

Abstract
Identification of the T cell immunoglobulin mucin-domain containing (Tim) gene family introduced a new family of cell surface molecules that is involved in the regulation of immune responses. We previously demonstrated that Tim-3 is expressed on terminally differentiated T helper (Th)1 cells, and serves to regulate Th1 immune responses. Here, we describe the identification and function of Tim-2, a novel member of the Tim gene family. In contrast with Tim-3, we demonstrate that Tim-2 is expressed preferentially in differentiated Th2 cells. Blockade of the Tim-2/Tim-2 ligand interaction, by administration of soluble Tim-2 fusion protein (Tim-2 immunoglobulin [Ig]), results in T cell hyperproliferation and the production of Th2 cytokines. Administration of Tim-2 Ig during the induction phase reduces the severity of experimental autoimmune encephalomyelitis, a Th1-mediated autoimmune disease model of multiple sclerosis. We propose that Tim-2, an orthologue of human Tim-1, is critical for the regulation of Th2 responses during autoimmune inflammation.
Activation of naive CD4+ T cells drives their differentiation into two phenotypically and functionally distinct subsets, Th1 or Th2, that differ in their cytokine production and effector functions (1–4). Th1 cells produce IFN-γ and TNF-β and are involved in the clearance of intracellular pathogens and the induction of autoimmune diseases (5). Th2 cells that produce cytokines, including IL-4 and IL-10, clear extracellular pathogens (e.g., helminths), regulate autoimmunity, and play critical roles in the induction of asthma and atopy (4, 6). Only recently have cell surface markers been identified that clearly distinguish Th1 and Th2 cells. Th1 and Th2 cells show a quantitative difference in chemokine and costimulatory receptor expression (7–9). Using an antibody screening approach, we identified cell surface molecule T cell immunoglobulin mucin-domain containing (Tim)-3 that is expressed specifically on the surface of Th1, but not Th2, cells (10). Tim-3 belongs to the novel Tim family of cell surface proteins. The mouse gene family consists of eight members (Tim-1 to Tim-8), whereas the human gene family consists of three members (Tim-1, Tim-3, and Tim-4; reference 11). All members share the characteristic structure of an IgV, mucin, transmembrane, and cytoplasmic domains. The gene family plays a critical role in the regulation of immune responses (10–14). Administration of soluble Tim-3 Ig fusion protein resulted in hyperproliferation of Th1 cells, exacerbation of autoimmunity, and prevention of transplantation and high-dose tolerance induction (10, 13, 14). Furthermore, the gene family has been linked genetically to murine airway hypersensitivity (a Th2-driven disease), and polymorphic alleles of Tim-1 play a role in susceptibility to human asthma (12). These findings suggested that other Tim family members have a role in regulating Th responses. Here, we have analyzed the expression and function of Tim-2. We demonstrate that Tim-2 is expressed preferentially in Th2 cells, and blockade of the Tim-2/Tim-2 ligand interaction results in expansion of a Th2 response and attenuation of a Th1-mediated autoimmune disease. We propose that Tim-2 plays an important role in the regulation of Th2 immunity.
RESULTS
Tim-2 is expressed preferentially in Th2 cells
Mouse Tim-2 shares the greatest degree of nucleotide sequence identity to mouse Tim-1 (85%). Tim-1 was identified originally on African green monkey kidney cells as a hepatitis A virus receptor, and later was found to be expressed on kidney epithelial cells (15, 16). More recently, it has been demonstrated that mouse Tim-1 also is expressed on the surface of T cells, with a higher expression on Th2 differentiated cells compared with Th1-type cells (17, 18). Given the sequence similarity, we were interested in investigating the expression profile of Tim-2. Using real-time quantitative PCR, we studied the expression pattern of Tim-2 in various cell populations. Low levels of Tim-2 were detected in unactivated and activated CD4+ and CD8+ T cells (Fig. 1 A). Tim-2 expression was increased only marginally after polyclonal stimulation of T cells with conalbumin A or anti-CD3/anti-CD28 stimulation (Fig. 1 B); no detectable level of Tim-2 expression was observed in other cell types, including B cells, macrophages, and dendritic cells (Fig. 1, A and B). Given the expression of Tim-2 in T cells, we investigated whether Tim-2, like Tim-3, was expressed differentially in Th1 versus Th2 cells. High levels of Tim-2 expression were observed in the long-term Th2 clone, D10G4, whereas little expression was detected in the Th1 clone, AE7 (Fig. 1 C). To establish the kinetics of Tim-2 expression during Th2 cell differentiation, we generated CD4+ Th1 and Th2 cell lines from DO11.10 TcR transgenic mice. We observed significant up-regulation of Tim-2 expression during late-stage Th2 differentiation in vitro, particularly after round III of stimulation under Th2-inducing conditions (Fig. 1 C). Although low levels of Tim-2 were detected in early rounds of Th1 polarization, by three rounds of polarization in vitro, Tim-2 expression was not detected in fully differentiated Th1 cells (Fig. 1 C). We next examined the pattern of Tim-2 expression in Th2 cells that were derived from BALB/c, C57BL/6, and SJL/J mouse strains, which are biased genetically to induce Th1 (SJL and C57BL/6) or Th2 (BALB/c) responses. Tim-2 was up-regulated preferentially in Th2 cells from all three strains (Fig. 1 D). Together, these data suggest that Tim-2 is specifically up-regulated in Th2 cells, and is down-regulated during Th1 differentiation. In the absence of a specific antibody to Tim-2, we determined whether Tim-2 is expressed as a protein. We generated Tim-2 hemagglutinin A (HA)–tagged T cell (TK-1 and EL-4) and non–T cell (CHO and HEK-293) transfectants, and demonstrated that Tim-2 is expressed on the cell surface (Fig. 1 E). Western blot analysis of transfectants using an anti-HA–specific antibody demonstrated that in contrast with normal rabbit IgG, the anti–HA-tagged–specific antibody immunoblotted a broad band of ∼65–70 kD, which was consistent with the heavily glycosylated Tim-2 molecule (Fig. 1 F; reference 19). In addition, we have expressed Tim-2 as an Ig fusion protein (see Materials and methods); this further supports the notion that Tim-2 is expressed as a functional protein.
Figure 1.

