

Abstract
Antiviral cell–mediated immunity is initiated by the dendritic cell (DC) network in lymph nodes (LNs). Plasmacytoid DCs (pDCs) are known to migrate to inflamed LNs and produce interferon (IFN)-α, but their other roles in antiviral T cell immunity are unclear. We report that LN-recruited pDCs are activated to create local immune fields that generate antiviral cytotoxic T lymphocytes (CTLs) in association with LNDCs, in a model of cutaneous herpes simplex virus (HSV) infection. Although pDCs alone failed to induce CTLs, in vivo depletion of pDCs impaired CTL-mediated virus eradication. LNDCs from pDC-depleted mice showed impaired cluster formation with T cells and antigen presentation to prime CTLs. Transferring circulating pDC precursors from wild-type, but not CXCR3-deficient, mice to pDC-depleted mice restored CTL induction by impaired LNDCs. In vitro co-culture experiments revealed that pDCs provided help signals that recovered impaired LNDCs in a CD2- and CD40L-dependent manner. pDC-derived IFN-α further stimulated the recovered LNDCs to induce CTLs. Therefore, the help provided by pDCs for LNDCs in primary immune responses seems to be pivotal to optimally inducing anti-HSV CTLs.
Viruses have evolved a variety of mechanisms to evade the immune system, invade host tissues, and, sometimes, establish persistent infections (1). One strategy by which hosts counter these assaults is the rapid response of the DC network to eliminate virally infected cells. In mice, there are at least three major functional subtypes of DCs in LNs: myeloid DCs (mDCs; CD11b+B220–CD11c+), CD8α+ DCs (CD8α+B220–CD11c+), and plasmacytoid DCs (pDCs; B220+CD11c+), which induce distinct types of antiviral T lymphocytes (2, 3). This heterogeneity in the DC network allows flexibility in the immune response, depending on the tissue environment and exogenous factors in LNs (2, 4). In HSV-2 infections of the vaginal submucosa, newly recruited mDCs induce virus-specific T helper cells (5), whereas in cutaneous HSV-1 infections, CD8α+ DCs, which mainly reside in LNs, are responsible for cross-priming antiviral CTLs (6). In contrast, pDCs function poorly as APCs for naive T cells, but produce large amounts of antiviral IFN-α. However, the contribution of pDCs to antiviral immunity in vivo remains controversial, as they have been reported to promote a variety of immune responses in vitro (3, 7).
Recent investigations have revealed that tissue-resident mDCs and pDCs in a normal, infection-free animal maintain peripheral tolerance (7, 8). In response to inflammation, these DCs are activated to induce T cell–mediated immunity, but the mechanism by which the DC system is converted from “tolerogenic” to “immunogenic” remains controversial (3, 8). We are particularly interested in the role of inflammation-associated circulating DC precursors in this conversion (9–11). In a cutaneous HSV-1 infection model, we recently demonstrated that large numbers of both mDC and pDC precursors appear de novo in the circulation in response to TNF-α (10). mDC precursors are preferentially recruited to sites of inflammation and remobilized into draining LNs, where they act as APCs (9, 10). In contrast, pDC precursors directly enter the LNs through activated high endothelial venules (HEVs) in a CXCL9- and E-selectin–dependent manner (10). This inflammation-dependent, CXCR3-driven trafficking pathway is distinct from that used by mDCs, but similar to that used by monocytes (12) and NK cells (13). This suggests that LN-recruited pDCs and mDCs have distinct functions, but the fate of pDCs after entering the LNs has not been established in vivo.
We have studied the contribution of LN-recruited pDCs to anti-HSV CTL-mediated immunity and have shown that they are associated with LNDCs as well as 5′-bromo-2′-deoxyuridine (BrdU)–positive T cells in vivo, but act as poor APCs for IFN-γ+ CTLs. In vivo depletion of LN pDCs using three distinct methods and in vivo reconstitution assays using freshly isolated pDC precursors showed that LN-recruited pDCs are able to convert virally impaired LNDC networks into completely activated ones that are able to induce anti-HSV CTLs. Although the DC–virus interplay is dependent on the type and dose of virus, this study provides novel insights into the role of pDCs in antiviral CTL immunity.
