

Abstract
Apoptosis-related genes play important roles in thymocyte maturation. We show that cellular FLICE-like inhibitory protein (c-FLIP), a procaspase-8–like apoptotic regulator, plays an essential role in the efficient development of mature T lymphocytes. Mice conditionally lacking c-FLIP in T lymphocytes display severe defects in the development of mature T cells, as indicated by a dramatically reduced number of CD4+ and CD8+ T cells in the spleen and lymph nodes of mutant mice. The impaired T lymphocyte maturation in c-FLIP conditional knockout mice occurs at the single-positive thymocyte stage and may be caused by enhanced apoptosis in vivo. Moreover, although c-FLIP has been implicated in T cell receptor signaling through nuclear factor (NF)-κB and Erk pathways, activation of NF-κB and Erk in c-FLIP–deficient thymocytes appears largely intact. Collectively, our data suggest that the primary role of c-FLIP in thymocyte maturation is to protect cells from apoptosis.
T lymphocyte development is tightly regulated by a plethora of signaling molecules and can be divided into distinct stages (for reviews see references 1–3). CD4−CD8− double-negative (DN) thymocytes represent the first step of thymocyte maturation. Upon TCRβ rearrangement and β-selection, DN cells proliferate and become CD4+CD8+ double positive (DP). DP thymocytes account for a majority of cells in the thymus. These cells rearrange TCRα genes and undergo a strict positive and negative selection process. DP thymocytes expressing an intermediate affinity for self-peptide/MHC are selected for further maturation and become either CD4+ or CD8+ single-positive (SP) thymocytes. SP thymocytes may undergo further functional maturation either within the thymus or after exit to the periphery.
The TNF family member Fas and its ligand FasL may regulate thymocyte maturation. Fas is expressed by DP, as well as CD4+ and CD8+ SP, but not DN thymocytes (4–7). FasL is detected in thymic stromal cells and in a fraction of thymocytes (8, 9). Several studies have implicated Fas and FasL in thymocyte maturation. For example, Fas has been shown to have a role in negatively selecting certain subsets of thymocytes (10, 11). FasL plays an important role in the selection of thymocyte subsets expressing moderate TCR affinity by signaling through itself (9). Although both Fas and FasL are expressed throughout the thymus, and DP thymocytes are sensitive to FasL-induced apoptosis, Fas-mediated thymocyte apoptosis does not appear to prevail in the thymus (12). This limited role of Fas in thymocyte apoptosis must be regulated by active mechanisms that prevent Fas signaling. One way to achieve this is by inhibiting the Fas-induced death signaling pathway through an apoptosis inhibitor named cellular FLICE-like inhibitory protein (c-FLIP).
c-FLIP (also called Casper, I-FLICE, CASH, FLAME-1, MRIT, CLARP, and usurpin) is a procaspase-8–like protein (13–20). Two isoforms of c-FLIP (c-FLIPL and c-FLIPS) derived from alternative splicing have been identified. c-FLIPL contains two tandem death effector domains at the amino terminus and a catalytically inactive caspase-like domain at the carboxy terminus. cFLIPS has only two death effector domains. Both isoforms of c-FLIP interact with an adaptor protein, Fas-associated death domain, and are recruited to the death-inducing signaling complex. The major function of c-FLIP in regulating apoptosis is to inhibit Fas-mediated caspase-8 activation (21). However, when expressed at a low concentration, c-FLIPL can act as an activator of procaspase-8 (22).
Additional studies also suggest that c-FLIP may be involved in T lymphocyte activation and proliferation (23), and transgenic expression of c-FLIPL in T cells results in increased CD3-induced proliferation (24). Furthermore, overexpression of c-FLIP in Jurkat T cells promotes activation of NF-κB and Erk signaling pathways (25). Nevertheless, the role of c-FLIP in T lymphocyte development and function has not been investigated in T cells lacking c-FLIP expression because c-FLIP–deficient mice die during embryogenesis (26).
To examine whether c-FLIP regulates T lymphocyte maturation, we generated c-FLIP conditional KO mice. T lineage–specific deletion of c-FLIP impairs thymocyte development at the CD4+ and CD8+ SP stage and results in severely reduced numbers of mature CD4+ and CD8+ T lymphocytes in the spleen and LN. c-FLIP−/− thymocytes exhibit enhanced sensitivity to TCR/CD3 and Fas-induced killing. Furthermore, freshly isolated CD4+ and CD8+ thymocytes from c-FLIP−/− mice display a higher rate of apoptosis than wild-type cells. Interestingly, activation of NF-κB and Erk pathways is largely intact in c-FLIP–deficient T lymphocytes. Collectively, our results demonstrate an essential role for c-FLIP in the development of mature T lymphocytes.
Results
Generation of c-FLIP conditional KO mice
To generate mice specifically lacking c-FLIP in T lymphocytes, we constructed a targeting vector with exon 1 encoding amino acids 1–98 of c-FLIP, flanked with two loxP sites (Fig. 1 A). The flanked exon is used by both c-FLIPL and c-FLIPS. A neomycin-resistant gene cassette located within the two loxP sites was flanked by two FRT sites (Fig. 1 A). We generated chimeric mice by microinjecting three correctly targeted embryonic stem (ES) cell clones (Fig. 1 B) into C57BL/6 blastocysts. Male chimeric mice were bred with FLPeR female mice to delete the neomycin cassette in vivo. The c-FLIP floxed mice (c-FLIPf/f) were then bred with Lck-cre transgenic mice to induce specific deletion of this gene in T lymphocytes. Restriction analysis of genomic DNA from total thymocytes of c-FLIPf/fLck-cre (c-FLIP−/−) and c-FLIPf/+Lck-cre (c-FLIP+/−) mice revealed that floxed c-FLIP exon 1 was deleted in >98% of the thymocytes (Fig. 1 C). c-FLIP protein expression in the thymus of these mice was also reduced >98% (Fig. 1 D). These results indicate that Lck-cre expression induced efficient deletion of c-FLIP in developing thymocytes.
Figure 1.

Generation of c-FLIP conditional KO mice. (A) Diagram of the c-FLIP targeting construct and targeted alleles. E1–E4, exon 1–4 of the c-FLIP gene; SI, SacI; EI, EcoRI; L, loxP site; F, FRT site; DT, diptheria toxin; Neo, neomycin-resistant gene. The probe used in DNA blot analysis is indicated. (B) Southern blot analysis of genomic DNA from targeted ES cells. Genomic DNA from targeted TC1 ES cells were digested with SacI and probed as indicated in A. (C) Southern blot analysis of genomic DNA from c-FLIP conditional KO mice. Total thymocyte DNA from c-FLIPf/fLck-cre and other control mice were digested with EcoRI and probed as in B. (D) Western blot analysis of c-FLIP protein expression in total thymocytes from c-FLIP−/− and c-FLIP+/− mice. Loading control is a nonspecific band on the same blot.
