neurons and increased gene transcription of tumor necrosis factor α and nitric oxide synthase-2, whereas overexpression of TREM2 increased phagocytosis and decreased microglial proinflammatory responses. Thus, TREM2 deficiency results in impaired clearance of apoptotic neurons and inflammation that might be responsible for the brain degeneration observed in patients with polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy/Nasu-Hakola disease.
Efficient removal of apoptotic cells during development and daily tissue homeostasis is essential in all multicellular organisms (1, 2). Clearance of apoptotic cells by phagocytes usually proceeds without any signs of inflammation. It has been demonstrated that apoptotic cells signal to phagocytes to act immunosuppressively (3, 4). In particular, macrophages that have ingested apoptotic cells inhibit proinflammatory cytokine production such as release of TNF-α. Furthermore, it was shown that the repression of proinflammatory phagocytes is directly coupled to the recognition of apoptotic cells (5, 6), but the molecular mechanism behind this antiinflammatory effect remains obscure.
In the central nervous tissue clearance of apoptotic cells has to be strictly controlled because inflammatory processes would be detrimental to the central nervous system (CNS; reference 7). Dysregulated chronic innate immunity of the CNS is likely to induce neurodegeneration. In particular, it has been suggested that inflammatory mediators of activated microglia and brain macrophages including proinflammatory cytokines, metalloproteinases, and nitric oxide are the primary cause of neurodegeneration and impaired regeneration during chronic infections or autoimmune processes (8–11).
Triggering receptor expressed on myeloid cells-2 (TREM2) is a member of the innate immune receptor TREM family (12). It is expressed on the cell membrane of monocyte-derived dendritic cells, osteoclasts, and microglia (12, 13). It is associated with the signaling molecule DAP12, and it was suggested that an unknown TREM2 ligand results in the phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM) in the cytoplasmic domain of DAP12 (12, 14). In dendritic cells, TREM2/DAP12 signaling induced phosphorylation of the extracellular signal–regulated kinase (ERK) and promoted up-regulation of the CC chemokine receptor 7 (CCR7; reference 14). Recently, mutations in TREM2 and DAP12 have been described in polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL)/Nasu-Hakola disease patients (15–17). It has been demonstrated that DAP12/TREM2 deficiency results in impaired osteoclast differentiation and function (18, 19), but the mechanism of neurodegeneration in DAP12/TREM2 deficiency is still unclear.
Here, we have analyzed microglial function in response to TREM2 stimulation or after knockdown of TREM2. Unexpectedly, we observed that TREM2 stimulation of microglia neither induces transcription of inflammatory mediators nor up-regulates immunoreceptors involved in antigen presentation. However, TREM2 stimulation increased migratory and phagocytic activity of microglia. Furthermore, TREM2 knockdown microglia showed impaired clearance of apoptotic neurons and increased gene transcription of TNF-α and nitric oxide synthase-2 (NOS2). Overexpression of TREM2 in microglia resulted in increased phagocytosis of apoptotic neurons and decreased gene transcription of TNF-α, IL-1β, and NOS2. These results demonstrate that TREM2 signaling participates in clearance of apoptotic neurons and down-regulation of inflammation to maintain the local immunosuppressive microenvironment in the CNS. Lack of TREM2 signaling in microglia results in impaired clearance of apoptotic neurons and inflammation.
Results
Gene transcription and protein expression of TREM2 in microglia
Primary microglia were isolated and purified from postnatal mouse brain tissue. Purity of microglial cultures was always >95% as determined by flow cytometry analysis with antibodies directed against the marker protein CD11b (Mac1). RNA was prepared from microglia, primary neurons, and splenocytes and analyzed by RT-PCR. Gene transcripts for TREM2 and DAP12 were detected in microglia and splenocytes but not in neuronal cultures (Fig. 1 A). Protein expression of TREM2 was analyzed by immunohistochemistry with a polyclonal antibody directed against TREM2. Expression of TREM2 was detected on isolated microglia identified by double labeling with antibodies directed against CD11b (Fig. 1 B). In total, 97.6 ± 2.4% (mean ± SEM) of microglia showed staining for TREM2. No staining of TREM2 was observed on neurons, astrocytes, or oligodendrocytes, which were double labeled by antibodies directed against β tubulin III, glial fibrillary acidic protein (GFAP), and the O4 antigen, respectively (Fig. 1 B).
Figure 1.
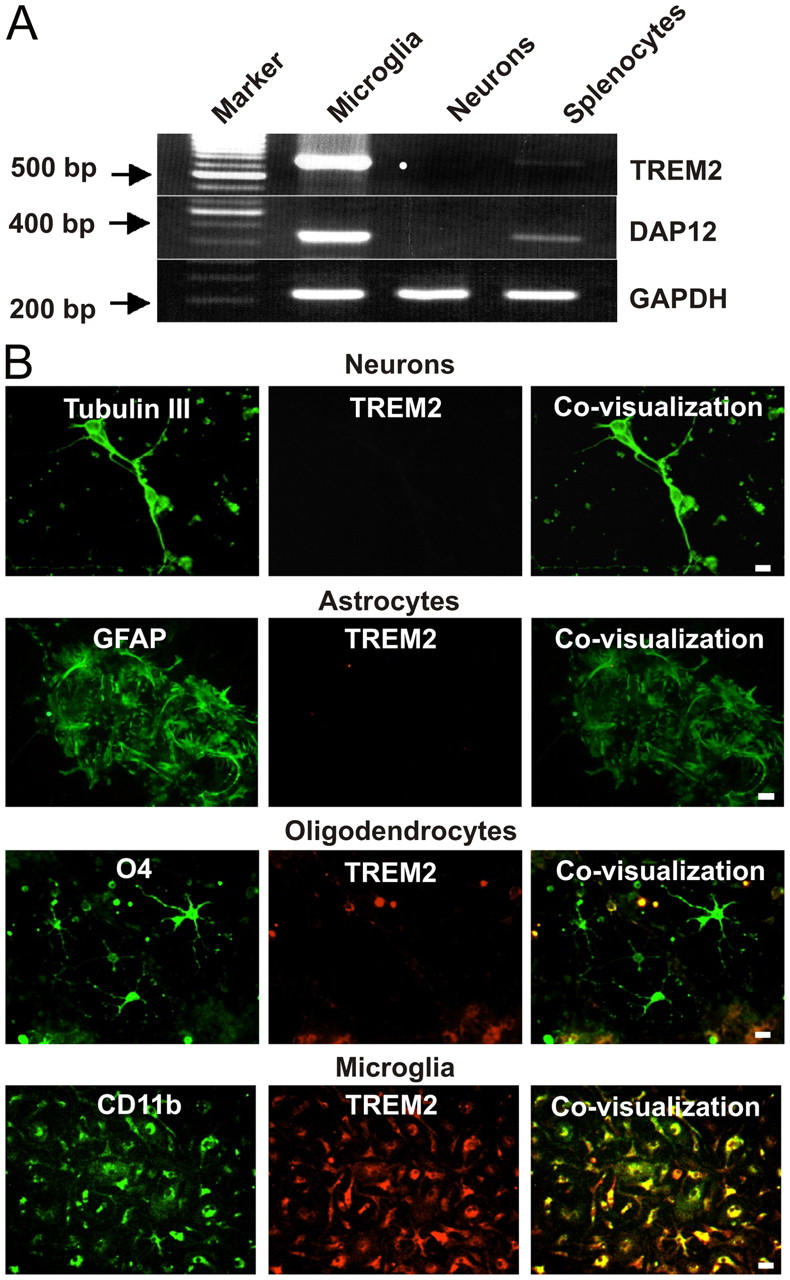
Gene transcription and protein expression of TREM2 in primary microglia. (A) RT-PCR for TREM2, DAP12, and GAPDH was performed from RNA of primary cultured microglia, neurons, and splenocytes. Gene transcripts of TREM2 and DAP12 were observed in microglia and weakly in splenocytes, but not in neurons. Molecular weight marker: 100-bp ladder. (B) Immunohistochemistry of cultured neurons, astrocytes, oligodendrocytes, and microglial cells. The distinct cell types were identified by specific antibodies (green fluorescence) directed against β tubulin–III, GFAP, O4, and CD11b. Cells were double labeled (red fluorescence) with specific polyclonal antibodies directed against TREM2. Expression of TREM2 was detected on 97.6 ± 2.4% (mean ± SEM) of microglial cells identified by double labeling with CD11b. No expression of TREM2 was detected on neurons, astrocytes, or oligodendrocytes. Bars, 10 μm.