Results
Recruitment and activation of pDCs in inflamed LNs
We have traced the fate of pDCs (B220+CD11c+CD3−–CD19−DX5−) in draining popliteal LNs (PLNs) after infecting mice with HSV-1 in the right footpad. The frequency and number of PLN pDCs peaked on day 2 and decreased thereafter (Fig. 1 A). The increase of PLN pDCs was largely caused by accelerated recruitment of pDC precursors directly from the circulation, as previously reported (10). Although blood pDC precursors (MHCII−B220+CD11c+CD3−CD19−DX5−) did not express CD86, CD40, and CD40 ligand (CD40L) on day 2 (Fig. 1 B), the expression of these activation markers was up-regulated on PLN pDCs on day 2 (Fig. 1 C). PLN pDCs isolated on day 2 and restimulated in vitro with irradiated HSV produced high levels of IFN-α, but uninfected PLN pDCs and blood pDC precursors did not (Fig. 1 D). These results suggested that pDCs were activated after entering the draining LNs and produced cytokines on encountering HSV-1.
Figure 1.

Recruitment and activation of pDCs in inflamed LNs. (A) The frequency (top) and the absolute numbers (bottom) of B220+CD11c+ pDCs in PLNs (rt. PLN) from uninfected (d0) or HSV-infected mice on day 2 (d2). (B) Expression of CD86, CD40, and CD40L on blood pDC precursors from uninfected (d0) or HSV-infected mice on day 2 (d2). (C) Expression of CD86, CD40, and CD40L on LN CD11c+ DCs from uninfected (d0) or HSV-infected mice on day 2 (d2). The percentages of cells are indicated. (D) IFN-α production by sorted pDC precursors from HSV-infected mice on day 2 (pDCpre) and PLN pDCs from uninfected (LN pDC d0) or HSV-infected mice on day 2 (LN pDC d2) after 16 h of incubation with irradiated HSV. (A and D) Representative values from three independent experiments are presented as the mean ± SD (n = 6).
Recruited pDCs contact physically with LNDCs
Before infection, only small numbers of pDCs were found in the paracortex, a T cell zone of PLNs (Fig. 2 A). However, a new influx of pDCs appeared in the paracortex on day 2 and colocalized with B220− LNDCs (mDCs and CD8α+ DCs; Fig. 2 B). To determine whether pDCs were attracted by LNDCs after entering LNs, we adoptively transferred carboxyfluorescein succinimidyl ester (CFSE)–labeled pDC precursors i.v. into HSV-infected mice on day 2. By 2 h, the transferred pDC precursors were in close contact with LNDCs in the paracortex, and some were associated with pDCs in the perifollicular zone (Fig. 2 C). 48 h later, transferred pDC precursors were still retained in these zones around HEVs (Fig. 2 D). The transferred pDCs did not always attach to CCL21+ stromal cells (Fig. 2 D), suggesting that other chemokines derived from LNDCs or pDCs might preferentially attract pDCs. Because LNDC-derived CXCL10 mediates DC–T cell interactions in inflamed LNs (14), the CXCL10–CXCR3 pathway must play an important role in inflammatory conditions. PLN pDCs on day 2 expressed high levels of CXCL10 (Fig. 2 E), which is known to attract Th1 precursor cells (14), as well as pDCs themselves (10).
Figure 2.