Tim-2 is up-regulated in Th2 cells. The expression pattern of Tim-2 was examined in various cell populations using real-time quantitative RT-PCR. Tim-2 expression was examined and expressed relative to GAPDH expression. (A) Ex vivo SJL CD4+, CD8+, B220+, CD11b+, and CD11c+ cells were purified from spleen cells. RNA was extracted and cDNA was generated for real-time quantitative PCR. (B) Ex vivo SJL CD4+, CD8+, B220+, CD11b+, and CD11c+ cells were purified from spleen cells. CD4+ and CD8+ cells were activated with plate-bound anti-CD3/anti-CD28; B220+, CD11b+, and CD11c+ were activated by LPS and IFN-γ. 48 h after activation, RNA was extracted and cDNA was generated for real-time quantitative PCR. (C) D011.10 CD4+ T cells were stimulated with OVA-peptide and APCs, in vitro under Th1 or Th2 polarizing conditions. At the end of each round of stimulation, RNA was extracted and cDNA was generated from Th1 and Th2 cells. (D) CD4+ T cells from SJL, C57BL/6, and BALB/c mice were stimulated with plate-bound anti-CD3/anti-CD28 in vitro under Th1 or Th2 polarizing conditions. At the end of the first round of stimulation, RNA was extracted and cDNA was generated from Th1 and Th2 cells. (E) Tim-2 is expressed as a cell surface protein. T cell (EL-4 and TK-1) and non–T cell (CHO and 293) transfectants were stained with biotinylated anti-HA antibody, and streptavidin-PE as a secondary detection reagent. Cells were analyzed by flow cytometry for surface expression of Tim-2. Solid line represents the staining of Tim-2 transfectants and the dotted line, empty vector. (F) Tim-2 expression by Western blot. Lysates from Tim-2–transfected CHO and HEK-293 cells were run on a 10% SDS-PAGE gel. Polyvinyldifluoride membranes were probed with anti-HA antibody or control normal rabbit IgG, followed by an HRP-conjugated anti–rabbit secondary antibody.
Tim-2 ligands are expressed on activated dendritic cells and macrophages
We next generated a Tim-2 Ig fusion protein to identify cell populations expressing Tim-2 ligands. Tim-2 Ig consisted of the IgV and mucin domains of Tim-2 fused to the human IgG Fc tail. Unactivated splenic-derived cells did not express the Tim-2 ligand. However, upon activation of APCs (B cells, macrophages, and dendritic cells) with LPS and IFN-γ, a marked up-regulation of Tim-2 ligand expression was observed (Fig. 2). A subset of activated B220+ and CD11b+ cells was positive for Tim-2 ligand expression, whereas all activated CD11c+ cells expressed the Tim-2 ligand (Fig. 2). The Tim-2 ligand was not detected on CD3+ T cells, either naive or activated by anti-CD3/anti-CD28 stimulation. These data suggest that Tim-2 expressed on Th2 cells binds a ligand that is expressed on activated APCs.
Figure 2.
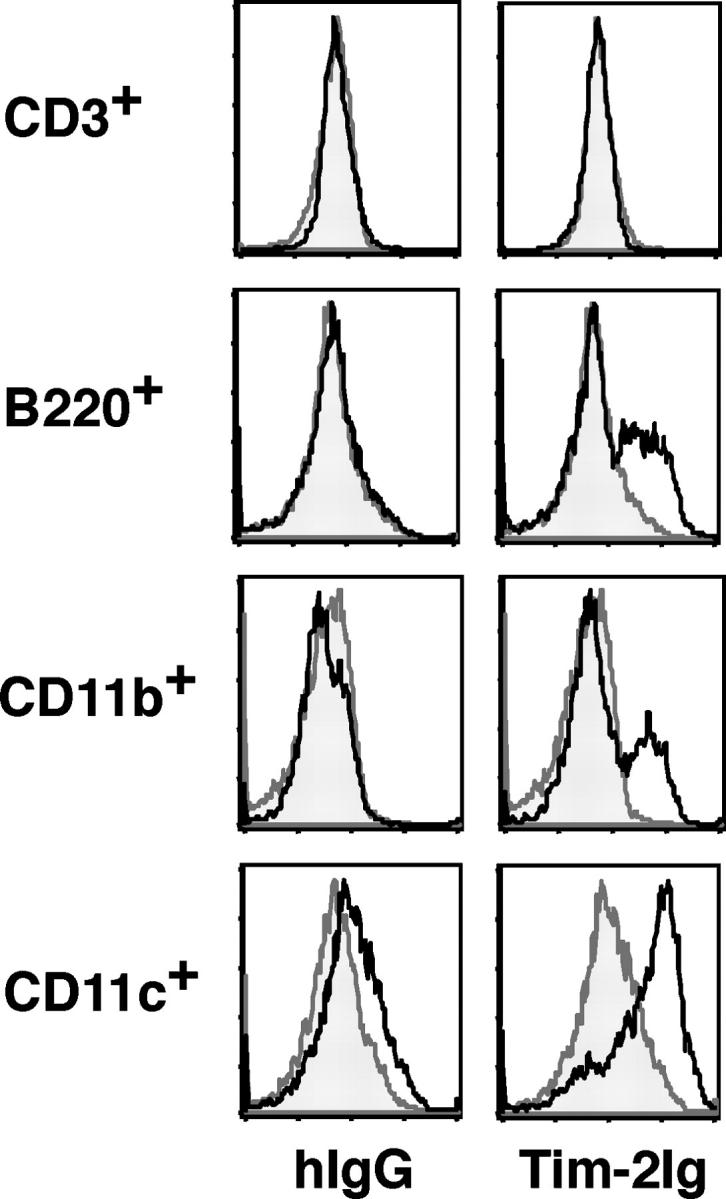
Tim-2 ligand is expressed on activated APCs. To determine the cell populations expressing Tim-2 ligands, we stained various cell populations with biotinylated Tim-2 Ig as follows: ex vivo BALB/c CD3+, B220+, CD11b+, or CD11c+ cells. Spleen cells from BALB/c mice were activated with conalbumin A (CD3+) or LPS and IFN-γ (B220+, CD11b+ and CD11c+), were column purified using negative selection (CD3+) or positive selection (B220+, CD11b+, and CD11c+) and stained for Tim-2 ligand expression with biotinylated Tim-2 Ig (solid black line), or biotinylated hIgG (shaded curve) as a control, followed by streptavidin-PE as a secondary reagent for detection.
Administration of Tim-2 Ig results in hyperproliferation and Th2 cytokine production
To determine the function of the Tim-2/Tim-2 ligand interaction during an autoimmune, inflammatory response, SJL/J mice were immunized with proteolipid protein (PLP) 139–151 peptide emulsified in CFA and treated with Tim-2 Ig, or controls (human IgG [hIgG] or PBS). 10 d after immunization, spleen cells harvested from Tim-2 Ig–treated mice demonstrated a pronounced level of basal proliferation in the presence of media without the addition of any exogenous antigen in vitro (Fig. 3 A). This proliferation was not observed for cells harvested from control mice (Fig. 3 A). Upon recall-stimulation with the immunizing antigen PLP 139–151, cells from control groups and the Tim-2 Ig–treated group demonstrated a similar proliferative dose-response curve (Fig. 3 B). These data suggest that Tim-2 Ig administration results in the hyperactivation of cells in vivo, such that they continue to proliferate in the absence of antigen in vitro.