TREM2 stimulation does not stimulate microglial inflammatory cytokine gene transcription
To analyze TREM2 signaling and function of primary microglia, we applied lentiviral vectors to transduce microglial cells. First, lentiviral vector expressing mouse TREM2 Flag tagged at the extracellular domain (Flag epitope–tagged TREM2 vector [fTREM2]), a GFP control vector (GFP1), a DAP12 vector tagged at the intracellular domain with GFP (DAP12-GFP), and a mutant DAP12 vector lacking the ITAM motif and tagged intracellularly with GFP (dominant negative DAP12[mtDAP12]-GFP vector) were used (Fig. 2 A). Western blotting was performed to confirm that stimulation of fTREM2 induces phosphorylation of DAP12. The 293 cell line was cotransfected with fTREM2 plus DAP12-GFP or fTREM2 plus mtDAP12-GFP. Cells were either stimulated with antibody directed against the Flag epitope or control antibody. Analysis of total protein lysates showed that stimulation of fTREM2 induced tyrosine phosphorylation of the DAP12-GFP (Fig. 2 B). No phosphorylation of DAP12 was detected under nonstimulated conditions or after cotransfection of the mutant DAP12. We then analyzed the usage of the lentiviral vectors for transduction of primary microglia. Lentiviral vectors were ideally suited to transfer genetic material into microglia because the transduction efficiency was regularly >90% (Table I). We first asked whether TREM2 signaling activates gene transcription of inflammatory cytokines and mediators in microglia. Microglia were lentivirally transduced with the fTREM2 vector or the GFP control vector. Microglia were then either cultured on plates coated with antibody directed against the Flag epitope or control antibody. After 24, 48, and 72 h of TREM2 stimulation, RNA was collected and analyzed by semiquantitative RT-PCR. Semiquantitative RT-PCR analysis showed TNF-α, IL-1β, and TGF-β gene transcription in nonstimulated microglia at levels comparable to TREM2-stimulated microglia at all analyzed time points (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20041611/DC1). No cytokine gene transcripts of NOS2, IFN-γ, IL-10, and IL-12p40 were detected in nonstimulated as well as TREM2-stimulated microglia (Fig. S1). Because we could not detect any difference in cytokine gene transcription by ordinary RT-PCR, we performed real-time PCR of gene transcripts for TNF-α, IL-1β, and TGF-β (Fig. S2). Again, despite the highly sensitive real-time PCR, no difference in the transcript levels for TNF-α, IL-1β, and TGF-β between nonstimulated and TREM2-stimulated microglia was observed (Fig. S2).
Figure 2.

Lentiviral vectors and phosphorylation of DAP12 after stimulation of TREM2. (A) Schematic drawing of lentiviral vectors for expression of fTREM2, GFP (GFP1), DAP12 tagged with GFP (DAP12-GFP), or mutant mtDAP12 tagged with GFP (mtDAP12-GFP). (B) Phosphorylated DAP12-GFP was detected after blotting with the antibody directed against tyrosine phosphate in fTREM2 plus DAP12-GFP–cotransfected cells and after cross-linking stimulation. DAP12-GFP or mtDAP12-GFP were detected after reblotting with the antibody directed against GFP in the samples, respectively.
Table I.
Lentiviral transduction efficiency
| Lentiviral vector | Transduction efficiency of microglia (mean ± SEM) |
|---|---|
| fTREM2 | 94.70 ± 0.81 |
| GFP | 93.85 ± 0.82 |
| fTREM2 1 GFP | 91.30 ± 1.10 |
| fTREM2 1 DAP12-GFP | 90.74 ± 0.56 |
| fTREM2 1 mtDAP12-GFP | 90.44 ± 1.91 |
| shTREM2 | 96.80 ± 1.20 |
| shControl | 92.15 ± 0.85 |
| wTREM2 | 93.20 ± 1.20 |
| GFP2 | 93.60 ± 0.30 |
Cross-linking fTREM2 of transduced microglia stimulates expression of CCR7 and migration toward CC chemokine ligand (CCL)19 and CCL21
To determine whether TREM2 signaling might stimulate antigen presentation or migratory capacity of microglia, we examined cell surface expression of several immune receptors by flow cytometry. Microglia were transduced with the fTREM2 vector or the GFP1 control vector. The microglia were then either cultured on plates coated with antibody directed against the Flag epitope or control antibody. Cells were collected after 72 h, immunolabeled with specific antibodies, and analyzed by flow cytometry. Stimulation of TREM2 signaling did not induce expression of MHC class II or CD11c (Fig. 3 A). Furthermore, expression levels of CD86 and CD36 were unaltered after stimulation of TREM2. Only the chemokine receptor CCR7 was slightly induced in microglia after stimulation of TREM2 (Fig. 3 A). To determine any functional consequences of increased CCR7 expression, a chemotaxis assay was performed. Microglial cells were stimulated via TREM2 and placed in a two-chamber system. The number of microglial cells migrating toward the chemokine ligands CCL19 and CCL21 was quantified (Fig. 3 B). Indeed, increased CCR7 expression after TREM2 stimulation had a strong functional impact. In detail, the number of microglia migrating toward CCL19 increased from 20.3 ± 0.9 (mean ± SEM) to 70.8 ± 6.8 and toward CCL21 increased from 18.0 ± 4.2 to 41.0 ± 3.8 (Fig. 3 B).
Figure 3.

TREM2 stimulation up-regulates CCR7 and increases migration of microglia toward CCL19 and CCL21. Primary murine microglia were transduced with fTREM2 or GFP control vector. Microglia were cultured on dishes coated with cross-linking antibody directed against the Flag epitope (+) or a control antibody (−). (A) Primary microglia were transduced with fTREM2 or GFP control vector and cultured on dishes coated with cross-linking antibody directed against the Flag epitope (red line) or control antibody (blue line) for 72 h. Cells were analyzed by flow cytometry after staining by specific antibodies directed against MHC class II, CD86, CD11c, CD36, and CCR7. Staining of cells with an isotype control antibody was performed (gray filled). A slight increase in CCR7 expression was observed in fTREM2-transduced microglia after stimulation of TREM2, which was not detected in the control vector–transduced microglia. (B) TREM2 stimulation directs migration of microglia toward the CCR7 ligands CCL19 and CCL21. fTREM2-transduced microglia stimulated for 24 h with precoated 10 μg/ml antibody directed against the Flag epitope (white bars), 10 μg/ml isotype control antibody (black bars), or LPS (gray bars) were tested in transwell chemotaxis assays for their ability to migrate toward medium alone or medium supplemented with CCL19 or CCL21. Microglia showed increased migration toward CCL19 and CCL21. *, P < 0.05; Mann-Whitney U test.
These results demonstrate that TREM2 stimulation does not stimulate antigen presentation but up-regulates expression of the chemokine receptor CCR7 and supports chemotaxis towards the corresponding ligands CCL19 and CCL21.
Cross-linking fTREM2 of transduced microglia stimulates phagocytosis
One important microglial function is the removal and clearance of apoptotic cells and debris. Because TREM2 is involved in bone resorption of osteoclasts, we speculated that TREM2 molecules of microglia might be involved in phagocytosis. To investigate this hypothesis, we performed a microsphere bead phagocytosis assay. Microglia isolated from postnatal mouse brains were transduced with the fTREM2 lentiviral vector or the control lentiviral vector. The transduced microglia were cultured for 24 h on plates coated with either Flag-specific antibodies or control antibodies. Microsphere beads were then added to the culture for 1 h, and phagocytosis of the beads was analyzed by fluorescence microscopy and flow cytometry. The fTREM2 microglia showed increased phagocytosis of multiple microsphere beads after cross-linking of TREM2 compared with microglia transduced with the control vector (Fig. 4 A). Furthermore, the increased phagocytosis was dependent on stimulation of TREM2 (Fig. 4 A). Phagocytosis of the red fluorescent microsphere beads was quantified by flow cytometry to determine the percentage of microglia that phagocytosed two or more beads. After the cross-linking of TREM2, the number of fTREM2-transduced microglia that phagocytosed multiple beads increased by 35 ± 11% (mean ± SEM) compared with no cross-linking of TREM2 (Fig. 4 B). No change in phagocytosis was observed after the cross-linking of TREM2 in control vector–transduced microglia (Fig. 4 B). Next, we investigated whether increased phagocytosis after TREM2 stimulation is mediated via the adaptor signaling molecule DAP12. Microglia were cotransduced with the fTREM2 lentiviral vector and an mtDAP12 lentiviral vector or the control vector. Again, we observed very high transduction efficiency showing cotransduction with both vectors in >90% of microglial cells (Table I). When the microglia were cotransduced with mtDAP12, stimulation of TREM2 failed to increase phagocytosis of microsphere beads (Fig. 4 C). No change of phagocytosis was observed for cotransduced fTREM2 and mtDAP12 microglia between the TREM2-stimulated and nonstimulated condition (Fig. 4 C). However, cotransduction of fTREM2 and the GFP control vector showed up-regulation of microglial phagocytosis by 29.1 ± 11.6% (mean ± SEM) after TREM2 stimulation compared with nonstimulated cells (Fig. 4 C). This result demonstrated that TREM2 signaling increased phagocytosis of microsphere beads via DAP12 adaptor molecule signaling.