Recruited pDCs contact LNDCs in a CXCL10- and CD2-dependent manner. (A and B) Distribution of B220−CD11c+ LNDCs (red) and B220+CD11c+ pDCs (yellow) in the T cell zones in normal (A, d0) and HSV-infected (B, d2) PLN. Arrows indicate contact between pDCs and LNDCs on day 2. Bars: (A and B, left) 40 μm; (B, right) 20 μm. (C) CFSE-labeled pDC precursors (green) were adoptively transferred into HSV-infected mice on day 2. PLNs 2 h after transfer (d2) are shown. CD11c is red and B220 is blue. Transferred pDCs (green/white) are in contact with B220−CD11c+ LNDCs (bottom right, red) and B220+CD11c+ pDCs (top right, pink). (D) PLNs 48 h after transfer (d4). CCL21 is red and B220 is blue. F, follicle; PC, paracortex. Bar, (C and D) 80 μm. (E) Relative expression levels of CXCL10 by freshly isolated pDC precursors from HSV-infected mice on day 2 (pDCpre) and PLN pDCs of uninfected (LN pDC d0) or HSV-infected mice on day 2 (LN pDC d2). The housekeeping enzyme hypoxanthine phosphoribosyl transferase (HPRT) was used as an internal standard. (F) Expression of CD2 on blood pDC precursors and PLN pDCs from uninfected (d0) or HSV-infected mice on day 2 (d2). (G) CD40L expression (red) on d2 LNDC–pDC conjugates. A CFSEhigh LNDC (asterisk) and three CFSElow pDCs are shown. High-contrast image shows that CD40L is expressed on the edge of pDCs (bottom right). Bar, 20 μm. (H) The effect of blocking Abs against CD2 and CD2 + CXCL10 on the percentage of LNDCs forming clusters, using PLN DCs from uninfected (d0) and HSV-infected mice on day 2 (d2). Representative data from three independent experiments are presented as the mean ± SD (n = 6). *, P < 0.05 by the Student's t test, comparing mice treated with control and blocking Abs.
Blood pDC precursors constitutively express high levels of adhesion molecules, including VLA-4/α4-integrin (97%) and CD11a/LFA-1 (99%; reference 10), but low levels of CD2/LFA-2. However, CD2 was up-regulated on day 2, but only on PLN pDCs (Fig. 2 F), indicating that activated pDCs up-regulate CD2 and CXCL10 rapidly after entering the LNs. To understand the mechanism by which recruited pDCs and LNDCs physically associate, we performed ex vivo clustering assays using pDCs and LNDCs isolated from PLNs. After 2 h of incubation at 40 revolutions per minute (rpm), the number of cluster-forming LNDCs was counted. Strong conjugates were seen only using DCs isolated from infected LNs, and CD40L was detected on the boundary (Fig. 2, G and H). Cell conjugates were significantly inhibited by treatment with anti-CD2 mAb alone or in combination with anti-CXCL10 mAb (P < 0.05; Fig. 2 H). This suggested that LNDC- and pDC-derived CXCL10 attracted pDCs, and that CXCR3 and CD2 on pDCs mediated their binding.
In vivo depletion of LN-recruited pDCs impairs CTL-mediated virus eradication
To investigate the contribution of LN-recruited pDCs to anti–HSV-1 CTL immunity, we used three independent approaches to deplete LN-recruited pDCs in vivo. We tested PLN CD8+ T cells on day 7 for CTL precursor frequency, using an IFN-γ ELISPOT assay, and for CTL function, using an in vitro cytotoxicity assay against HSV-1–infected syngeneic splenocytes.
First, CXCR3-driven LN recruitment of pDCs was inhibited by the combined blockade of CXCL9 and E-selectin (10). A single injection of 100 μg of blocking anti-CXCL9 and anti–E-selectin antibodies (Abs) on day 0 almost completely blocked the increase in pDCs, but not LNDCs, in the PLNs (Fig. 3 A). This was mostly because of the selective inhibition of pDC precursor transmigration across inflamed LN HEVs (10). Both the induction of IFN-γ+ CTLs and CTL function were detected in CD8+ T cells isolated from PLNs from control mice, but reduced in mice treated with blocking Abs (Fig. 3, B and C). Control mice were able to eliminate HSV-1 to a significant extent, but the virus persisted in the anti-CXCL9 and anti–E-selectin Ab–treated mice (P < 0.05; Fig. 3 D). Although these findings showed that the early recruitment (Fig. 3 A) of pDC precursors into the inflamed LNs was crucial for generating functional anti–HSV-1 CTLs, we performed further experiments to rule out the possibility that a CXCL9 blockade was affecting late T cell responses, as well as early pDC effects.
Figure 3.