Figure 3.
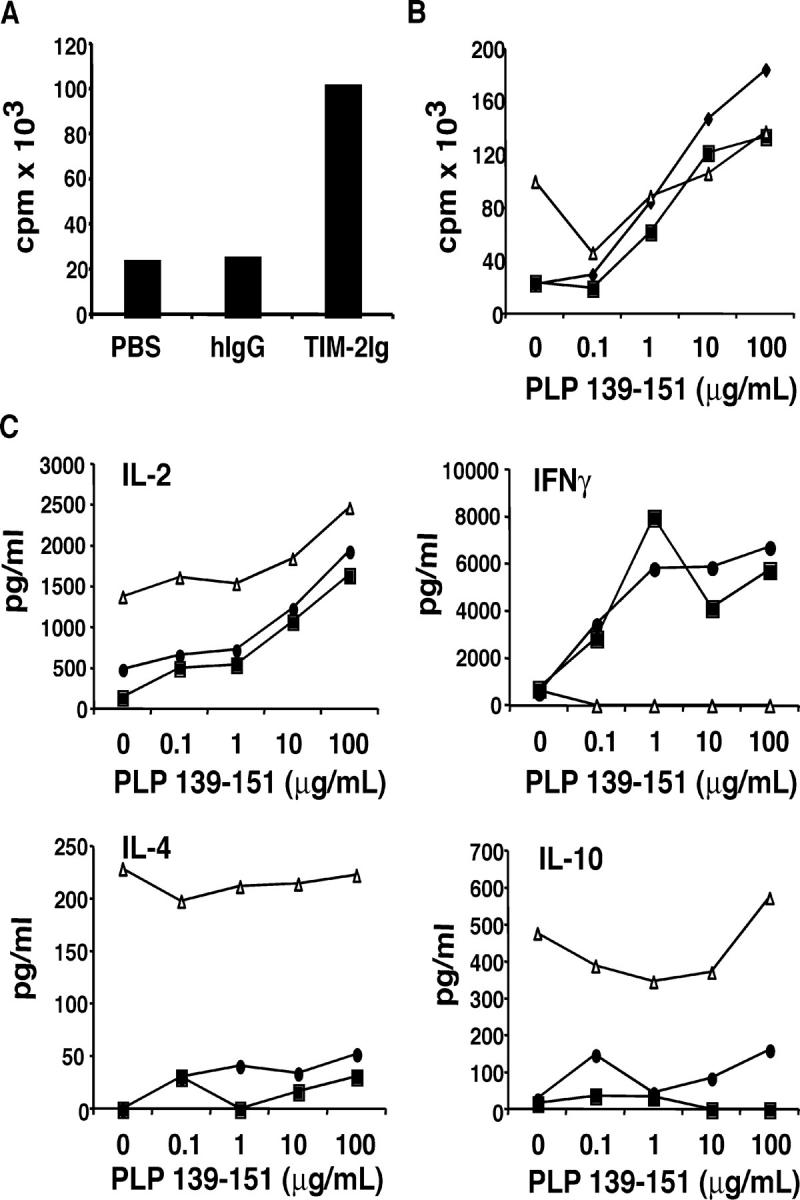
Administration of Tim-2 Ig induces hyperproliferation and production of Th2 cytokines. (A) Spleen cells from SJL/J mice immunized with PLP 139–151 peptide emulsified in CFA, and treated in vivo with Tim-2 Ig, or hIgG or PBS as controls, were cultured in vitro for 48 h without peptide restimulation. Proliferation was measured in triplicate wells after 48 h by 3[H]-thymidine incorporation. Data shown are for individual mice, representative of seven experiments for proliferation. (B) Spleen cells taken from immunized, fusion-protein–treated mice were stimulated in vitro with 0–100 μg of PLP 139–151 peptide. Proliferation was measured after 48 h. ♦, PBS; ▪, hIgG; ▵, Tim-2 Ig treated. (C) Supernatants were taken at 48 h from in vitro cultures of spleen cells restimulated with PLP 139–151 peptide, and cytokine ELISAs for IL-2, IL-4, IL-10, and IFN-γ were performed. •, PBS; ▪, hIgG; ▵, Tim-2 Ig treated. Data shown for individual mice; representative of five experiments for cytokines.
Supernatants from these cell cultures were analyzed 48 h after restimulation by cytokine ELISA for the production of IL-2, IFN-γ, IL-4, and IL-10. Spleen cells from control groups—treated with hIgG or PBS—demonstrated the predicted Th1 type response with production of IL-2 and IFN-γ (Fig. 3 C). Spleen cells from Tim-2 Ig–treated mice secreted high levels of IL-2, which was consistent with the high level of basal proliferation that was observed. However, in contrast with control groups, spleen cells from Tim-2 Ig–treated mice demonstrated little or no IFN-γ production (Fig. 3 C). Surprisingly, there was increased production of hallmark Th2 cytokines, IL-4 and IL-10, in the Tim-2 Ig–treated groups, even when immunized with PLP emulsified in CFA, a known inducer of Th1 (IFN-γ) responses (Fig. 3 C). We previously proposed that an interaction between Tim-3 and its ligand serves to down-regulate the Th1 response, and that administration of Tim-3 Ig blocks this inhibitory interaction, and results in the expansion and hyperproliferation of Th1 cells (13). Administration of Tim-2 Ig results in similar hyperproliferation as observed with Tim-3 Ig. However, in contrast with Tim-3 Ig, Tim-2 Ig results in the expansion of a Th2 cytokine response.
Because spleen cells from Tim-2 Ig–treated mice showed higher basal proliferation (Fig. 3 A), we wanted to analyze which cells were involved in the enhanced proliferative and cytokine response. T cells and APCs from immunized control and Tim-2 Ig–treated mice were purified, cultured separately, or recombined. Separation of cell subsets and recombination of T cells and APCs from Tim-2 Ig–treated or control mice demonstrated that the hyperproliferative response was dependent on T cells from Tim-2 Ig–treated mice and not macrophages or B cells. T cells from Tim-2 Ig–treated mice, even when recombined with B cells or macrophages from control animals, reconstituted the hyperproliferative phenotype (Fig. 4 B). Furthermore, recombination of T cells from control mice with APCs from Tim-2 Ig–treated mice did not reconstitute the proliferative response. This indicated that the hyperproliferative response is dependent on T cells from Tim-2 Ig–treated mice (Fig. 4 B).