Figure 4.
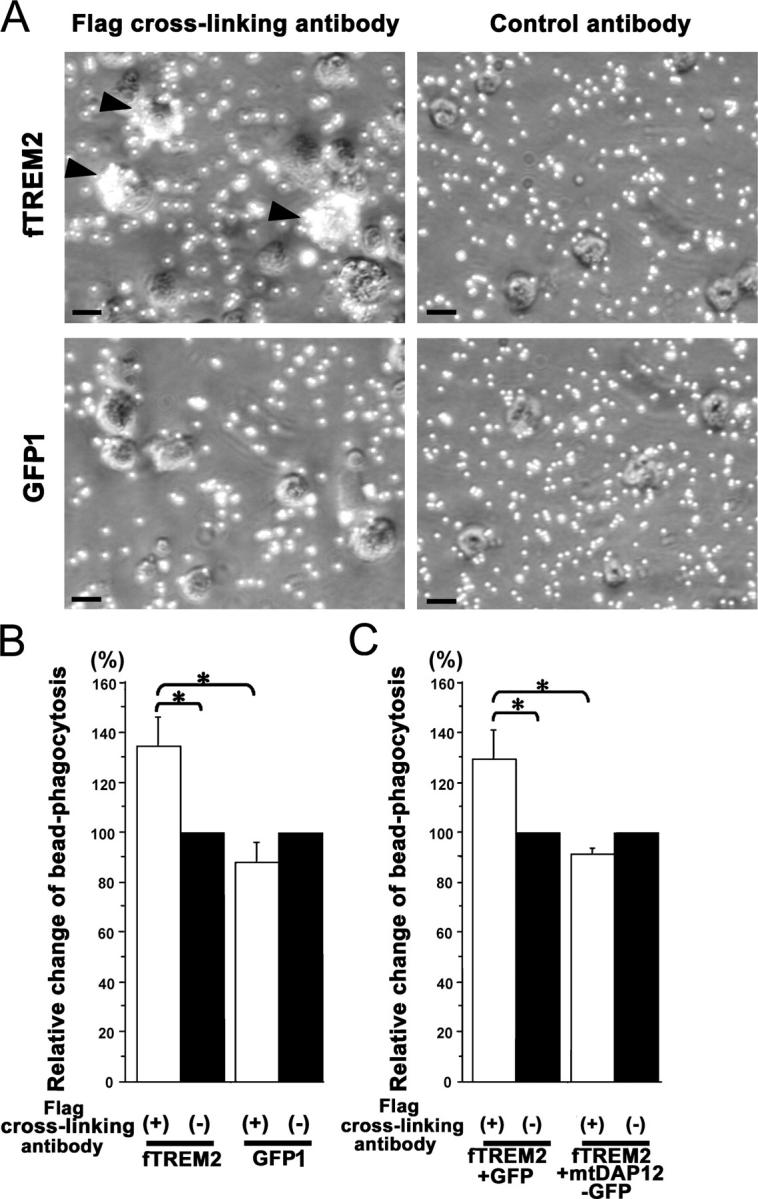
Increased bead phagocytosis after stimulation of TREM2. (A) Primary murine microglia were transduced either with fTREM2 vector (fTREM2) or GFP1 control vector. Microglia were cultured on dishes coated with antibodies directed against the Flag epitope to cross-link the fTREM2 or control antibodies. Microglia were stimulated with antibodies for 24 h. Microsphere beads were then added for 1 h. Phase contrast images are shown, demonstrating visible phagocytosis of microshpere beads after stimulation of fTREM2-transduced microglia by cross-linking antibodies. Bars, 10 μm. (B) Phagocytosis was quantified by flow cytometry. The relative change in bead phagocytosis after fTREM2 stimulation was compared with nonstimulated bead phagocytosis. Microglia transduced with fTREM2 showed a significant increase in bead phagocytosis after cross-link stimulation, whereas no significant change in bead phagocytosis was observed in GFP1-transduced microglia after cross-link stimulation. Data are presented as mean ± SEM of at least three independent experiments. *, P < 0.05; Mann-Whitney U test. (C) The relative change in bead phagocytosis after fTREM2 stimulation compared with the nonstimulated situation by flow cytometry. Microglia was transduced with fTREM2 plus GIPγ or fTREM2 plus mutant DAP12 (mtDAP12-GFP). The mtDAP12 transduction prevented the increase in bead phagocytosis after fTREM2 stimulation. Data are presented as mean ± SEM of at least three independent experiments. *, P < 0.01; Mann-Whitney U test.
Cross-linking fTREM2 of transduced microglia leads to reorganization of F-actin and phosphorylation of ERK
Phagocytosis requires kinase signaling and reorganization of actin cytoskeleton. We analyzed whether TREM2 stimulation might induce actin cytoskeleton reorganization in microglia. Therefore, microglia transduced with the fTREM2 vector were added to culture plates coated with the Flag-specific antibody or control antibody. Cells were fixed after 1 h and F-actin was labeled with a fluorescence dye. Serial confocal images demonstrated F-actin staining exactly at the site of TREM2 stimulation (Fig. 5 A). In total, 72.5 ± 2.5% (mean ± SEM) of microglia showed polarized F-actin staining at the site of TREM2 stimulation (Fig. 5 B). Thus, cross-linking of TREM2 induced polarization and reorganization of F-actin in microglia as a first sign of phagocytosis (Fig. 5, A and B). Because TREM2/DAP12 signaling induced ERK phosphorylation in immature dendritic cells, we analyzed phosphorylation of ERK after cross-linking stimulation of fTREM2 transduced microglia by Western blotting. Stimulation of TREM2 of microglia induced phosphorylation of ERK as demonstrated by a specific antibody recognizing the phosphorylated form of ERK (Fig. 5 C). To determine the involvement of ERK signaling in increased phagocytosis, the microglia were pretreated with the ERK inhibitor PD98059. Indeed, increased bead phagocytosis after TREM2 stimulation was neutralized by treatment with the ERK inhibitor (Fig. 5 D).
Figure 5.
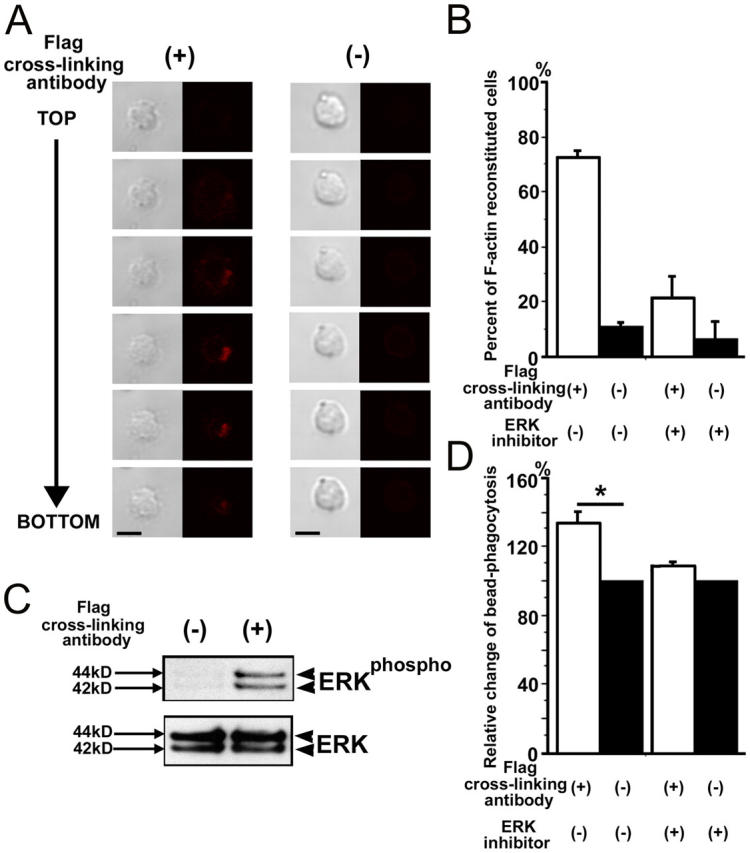
Cytoskeleton reorganization and ERK signaling after stimulation of microglial phagocytosis by TREM2 cross-linking. (A) Primary murine microglia were transduced with fTREM2 vector. Microglia were cultured on dishes coated with antibodies directed against the Flag epitope (+) to cross-link the fTREM2 or control antibodies (−) for 1 h. Microglia were fixed and then stained with Alexa Fluor 546–conjugated phalloidin. Individual microglial cells were scanned by a confocal microscope from top (TOP) to bottom (BOTTOM) with a step of 1.6 μm between each optical section. Strong F-actin staining was observed at the bottom of the cells after TREM2 stimulation. Phase contrast and confocal images are shown. Bars, 10 μm. (B) Cytoskeleton reorganization of F-actin was quantified by counting the number of cells showing F-actin at the bottom of the cells. Data are presented as mean ± SEM. (C) Western blot analysis of protein lysates of primary microglial cells. Phosphorylated ERK was detected after blotting with a specific antibody directed against phosphorylated ERK in fTREM2-transfected cells after stimulation with a cross-linking antibody (+), but not the control antibody (−). The total protein amount of ERK was detected after reblotting with an antibody directed against total ERK. (D) Increased bead phagocytosis after fTREM2 stimulation is neutralized by ERK inhibitor. The relative change in bead phagocytosis after fTREM2 stimulation was compared between the stimulated (open bars) and nonstimulated (closed bars) situation by flow cytometry. Microglia were transduced with fTREM2 vector and treated with or without ERK inhibitor. ERK inhibitor reverted the increase of phagocytosis stimulated by TREM2. Data are presented as mean ± SEM of at least three independent experiments. *, P < 0.05; Mann-Whitney U test.