In vivo depletion of pDCs impairs CTL-mediated virus eradication. The effect of anti-CXCL9 and anti–E-selectin Abs on (A) the numbers of B220− LNDCs (mDCs and CD8α+ DCs) and B220+ pDCs in PLNs after HSV infection, (B) the numbers of IFN-γ+ spots produced by PLN CD8+ T cells, (C) the specific lysis in vitro by PLN CD8+ T cells, and (D) the virus titer in PLNs. The effect of anti-Ly6G/C Ab on (E) the numbers of B220− LNDCs (mDCs and CD8α+ DCs) and B220+ pDCs in PLNs after HSV infection, (F) the numbers of IFN-γ+ spots produced by PLN CD8+ T cells, (G) the specific lysis in vitro by PLN CD8+ T cells, and (H) the virus titer in PLNs. The effect of anti–PDCA-1 mAb on (I) the numbers of B220− LNDCs (mDCs and CD8α+ DCs) and B220+ pDCs in PLNs after HSV infection, (J) the numbers of IFN-γ+ spots produced by PLN CD8+ T cells, and (K) the specific lysis in vitro by PLN CD8+ T cells. Representative data from three independent experiments are presented as the mean ± SD (n = 6 or n = 5 for anti–PDCA-1 mAb experiments. *, P < 0.05 by Student's t test, comparing mice treated with control and blocking Abs.
Our second approach used anti-Ly6G/C Ab to deplete the pDC population without affecting other APCs, B cells, and macrophages (15). Injecting mice twice with this antibody depleted ∼70% of PLN-resident pDCs (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20041961/DC1) and inhibited the influx of new pDCs during the experiment without affecting the numbers of LNDCs (Fig. 3 E) or the expression of CXCL9 and E-selectin on inflamed HEVs (Fig. S1 B). On day 7 after infection, mice treated with anti-Ly6G/C Ab showed impaired CTL induction and function (Fig. 3, F and G) and poor viral elimination (Fig. 3 H), compared with control mice.
The third method used anti–pDC antigen (PDCA)-1 Ab, which is more specific for pDCs (16). Injecting mice twice with 100 μg of anti–PDCA-1 Ab on days –1.5 and 0.5 depleted ∼80% of PLN-resident pDCs (Fig. S1 C) without affecting LNDCs. This protocol also inhibited the increase in pDCs, but not LNDCs, in the PLNs (Fig. 3 I). The number of IFN-γ+ CTLs and the CTL activity were again reduced in anti–PDCA-1 Ab–treated mice, compared with control mice (Fig. 3, J and K).
Impaired APC function of LNDCs in anti-Ly6G/C Ab–treated mice
We next investigated the stage at which CTL generation was affected by the reduction in recruited pDCs in anti-Ly6G/C mAb–treated mice. To distinguish the APC activities of different LN DC subtypes, we isolated them from HSV-infected LNs and tested their abilities to stimulate IFN-γ production by HSV-infected LN T cells in vitro, without any further restimulation. To our surprise, LN pDCs alone poorly induced HSV-specific IFN-γ production (Fig. 4 A) by both CD4+ and CD8+ T cells, indicating that they were not themselves acting as APCs in vivo. In contrast, mDCs preferentially primed HSV-specific CD4+ T cells, and CD8α+ DCs selectively primed HSV-specific CTLs (Fig. 4 A), which was consistent with previous reports (6, 7). However, mDCs and CD8α+ DCs isolated from anti-Ly6G/C Ab–treated mice on day 2 failed to prime anti-HSV CD4+ or CD8+ T cells (Fig. 4 A).
Figure 4.