Thymocyte development in c-FLIP conditional KO mice
c-FLIP−/− mice had normal growth and development. We examined thymocyte development in 3–5-wk-old c-FLIP−/− and age-matched wild-type (c-FLIP+/+) mice. Total thymocyte numbers in c-FLIP−/− mice were not significantly different from those in c-FLIP+/+ mice (KO, 263 ± 99 × 106, n = 19; control, 312 ± 94 × 106, n = 21; P = 0.126). Thymocyte development was further analyzed by the expression of CD4 and CD8. c-FLIP−/− thymocytes developed successfully from the DN to the DP stage (Fig. 2 A). However, the percentages of CD4+ and CD8+ SP thymocytes in c-FLIP−/− mice were reduced by 50–70% when compared with wild-type control cells (Fig. 2 A). Furthermore, these cells did not completely down-regulate their coreceptors. The mean fluorescence intensity (MFI) of CD8 expression on c-FLIP−/− CD4+ SP was higher than that of control CD4+ SP (10.2 vs. 2.9), whereas the MFI of CD4 expression on c-FLIP−/− CD8+ SP thymocytes was also higher than that of control CD8+ SP (50.1 vs. 31.3; Fig. 2 A), indicating that these CD4+CD8low and CD8+CD4low thymocytes are on their way to complete maturation (27). Correlating with the incomplete down-regulation of coreceptors in CD4+ and CD8+ SP thymocytes in c-FLIP−/− mice, the expression of heat-stable antigen (CD24) on CD4+ and CD8+ SP cells was not obviously down-regulated when compared with control cells (Fig. 2 C and not depicted).
Figure 2.
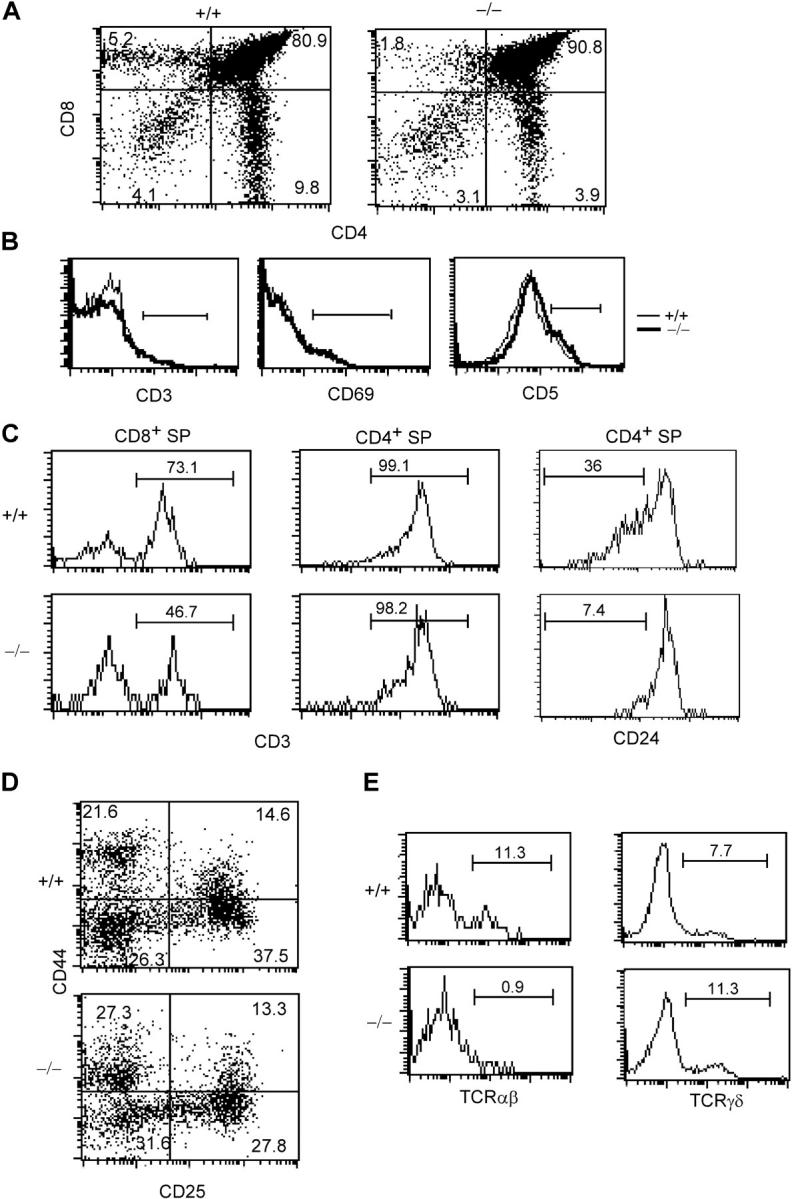
Thymocyte maturation in c-FLIP conditional KO mice. (A) FACS analysis of thymocytes from 3-wk-old c-FLIP−/− and wild-type mice (+/+). The percentages of CD8 SP (top left), DP (top right), DN (bottom left), and CD4 SP (bottom right) cells are shown. (B) Histograms of CD3, CD69, and CD5 expression on CD4+CD8+ DP thymocytes from c-FLIP−/− and control mice. (C) CD3 and CD24 expression on CD8+ or CD4+ SP thymocytes of c-FLIP−/− and control mice. (D) Development of pro–T cells in c-FLIP−/− mice. CD3−CD4−CD8− triple-negative thymocytes were analyzed for the expression of CD44 (top) and CD25 (right). (E) Expression of TCRαβ and TCRγδ in the CD4−CD8− DN compartment of c-FLIP−/− mice. DN thymocytes were analyzed for the expression of TCRαβ and TCRγδ. The percentages of total DN thymocytes are shown. The horizontal lines in B, C, and E indicate the flow cytometry analysis gates and mark the respective cell populations.
To determine whether the reduced number of CD4+ and CD8+ SP thymocytes in c-FLIP−/− mice was caused by an impaired positive selection of DP thymocytes, we examined the expression of CD3, CD69, and CD5 on c-FLIP−/− DP thymocytes. Up-regulation of these surface molecules on DP thymocytes occurs after successful positive selection. As shown in Fig. 2 B, the expression of CD3, CD69, and CD5 was indistinguishable between c-FLIP−/− and c-FLIP+/+ DP cells, suggesting that thymocyte positive selection is not impaired in the absence of c-FLIP. We then determined the expression of TCR on c-FLIP−/− CD4+ and CD8+ SP thymocytes. Both c-FLIP−/− CD4+ and CD8+ SP thymocytes expressed similar levels of surface TCR to those on control cells (Fig. 2 C). However, the percentage of c-FLIP−/− CD8+CD3+ SP thymocytes was lower than that in the control cells (46.7% vs. 73.1%), indicating fewer mature CD8+ SP and a relatively more immature SP thymocytes among CD8+ thymocytes (Fig. 2 C).
We next examined the DN compartment in the c-FLIP−/− thymus. CD4−CD8−CD3− triple negative thymocytes were gated, and the expression of CD25 and CD44 was analyzed. Pro–T cell development as defined by the expression of CD25 and CD44 was comparable in both c-FLIP−/− and c-FLIP+/+ mice (Fig. 2 D). However, when the expression of TCRαβ and TCRγδ was assessed in the CD4−CD8− DN compartment, we found that the percentage of TCRαβ+ DN thymocytes in c-FLIP−/− mice was reduced >90%, whereas the percentage of TCRγδ+ DN thymocytes was slightly increased (Fig. 2 E). Collectively, these results demonstrate that the development of CD4+ and CD8+ SP thymocytes and TCRαβ DN cells was impaired in c-FLIP−/− mice.