Figure 4.
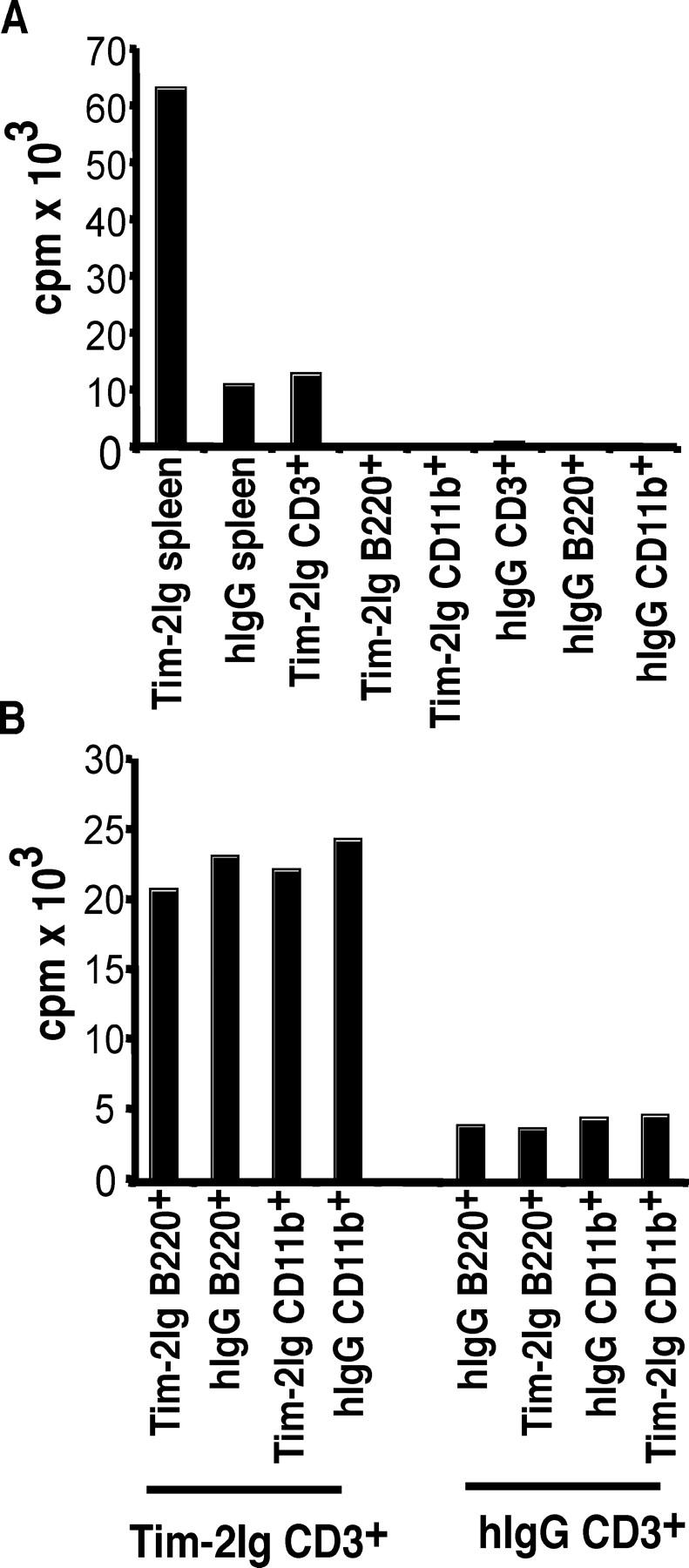
Hyperproliferation of cells from Tim-2 Ig–treated mice is mediated by an interaction between T cells from Tim-2 Ig–treated mice and APCs. (A) Individual cell populations: CD3+ T, B220+ B, and CD11b+ cells were purified from the spleens of immunized, and Tim-2 Ig or hIgG–treated SJL/J mice. Splenocytes (5 × 105), T cells (105), B cells (2 × 105), and CD11b+ cells (2 × 105) were cultured individually, and the proliferative responses were measured 48 h later by 3[H]-thymidine incorporation in triplicate wells. Results are represented as average for four mice combined, representative of three experiments. (B) Recombined cell populations: CD3+ T, B220+ B, and CD11b+ cells were purified from the spleens of immunized, and Tim-2 Ig or hIgG–treated SJL/J mice. T cells (105) from Tim-2 Ig or hIgG–treated mice were incubated in the presence of 2 × 105 B cells or 2 × 105 CD11b+ cells purified from Tim-2 Ig and hIgG–treated mice; the proliferative response was measured 48 h later by 3[H]-thymidine incorporation in triplicate wells. Results are represented as average for four mice combined, representative of three experiments.
Blockade of the Tim-2/Tim-2 ligand interaction during the induction phase of experimental autoimmune encephalomyelitis
The production of Th1 cytokines by myelin-specific T cells has been associated with the development of experimental autoimmune encephalomyelitis (EAE), whereas the secretion of Th2 cytokines has been associated with disease regulation and recovery (20, 21). Thus, the onset and progression of disease is dependent, in part, on a balance between the two responses. Given that Tim-2 Ig administration resulted in the skewing of an immune response toward a Th2 phenotype, we hypothesized that Tim-2 Ig treatment may promote expansion of Th2 cells which could regulate the development of EAE. We immunized SJL/J mice with the encephalitogenic peptide, PLP 139–151, in CFA to induce EAE (22). Immunized mice were treated with four doses of Tim-2 Ig, hIgG, or PBS, every other day from day 0 to day 8. Immunized mice that were treated with hIgG or PBS demonstrated a typical EAE disease course with the onset of clinical symptoms between days 10 and 12, followed by peak clinical disease around day 17 (Fig. 5 A). In contrast, treatment with Tim-2 Ig resulted in a delay in the onset of clinical signs; these Tim-2 Ig–treated mice displayed only mild signs of paralysis, and demonstrated peak clinical disease only after day 22 (Fig. 5 A). The administration of a single dose of Tim-2 Ig just before the onset of disease also was sufficient to reduce the clinical signs of EAE (Fig. 5 B). Histologic examination of brain and spinal cords demonstrated a significant reduction in the number of inflammatory lesions in the meninges and parenchyma in mice that were treated with Tim-2 Ig (mean total inflammatory foci ± SD = 53 ± 17) in comparison with control animals (108 ± 30; Fig. 5 C). Taken together, these results suggest that blockade of the Tim-2/Tim-2 ligand interaction results in the expansion of a Th2 response, even within a Th1-immune setting.
Figure 5.