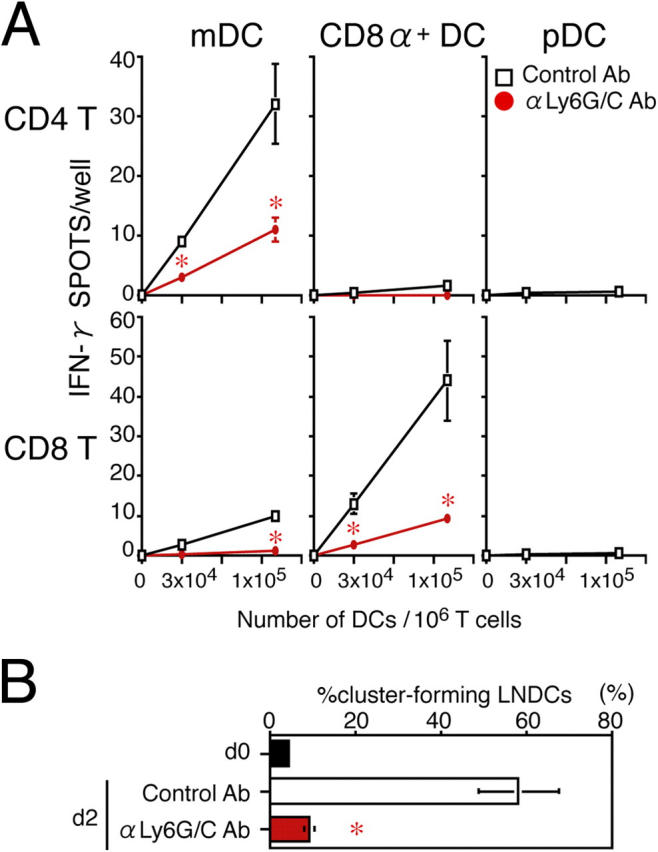
Impaired APC function of LNDCs in pDC-depleted mice. (A) The number of IFN-γ+ spots produced by CD4+ or CD8+ T cells obtained from control PLNs on day 2, after 16-h incubations with the indicated numbers of DCs with no in vitro restimulation. DCs alone formed no spots. (B) The percentage of cluster-forming LNDCs from uninfected mice (d0), HSV-infected, control Ab–treated mice on day 2, and HSV-infected, anti-Ly6G/C Ab–treated mice on day 2. Indicated LNDCs were incubated for 2 h with LN CD3+ T cells from HSV-infected mice on day 2. T cells alone rarely formed clusters (<5%) in all groups tested. Representative data from three independent experiments are presented as the mean ± SD (n = 3). *, P < 0.05 by Student's t test, comparing mice treated with control or anti-Ly6C/G Ab.
LNDCs from anti-Ly6G/C Ab–treated mice did not show reduced HSV uptake (5.3 ± 1.1% in anti-Ly6G/C Ab–treated mice vs. 5.9 ± 1.4% in control mice, on cytosmear preparations) or accumulation in LNs (Fig. 3 E). To examine the ability of LNDCs to attract T cells, we performed ex vivo clustering assays using B220−CD11c+ LNDCs and CD3+ T cells isolated from PLNs from infected mice. Cluster formation was significantly higher with cells isolated from mice treated with control Ab, compared with anti-Ly6G/C Ab (P < 0.05; Fig. 4 B). As a result, mDCs and CD8α+ DCs from the anti-Ly6G/C Ab–treated mice also failed to form clusters with T cells and to induce IFN-γ–producing CD4+ or CD8+ T cells, respectively (Fig. 4 A).
Reconstitution of anti-Ly6G/C Ab–treated mice with pDC precursors restores CTL induction by LNDCs in vivo
To find out whether the impaired CTL responses observed in anti-Ly6G/C Ab–treated mice were actually dependent on LN-recruited pDCs, we performed in vivo reconstitution assays by transferring blood pDC precursors i.v. into HSV-infected, anti-Ly6G/C Ab–treated mice (Fig. 5 A). We used freshly isolated blood pDC precursors because maturing pDCs, restimulated in vitro, poorly enter LNs when transferred (10). The donor pDC precursors were characterized as nonantigen-pulsed, nonactivated cells (Fig. 1 B), which did not produce cytokines or chemokines unless they entered infected LNs (Fig. 1 D and Fig. 2 F). Blood pDC precursors from Propionibacterium acnes–primed mice were also used to see whether recruitment to LNs was antigen-dependent or not.
Figure 5.