Lack of mature T lymphocytes in c-FLIP−/− mice
We first examined the peripheral T cell compartment in 3-wk-old c-FLIP−/− mice. Strikingly, although control mice had a filled peripheral T cell compartment, c-FLIP−/− mice essentially lacked peripheral CD4+ and CD8+ T lymphocytes (Fig. 3 A). The numbers of CD4+ and CD8+ T cells in the spleen and LN of c-FLIP−/− mice were in the range of 0.1–2% of those in control mice (Fig. 3 B). Because the lack of mature T cells in c-FLIP−/− mice may be caused by a delayed filling of the peripheral lymphoid compartment, we examined the peripheral T cell compartment in 5–10-wk-old mice. The percentages of CD4+ and CD8+ peripheral T lymphocytes in 5–10-wk-old c-FLIP−/− mice were higher than those in 3-wk-old mutant mice but still dramatically lower than those in age-matched controls (Fig. 3 A). Interestingly, the few CD4+ and CD8+ T cells in c-FLIP−/− mice expressed higher levels of CD44 and lower levels of CD62L (Fig. 3 C). In addition, when compared with controls, a higher fraction of CD4+ and CD8+ mature T cells in c-FLIP−/− mice expressed the T cell activation markers CD25 and CD69 (Fig. 3 C). Interestingly, TCRβ expression on both c-FLIP−/− CD4+ and CD8+ peripheral T cells was lower than that on control cells (Fig. 3 C). Notably, a fraction of c-FLIP−/− CD8+ T cells were also TCRβ− (Fig. 3 C). This CD8+TCRβ− population expressed CD11c and accounted for 0.42% of the total splenocytes, compared with a similar population in control spleen that accounted for 0.36% of the total splenocytes. Therefore, these cells are splenic dendritic cells that are enriched because of the markedly lower number of CD8+ T cells in c-FLIP−/− mice. Collectively, these phenotypic characteristics suggest that a fraction of mature T cells in c-FLIP−/− mice were undergoing active proliferation. However, it remains to be determined whether all these cells that represent mature T cells escaped Lck-Cre–mediated deletion or cells differentiated in the c-FLIP–independent pathway.
Figure 3.


Lack of mature T cells in c-FLIP conditional KO mice. (A) FACS analyses of CD4 and CD8 expression in the spleen and LN of 3- or 5-wk-old c-FLIP−/− and control mice. The percentages of CD4+ or CD8+ mature T cells in these lymphoid organs are shown. (B) Total cell numbers of CD4+ and CD8+ T cells in c-FLIP−/− and age- and sex- matched control mice that were 3–5-wk old. (C) FACS analyses of surface molecule expression on CD4+ and CD8+ T cells from c-FLIP−/− and control mice (8-wk old). Numbers for CD25, CD69, CD62L, and CD44 staining represent the percentages of gated cells. Numbers for TCRβ staining are MFI of all cells in each file. (D) PCR analysis of floxed c-FLIP allele in CD4+ T cells from the spleen of c-FLIP−/− mice as described in Materials and methods. Thymocytes from c-FLIPf/+, c-FLIP−/−, and wild-type mice serve as controls. Bcl-2 functions as a PCR template loading control.
The age-related accumulation of CD4+ and CD8+ T lymphocytes in the spleen and LN of c-FLIP−/− mice raised the possibilities that either these cells underwent maturation independent of c-FLIP or these cells derived from a few cells that escaped cre-mediated deletion in the thymus. To distinguish these possibilities, we purified CD4+ and CD8+ T lymphocytes from the spleen and LN of c-FLIP−/− mice by double-FACS sorting and tested the deletion of c-FLIP alleles in these cells. As shown in Fig. 3 D, the floxed c-FLIP allele was readily detected in the sorted CD4+ peripheral T cells from c-FLIP−/− mice, indicating that c-FLIP allele deletion was incomplete in at least some of these cells. In contrast, we did not detect the floxed c-FLIP allele from total thymocytes of c-FLIP−/− mice under this PCR condition, suggesting a high deletion efficiency of the c-FLIP allele in the thymus. Collectively, our results demonstrate that c-FLIP is essential for the efficient development of mature T lymphocytes.
Enhanced apoptosis to TCR/CD3 and Fas stimulation in thymocytes from c-FLIP−/− mice
The lack of mature T lymphocytes in c-FLIP−/− mice may result from impaired survival and/or expansion of these cells. Given the extensive evidence showing that c-FLIP modulates TCR signaling, we examined thymocyte proliferation after TCR-mediated stimulation. Total thymocytes from c-FLIP−/− and control mice were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and stimulated with plate-bound CD3 for 4 d. The proliferation of CD4+ and CD8+ SP thymocytes was determined by a FACS analysis of CFSE dilution after gating on 7-amino-actinomycin D (7-AAD)−Annexin V− live cells. As expected, wild-type CD4+ and CD8+ SP but not DP thymocytes proliferated extensively 4 d after stimulation (Fig. 4 A). In contrast, we did not observe any noticeable number of CD4+ or CD8+ SP thymocytes from c-FLIP−/− mice with diluted CFSE labeling (Fig. 4 A). The lack of CFSElow cells in the c-FLIP−/− thymocyte culture is further supported by a lack of IL-2 production in the culture stimulated either by anti-CD3 or anti-CD3 plus anti-CD28 (Fig. 4 B). An identical result was seen when c-FLIP−/− SP thymocytes were stimulated by PMA plus ionomycin, which activates T cells by circumventing surface TCR (unpublished data).
Figure 4.

Anti-CD3–induced proliferation and activation of c-FLIP−/− thymocytes. (A) Proliferation of thymocytes from c-FLIP−/− and control mice. CFSE-labeled thymocytes were stimulated with plate-bound anti-CD3 and anti-CD28 for 4 d, and the proliferation was analyzed by four-color FACS. The percentages of proliferative cells are shown. (B) IL-2 production by thymocytes from c-FLIP−/− and control mice. Purified CD4+ SP thymocytes were stimulated with plate-bound antibodies as indicated and measured for IL-2 production with ELISA. (C) Expression of CD25 on CD4+ SP thymocytes after anti-CD3 and anti-CD28 stimulation. Thymocytes were stimulated with plate-bound anti-CD3/CD28 for 2 d, and CD4+ SP thymocytes were analyzed for the expression of CD25.
The lack of proliferating c-FLIP−/− SP thymocytes may reflect a defect in TCR-mediated activation in these cells. We then examined CD25 up-regulation on SP thymocytes after anti-CD3 plus anti-CD28 stimulation. Unlike peripheral T cells, up-regulation of CD25 in wild-type SP thymocytes was at a minimal level after a short period (≥10 h) of stimulation and at a modest level after 24 h of stimulation (unpublished data). We therefore stimulated SP thymocytes for 48 h and analyzed CD25 expression on 7-AAD−Annexin V− live cells. As shown in Fig. 4 C, 48 h of stimulation by anti-CD3/CD28 resulted in the expression of CD25 on 58% of wild-type CD4+ SP thymocytes. However, no increase of CD25 expression in c-FLIP−/− CD4+ SP thymocytes was observed. A similar defect was observed for CD25+ expression on c-FLIP−/− CD8+ SP thymocytes and for CD69 up-regulation on c-FLIP−/− SP cells (unpublished data).