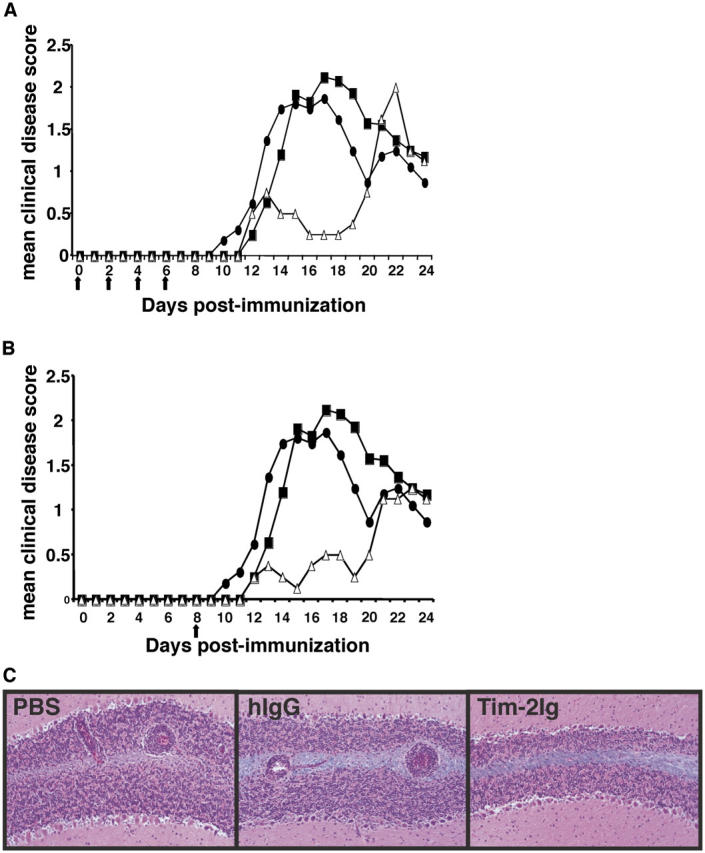
Tim-2 Ig delays the onset and decreases the severity of EAE. (A) SJL/J mice were immunized for the induction of EAE with PLP 139–151 emulsified in CFA, and treated with five injections of Tim-2 Ig every other day for 8 d with Tim-2 Ig (▵), hIgG (▪), or PBS (•) as controls. Mice were examined daily for signs of EAE. Mice treated with Tim-2 Ig demonstrated significantly lower (P < 0.01) cumulative disease scores when compared with PBS and hIgG controls. (B) SJL/J mice were immunized for the induction of EAE with PLP 139–151 emulsified in CFA, and treated with only one injection on day 8 with Tim-2 Ig (▵), hIgG (▪), or PBS (•) as controls. Mice were examined daily for signs of EAE. Mice treated with Tim-2 Ig demonstrated significantly lower (P < 0.01) cumulative disease scores when compared with PBS and hIgG controls. (C) Representative fields of cerebellar white matter demonstrate typical perivascular mononuclear cell infiltrates in control PBS- and hIgG-treated, but not the Tim-2 Ig–treated, SJL mice immunized for EAE. Luxol fast blue-hematoxylin and eosin stains. Original magnification, 80.
DISCUSSION
The identification of Tim-3 not only identified a Th1-specific cell surface molecule, but also introduced a new family of genes, the Tim gene family. Here we begin to elucidate the role of a novel member of the Tim gene family, Tim-2. We demonstrate that in contrast with Tim-3, a known Th1 cell surface marker, Tim-2 seems to be up-regulated preferentially in Th2 cells and down-regulated in Th1 cells. The use of a soluble Tim-2 fusion protein explores the in vivo role of the Tim-2/Tim-2 ligand interaction in an autoimmune setting.
Quantitative mRNA analysis of lymphoid cell populations from mice demonstrated that Tim-2 expression was detected at low levels in naive CD4+ and CD8+ T cells. After activation, CD4+ and CD8+ T cells demonstrated up-regulation in Tim-2 expression. Further examination of Th1 and Th2 differentiated cells revealed that Tim-2 was expressed significantly more in Th2 clones and lines than in Th1 clones and lines. Thus, in contrast with Tim-3, which is expressed on terminally differentiated Th1 cells, Tim-2 is expressed at low levels in naive and activated T cells and preferentially up-regulated during Th2 differentiation. Tim-2 demonstrates a far more restricted expression pattern in comparison with mouse Tim-1 which is found on all activated T cells, Th1 and Th2 cells (although at higher levels on Th2 cells), and epithelial cells (15–17, 23). Therefore, although mouse Tim-1 shares the greatest sequence identity with human Tim-1 (as suggested by the nomenclature), mouse Tim-2 may share more functional characteristics with human Tim-1. Therefore, mouse Tim-2 also could be considered as an orthologue of human Tim-1. The high expression of Tim-2 in late Th2 differentiation also is interesting given the association of the Tim gene family with the regulation of Th1 versus Th2 responses. Given the late expression of Tim-2 during Th2 differentiation, it is probable that the in vivo role of Tim-2 is more important during the effector phase of an immune response rather than the initial differentiation phase.
Using an expression cloning approach, Tim-2 was identified as a potential ligand for the semaphorin family member Sema4a (19). Using anti-Sema4a antibody, Kumanogoh et al. demonstrated expression of Sema4a predominantly on dendritic cells and B cells, with up-regulation upon activation. They also identified low levels of Sema4a expression on the surface of activated T cells (19). Although we have demonstrated that Tim-2 ligand is expressed on activated APCs by staining with Tim-2 Ig, in contrast with Kumanogoh et al., we did not detect any expression of Tim-2 ligand on T cell populations (naive or activated). This could reflect conformational or expression differences of Sema4a in T cells that are not recognized by Tim-2 Ig. Also, it is highly likely that Tim-2 binds a variety of ligands and that Tim-2 Ig is more specific for ligands other than Sema4a on the surface of activated APCs. Our data on the expression pattern of Tim-2 on differentiated Th2 cells suggests that the Tim-2/Tim-2 ligand interaction predominantly involves associations between Th2 cells and activated APCs.