Reconstitution of pDC-depleted mice with pDC precursors restores CTL induction. (A) Donor precursor cells isolated from the peripheral blood of HSV-infected WT mice (WT pDC), HSV-infected CXCR3-deficient mice (CXCR3−/− pDC), and P. acnes–primed WT mice (WT pDC [p.a.]) on day 2 were labeled with fluorescence dyes and adoptively transferred into HSV-infected, anti-Ly6G/C Ab–treated mice. LNDCs were isolated from reconstituted mice on day 2 of infection, and LN CD8+ T cells were isolated from reconstituted mice on day 7 of infection, for functional tests. The letters in parentheses correspond to panels in the figure. (B) Effective recruitment of WT, but not CXCR3−/−, pDC precursors on day 2 of infection. (left and middle) Entry of CMTMR-labeled pDC precursors (orange) in the T cell zone (blue, CD3; green, B220) of PLN 2 h after cell transfer. Magnification, 200. (right) The number of transferred pDCs observed in the T cell zone of PLN on day 2 of infection. Representative data from three independent experiments are presented as the mean ± SD (n = 3 for 15-mm2 sections). *, P < 0.05 by the Student's t test, comparing WT with CXCR3−/− pDCs. (C) CFSE-labeled WT pDC precursors (green) in contact with DEC-205+ (red) LNDCs in the T cell zone of PLN 2 h after cell transfer (day 2 of infection) at a magnification of 200. The inset shows a higher magnification (400). (D) CFSE-labeled WT pDC precursors in contact with BrdU+ (red) and CD4+ (blue) T cells of PLN 48 h after cell transfer (day 4 of infection) at a magnification of 200. A higher magnification is indicated by the box (400). F, follicle; PC, paracortex; S, sinus. (E) The number of IFN-γ+ spots produced by PLN CD4+ or CD8+ T cells (106 cells/well) from HSV-infected mice on day 2, after 16 h of incubation with LNDCs with no in vitro restimulation. LNDCs (mDCs or CD8α+ DCs; 105 cells/well) were obtained from anti-Ly6G/C mAb–treated mice reconstituted with WT pDC or CXCR3−/− pDCs on day 2. DCs alone produced no spots. (F) The percentage of cluster-forming LNDCs obtained from anti-Ly6G/C mAb–treated mice reconstituted with WT pDC, CXCR3−/− pDC, or WT pDC (p.a.) on day 2. Freshly isolated LNDCs were incubated with LN CD3+ T cells from HSV-infected mice for 2 h. (G) Specific lysis in vitro of PLN CD8+ T cells and virus titers of PLNs obtained from pDC-depleted mice reconstituted with WT pDCs, CXCR3−/− pDCs, or WT pDCs (p.a.) on day 7. Values for mice treated with control Ab (black dashed lines) and anti-Ly6G/C Ab (red dashed lines) on days 2 (F) and 7 (G) of infection are also shown to compare reconstituted mice with nonreconstituted mice as described in Figs. 3, G and H and Fig. 4 B. Representative data from three independent experiments are presented as the mean ± SD (n = 3). *, P < 0.05 by Student's t test, comparing WT with CXCR3−/− pDCs.
In anti-Ly6G/C Ab–treated mice reconstituted with WT pDC precursors, significant numbers of pDCs were recruited to the paracortex of the PLNs (P < 0.05; Fig. 5 B) and made contact with DEC-205+ LNDCs (Fig. 5 C) on day 2 of infection. Recruited pDCs were still detectable on day 4 of the infection (48 h after transfer) and were associated with increased numbers of BrdU+CD4+ T cells in the T cell zone (Fig. 5 D). To investigate whether the function of LNDCs was restored, LNDCs from reconstituted mice were tested for in vivo APC activity and the ability to attract T cells. mDCs and CD8α+ DCs isolated from reconstituted mice primed anti-HSV CD4+ or CD8+ T cells (compare Fig. 5 E with Fig. 4A). The ability of LNDCs to form clusters with T cells was also restored (Fig. 5 F). On day 7, the functional CTL responses were restored, and HSV was effectively eliminated after reconstitution with blood pDC precursors (Fig. 5 G). Interestingly, blood pDC precursors obtained from P. acnes–primed mice could restore the host responses (Fig. 5, B, F, and G), suggesting that pDC precursors could enter the inflamed LNs in an antigen-independent manner but flexibly respond to an antigen-bearing environment after entry.