We considered two possibilities for the absence of CD25+ and CD69+ c-FLIP−/− CD4+ SP thymocytes after anti-CD3/CD28 stimulation. First, c-FLIP might be essential for TCR-mediated activation, and impaired TCR signaling results in a complete lack of activation and proliferation in c-FLIP−/− SP thymocytes. Second, TCR signaling may not be impaired in c-FLIP−/− thymocytes. Instead, activated SP thymocytes undergo rapid cell death caused by a lack of protection by c-FLIP, whereas unactivated thymocytes survive. The observation that a fraction of wild-type CD4+ (59%) and CD8+ (28.8%) SP thymocytes remained CFSEhigh after anti-CD3 stimulation (Fig. 4 A) suggests that these cells did not receive sufficient activating signals from plate-bound anti-CD3. This further suggests that the CFSEhigh c-FLIP−/− CD4+ and CD8+ SP thymocytes (Fig. 4 A) represent cells that did not receive sufficient activating signals and, therefore, survived. To directly examine the effect of TCR/CD3 signaling on c-FLIP−/− thymocytes, we stimulated FACS-sorted CD4+ SP thymocytes with plate-bound anti-CD3 and examined the apoptosis of these cells at different time points. Anti-CD3 stimulation for as short as 2 h resulted in apoptosis of ∼40% of c-FLIP−/− CD4+ thymocytes, whereas only 12% of control cells were apoptotic (Fig. 5 A). Furthermore, >80% of c-FLIP−/− CD4+ thymocytes underwent apoptosis by 18 h of stimulation, whereas the apoptosis rate for control cells remained relatively constant (Fig. 5 A). These results demonstrate that anti-CD3 stimulation of c-FLIP−/− SP thymocytes induced rapid cell death instead of proliferation as in the case for wild-type CD4+ T cells.
Figure 5.

Enhanced apoptosis to TCR/CD3 and Fas stimulation by c-FLIP−/− thymocytes. (A) Percentages of surviving cells after anti-CD3 stimulation. FACS-sorted CD4+ SP thymocytes from c-FLIP−/− and control mice were stimulated with 5 μg/ml of plate-bound anti-CD3 for the indicated time and analyzed for apoptosis by 7-AAD and Annexin V staining. The percentages of 7-AAD−Annexin V− cells are shown. (B) Percentages of apoptotic cells after anti-Fas stimulation for 5 h. Total thymocytes from c-FLIP−/− and control mice were stimulated with 5 μg/ml of plate-bound anti-Fas for 5 h, and the apoptosis was analyzed by four-color staining of CD4, CD8, 7-AAD, and Annexin V. The percentages of Annexin V+ cells among each thymocyte subset are shown. Values are mean ± SD.
Given the role of c-FLIP in inhibiting caspase-8 activation, the increased apoptosis in c-FLIP−/− SP thymocytes to TCR/CD3 stimulation may be caused by their enhanced sensitivity to Fas-mediated killing. To test this, we incubated total thymocytes from c-FLIP−/− and wild-type mice with plate-bound anti-Fas for 5 h and examined apoptotic cells using Annexin V staining. Strikingly, 5 h of anti-Fas treatment induced a majority (85–95%) of c-FLIP−/− CD4+ and CD8+ SP to undergo apoptosis as compared with only a small fraction (25–30%) of control cells (Fig. 5 B). Interestingly, c-FLIP−/− DP thymocytes also exhibited similarly high sensitivity to Fas-induced killing (Fig. 5 B). It was reported that anti-Fas antibody induces selective killing of DP thymocytes after a 16-h stimulation (4). We observed only ∼45% of wild-type DP cells positive for Annexin V after 5 h of treatment (Fig. 5 B). However, treatment of thymocytes with anti-Fas antibody for 16 h resulted in ∼99% cell death in c-FLIP−/− DP and CD4+ and CD8+ SP thymocytes, and ∼99% cell death in wild-type DP thymocytes, but only ∼30% cell death in wild-type CD4+ and CD8+ SP thymocytes (unpublished data). These results are in agreement with previous data (4) and suggest that c-FLIP regulates the sensitivity of DP and CD4+ and CD8+ SP thymocytes to TCR/CD3 and Fas-induced apoptosis.
Enhanced apoptosis in freshly isolated SP thymocytes from c-FLIP−/− mice
The fact that the DP compartment in c-FLIP−/− mice is normal even though these cells are just as sensitive as CD4+ and CD8+ SP c-FLIP−/− thymocytes to Fas-induced death in vitro (Fig. 5 B) suggests that DP and SP thymocytes may have received different levels of Fas signaling in vivo. If this is the case, c-FLIP−/− SP thymocytes may exhibit a higher rate of apoptosis than controls without stimulation. We examined apoptosis in freshly isolated thymocytes from c-FLIP−/− and wild-type mice by Annexin V staining. We detected small fractions of apoptotic DP and SP thymocytes from wild-type mice (Fig. 6 A). Whereas we observed three- to fourfold increases of apoptotic cells in c-FLIP−/− CD4+ and CD8+ SP thymocytes over the control cells, there was no such increase for c-FLIP−/− DP cells (Fig. 6 A). Interestingly, when c-FLIP−/− thymocytes were cultured in vitro for 5 h without any stimulation, a dramatic increase in the apoptosis of DP, as well as CD4+ and CD8+ SP, thymocytes was observed (Fig. 6 B). To determine whether the enhanced apoptosis is caused by altered Fas expression, we examined Fas expression on these cells. Fas expression on c-FLIP−/− DN, DP, and CD4+ SP thymocytes was comparable to that on control cells (Fig. 6 C). The lower level of Fas expression on c-FLIP−/− CD8+ SP thymocytes may reflect an increased proportion of immature SP thymocytes in this subset (Fig. 6 C). Collectively, these results demonstrate elevated apoptosis in freshly isolated c-FLIP−/− SP thymocytes.
Figure 6.

Enhanced apoptosis of freshly isolated c-FLIP−/− thymocytes. (A) Percentages of apoptotic cells in freshly isolated c-FLIP−/− and control thymocytes. Freshly isolated thymocytes were stained with anti-CD4, CD8, and Annexin V. Annexin V+ cells in each subset are shown. Data represent five mice in each group and are representative of three independent experiments. (B) Percentages of apoptotic cells in c-FLIP−/− and control thymocytes cultured for 5 h (n = 3). Values shown are mean ± SD. (C) Fas expression on c-FLIP−/− and control thymocytes. Numbers are MFI. (D) Semiquantitative RT-PCR analysis of c-FLIPL in developing T lymphocytes. RNA from FACS-sorted T lymphocyte subsets at different developing stages were analyzed for the expression of c-FLIPL. Samples are in 1:5 serial dilution. Hypoxanthine phosphoribosyltransferase (HPRT) serves as a template quantity control.
To better explain the defective thymocyte development in c-FLIP−/− mice, we examined c-FLIP expression in developing T lymphocytes. Total RNA from FACS-sorted DP and CD4+ and CD8+ SP thymocytes, as well as CD4+ and CD8+ mature T cells of normal C57BL/6 mice, were subjected to semiquantitative RT-PCR analyses. We used total thymocytes from Rag-2–deficient mice as DN cells. As shown in Fig. 6 D, c-FLIP was expressed at a low level in DN cells and up-regulated in DP cells. c-FLIP expression was detected at similar levels in CD4+ and CD8+ SP thymocytes and peripheral mature T cells (Fig. 6 D). These data indicate that c-FLIP expression is developmentally regulated. Furthermore, the expression pattern of c-FLIP in developing T lymphocytes is highly correlated with that of Fas expression (Fig. 6 C; reference 5).