TREM2 expression of microglia determines phagocytosis of apoptotic neurons
Up-regulation of phagocytosis without induction of inflammatory cytokines and cytotoxic mediators implies clearance without an inflammatory reaction. Because microglia play an important role in the removal of apoptotic neurons during development and aging, TREM2 might recognize apoptotic neuronal membranes and participate in their clearance. To study the pathophysiological function of TREM2, we used a lentiviral strategy to either knock down TREM2 by RNA interference or to overexpress TREM2 (Fig. 6). Microglia were transduced with a lentiviral vector expressing either short hairpin TREM2 (shTREM2) RNA, a control vector that expressed a short hairpin scrambled sequence of TREM2 (short hairpin control [shControl]), wild-type TREM2 (wTREM2) to overexpress the receptor, or GFP as a control (GFP2). To confirm knockdown of TREM2 gene transcripts, microglial RNA was analyzed at 72 h after transduction with the lentiviral RNA interference vector. Indeed, no gene transcripts for TREM2 were detected after 40 PCR cycles in reverse-transcribed RNA of microglia after knockdown of TREM2 (Fig. 7 A). The control vectors did not knock down TREM2. Immunohistochemistry was performed to confirm absence of TREM2 expression on microglia after knockdown. Although TREM2 was detected by specific antibodies on control vector–transduced microglia, no expression of TREM2 was observed after transduction of the microglia by the shTREM2 vector (Fig. 7 B). Again, transduction efficiency was always >92%, leading to a loss of TREM2 in almost all microglial cells after transduction with the shTREM2 vector (Table I). Flow cytometry analysis of microglia transduced with shTREM2 by polyclonal antibodies directed against TREM2 confirmed knockdown of TREM2, and it demonstrated increased levels of TREM2 expression after transduction of microglia with the vector wTREM2 to overexpress TREM2 (Fig. 7 C).
Figure 6.

Schematic drawing of lentiviral vectors for knocking down TREM2 (shTREM2), shControl vector, wTREM2 vector, and GFP control vector (GFP2).
Figure 7.
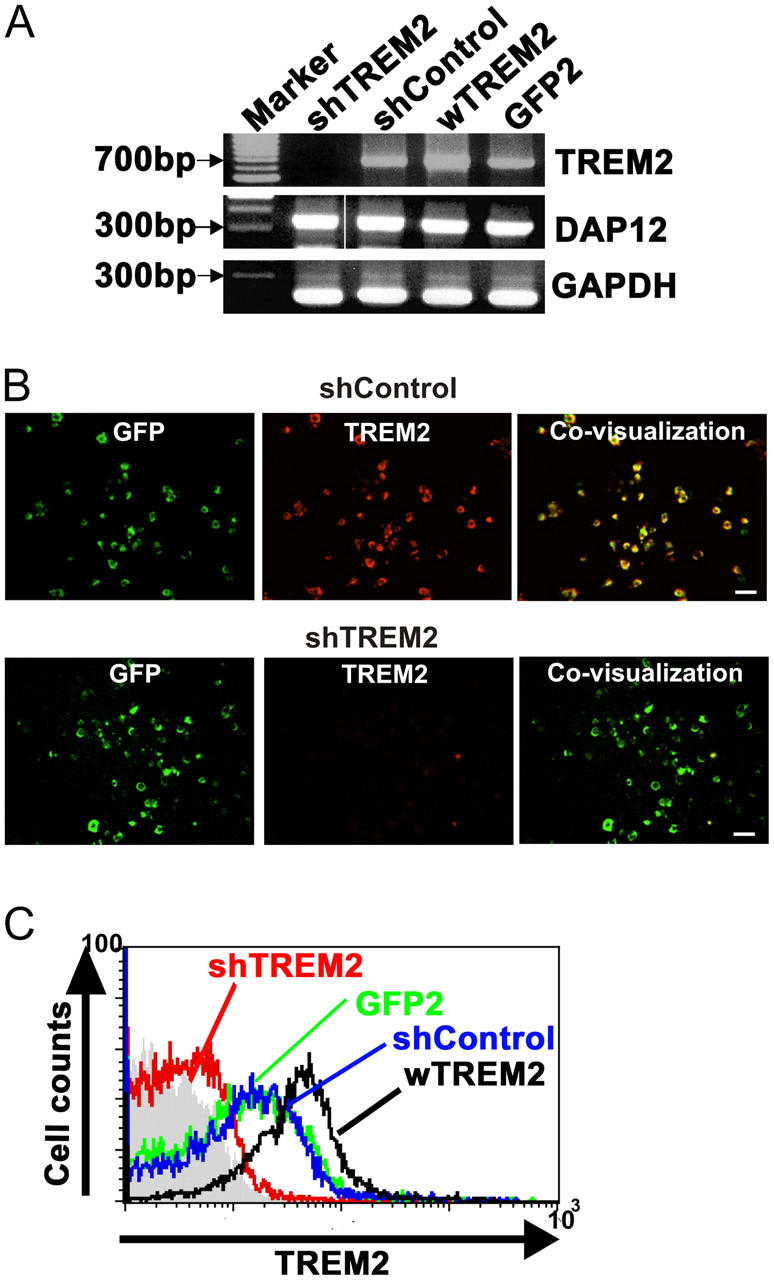
Knockdown and overexpression of TREM2 receptors in microglia by lentiviral vectors. (A) Gene transcript analysis of cultured microglia transduced with the shTREM2, shControl, wTREM2, or GFP2 vector. Gene transcripts for TREM2 and GAPDH were analyzed by RT-PCR. The lentiviral vector producing short hairpin RNAs specific for TREM2 induced complete knockdown of TREM2 gene transcripts. Gene transcripts for GAPDH and DAP12 served as an internal control and were not affected by the shTREM2 vector. Molecular weight marker: 100-bp ladder. (B) Protein expression of TREM2 was analyzed by a polyclonal antibody directed against TREM2 of cultured microglia transduced with the shTREM2 or shControl vector. Expression of TREM2 was not detectable by immunohistochemistry in microglia transduced with the shTREM2 vector. Bars, 20 μm. (C) Flow cytometry analysis of cultured microglial cells transduced either with a lentiviral vector producing short hairpin RNAs specific for TREM2 (shTREM2), irrelevant short hairpin RNAs (shControl), wTREM2, or control GFP (GFP2). shTREM2 transduction expression of TREM2 was almost undetectable, whereas wTREM2 transduction increased levels of TREM2 expression on microglia.
Phagocytosis of apoptotic neurons by microglia was analyzed after knockdown or overexpression of TREM2. Neurons were labeled by a red fluorescent membrane dye and pretreated with okaidic acid to induce apoptosis. Microglia transduced with the control vector (shControl) phagocytosed the red fluorescent–labeled apoptotic neuronal membranes within 1 h (Fig. 8, A and C). However, phagocytosis of apoptotic membrane fragments was barely detected under the fluorescence microscope after 1 h, when the TREM2 receptor was knocked down in microglia (Fig. 8 B). The number of microglia having phagocytosed membrane material derived from apoptotic neurons was quantified by flow cytometry. In detail, only 9.5 ± 1.6% (mean ± SEM) of microglia showed phagocytosed neuronal material after knockdown of TREM2, whereas 22.2 ± 2.4% of microglia transduced with the control vector showed phagocytosis. Even after 24 h, only 11.9 ± 1.5% of microglia phagocytosed apoptotic neuronal material if TREM2 was knocked down (Fig. 8 D). Next, we analyzed whether the mtDAP12 impairs phagocytosis of microglia. After transduction of mtDAP12, phagocytosis of apoptotic neuronal membranes was reduced from 24.5 ± 0.9% to 8.0 ± 1.8% (Fig. 8 D). Interestingly, overexpression of TREM2 increased phagocytosis of apoptotic neuronal material. In total, 31.8 ± 2.2% (mean ± SEM) of microglia showed phagocytosis of apoptotic neuronal material after overexpression of TREM2, whereas only 19.4 ± 1.3% of microglia transduced with the control vector showed phagocytosis (Fig. 8 D).