In contrast, when anti-Ly6G/C Ab–treated mice were reconstituted with blood pDC precursors from CXCR3–/– mice, donor pDCs did not increase in the LNs (Fig. 5 B), and neither LNDC function nor anti-HSV host responses were restored (Fig. 5, E–G). These results demonstrated that CXCR3-mediated recruitment of pDC precursors to LNs is required to induce an anti-HSV CTL response in vivo.
LN-recruited pDCs restore CTL induction by impaired LNDCs in vitro
The ability of LN-recruited pDCs to restore the impaired induction of anti-HSV CTLs by LNDCs was next examined in co-culture experiments (Fig. 6 A). LN pDCs from mice treated with control Ab (pDC) and LNDCs from anti-Ly6G/C Ab–treated mice (impaired LNDC) on day 2 of infection failed to induce anti-HSV CTLs in vitro (Fig. 6 B). Because pDCs are major producers of cytokines (references 7, 15, 17, 18; Fig. 1 D), we tested the effect of several cytokines on CTL induction by impaired LNDCs in this system. However, adding IFN-α (Fig. 6 B) or other pDC cytokines, including TNF-α, IL-10, and IL-12 (unpublished data) was not sufficient to restore the impaired LNDCs in co-culture. In fact, co-culture with LN pDCs was essential to restore the ability of impaired LNDCs to generate anti-HSV CTLs, and this process was dose dependent (Fig. 6 C). In addition, when pDCs and impaired LNDCs were co-cultured in separate compartments of transwells, anti-HSV CTL activity was not restored (Fig. 6 C), demonstrating that the help provided by LN-recruited pDCs to completely activate anti-HSV CTLs depends on cell–cell contact.
Figure 6.

CTL induction was restored using in vitro co-cultures with LN pDCs. (A) Diagram showing the protocol for co-culture experiments. CTL activities were estimated after 48 h of co-culture. The letters in parentheses correspond to panels in the figure. Ctl LN, HSV-infected PLN treated with control Ab at day 2. (B) In vitro CTL activity induced by pDCs (purple line), LNDCs (pink line), and impaired LNDCs in the presence (orange line) or absence (green line) of rIFN-α. (C) In vitro CTL activities induced by impaired LNDCs in the presence of pDCs, at ratios between 1:1 and 1:10, or with pDCs separated by transwells (red line, 1:1 ratio). (D, top) Diagram showing the protocol for two-step co-culture experiments. (top row) Step one, co-culture of impaired LNDCs and pDCs for 16 h. (bottom row) Addition of LN CD8+ T cells for a further 48 h. (bottom) In vitro CTL activities induced by impaired LNDCs in the presence of equal numbers of pDCs, treated as indicated in the figure. Effector/target (E:T) ratio, 30:1. Representative data from three independent experiments are presented as the mean ± SD (n = 3). *, P < 0.05 by Student's t test, comparing cells treated with control and blocking Abs. (E) in vitro CTL activities induced by impaired LNDCs in the presence of equal numbers of WT or CD40L−/− pDCs. E:T ratio, 30:1. Representative data from three independent experiments are presented as the mean ± SD (n = 3).
To investigate which molecules are involved in functional recovery of impaired LNDCs in the presence of pDCs, we set up a two-step co-culture system (Fig. 6 D). In step one, impaired LNDCs and pDCs were co-cultured for 16 h. After washing, LN CD8+ T cells were added and co-cultured for a further 48 h as step two. Impaired LNDC-mediated CTL induction was restored in these culture conditions (Fig. 6 D, left), indicating that the initial interaction with pDCs provided sufficient signals to activate LNDCs to induce CTLs. When different blocking Abs were added during step one, pDC-mediated recovery was not inhibited by anti-CD86, -CD80, or -CD54 Abs (unpublished data), but was significantly inhibited by anti-CD2 and -CD40L Abs (P < 0.05; Fig. 6 D, middle). Although LN pDCs readily produce IFN-α, even in short-term culture (Fig. 1 D), adding anti–IFN-α Ab during step one did not affect the restoration of LNDC function (Fig. 6 D, middle). However, when anti–IFN-α Ab was added during step two, CTL activity was inhibited (Fig. 6 D, right). Moreover, pDCs from CD40L−/− mice failed to restore the function of LNDCs (Fig. 6 E). Collectively, these results provide evidence that LNDC–pDC contact, involving the CD2 and CD40L pathways, is initially required to restore impaired LNDC function, and IFN-α promotes CTL priming by LNDCs, once the provision of pDC help has converted their status from “impaired” into “recovered.”