Erk and NF-κB signaling in c-FLIP−/− SP thymocytes
Previous data showed that c-FLIP interacts with TRAF1, TRAF2, RIP, and Raf-1 and promotes the activation of NF-κB and Erk signaling pathways (25). To determine whether c-FLIP is required for activation of these pathways, we first tested Erk phosphorylation in purified c-FLIP−/− CD4+ SP thymocytes stimulated with anti-CD3. The phosphorylation of Erk after anti-CD3 stimulation was comparable in c-FLIP−/− and control CD4+ SP thymocytes (Fig. 7 A). We also observed a similar pattern of Erk phosphorylation in c-FLIP−/− thymocytes stimulated with PMA plus ionomycin (Fig. 7 B). To examine NK-κB signaling, we tested IκBα degradation in CD4+ SP thymocytes from c-FLIP−/− and control mice. IκBα degradation in c-FLIP−/− SP thymocytes was comparable to that in control cells after stimulation by either anti-CD3 or PMA plus ionomycin (Fig. 7 C). We also measured Ca2+ flux in c-FLIP−/− CD4+ SP thymocytes after anti-CD3 and CD4 cross-linking. The overall Ca2+ flux of c-FLIP−/− CD4+ SP thymocytes was not obviously changed except for a slight delay in the peak response (Fig. 7 D). Collectively, these results demonstrate that the Erk and NF-κB signaling pathways in c-FLIP−/− SP thymocytes are largely intact and suggest that c-FLIP is not essential for the activation of these pathways on TCR/CD3 stimulation.
Figure 7.
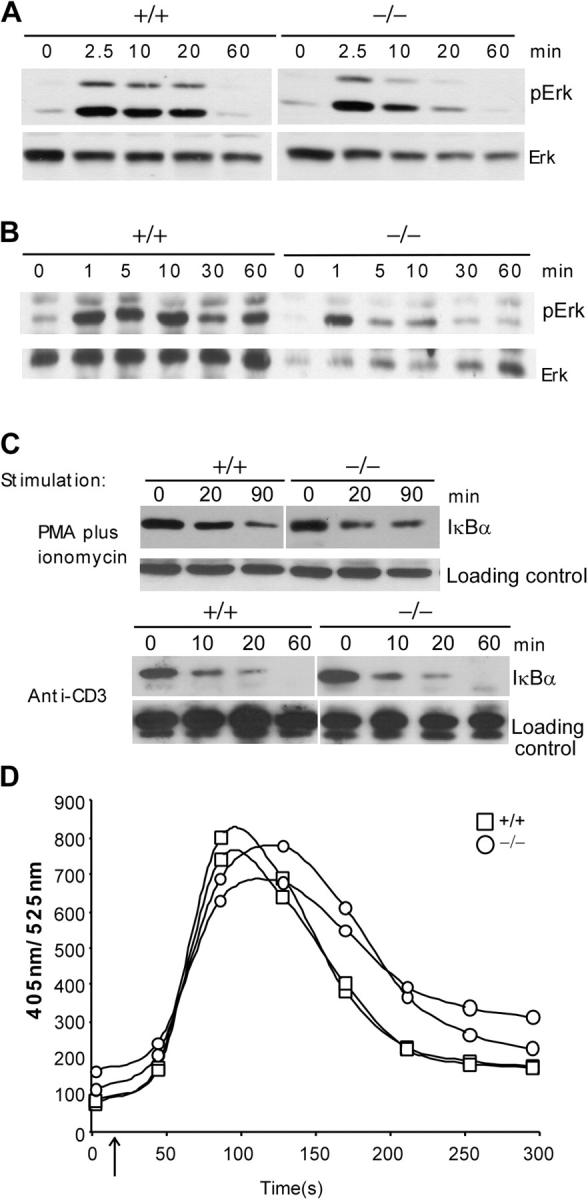
TCR-induced signaling in c-FLIP−/− thymocytes. (A) Erk phosphorylation of CD4+ SP thymocytes from c-FLIP−/− and control mice after anti-CD3 stimulation. FACS-purified CD4+ SP thymocytes were stimulated with anti-CD3 and lysed for anti-pERK or -ERK blot analyses. (B) Erk phosphorylation of CD4+ SP thymocytes from c-FLIP−/− and control mice after stimulation with 10 ng/ml PMA plus 1 μg/ml ionomycin. (C) IκBα degradation in c-FLIP−/− CD4+ SP thymocytes. Thymocytes were stimulated as in A and B. Loading controls are nonspecific bands that appeared on the same blot. (D) Ca2+ flux in CD4+ SP thymocytes from c-FLIP−/− and control mice.
Discussion
By analyzing T lymphocyte development in c-FLIP conditional KO mice, we have demonstrated that c-FLIP is essential for the efficient development of mature T lymphocytes. The almost complete lack of mature T lymphocytes in the LN and spleen of c-FLIP-deficient mice is because of impairment at the SP stage in the thymus. Although CD4+ and CD8+ mature T cells gradually accumulate in the spleen and LN of 5-wk-old and older c-FLIP mutant mice, at least a fraction of these cells are likely derived from the homeostatic expansion of a few cells that escaped Lck-cre–mediated deletion of c-FLIP in the thymus. These cells contain the floxed c-FLIP allele. These results suggest a critical requirement for c-FLIP in the maturation of conventional TCRαβ+ T lymphocytes. In addition, the reduced number of TCRαβ DN T cells in the thymus of c-FLIP−/− mice suggests that the development of this T cell lineage also depends on c-FLIP. In contrast, TCRγδ T cells differentiate normally in c-FLIP−/− mice. These results may be because of a differential requirement for c-FLIP in the development of these two lineages. Alternatively, the floxed c-FLIP alleles may be differentially deleted in these two subsets of T cells. Furthermore, our results do not rule out that c-FLIP may also be required for efficient development of DN thymocytes. The Lck promoter–driven, Cre-mediated deletion of floxed genes starts at the DN3 stage or later (28). Residual c-FLIP in developing thymocytes may be sufficient for DN cells to differentiate into the DP stage. In fact, a severe defective DN3 to DN4 transition was observed in c-FLIP−/−/Rag2−/− chimeric mice (see Chau et al. [29] on p. 405 of this issue), suggesting that c-FLIP plays critical roles at multiple stages of thymocyte development.
DP thymocytes in c-FLIP–deficient mice appear to be normal by several criteria. First, DP thymocyte numbers in c-FLIP−/− mice are comparable to those in control mice. Second, DP thymocytes in the mutant mice up-regulate several surface markers, such as CD69 and CD5, at similar levels to those of control thymocytes, suggesting normal positive selection in these cells. Third, similar expression levels of TCR/CD3 on CD4+ and CD8+ SP thymocytes are found in c-FLIP−/− and control mice, further suggesting normal TCRαβ rearrangement and selection. In contrast, the number of CD4+ and CD8+ SP thymocytes in c-FLIP−/− mice is 50–70% lower than that of controls. Moreover, these cells have not down-regulated one coreceptor, as well as CD24, indicating that these cells have not completed the last steps of maturation at the SP stage.