Figure 8.

TREM2 expression of microglia determines phagocytosis of apoptotic neuronal membranes. Cultured neurons were labeled with a red fluorescent membrane dye, treated with okadaic acid to induce apoptosis, and cocultured with microglia transduced either with a lentiviral vector producing short hairpin RNAs specific for TREM2 (shTREM2), irrelevant short hairpin RNAs (shControl), wTREM2, control GFP2 vector, mtDAP12 (mtDAP12-GFP), or control GFP1 vector. Cells were analyzed by fluorescence microscopy and flow cytometry. (A) Microglia (green fluorescent protein) lentivirally transduced with the control vector (shControl) phagocytosed apoptotic neuronal membrane fragments (red fluorescent dye) as shown by confocal images. Inset as indicated. Bar, 10 μm. (B) No obvious phagocytosis was observed in TREM2 knockdown microglia. Bar, 10 μm. (C) High magnification of the shControl image. Bar, 10 μm. (D) The percentage of cells having phagocytosed apoptotic neuronal membrane fragments was quantified by flow cytometry at 1 and 24 h of coculture. Microglia were transduced with lentiviral vectors producing short hairpin RNAs specific for TREM2 (shTREM2), irrelevant short hairpin RNAs (shControl), wTREM2, control GFP2, mtDAP12 (mtDAP12-GFP), or control GFP1. wTREM2 vector (wTREM2)-transduced microglia phagocytosed significantly more apoptotic neuronal material after 1 and 24 h compared with GFP- (GFP1 or GFP2) or control vector (shControl)-transduced microglia. Control vector (shControl)- and GFP (GFP1 or GFP2)-transduced microglia phagocytosed more apoptotic neuronal material after 1 and 24 h compared with microglia transduced with the TREM2 knockdown (shTREM2) or mtDAP12 (mtDAP12-GFP) vector. Data are presented as mean ± SEM. *, P < 0.02; **, P < 0.05; ***, P < 0.01; Mann-Whitney U test.
TREM2 determines gene transcription of inflammatory mediators during phagocytosis of apoptotic neurons
After having demonstrated that TREM2 knockdown microglia showed reduced phagocytosis and TREM2 overexpressing microglia showed increased phagocytosis compared with control vector–transduced microglia, we asked whether the impaired phagocytosis is associated with a dysregulated cytokine transcription. To study this question, we investigated gene transcripts of proinflammatory cytokines and mediators in microglia cocultured together for 48 h with apoptotic neuronal cells.
Gene transcripts for TNF-α, IL-1β, TGF-β, and NOS2 were detected in the cultured cells and quantified by real-time PCR (Fig. 9). Gene transcripts for IL-12p40 were only detected in microglia after knockdown of TREM2, but no other samples (not depicted). No gene transcripts of IFN-γ and IL-10 were detected in any experimental group. Knockdown of TREM2 in microglia increased the relative level of TNF-α from 1 to 1.9 ± 0.3 (mean ± SEM), of IL-1β to 1.3 ± 0.3, and of NOS2 to 1.8 ± 0.5 compared with the shControl-transduced microglia. In contrast, overexpression of TREM2 in microglia reduced the relative level of inflammatory gene transcripts after 48 h of coculture with apoptotic neurons. In detail, microglia cells transduced with wTREM2 showed a reduction of relative gene transcript levels for TNF-α from 1 to 0.7 ± 0.1 (mean ± SEM), for IL-1β to 0.6 ± 0.2 SEM, and for NOS2 to 0.4 ± 0.2 (Fig. 9). Gene transcript levels of TGF-β were not modulated by knockdown or overexpression of TREM2. Surprisingly, expression of mtDAP12 in microglia did not change the relative level of TNF-α, IL-1β, TGF-β, or NOS2 gene transcripts. Thus, gene transcripts of microglial inflammatory mediators were significantly modulated by the presence or absence of TREM2 on microglia and even by the level of TREM2 expression.
Figure 9.

TREM2 expression determines inflammatory response of microglia during phagocytosis of apoptotic neuronal material. Gene transcription of inflammatory mediators was analyzed by real-time RT-PCR of microglia cocultured with apoptotic neurons. Microglia were transduced with lentiviral vectors producing short hairpin RNAs specific for TREM2 (shTREM2), irrelevant short hairpin RNAs (shControl), wTREM2, control GFP2, mtDAP12 (mtDAP12-GFP), or control GFP1 vector. Relative level of gene transcripts for TNF-α, IL-1β, and NOS2 were increased in microglia transduced with the TREM2 knockdown vector compared with the control vector and decreased in microglia-overexpressing TREM2 compared with the control vector. Data are presented as mean ± SEM. *, P < 0.02; Mann-Whitney U test.
Discussion
Gene transfer into primary cultured microglia or macrophages by classical transfection procedures is very poor and results in very low transfection efficiency (20). We applied vesicular stomatitis virus glycoprotein–pseudotyped lentiviral vectors to transduce primary microglia. Transduction efficiency was very high (>90%). The expression was stable and transduction was reproducible. Furthermore, lentiviral vectors were successfully used for specific knockdown of gene transcripts by small hairpin RNAs. RNA interference for TREM2 by lentiviral small hairpin RNAs reduced TREM2 gene transcripts to levels undetectable by 40 cycles of RT-PCR, whereas the gene transcripts of GAPDH or DAP12 were not affected. Knockdown of TREM2 in microglia by RNA interference allowed us to study the role of TREM2 in mature and differentiated microglia. Although we have not observed any effect of the transduction procedure or the expression of nonrelated small hairpin RNAs in our control experiments, we cannot completely exclude that the lentiviral transduction procedure per se might influence the phenotype of microglia.
Function of TREM2 has been studied before in dendritic cells and osteoclasts. When immature human dendritic cells were stimulated by a cross-linking monoclonal antibody directed against human TREM2, MHC class II and the costimulatory CD86 and CD40 molecules were up-regulated (14). Moreover, TREM2 stimulation of immature dendritic cells strongly induced expression of CCR7, a chemokine receptor binding the ligands CCL19 and CCL21 (14). In contrast, in our study we observed that TREM2 signaling of microglial cells neither stimulated MHC class II nor costimulatory molecules. Only an up-regulation of CCR7 was observed after triggering TREM2 in microglia. The up-regulation of CCR7 after TREM2 stimulation appeared to be functional because the TREM2-stimulated microglia showed increased chemotaxis toward CCL19 and CCL21, the ligands of CCR7. Furthermore, in our study the effect of TREM2 stimulation on microglia was relatively selective for phagocytosis, indicating that TREM2 might activate intracellular pathways distinct from other innate immune receptors. Phagocytosis has been shown to require actin cytoskeleton reorganization (21) and kinase signaling (22). We observed that TREM2 stimulation induced polarization of F-actin and ERK/MAPK phosphorylation, but not phosphatidylinositol 3-kinase (PI3K) signaling (not depicted). In this respect, microglia behave like dendritic cells, where it was shown that TREM2 stimulates ERK, but not PI3K signaling (14). There are several similarities between TREM2 receptor and Toll-like receptors. Both Toll-like receptors and TREM2 receptor are innate immune receptors involved in phagocytosis (23) and actin remodeling (24). However, TREM2 stimulates phagocytosis without inflammation, whereas Toll-like receptor–mediated signaling is known to activate NF-κB and inflammatory cytokine gene transcription. Interestingly, in dendritic cells, TREM2 stimulation was also independent of NF-κB activity (14).
Recently, it was demonstrated that DAP12/TREM2 deficiency results in impaired and delayed osteoclast differentiation with a reduced bone resorption capability in vitro (18, 19). Principally, this observed defect in bone resorption could be due to impaired phagocytosis, although no direct evidence has been provided for this mechanism in osteoclasts. We now have shown impaired phagocytosis of apoptotic neurons after knockdown of TREM2 in microglia.