Based on the mechanistic paradigm proposed for Tim-3, it is possible that the Tim-2/Tim-2 ligand interaction also may be inhibitory, and thus, cross-linking of Tim-2 on Th2 cells may down-regulate the Th2 response. This would suggest that the administration of Tim-2 Ig would block the endogenous interaction between Tim-2 and its ligand, and allow for the expansion of Th2 cells and cytokines. In keeping with this, administration of Tim-2 Ig during a Th1-biased immune response resulted in the hyperproliferation of T cells and the production of Th2 cytokines. If, as our data suggest, the Tim-2/Tim-2 ligand interaction is inhibitory, blockade of this interaction during the induction phase would allow for the expansion of Th2 cells, and therefore, affect the EAE disease course. Treatment of mice with Tim-2 Ig during the induction phase of EAE reduced the severity of clinical and histologic disease. Consistent with this, administration of a blocking antibody to Sema4a (a Tim-2 ligand) resulted in a reduced Th1 response and inhibition of myelin oligodendrocyte glycoprotein–induced EAE (19). Furthermore, recent work with the Sema4a knockout mouse demonstrated a defect in establishing Th1, but not Th2, responses (24). Thus, if the Tim-2/Sema4a interaction served to down-regulate a Th2 response, blocking this interaction would result in inhibition of a Th1 response, as observed with anti-Sema4a antibody or by administration of Tim-2 Ig during EAE. Like Tim2, Sema4a may have other ligands, and the blockade of Sema4a and Tim-2 may not be equivalent. We recently demonstrated that in contrast with Tim-2, Tim-1 interacts specifically with another Tim family member, Tim-4 (18). Umetsu and colleagues demonstrated that cross-linking Tim-1 on the surface of T cells stimulates T cell expansion (17). Furthermore, we show that administration of high amounts of Tim-4 Ig in the presence of low levels of anti-CD3/anti-CD28 stimulation enhances T cell proliferation (18). Taken together, we suggest that the Tim-1/Tim-4 interaction has the potential to costimulate T cell expansion (17, 18). Although Tim-1 and Tim-2 are very similar molecules and closely related, their intracellular tails are significantly different. Tim-2 shares only 56% identity with Tim-1 in the cytoplasmic tail, and contains six extra amino acids in the COOH-terminal end of the tail, which contains an additional tyrosine phosphorylation motif. If Tim-1 and Tim-2 turn out to be functionally different, we suggest that the differences in the intracellular tail may explain, in part, their different mechanisms of action. Our data and those of Kumanogoh and colleagues support our proposal that Tim-2 serves to regulate a Th2 response negatively in an autoimmune setting. However, further study is required to determine conclusively that Tim-2 Ig is not an agonist promoting a Th2 response.
In addition to our observations of the role of Tim-2 in an autoimmune Th1 setting, Tim-2 also may have a role in pathogenic Th2 responses. Human Tim-1 initially was identified as a hepatitis A viral cellular receptor, and epidemiologic studies have shown that infection with hepatitis A virus (HAV) is associated with protection against asthma and allergies (23). If, like Tim-2, human Tim-1 is expressed on the surface of Th2 cells, HAV might bind to Tim-1 and eliminate or block Th2 effector functions. Consistent with this hypothesis, polymorphic alleles of human Tim-1 have been identified, and a deletion of six amino acid residues in the mucin domain of Tim-1 has demonstrated strong linkage with HAV infection and protection against asthma (25). Further investigation will be required to identify the role of Tim-2 in productive and pathogenic Th2 immune responses.
These data, in conjunction with our understanding of Tim-3 and Tim-1, further support a crucial role for the Tim family of genes in the differential regulation of Th1 and Th2 responses, and hence, their potential as therapeutic targets in various immune-mediated diseases.
MATERIALS AND METHODS
Th1 and Th2 cell lines and clones.
AE7, a pigeon cytochrome c–specific, I-Ek–restricted Th1 clone, and D10G4, a conalbumin A–specific, I-A-k–restricted Th2 clone, were maintained as described previously (10). Cells were maintained in DMEM supplemented with 0.1 mM nonessential amino acids, sodium pyruvate (1 mM), L-glutamine (2 mM), MEM essential vitamin mixture (1×), penicillin (100 U/ml), streptomycin (100 U/ml), gentamicin (0.1 mg/ml), 10% heat-inactivated FBS (BioWhittaker, Inc.), asparagine (0.1 mM), folic acid (0.1 mg/ml), 2-mercaptoethanol (5 × 10−5 M) (Sigma-Aldrich), 0.6% T cell growth factor (T-Stim, Collaborative Biomedical Research), and 0.06% recombinant IL-2. Cells were maintained in a rest-stimulation protocol (stimulation media is complete medium without T-Stim and recombinant IL-2) as described. DO11.10 TcR transgenic Th1/Th2 cell lines were maintained in the same complete medium, and were restimulated with irradiated BALB/cJ spleen cells and OVA 323–339 peptide (10 μg/ml, Quality Controlled Biochemicals) in the presence of recombinant IL-12 and anti–IL-4 (Th1), or IL-4 and anti–IL-12 (Th2). RNA was extracted from cells and converted to cDNA for use in quantitative Taqman RT-PCR.
Quantitative TaqMan RT-PCR.
RNA was reverse transcribed to cDNA. Real-time RT-QPCR was performed using the TaqMan strategy (Applied Biosystems). The expression levels of Tim-2 and internal reference GAPDH were measured by multiplex PCR using probes labeled with 6-carboxyfluorescein (FAM) or 6-carboxyrhodamine (VIC; Applied Biosystems), respectively. The primers and probes were designed using Primer Express v1.0 software (Applied Biosystems). The Tim-2 primer/probe sequences are as follows: FAM probe: 5′-ACAGCTGCCTGCCCAGTGCCC-3′; forward primer: 5′-GCCGGTGGACCTCAGTTTC-3′; reverse primer: 5′-TGGGAGCCAGCACAGATCA-3′ (Applied Biosystems). The simultaneous measurement of Tim-2–FAM and GAPDH–VIC permitted normalization of the amount of cDNA added per sample. PCRs were performed using the Taqman Universal PCR Master Mix and the ABI PRISM 7700 Sequence Detection System. Relative gene expression levels were determined as described previously (13).
Generation of cell lines expressing hemagglutinin A–tagged Tim-2.
Splenocytes from an SJL/J mouse were activated with concanavalin A, 1 μg/ml (Sigma-Aldrich), for 48 h. Total RNA was extracted with TRIzol reagent (Invitrogen), and cDNA was synthesized using Superscript II reverse transcriptase enzyme (Invitrogen). The Tim-2 cDNA lacking the first 63 nucleotides that encode signal peptide was generated by PCR using PfuUltra High-Fidelity DNA polymerase (Stratagene). The primers used were: forward, 5′-CATACAGCAGTGCAGGGGCTGG-3′; reverse, GTAGGTCGACTAGGACTCTTCTTCGGGGTAAGG-3′ with SalI site as overhang at the 5′ end. After confirming the DNA sequence, the PCR product was cloned between SmaI and SalI sites into the pDisplay vector containing a signal peptide and HA-tag (Invitrogen), and the open reading frame was confirmed. CHO and 293 cells were transfected with the above construct using Genejuice transfection reagent (Novagen), whereas TK-1 and EL-4 cells were transfected by electroporation. 48 h after transfection, the transfectants were cultured in medium containing 1.0 mg/ml G418. Cells that survived the G418 selection were analyzed by flow cytometry using a biotinylated anti-HA antibody and streptavidin-PE as a detection reagent. Sorted cells were expanded in G418-containing medium and examined for surface expression of Tim-2.