Our results suggest that the primary role of c-FLIP in thymocyte maturation is to protect SP thymocytes from apoptosis. It is well established that Fas expression is up-regulated at the DP stage and remains high at the SP stage (4–7). FasL has also been detected on thymic stromal cells and a fraction of thymocytes (8, 9). The expression pattern of Fas and FasL in the thymus raises the question of why Fas does not appear to induce large-scale thymocyte death in the normal thymus. Given the role of c-FLIP in inhibiting Fas-induced caspase-8 activation in other types of cells (21), c-FLIP is an attractive candidate to regulate thymocyte sensitivity to Fas–FasL interaction in vivo. Several lines of evidence support such a role for c-FLIP in thymocyte maturation. First, c-FLIP is up-regulated in DP thymocytes and expressed in CD4+ and CD8+ SP thymocytes, an almost identical expression pattern as that of Fas (Fig. 6, C and D; reference 5). Second, DP and SP thymocytes from c-FLIP−/− mice exhibit a dramatically enhanced sensitivity to Fas-induced killing. Third, instead of being activated by TCR/CD3 stimulation, CD4+ SP thymocytes from c-FLIP−/− mice undergo rapid apoptosis on stimulation. This result also supports a long-held notion that primary T cells are resistant to activation-induced cell death caused by protection conferred by c-FLIP (21). Fourth, freshly isolated CD4+ and CD8+ SP thymocytes have higher apoptotic rates. It is important to note that c-FLIP−/− thymocytes displayed higher apoptosis rates in culture without stimulation. The increased death might be caused by the loss of its inhibiting effect of c-FLIP on caspase activation induced by in vitro culture. Alternatively, c-FLIP−/− thymocytes might have already received death signals in vivo. In either case, the increased “spontaneous” apoptosis in c-FLIP−/− thymocytes seems only to contribute partially to the increased TCR/CD3 and Fas-induced apoptosis based on two pieces of our data. First, ∼70–95% of c-FLIP−/− SP thymocytes undergo apoptosis after TCR/CD3 and Fas stimulation for 5 h, compared with ∼50% spontaneous apoptosis (Figs. 5 and 6), suggesting a further increase in cell death on these stimulations. Second, no live cells were detected among CFSElow CD4+ and CD8+ SP thymocytes from c-FLIP−/− mice after TCR/CD3 stimulation. This suggests that activated cells are more prone to apoptosis than resting thymocytes (Fig. 4).
Why are CD4+ and CD8+ SP compartments in c-FLIP−/− mice selectively impaired even though DP thymocytes also exhibit dramatically increased sensitivity to Fas-induced killing in vitro? The answer to this may lie in the expression pattern of FasL in the thymus. FasL expression is detected in thymic epithelial cells and dendritic cells in the medulla (8), a region to which thymocytes migrate after positive selection (2). The interaction of Fas-expressing SP thymocytes with FasL-expressing thymic stromal elements in this region may result in apoptosis of CD4+ and CD8+ thymocytes if these cells are not protected by c-FLIP. Consistent with this notion, freshly isolated CD4+ and CD8+ SP thymocytes from c-FLIP−/− mice exhibit a higher degree of apoptosis than cells from control mice, suggesting that these cells receive FasL signals in the medulla in vivo.
However, FasL is also detected in a fraction of cells of each thymic subset (9). This observation raises an intriguing question regarding the role of FasL expressed by thymocytes: is thymocyte FasL able to trigger Fas by an autocrine manner? FasL expression on thymocytes is extremely low (8, 9). Low-level expression may mediate a positive signaling process through its own cytoplasmic tail in thymocyte selection, but it may not be sufficient to trigger Fas. In support of this, enhanced expression of thymocyte FasL in a transgenic model induced thymocyte apoptosis (30). Although we favor the view that the increased apoptosis of thymocytes in c-FLIP−/− mice is mediated by Fas–FasL interaction, other death receptors may also be responsible for the enhanced apoptosis. Future analysis of c-FLIP−/− mice in gld (FasL) or lpr (Fas) mutant backgrounds will address this issue. Nonetheless, our results suggest that expression of c-FLIP in SP thymocytes represents a key mechanism to ensure the completion of T cell maturation. Other mechanisms may also exist to regulate the difference in sensitivity to Fas-induced killing by normal DP and SP thymocytes.
Many studies have suggested the involvement of c-FLIP in T lymphocyte activation and proliferation (24, 25, 31–33). Transgenic expression of c-FLIPL in T cells results in increased CD3-induced proliferation (24). Furthermore, overexpression of c-FLIPL in Jurkat T cells promotes activation of NF-κB and Erk signaling pathways (25). However, several other studies reported different effects on T cell activation by overexpressed c-FLIPL. In one report, retrovirally introduced c-FLIPL did not have any effect on T lymphocyte proliferation (34). In another study, transgenic expression of c-FLIPL in T cells inhibits their CD3-induced proliferation and activation of Erk and NF-κB (31). In addition, c-FLIPL has been reported to inhibit activation of p38 mitogen-activated protein kinase and NF-κB in other cell types (35–37). These contradictory results are likely caused by the differences in the overexpression levels of c-FLIPL in each system. Our results show that the phosphorylation of Erk and activation of NF-κB in c-FLIP−/− thymocytes stimulated through TCR/CD3 are largely intact. However, we have observed certain degrees of defects, such as a delayed peak of Ca2+ flux in c-FLIP−/− thymocytes after TCR/CD3 activation. Furthermore, our data show an almost complete lack of activated and proliferating T cells, as well as reduced production of IL-2 in c-FLIP−/− thymocytes after TCR/CD3 stimulation. This may likely be caused by rapid apoptosis of c-FLIP−/− thymocytes on stimulation. However, in light of the role of c-FLIP in mature T cell proliferation (see Chau et al. [29] on p. 405 of this issue), both defective SP thymocyte proliferation and rapid apoptosis may contribute to the lack of proliferating T cells in c-FLIP−/− SP thymocytes stimulated by anti-CD3.
It is interesting to note that mice lacking caspase-8 exhibit a similar defect in the mature T lymphocyte compartment (38). Caspase-8 conditional KO mice have a dramatically reduced number of CD4+ and CD8+ T cells in the LN and spleen, whereas thymocyte development appears to be normal. Although the caspase-8 gene is located in close proximity to that of c-FLIP (∼40 kb), the defective mature T cell development in c-FLIP−/− mice is not caused by altered caspase-8 expression (unpublished data). Furthermore, the defective mature T cell compartment in caspase-8 conditional KO mice may be caused by a role of caspase-8 in T cell homeostatic proliferation.
Finally, it is important to point out that the impaired T cell maturation in c-FLIP−/− mice results from deletion of both c-FLIPL and c-FLIPS. In contrast, most of the overexpression studies have only used c-FLIPL. Although it has been shown that both c-FLIPL and c-FLIPS inhibit caspase-8 activation, these two isoforms do have different functions (21). Our c-FLIP conditional mice will provide a good model to dissect the in vivo role of these isoforms in T cell development and activation.
Materials and Methods
Generation of c-FLIP conditional KO mice.
To generate c-FLIP conditional KO mice, genomic fragments from a c-FLIP bacterial artificial chromosome clone (RPCI) were cloned into the pGKneoF2L2DTA targeting vector so that exon 1 of the c-FLIP gene was flanked by two loxP sites. The targeting construct was linealized by NotI and transfected into the TC1 ES cells (39). ES cell clones with homologous recombination were identified by PCR and Southern blot. Three ES cell clones with the correct targeting events were injected into C57BL/6 blastocysts. Chimeric male founder mice (c-FLIPfl/+) were crossed with the FLPeR female mice (provided by P. Soriano, Fred Hutchinson Cancer Research Center, Seattle, WA; reference 40) to delete the neomycin cassette flanked by two FRT sites. c-FLIPfl/+ mice were bred with Lck-cre transgenic mice (The Jackson Laboratory; reference 28) to generate c-FLIPfl/flLck-cre, c-FLIPfl/+Lck-cre, and c-FLIPfl/fl mice. The phenotypes of c-FLIPfl/+Lck-cre mice are indistinguishable from those of wild-type C57BL/6 × 129 mice (c-FLIP+/+). These two types of mice were used as controls throughout the experiments. Animal usage was conducted according to protocols approved by the Duke University institutional animal care and use committee.