A recent publication suggested that microglia as well as oligodendrocytes express DAP12 molecules, which might participate in myelin loss and neurodegeneration of DAP12 mutant mice (25). In our immunohistochemistry analysis, we did not detect TREM2 expression in oligodendrocytes and found TREM2 expression solely on microglia and no other brain cell types. Furthermore, we demonstrated in this study that intact signaling via DAP12 in microglia is required for increasing phagocytic activity after TREM2 stimulation. Furthermore, we demonstrate here that expression of mtDAP12 in microglia impairs phagocytosis of apoptotic neurons. PLOSL, also named Nasu-Hakola, is a recessively inherited disease due to functional deficiency of TREM2 or DAP12 (16, 26). DAP12/KARAP has been shown to be involved in dendritic cell function and development of autoimmunity (27–29). Clinically and histopathologically, no difference has been described between patients with TREM2 and DAP12 mutations. Thus, our data suggest microglial impairment to be responsible for the CNS phenotype of PLOSL/Nasu-Hakola disease. Because no function of DAP12 in oligodendrocytes has been described so far, it is unclear whether loss of DAP12 function in oligodendrocytes has any pathophysiological consequences.
Cells that die in a physiological context are typically removed rapidly by phagocytic cells including macrophages and microglia (1, 2). In contrast to removal of necrotic cells, apoptotic cells are eliminated without inflammation or pathology. Interestingly, lack of inflammatory responses associated with engulfment of apoptotic debris is more than a passive avoidance of stimulation. Apoptotic cells affirmatively inhibit inflammatory responses and cytokine production including TNF-α from the phagocytic cells that ingest them (4). The ingestion of apoptotic cells even attenuates the stimulatory response of the Toll-like receptor complex (5), demonstrating that the inhibitory effect of apoptotic targets is dominant in trans to the stimulatory effect of Toll-like receptors (5). Furthermore, the inhibitory effect does not modulate the NF-κB activation by stimulation of the Toll-like receptors via LPS (6). So far, the receptors that could signal this inhibitory effect remained unresolved. In this study, we found that the TREM2 receptor fulfills most of the features of such a postulated inhibitory receptor on phagocytic cells stimulated by apoptotic cells. Particularly, TREM2 stimulation selectively increased phagocytic activity without the stimulation of cytokine production by microglia. More strikingly, knockdown of TREM2 in microglia increased the gene transcription of the proinflammatory mediators TNF-α and NOS2 and induced gene transcription of IL12p40 after challenge of microglia with apoptotic neurons. Surprisingly, microglia with functionally impaired DAP12 signaling did not change gene transcript level of proinflammatory mediators after challenge with apoptotic neurons. Thus, mtDAP12 might interfere with additional DAP12-associated molecules.
In the nervous tissue, microglia have been shown to express TREM2 (13). It was speculated that microglia might have a primary role in the pathogenesis of the CNS phenotype in PLOSL/Nasu-Hakola disease (12). The characteristic hallmarks of PLOSL/Nasu-Hakola disease are neuropsychiatric symptoms followed by neurodegeneration and cystic bone lesions becoming symptomatic during dulthood. The pathological changes lead to bone fractures and profound presenile dementia and premature death. Interestingly, patients with PLOSL/Nasu-Hakola disease show massive gliosis including activated microglia in the CNS despite impaired ITAM signaling via DAP12/TREM2. The TREM2 receptor signals via DAP12, which contains ITAM signaling molecules that are supposed to activate microglia. Thus, one would expect that mutation of TREM2 or DAP12 leads to inactivation of microglia. Surprisingly, the opposite effect was observed. Our in vitro results now demonstrate that TREM2 and DAP12 signaling play an important role in the clearance of apoptotic neurons. Furthermore, the data show that microglia with functionally impaired TREM2 have increased gene transcription of inflammatory mediators after confrontation with apoptotic neurons. This allows us to suggest that TREM2 deficiency dysregulates microglial phagocytosis and release of inflammatory mediators. Because neuronal cell death is a regular but relatively rare event, the inflammatory lesions and the associated neurodegeneration might develop over several years in PLOSL patients and might be difficult to detect in the short life span of the DAP12 mutant mice, which have synaptic degeneration, myelin abnormalities, and microglial activation (25).
In summary, the data demonstrate that TREM2 and its signaling molecule DAP12 of microglia are involved in phagocytosis. Deficiency of TREM2 results in impaired uptake of apoptotic neurons and increased production of inflammatory mediators. Thus, impaired microglia might induce inflammatory lesions in PLOSL/Nasu-Hakola disease.
Materials and Methods
Primary cell culture
Microglia were prepared from the brains of postnatal day 3 to 5 (P3 to P5) C57BL/6 mice as described previously (30). In brief, meninges were removed mechanically, and the cells were dissociated by trituration and cultured in basal medium (BME; GIBCO BRL), 10% FCS (PAN Biotech GmbH), 1% glucose (Sigma-Aldrich), 1% l-glutamine (GIBCO BRL), and 1% penicillin/streptomycin (GIBCO BRL) for 14 d to form a confluent glial monolayer. To collect microglial cells, the cultures were shaken on a rotary shaker (200 rpm) for 2 h. The attached astrocytes were used for immunohistochemistry. The detached microglial cells were seeded in normal culture dishes for 1 h, and then all nonadherent cells were removed and discarded. Purity of the isolated microglia was >95% as determined by flow cytometry analysis with antibody directed against CD11b (BD Biosciences). Microglial cells were cultured in basal medium as described above.
Oligodendrocytes and neuron-enriched cells were prepared from the brain of C57BL/6 mouse embryos (E15-16) as previously described (31). In brief, brain tissue was isolated and mechanically dispersed and seeded in culture dishes precoated with 0.01 mg/ml poly-l-ornithin (Sigma-Aldrich) and 10 μg/ml laminin (Sigma-Aldrich). Cells were cultured in neuronal condition medium (BME; GIBCO BRL) supplemented with 2% B-27 supplement (GIBCO BRL), 1% glucose (Sigma-Aldrich), and 1% FCS (PAN Biotech GmbH). Cells were cultured for 5–10 d to obtain morphologically mature neurons or oligodendrocytes.
RT-PCR analysis for TREM2 gene transcripts
RNA was isolated from microglia, neuronal cultures, or splenocytes by the RNeasy Mini Kit (QIAGEN). Reverse transcription of RNA was performed with SuperScript III reverse transcriptase (Invitrogen) and hexamer random primers (Roche Molecular Biochemicals). For semiquantification, all samples were normalized with GAPDH amplification. The following oligonucleotides were used for PCR amplification: GAPDH forward: 5′-AACTTTGGCATTGTGGAAGG-3′, reverse: 5′-ACACATTGGGGGTAGGAACA-3′; TREM2 forward: 5′-ATGGGACCTCTCCACCAGTT-3′, reverse: 5′-TCACGTACCTCCGGGTCCA-3′; and DAP12 forward: 5′-ATGGGGGCTCTGGACCCCT-3′, reverse: 5′-TCATCTGTAATATTGCCTCTGTGT.
Immunohistochemistry for TREM2
Isolated microglia and astrocytes as well as neuronal cultures containing neurons and oligodendrocytes were fixed in 4% paraformaldehyde for 1 h, blocked by 1% BSA/PBS for 2 h, and then immunostained with a purified polyclonal rabbit antibody directed against TREM2 (R&D Systems) and a secondary fluorescence Cy3-conjugated antibody directed against rabbit IgG (1:200; Dianova). To identify the cell type, cells were double labeled with monoclonal mouse antibody directed against β tubulin III (Sigma-Aldrich), GFAP (DakoCytomation), O4 (R&D Systems), and CD11b followed by a secondary fluorescence FITC-conjugated antibody directed against mouse IgG. Images were collected by confocal laser scanning microscopy with a 40× objective (Leica).
Lentivirus vector system and microglial transduction
Lentiviral vectors of the third generation, PLL3.7 (provided by L. van Parijs, MIT, Cambridge, MA) and pLenti6/V5 (Invitrogen), were used for the transduction of microglia. The mouse TREM2 gene was derived from reverse-transcribed RNA obtained from primary cultured microglia and was tagged three times at the NH2 terminus with the Flag epitope. fTREM2 and the GFP gene (BD Biosciences) were cloned into the pLenti6/V5 vector. The mouse DAP12 (provided by E. Vivier, Centre d'Immunologie, Marseille, France) and deletion mutant mouse DAP12 (amino acids 1–88) were tagged with the GFP gene and cloned into the pLenti6/V5 vector. The correct nature of all cloned sequences was confirmed by automated sequencing (Seqlab) of the vectors. wTREM2 short hairpin RNA sequence (5′-TGA TGC TGG AGA TCT CTG GGT TCA AGA GAC CCA GAG ATC TCC AGC ATC TTT TTT C-3′) and mutant-type mouse TREM2 short hairpin RNA sequence for control study (5′-TGA TGC TGA AGG TCG CTT GGT TCA AGA GAC CAA GCG ACC TCC AGC ATC TTT TTT C-3′) were inserted into the PLL3.7 vector as described by Rubinson et al. (32). For overexpression of TREM2, we replaced the blastcidin resistance gene in the pLenti6/V5 vector by the GFP gene under a second CMV promoter and subcloned the wTREM2 gene under the first CMV promoter.