Western blot analysis.
Cells were harvested and lysed in lysis buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton, 1% glycerol, 2 mM NaVO4, 2 mM leupeptin, 2 mM pepstatin A, 2 mM chymostatin). Protein concentration was determined using Coomassie Plus Protein Assay Reagent (Pierce Chemical Co.) and ∼50 μg was separated by 10% SDS-PAGE. Polyvinyldifluoride blots were blocked with 5% (wt/vol) nonfat dry milk and probed with anti-HA tag rabbit polyclonal IgG or normal rabbit IgG (Upstate Biotechnology) followed by HRP-conjugated goat anti–rabbit IgG secondary antibody (Upstate Biotechnology). Blots were visualized with ECL Western Blotting Detection reagents (GE Healthcare).
APC purification and Tim-2 ligand staining.
Spleen and lymph node cells were harvested from naive SJL/J, BALB/c, NOD, or C57BL/6 mice (Jackson ImmunoResearch Laboratories); and activated with concanavalin A (CD3+), LPS (Sigma-Aldrich), and IFN-γ (B220+, CD11b+, CD11c+); and immediately used for purification. Subsets were purified using column based positive selection magnetic-activated cell sorting beads (Miltenyi Biotec). B220+ beads were used for the purification of B cells, CD11b+ beads were used for the purification of macrophage and dendritic populations, and CD11c+ beads were used for the purification of dendritic cells. CD4+ T cells were column purified by negative selection (R&D Systems). Cells were stained with biotinylated hIgG (as a negative control), or biotinylated Tim-2. Streptavidin-PE was used as the secondary detection reagent. All cells were analyzed by flow cytometry using a Becton Dickinson FACSCalibur.
Proliferation assays.
Female SJL/J mice (6–12 wk old) were injected s.c. in each flank with 50 μg PLP 139–151 peptide (HSLGKWLGHPDKF; Quality Controlled Biochemicals) emulsified in CFA (Difco). Mice were injected i.p. every other day (beginning day 0, and continuing through day 8) with 100 μg Tim-2 Ig, 100 μg control hIgG, or 100 μl PBS. Mice were killed on day 10, and spleens harvested. Cells were plated at 5 × 105 cells/well in 96-well plates (Falcon, Becton Dickinson) with PLP 139–151 added at 0–100 μg/ml for 48 h. Plates were pulsed with 1 μCi 3[H]-thymidine/well for 16–18 h. Incorporation was measured using a Beta Plate scintillation counter (PerkinElmer). The data are shown as mean cpm in triplicate wells.
For cell separation experiments, CD11b+ and B220+ cells were purified through positive selection as described above, and CD3+ T cells were purified using negative selection columns (R&D Systems) after depletion of CD11b+ and B220+ cells. CD3+ T cells were plated at 105 cells/well, and CD11b+ and B220+ cells were plated at 2 × 105 cells/well.
Cytokine ELISAs.
Cytokine production was measured for IL-2, IL-4, IL-10, IFN-γ, and TNF-α by quantitative capture ELISA. In brief, capture mAbs to mouse IL-2 (clone JES-1A12), IL-4 (clone BVD4-1D11), IL-10 (clone JES5-2A5), and IFN-γ (clone R4-6A2) were from BD Biosciences. Recombinant mouse cytokines (BD Biosciences) were used as standards. Biotinylated rat mAbs to mouse IL-2 (clone JES6-5H4), IL-4 (clone BVD6-24G2), IL-10 (clone SXC-1), IFN-γ (clone XMG1.2), and TNF-α (clone MP6-XT3) were used as the second antibody. Assays were developed with TMB microwell peroxidase substrate (Kirkegaard and Perry Laboratories) and read at 450 nm using a Benchmark microplate reader (Bio-Rad Laboratories).
Generation of Tim2 Ig fusion protein.
The DNA sequence containing the extracellular domain of Tim-2 was amplified by PCR and cloned into a vector containing the CD5 signal sequence and the human IgG1 constant region. NS.1 cells were transfected stably, and recombinant protein was purified from supernatant using protein A chromatography.
Induction of experimental autoimmune encephalomyelitis.
Female SJL mice (4–8 wk old; The Jackson Laboratory) were injected s.c. in each flank with 50–75 μg of PLP 139–151 peptide in CFA supplemented with 400 μg of Mycobacterium tuberculosis (Difco). Each mouse also was injected i.v. with 100 ng of pertussis toxin (List Biological Laboratories) in 0.1 ml of PBS. Mice were injected i.p. on alternate days beginning at day 0 and continuing for 8 d with 100 μg Tim-2 Ig (endotoxin activity of 0.2 to 0.8 EU/mg) or 100 μg control hIgG or 100 μL PBS. Mice were examined daily for signs of EAE, which were graded as follows: flaccid tail, 1; uneven gait and impaired righting reflex, 2; total hindlimb paralysis, 3; fore- and hindlimb paralysis, 4; and moribund, 5. Disease scores over the course of the experiment (25 d) were totaled for each animal, and the mean for the experimental group was expressed as a cumulative score. Significance was established using the Student's t test. At the peak of the disease or at the end of the experiment, brains and spinal cords were removed, fixed in 10% formalin, and examined histopathologically for inflammation and demyelination. Inflammatory foci in meninges and parenchyma were counted by a blinded observer as described previously (20). Mice were housed in a specific pathogen-free, viral antibody–free animal facility at the Harvard Institutes of Medicine. All breeding and experiments were performed in accordance with the guidelines of the Committee on Animals of Harvard Medical School.
Acknowledgments
This work was supported by the National Multiple Sclerosis Society (RG-2571-D-9 and FG-1478-A-1), and the National Institutes of Health (NS056141-02, 1R01NS045937-01, 2R01NS35685-06, 2R37NS30843-11, 1R01A144880-03, 2P01A139671-07, 1P01NS38037-04, 1F31GM20927-01). S. Chakravarti is a recipient of a National Multiple Sclerosis Society Postdoctoral Fellowship. V.K. Kuchroo is the recipient of a Javits Neuroscience Investigator Award from the National Institutes of Health. Research in the laboratory is funded, in part, by Forest Health, Inc.
The authors have no conflicting financial interests.
Abbreviations used: EAE, experimental autoimmune encephalomyelitis; HA, hemagglutinin A; HAV, hepatitis A virus; hIgG, human IgG; PLP, proteolipid protein; Tim, T cell immunoglobulin mucin-domain containing.