Flow cytometric analysis.
Single-cell suspensions of the thymus, spleen, and lymph nodes were lysed of RBCs, incubated with an FcR blocker (2.4G2; eBioscience), followed by biotinylated mAb, PE-streptavidin, and FITC-, CyChrome-, or APC-labeled mAb on ice for 30 min, and washed with PBS containing 2% FCS. Data for 1–5 × 104 cells were collected on a FACScan flow cytometer (BD Biosciences) and analyzed using CellQuest (Becton Dickinson) software. All fluorescence-labeled antibodies, including anti-CD3, -CD4, -CD5, -CD8, -CD24, -CD25, -CD69, -CD44, -CD62L, -TCRαβ, -TCRγδ, and -Fas, were obtained from eBioscience, Biolegend, or BD Biosciences. Apoptotic cells were determined by Annexin V and 7-AAD staining using an Annexin V–PE kit (BD Biosciences).
PCR and Southern blot analysis.
PCR to determine the floxed and wild-type c-FLIP alleles was performed at 93°C for 30 s, 56°C for 30 s, and 65°C for 90 s for 40 cycles. The primers for the wild-type and floxed c-FLIP alleles were the same: forward, 5′-TAGCTGATGCATGAGCCTGAGC-3′; reverse, 5′-GTACCAGAACTCTCCAGTCAT ACTTG-3′. Southern blot analysis was performed as described previously (41).
Lymphocyte activation and Western blot assay.
Total or purified thymocytes were incubated with 10 μg/ml anti-CD3 (2C11) on ice for 30 min, washed with ice-cold RPMI 1640 with 10% FBS, and cross-linked with 75 μg/ml rabbit anti–hamster IgG (Sigma-Aldrich) at 37°C for the time indicated in the figures. Total cell lysates were generated after TCR stimulation and subjected to Western blot assay. Anti–c-FLIP was from Qbiogene. Anti-Erk and -Erkp were from Santa Cruz Biotechnology, Inc.
Cell proliferation assays.
CFSE-labeled (Molecular Probes) thymocytes were stimulated with plate-bound anti-CD3 (2C11; eBioscience) and/or anti-CD28 (clone 37.51; BioLegend) for 3 d, and the proliferation was determined by FACS analysis of CD4+ or CD8+ T cells.
Ca2+ flux assay.
107 thymocytes were harvested and diluted in 1 ml of loading buffer (10 mM Hepes, pH 7.5, and 1% FBS). Thymocytes were incubated with 8 μl Indo-1 mix, containing 0.25 μg/μl Indo-1 (Molecular Probes) and 2.5% Pluronic (Molecular Probes) in DMSO, and 65% FBS for 30 min at 30°C. Cells were washed, resuspended in loading buffer, and incubated with PE-CD4 and FITC-CD8 for 15 min. 106 labeled cells were resuspended in 500 μl of loading buffer and incubated with 6 μl biotin–anti-CD3/biotin–anti-CD4 (5:1; 0.5 mg/ml each) for 1 min, then subjected to FACS analysis. After 15 s, 12.5 μl of 1 mg/ml streptavidin was added to induce Ca2+ flux, and the analysis was kept for another 5 min.
Fas- or TCR/CD3-induced apoptosis.
Total or purified thymocytes, as indicated in the figures, were incubated on plate-bound anti-Fas (clone Jo2; BD Biosciences) or anti-CD3 (2C11) for the indicated times. Apoptotic cells were determined by Annexin V and 7-AAD staining.
Acknowledgments
We thank Dr. Motonari Kondo for help in cell sorting and Dr. Michael Krangel and Heather Hartig for critical reviews of this manuscript.
This work was supported by National Institutes of Health grant CA92123 and American Cancer Society grant RSG-0125201 (to Y.-W. He).
The authors have no conflicting financial interests.
Abbreviations used: 7-AAD, 7-amino-actinomycin D; c-FLIP, cellular FLICE-like inhibitory protein; CFSE, carboxyfluorescein diacetate succinimidyl ester; DN, double negative; DP, double positive; ES, embryonic stem; MFI, mean fluorescence intensity; SP, single positive.
References
- 1.Goldrath, A.W., and M.J. Bevan. 1999. Selecting and maintaining a diverse T-cell repertoire. Nature. 402:255–262. [DOI] [PubMed] [Google Scholar]
- 2.Starr, T.K., S.C. Jameson, and K.A. Hogquist. 2003. Positive and negative selection of T cells. Annu. Rev. Immunol. 21:139–176. [DOI] [PubMed] [Google Scholar]
- 3.Godfrey, D.I., and A. Zlotnik. 1993. Control points in early T-cell development. Immunol. Today. 14:547–553. [DOI] [PubMed] [Google Scholar]
- 4.Ogasawara, J., T. Suda, and S. Nagata. 1995. Selective apoptosis of CD4+CD8+ thymocytes by the anti-Fas antibody. J. Exp. Med. 181:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andjelic, S., J. Drappa, E. Lacy, K.B. Elkon, and J. Nikolic-Zugic. 1994. The onset of Fas expression parallels the acquisition of CD8 and CD4 in fetal and adult alpha beta thymocytes. Int. Immunol. 6:73–79. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura, Y., A. Ishii, Y. Kobayashi, Y. Yamasaki, and S. Yonehara. 1995. Expression and function of mouse Fas antigen on immature and mature T cells. J. Immunol. 154:4395–4403. [PubMed] [Google Scholar]
- 7.Drappa, J., N. Brot, and K.B. Elkon. 1993. The Fas protein is expressed at high levels on CD4+CD8+ thymocytes and activated mature lymphocytes in normal mice but not in the lupus-prone strain, MRL lpr/lpr. Proc. Natl. Acad. Sci. USA. 90:10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.French, L.E., A. Wilson, M. Hahne, I. Viard, J. Tschopp, and H.R. MacDonald. 1997. Fas ligand expression is restricted to nonlymphoid thymic components in situ. J. Immunol. 159:2196–2202. [PubMed] [Google Scholar]
- 9.Boursalian, T.E., and P.J. Fink. 2003. Mutation in Fas ligand impairs maturation of thymocytes bearing moderate affinity T cell receptors. J. Exp. Med. 198:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kishimoto, H., C.D. Surh, and J. Sprent. 1998. A role for Fas in negative selection of thymocytes in vivo. J. Exp. Med. 187:1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castro, J.E., J.A. Listman, B.A. Jacobson, Y. Wang, P.A. Lopez, S. Ju, P.W. Finn, and D.L. Perkins. 1996. Fas modulation of apoptosis during negative selection of thymocytes. Immunity. 5:617–627. [DOI] [PubMed] [Google Scholar]
- 12.Nagata, S., and T. Suda. 1995. Fas and Fas ligand: lpr and gld mutations. Immunol. Today. 16:39–43. [DOI] [PubMed] [Google Scholar]
- 13.Irmler, M., M. Thome, M. Hahne, P. Schneider, K. Hofmann, V. Steiner, J.L. Bodmer, M. Schroter, K. Burns, C. Mattmann, et al. 1997. Inhibition of death receptor signals by cellular FLIP. Nature. 388:190–195. [DOI] [PubMed] [Google Scholar]
- 14.Shu, H.B., D.R. Halpin, and D.V. Goeddel. 1997. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 6:751–763. [DOI] [PubMed] [Google Scholar]