Lentiviral transduction was performed as described previously (32). In brief, PLL3.7 or pLenti6/V5 vectors were purified and then cotransfected together with packaging vectors (Invitrogen) into 293FT cells (Invitrogen). Supernatant was collected after 48 h, and viral particles in the supernatant were concentrated at 1:100 by ultracentrifugation for 90 min at 25,000 rpm (SW28 rotor; Beckman Coulter) and recovered by suspension in PBS. Titers of viral particles ranged between 106 and 107 multiplicity of infection. Purified microglia were seeded at 2 × 105 cells/ml into 24-well plates. Lentiviral particles and 8 μg/ml polybrene (Sigma-Aldrich) were added to the culture and centrifuged for 90 min at 30°C. Supernatant was removed immediately after infection and replaced with BME medium containing 10% FCS and 50% glial culture supernatant. In all experiments the efficiency of microglia transduction was at least >90% as determined by the number of microglia expressing the GFP molecule and by immunohistochemistry.
Western blot
For Western blotting, 293 cells (Invitrogen) were cotransfected with the fTREM2 plus DAP12-GFP plasmids or the fTREM2 plus mtDAP12-GFP plasmids. Cells were added to culture dishes coated with the antibody directed against the Flag epitope or control antibody and shortly centrifuged. After 10 min the cells were lysed, and the total protein lysate was analyzed with antibody specific for tyrosine phosphate (4G10; Upstate Biotechnology) by NuPAGE electrophoresis system (Invitrogen) and ECL Advance Western Blotting Detection Kit (Amersham Biosciences). In a second step, the membrane was reblotted with antibody directed against GFP (BD Biosciences).
RT-PCR for analysis of inflammatory gene transcripts
Microglia were transduced with the fTREM2 vector or the GFP1 (refer to Fig. 2) control vector. Cells were then cultured on dishes and precoated with 10 μg/ml antibody directed against the Flag epitope (Sigma-Aldrich) or 10 μg/ml isotype control antibody (Sigma-Aldrich). After 24, 48, and 72 h, RNA was isolated from microglia by the RNeasy Mini Kit (QIAGEN). RNA was also collected from microglia that have been transduced with shTREM2, shControl, wTREM2, GFP2 (refer to Fig. 6), mtDAP12-GFP, and GFP1 (refer to Fig. 2) vector and cocultured with apoptotic neurons for 48 h as described below. Reverse transcription of RNA was performed as described above. Quantitative RT-PCR by SYBR Green was performed on an ABI Prism 5700 Sequence Detection System (PerkinElmer). Amplification of GAPDH was used for sample normalization. The amplification protocol followed the GeneAmp 5700 Sequence Detection System Software (version 1.3). For detection of GAPDH, TNF-α, IL-1β, NOS2, and TGF-β transcripts, the following forward and reverse primers were used at final concentrations of 200 nM: GAPDH forward: 5′-CTCCACTCACGGCAAATTCAA-3′, reverse: 5′-GATGACAAGCTTCCCATTCTCG-3′; TNF-α forward: 5′-CCGTCAGCCGATTTGCTATCT-3′, reverse: 5′-ACGGCAGAGAGGAGGTTGACTT-3′; IL-1β forward: 5′-ACAACAAAAAAGCCTCGTGCTG-3′, reverse: 5′-CCATTGAGGTGGAGAGCTTTCA-3′; NOS2 forward: 5′-GGCAAACCCAAGGTCTACGTTC-3′, reverse: 5′-TACCTCATTGGCCAGCTGCTT-3′; and TGF-β1 forward: 5′-AGGACCTGGGTTGGAAGTGG-3′, reverse: 5′-AGTTGGCATGGTAGCCCTTG-3′.
Flow cytometry analysis
Microglia were transduced with the fTREM2 vector or the GFP control vector. Cells were then cross-linked as mentioned above. After 72 h, microglia were first incubated for Fc receptor blockade by CD16/CD32 antibody (BD Biosciences) and then stained with either biotin-conjugated anti-CD11b, anti-CD11c, anti-CD86, anti–I-Ab, anti-CD36 (BD Biosciences), or anti–mouse CCR7 (ALEXIS Corporation) followed by CyChrome-conjugated streptavidin (BD Biosciences) or biotin-conjugated anti–goat IgG and Cy5-conjugated streptavidin (Dianova). Analysis was performed with a FACSCalibur flow cytometer (BD Biosciences). Live gating was performed with propidium iodide (Sigma-Aldrich).
Microsphere bead phagocytosis assay
Microglia were transduced with the fTREM2 vector or the GFP control vector, or cotransduced with the fTREM2 and mtDAP12 vector or the fTREM2 and the GFP control vector. Cells were then cross-linked as mentioned above. After 24 h, 1.00 μm of red fluorescent microsphere beads (Fluoresbrite Polychromatic Red Microspheres; Polysciences Inc.) were added for 1 h. Phagocytosis of microsphere beads by microglia was analyzed by fluorescence microscopy. Furthermore, microglia were collected from the culture plates and analyzed by flow cytometry. The percentage of microglia having phagocytosed beads was determined. Because phagocytosis varied from one experiment to the other, the relative change in phagocytosis was determined. Data are shown as the relative change in phagocytosis between microglia cultured on antibody directed against the Flag epitope and control antibody.
Reorganization of the actin cytoskeleton
Microglia were transduced with the fTREM2 vector. Cells were then cross-linked for 1 h as mentioned above. Cells were fixed, blocked, and then stained with Alexa Fluor 546–conjugated phalloidin (Molecular Probes). Images were collected by confocal laser scanning microscopy with a 40× objective (Leica).
Pharmacological inhibition
In blocking experiments, 20 μM ERK inhibitor PD98059 and 10 μM PI3K inhibitor LY294002 (both from Calbiochem) were added 60 min before stimulation of TREM2.
Chemotaxis assay
Microglia were transduced with the fTREM2 vector. Cells were then cross-linked for 24 h as mentioned above and treated with 1 μg/ml LPS as indicated in Fig. 3. Microglia were transferred into the upper chamber of a transwell system (3-μm pore filter; Millipore) containing 450 μl medium with 100 ng/ml CCL19 or CCL21 (both from PeproTech) in the lower chamber. After a 1-h incubation period, the number of microglia that had migrated to the lower chamber was counted in three independent areas by microscopy.
Western blot analysis of ERK activation
Microglia were transduced with fTREM2 vector and 2 × 105 cells were then cross-linked for 1 h as mentioned above. After stimulation, cells were lysed in reducing sample buffer for Western blot analysis. Phosphorylation of ERK and total amount of ERK were determined by immunodetection with anti–phospho-ERK and anti-ERK antibodies, respectively (both from Cell Signaling Technology).
Phagocytosis assay of apoptotic neurons
Microglia were transduced with shTREM2, shControl, wTREM2, GFP1 control (refer to Fig. 2), mtDAP12-GFP, and GFP2 control (refer to Fig. 6) vector. After transduction, microglia were cultured for 72 h to achieve effective knockdown of TREM2 by RNA interference. Neurons were cultured for 5–10 d, and okadaic acid was then added at the final concentration of 30 nM for 3 h to induce apoptosis. Neuronal cell membranes were labeled with CellTracker CM-DiI membrane dye (Molecular Probes). After incubation, apoptotic neurons were washed two times and added to the transduced microglial culture at an effector/target ratio of 1:20. At 1 and 24 h after addition of apoptotic neurons, the number of microglia having phagocytosed neuronal cell membranes were counted under a confocal fluorescence microscope (Leica). Apoptotic cells were counted in three different areas at a magnification of 60. The amount of phagocytosis was confirmed by flow cytometry. Moreover, 24, 48, or 72 h after the addition of apoptotic neurons, cells were collected and used for RT-PCR of cytokines.
Statistical analysis
Data are presented as mean ± SEM of at least three independent experiments. Data were analyzed by the Mann-Whitney U test to determine significant differences.
Online supplemental material.
Gene transcripts of TNF-α, IL-1β, TGF-β, NOS2, IFN-γ, IL-12p40, IL-10, and GAPDH were analyzed by semiquantitative and real-time RT-PCR. Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20041611/DC1.