References
- 1.Constant, S.L., and K. Bottomly. 1997. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 15:297–322. [DOI] [PubMed] [Google Scholar]
- 2.O'Garra, A., and N. Arai. 2000. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 10:542–550. [DOI] [PubMed] [Google Scholar]
- 3.Szabo, S.J., B.M. Sullivan, S.L. Peng, and L.H. Glimcher. 2003. Molecular mechanisms regulating Th1 immune responses. Annu. Rev. Immunol. 21:713–758. [DOI] [PubMed] [Google Scholar]
- 4.Abbas, A.K., K.M. Murphy, and A. Sher. 1996. Functional diversity of helper T lymphocytes. Nature. 383:787–793. [DOI] [PubMed] [Google Scholar]
- 5.Liblau, R.S., S.M. Singer, and H.O. McDevitt. 1995. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol. Today. 16:34–38. [DOI] [PubMed] [Google Scholar]
- 6.Sher, A., and R.L. Coffman. 1992. Regulation of immunity to parasites by T cells and T cell-derived cytokines. Annu. Rev. Immunol. 10:385–409. [DOI] [PubMed] [Google Scholar]
- 7.Loetscher, P., M. Uguccioni, L. Bordoli, M. Baggiolini, B. Moser, C. Chizzolini, and J.M. Dayer. 1998. CCR5 is characteristic of Th1 lymphocytes. Nature. 391:344–345. [DOI] [PubMed] [Google Scholar]
- 8.Sallusto, F., A. Lanzavecchia, and C.R. Mackay. 1998. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today. 19:568–574. [DOI] [PubMed] [Google Scholar]
- 9.Bonecchi, R., G. Bianchi, P.P. Bordignon, D. D'Ambrosio, R. Lang, A. Borsatti, S. Sozzani, P. Allavena, P.A. Gray, A. Mantovani, and F. Sinigaglia. 1998. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monney, L., C.A. Sabatos, J.L. Gaglia, A. Ryu, H. Waldner, T. Chernova, S. Manning, E.A. Greenfield, A.J. Coyle, R.A. Sobel, et al. 2002. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 415:536–541. [DOI] [PubMed] [Google Scholar]
- 11.Kuchroo, V.K., D.T. Umetsu, R.H. DeKruyff, and G.J. Freeman. 2003. The TIM gene family: emerging roles in immunity and disease. Nat. Rev. Immunol. 3:454–462. [DOI] [PubMed] [Google Scholar]
- 12.McIntire, J.J., S.E. Umetsu, O. Akbari, M. Potter, V.K. Kuchroo, G.S. Barsh, G.J. Freeman, D.T. Umetsu, and R.H. DeKruyff. 2001. Identification of Tapr (an airway hyperreactivity regulatory locus) and the linked Tim gene family. Nat. Immunol. 2:1109–1116. [DOI] [PubMed] [Google Scholar]
- 13.Sabatos, C.A., S. Chakravarti, E. Cha, A. Schubart, A. Sanchez-Fueyo, X.X. Zheng, A.J. Coyle, T.B. Strom, G.J. Freeman, and V.K. Kuchroo. 2003. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 4:1102–1110. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Fueyo, A., J. Tian, D. Picarella, C. Domenig, X.X. Zheng, C.A. Sabatos, N. Manlongat, O. Bender, T. Kamradt, V.K. Kuchroo, et al. 2003. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 4:1093–1101. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan, G., A. Totsuka, P. Thompson, T. Akatsuka, Y. Moritsugu, and S.M. Feinstone. 1996. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 15:4282–4296. [PMC free article] [PubMed] [Google Scholar]
- 16.Ichimura, T., J.V. Bonventre, V. Bailly, H. Wei, C.A. Hession, R.L. Cate, and M. Sanicola. 1998. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J. Biol. Chem. 273:4135–4142. [DOI] [PubMed] [Google Scholar]
- 17.Umetsu, S.E., W.L. Lee, J.J. McIntire, L. Downey, B. Sanjanwala, O. Akbari, G.J. Berry, H. Nagumo, G.J. Freeman, D.T. Umetsu, and R.H. DeKruyff. 2005. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat. Immunol. 6:447–454. [DOI] [PubMed] [Google Scholar]
- 18.Meyers, J.H., S. Chakravarti, D. Schlesinger, Z. Illes, H. Waldner, S.E. Umetsu, J. Kenny, X.X. Zheng, D.T. Umetsu, R.H. DeKruyff, et al. 2005. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat. Immunol. 6:455–464. [DOI] [PubMed] [Google Scholar]
- 19.Kumanogoh, A., S. Marukawa, K. Suzuki, N. Takegahara, C. Watanabe, E. Ch'ng, I. Ishida, H. Fujimura, S. Sakoda, K. Yoshida, and H. Kikutani. 2002. Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature. 419:629–633. [DOI] [PubMed] [Google Scholar]
- 20.Kuchroo, V.K., M.P. Das, J.A. Brown, A.M. Ranger, S.S. Zamvil, R.A. Sobel, H.L. Weiner, N. Nabavi, and L.H. Glimcher. 1995. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 80:707–718. [DOI] [PubMed] [Google Scholar]
- 21.Nicholson, L.B., J.M. Greer, R.A. Sobel, M.B. Lees, and V.K. Kuchroo. 1995. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 3:397–405. [DOI] [PubMed] [Google Scholar]
- 22.Zamvil, S.S., and L. Steinman. 1990. The T lymphocyte in experimental allergic encephalomyelitis. Annu. Rev. Immunol. 8:579–621. [DOI] [PubMed] [Google Scholar]
- 23.Feigelstock, D., P. Thompson, P. Mattoo, Y. Zhang, and G.G. Kaplan. 1998. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J. Virol. 72:6621–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumanogoh, A., T. Shikina, K. Suzuki, S. Uematsu, K. Yukawa, S. Kashiwamura, H. Tsutsui, M. Yamamoto, H. Takamatsu, E.P. Ko-Mitamura, et al. 2005. Nonredundant roles of Sema4A in the immune system: defective T cell priming and Th1/Th2 regulation in Sema4A-deficient mice. Immunity. 22:305–316. [DOI] [PubMed] [Google Scholar]
- 25.McIntire, J.J., S.E. Umetsu, C. Macaubas, E.G. Hoyte, C. Cinnioglu, L.L. Cavalli-Sforza, G.S. Barsh, J.F. Hallmayer, P.A. Underhill, N.J. Risch, et al. 2003. Immunology: hepatitis A virus link to atopic disease. Nature. 425:576. [DOI] [PubMed] [Google Scholar]