- 15.Hu, S., C. Vincenz, J. Ni, R. Gentz, and V.M. Dixit. 1997. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J. Biol. Chem. 272:17255–17257. [DOI] [PubMed] [Google Scholar]
- 16.Goltsev, Y.V., A.V. Kovalenko, E. Arnold, E.E. Varfolomeev, V.M. Brodianskii, and D. Wallach. 1997. CASH, a novel caspase homologue with death effector domains. J. Biol. Chem. 272:19641–19644. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasula, S.M., M. Ahmad, S. Ottilie, F. Bullrich, S. Banks, Y. Wang, T. Fernandes-Alnemri, C.M. Croce, G. Litwack, K.J. Tomaselli, et al. 1997. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J. Biol. Chem. 272:18542–18545. [DOI] [PubMed] [Google Scholar]
- 18.Han, D.K., P.M. Chaudhary, M.E. Wright, C. Friedman, B.J. Trask, R.T. Riedel, D.G. Baskin, S.M. Schwartz, and L. Hood. 1997. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc. Natl. Acad. Sci. USA. 94:11333–11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inohara, N., T. Koseki, Y. Hu, S. Chen, and G. Nunez. 1997. CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc. Natl. Acad. Sci. USA. 94:10717–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasper, D.M., J.P. Vaillancourt, S. Hadano, V.M. Houtzager, I. Seiden, S.L. Keen, P. Tawa, S. Xanthoudakis, J. Nasir, D. Martindale, et al. 1998. Cell death attenuation by “Usurpin”, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 5:271–288. [DOI] [PubMed] [Google Scholar]
- 21.Thome, M., and J. Tschopp. 2001. Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 1:50–58. [DOI] [PubMed] [Google Scholar]
- 22.Chang, D.W., Z. Xing, Y. Pan, A. Algeciras-Schimnich, B.C. Barnhart, S. Yaish-Ohad, M.E. Peter, and X. Yang. 2002. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 21:3704–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Budd, R.C. 2002. Death receptors couple to both cell proliferation and apoptosis. J. Clin. Invest. 109:437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lens, S.M., T. Kataoka, K.A. Fortner, A. Tinel, I. Ferrero, R.H. MacDonald, M. Hahne, F. Beermann, A. Attinger, H.A. Orbea, et al. 2002. The caspase 8 inhibitor c-FLIP(L) modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol. Cell. Biol. 22:5419–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kataoka, T., R.C. Budd, N. Holler, M. Thome, F. Martinon, M. Irmler, K. Burns, M. Hahne, N. Kennedy, M. Kovacsovics, and J. Tschopp. 2000. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr. Biol. 10:640–648. [DOI] [PubMed] [Google Scholar]
- 26.Yeh, W.C., A. Itie, A.J. Elia, M. Ng, H.B. Shu, A. Wakeham, C. Mirtsos, N. Suzuki, M. Bonnard, D.V. Goeddel, and T.W. Mak. 2000. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 12:633–642. [DOI] [PubMed] [Google Scholar]
- 27.Lucas, B., and R.N. Germain. 1996. Unexpectedly complex regulation of CD4/CD8 coreceptor expression supports a revised model for CD4+CD8+ thymocyte differentiation. Immunity. 5:461–477. [DOI] [PubMed] [Google Scholar]
- 28.Hennet, T., F.K. Hagen, L.A. Tabak, and J.D. Marth. 1995. T-cell-specific deletion of a polypeptide N-acetylgalactosaminyl-transferase gene by site-directed recombination. Proc. Natl. Acad. Sci. USA. 92:12070–12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chau, H., V. Wong, N.-J. Chen, H.-L. Huang, W.-J. Lin, C. Mirtsos, A. Elford, M. Bonnard, A. Wakeham, A.I. You-Ten, et al. 2005. Cellular FLICE-inhibitory protein (cFLIP) is required for T cell survival and cycling. J. Exp. Med. 202:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng, J., C. Liu, P. Yang, T. Zhou, and J.D. Mountz. 1997. Increased lymphocyte apoptosis in Fas ligand transgenic mice. J. Immunol. 159:674–684. [PubMed] [Google Scholar]
- 31.Tai, T.S., L.W. Fang, and M.Z. Lai. 2004. c-FLICE inhibitory protein expression inhibits T-cell activation. Cell Death Differ. 11:69–79. [DOI] [PubMed] [Google Scholar]
- 32.Wu, W., L. Rinaldi, K.A. Fortner, J.Q. Russell, J. Tschopp, C. Irvin, and R.C. Budd. 2004. Cellular FLIP long form-transgenic mice manifest a Th2 cytokine bias and enhanced allergic airway inflammation. J. Immunol. 172:4724–4732. [DOI] [PubMed] [Google Scholar]
- 33.Tseveleki, V., J. Bauer, E. Taoufik, C. Ruan, L. Leondiadis, S. Haralambous, H. Lassmann, and L. Probert. 2004. Cellular FLIP (long isoform) overexpression in T cells drives Th2 effector responses and promotes immunoregulation in experimental autoimmune encephalomyelitis. J. Immunol. 173:6619–6626. [DOI] [PubMed] [Google Scholar]
- 34.Van Parijs, L., Y. Refaeli, A.K. Abbas, and D. Baltimore. 1999. Autoimmunity as a consequence of retrovirus-mediated expression of C-FLIP in lymphocytes. Immunity. 11:763–770. [DOI] [PubMed] [Google Scholar]
- 35.Grambihler, A., H. Higuchi, S.F. Bronk, and G.J. Gores. 2003. cFLIP-L inhibits p38 MAPK activation: an additional anti-apoptotic mechanism in bile acid-mediated apoptosis. J. Biol. Chem. 278:26831–26837. [DOI] [PubMed] [Google Scholar]
- 36.Bannerman, D.D., K.T. Eiting, R.K. Winn, and J.M. Harlan. 2004. FLICE-like inhibitory protein (FLIP) protects against apoptosis and suppresses NF-kappaB activation induced by bacterial lipopolysaccharide. Am. J. Pathol. 165:1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kreuz, S., D. Siegmund, J.J. Rumpf, D. Samel, M. Leverkus, O. Janssen, G. Hacker, O. Dittrich-Breiholz, M. Kracht, P. Scheurich, and H. Wajant. 2004. NFκB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J. Cell Biol. 166:369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salmena, L., B. Lemmers, A. Hakem, E. Matysiak-Zablocki, K. Murakami, P.Y. Au, D.M. Berry, L. Tamblyn, A. Shehabeldin, E. Migon, et al. 2003. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17:883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng, C., A. Wynshaw-Boris, F. Zhou, A. Kuo, and P. Leder. 1996. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 84:911–921. [DOI] [PubMed] [Google Scholar]
- 40.Farley, F.W., P. Soriano, L.S. Steffen, and S.M. Dymecki. 2000. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 28:106–110. [PubMed] [Google Scholar]
- 41.Guo, J., A. Hawwari, H. Li, Z. Sun, S.K. Mahanta, D.R. Littman, M.S. Krangel, and Y.W. He. 2002. Regulation of the TCRalpha repertoire by the survival window of CD4(+)CD8(+) thymocytes. Nat. Immunol. 3:469–476. [DOI] [PubMed] [Google Scholar]