Acknowledgments
We thank Massimiliano Stagi and Philippe Gorlovoi for their technical assistance with confocal microscopy. We are grateful to Alexandra Bohl and Heiko Röhse for excellent technical support of cultures and molecular biology. We thank Dr. Luc van Parijs for the lentiviral RNAi vector system, Dr. Eric Vivier for the DAP12 plasmid, and Gerald Ponath for cloning of DAP12.
The Neuroimmunology Group is supported by the University of Göttingen. This project was supported by the Deutsche Forschungsgemeinschaft and Hertie-Foundation. C.D.P. Rochford is a fellow of the Boehringer Ingelheim Fonds.
The authors have no conflicting financial interests.
Abbreviations used: CCL, CC chemokine ligand; CCR7, CC chemokine receptor 7; CNS, central nervous system; ERK, extracellular signal–regulated kinase; fTREM2, Flag epitope–tagged triggering receptor expressed on myeloid cells-2 vector; GFAP, glial fibrillary acidic protein; ITAM, immunoreceptor tyrosine-based activation motif; mtDAP12, dominant negative DAP12; NOS2, nitric oxide synthase-2; PI3K, phosphatidylinositol 3-kinase; PLOSL, polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy; shControl, short hairpin control; shTREM2, short hairpin triggering receptor expressed on myeloid cells-2; TREM2, triggering receptor expressed on myeloid cells-2; wTREM2, wild-type TREM2.
References
- 1.Savill, J., I. Dransfield, C. Gregory, and C. Haslett. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2:965–975. [DOI] [PubMed] [Google Scholar]
- 2.Lauber, K., S.G. Blumenthal, M. Waibel, and S. Wesselborg. 2004. Clearance of apoptotic cells: getting rid of the corpses. Mol. Cell. 14:277–287. [DOI] [PubMed] [Google Scholar]
- 3.Voll, R.E., M. Herrmann, E.A. Roth, C. Stach, J.R. Kalden, and I. Girkontaite. 1997. Immunosuppressive effects of apoptotic cells. Nature. 390:350–351. [DOI] [PubMed] [Google Scholar]
- 4.Fadok, V.A., D.L. Bratton, A. Konowal, P.W. Freed, J.Y. Westcott, and P.M. Henson. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101:890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cocco, R.E., and D.S. Ucker. 2001. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell. 12:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cvetanovic, M., and D.S. Ucker. 2004. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J. Immunol. 172:880–889. [DOI] [PubMed] [Google Scholar]
- 7.Neumann, H., and H. Wekerle. 1998. Neuronal control of the immune response in the central nervous system: linking brain immunity to neurodegeneration. J. Neuropathol. Exp. Neurol. 57:1–9. [DOI] [PubMed] [Google Scholar]
- 8.Perry, V.H., T.A. Newman, and C. Cunningham. 2003. The impact of systemic infection on the progression of neurodegenerative disease. Nat. Rev. Neurosci. 4:103–112. [DOI] [PubMed] [Google Scholar]
- 9.Smith, K.J., and H. Lassmann. 2002. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 1:232–241. [DOI] [PubMed] [Google Scholar]
- 10.Ransohoff, R.M. 2003. Snip-snip, kill-kill: truncated SDF-1 and HIV-associated neurodegeneration. Nat. Neurosci. 6:1009–1011. [DOI] [PubMed] [Google Scholar]
- 11.Kempermann, G., and H. Neumann. 2003. Neuroscience. Microglia: the enemy within? Science. 302:1689–1690. [DOI] [PubMed] [Google Scholar]
- 12.Colonna, M. 2003. TREMs in the immune system and beyond. Nat. Rev. Immunol. 3:445–453. [DOI] [PubMed] [Google Scholar]
- 13.Schmid, C.D., L.N. Sautkulis, P.E. Danielson, J. Cooper, K.W. Hasel, B.S. Hilbush, J.G. Sutcliffe, and M.J. Carson. 2002. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 83:1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouchon, A., C. Hernandez-Munain, M. Cella, and M. Colonna. 2001. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paloneva, J., M. Kestila, J. Wu, A. Salminen, T. Bohling, V. Ruotsalainen, P. Hakola, A.B. Bakker, J.H. Phillips, P. Pekkarinen, et al. 2000. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25:357–361. [DOI] [PubMed] [Google Scholar]
- 16.Paloneva, J., T. Autti, R. Raininko, J. Partanen, O. Salonen, M. Puranen, P. Hakola, and M. Haltia. 2001. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology. 56:1552–1558. [DOI] [PubMed] [Google Scholar]
- 17.Paloneva, J., T. Manninen, G. Christman, K. Hovanes, J. Mandelin, R. Adolfsson, M. Bianchin, T. Bird, R. Miranda, A. Salmaggi, et al. 2002. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paloneva, J., J. Mandelin, A. Kiialainen, T. Bohling, J. Prudlo, P. Hakola, M. Haltia, Y.T. Konttinen, and L. Peltonen. 2003. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J. Exp. Med. 198:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cella, M., C. Buonsanti, C. Strader, T. Kondo, A. Salmaggi, and M. Colonna. 2003. Impaired differentiation of osteoclasts in TREM-2–deficient individuals. J. Exp. Med. 198:645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mack, K.D., R. Wei, A. Elbagarri, N. Abbey, and M.S. McGrath. 1998. A novel method for DEAE-dextran mediated transfection of adherent primary cultured human macrophages. J. Immunol. Methods. 211:79–86. [DOI] [PubMed] [Google Scholar]
- 21.Vicente-Manzanares, M., and F. Sanchez-Madrid. 2004. Role of the cytoskeleton during leukocyte responses. Nat. Rev. Immunol. 4:110–122. [DOI] [PubMed] [Google Scholar]
- 22.Rosenberger, C.M., and B.B. Finlay. 2003. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 4:385–396. [DOI] [PubMed] [Google Scholar]
- 23.Blander, J.M., and R. Medzhitov. 2004. Regulation of phagosome maturation by signals from Toll-like receptors. Science. 304:1014–1018. [DOI] [PubMed] [Google Scholar]
- 24.West, M.A., R.P. Wallin, S.P. Matthews, H.G. Svensson, R. Zaru, H.G. Ljunggren, A.R. Prescott, and C. Watts. 2004. Enhanced dendritic cell antigen capture via Toll-like receptor-induced actin remodeling. Science. 305:1153–1157. [DOI] [PubMed] [Google Scholar]
- 25.Kaifu, T., J. Nakahara, M. Inui, K. Mishima, T. Momiyama, M. Kaji, A. Sugahara, H. Koito, A. Ujike-Asai, A. Nakamura, et al. 2003. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J. Clin. Invest. 111:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondo, T., K. Takahashi, N. Kohara, Y. Takahashi, S. Hayashi, H. Takahashi, H. Matsuo, M. Yamazaki, K. Inoue, K. Miyamoto, and T. Yamamura. 2002. Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology. 59:1105–1107. [DOI] [PubMed] [Google Scholar]
- 27.Lucas, M., L. Daniel, E. Tomasello, S. Guia, N. Horschowski, N. Aoki, D. Figarella-Branger, S. Gomez, and E. Vivier. 2002. Massive inflammatory syndrome and lymphocytic immunodeficiency in KARAP/DAP12-transgenic mice. Eur. J. Immunol. 32:2653–2663. [DOI] [PubMed] [Google Scholar]
- 28.Tomasello, E., P.O. Desmoulins, K. Chemin, S. Guia, H. Cremer, J. Ortaldo, P. Love, D. Kaiserlian, and E. Vivier. 2000. Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity. 13:355–364. [DOI] [PubMed] [Google Scholar]
- 29.Bakker, A.B., R.M. Hoek, A. Cerwenka, B. Blom, L. Lucian, T. McNeil, R. Murray, L.H. Phillips, J.D. Sedgwick, and L.L. Lanier. 2000. DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity. 13:345–353. [DOI] [PubMed] [Google Scholar]
- 30.Iliev, A.I., A.K. Stringaris, R. Nau, and H. Neumann. 2004. Neuronal injury mediated via stimulation of microglial Toll-like receptor-9 (TLR9). FASEB J. 18:412–414. [DOI] [PubMed] [Google Scholar]
- 31.Neumann, H., R. Schweigreiter, T. Yamashita, K. Rosenkranz, H. Wekerle, and Y.A. Barde. 2002. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J. Neurosci. 22:854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rubinson, D.A., C.P. Dillon, A.V. Kwiatkowski, C. Sievers, L. Yang, J. Kopinja, D.L. Rooney, M.M. Ihrig, M.T. McManus, F.B. Gertler, et al. 2003. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 33:401–406. [DOI] [PubMed] [Google Scholar]