Discussion
Viruses, including HSV-1, can target DCs and disrupt the antigen-presenting pathway at multiple levels, leading to impairment of DC-mediated CTL immunity (1). Indeed, HSV-infected hosts have both activated DCs, responsible for T cell priming, and impaired DCs, showing reduced transport of MHC–peptide complexes, T cell stimulatory capacity, and cytokine production (1, 19–22). Therefore, the balance between the host DC networks and viruses, that is, the balance between DC activation and impairment, would determine the extent of antiviral host immunity. The mechanisms that skew the DC network toward activation are not completely understood but require the host to mount functional CTL immunity and to prevent viral escape. We have shown that pDCs contribute to the activation of the LNDC network after migrating to inflamed LNs. Although pDCs do not directly prime anti-HSV T cells, they help LNDCs to prime anti-HSV CTLs by multiple mechanisms.
In response to local HSV-1 infection, pDC precursors are rapidly mobilized into the circulation and recruited to inflamed LNs (10). Here, pDCs up-regulate activation markers and T cell–attracting chemokines, change the integrins they express, and produce cytokines in response to encountering HSV. LN-recruited pDCs are retained for at least 48 h in the paracortex and associate with LNDCs. Thus, pDCs actively participate in ongoing T cell responses by creating appropriate cytokine fields in their vicinity and attracting immune cells by secreting chemokines. CXCR3 on pDCs plays an important role in mediating both trans-HEV migration (10) and DC–DC interactions in this model.
To inhibit the early, short-term accumulation of LN pDCs (Fig. 1 A), we injected anti-CXCL9 and E-selectin Abs or anti-Ly6G/C Ab at the time of HSV inoculation. LN T cells isolated on day 2 from HSV-infected mice, treated with either blocking Ab regime, could proliferate when stimulated ex vivo with HSV-pulsed splenic DCs and migrate to HSV-infected PLNs when adoptively transferred (unpublished data). Although these findings suggested that the blocking Abs affected LN pDCs selectively at an early stage, we could not completely rule out the possibility that the blocking Abs had a direct affect on activated T cells at a later phase of infection. To rule this out, we performed in vivo reconstitution assays. Taking advantages of CXCR3-mediated trans-HEV migration of circulating pDC precursors (10), we intravenously transferred 2 × 106 freshly isolated blood pDC precursors per mouse, after treatment with anti-Ly6G/C Ab (Fig. 5 A). This was a greater number of blood pDC precursor cells than is present in an uninfected mouse (∼104) or an HSV-infected mouse (∼1–3 × 105; reference 10). Because depletion of pDCs by anti-Ly6G/C Ab treatment was incomplete, reducing the numbers by ∼70%, as described above, it is possible that sufficient numbers of adoptively transferred pDCs survived, even in the presence of anti-Ly6G/C Ab in the circulation. We actually detected a significant (P < 0.05) number of transferred pDCs in inflamed LNs (Fig. 5, B–D) from day 2 to 4 of infection, but this temporal presence seems to be enough for these cells to function, as accumulation of endogenous pDCs is also short-term (Fig. 1 A) in this model. In addition, a complete anti-HSV CTL response was mounted even in reconstituted, anti-Ly6G/C Ab–treated mice (Fig. 5, F and G), ruling out the possibility that this Ab directly affected activated CD8+ T cells. Furthermore, the impairment of the CTL response in mice treated with anti–PDCA-1 Ab (Fig. 3, I–K), strongly suggested that pDCs are required in anti-HSV, CTL-mediated immunity.
One mechanism for the effect of pDCs may be the promotion of DC–T cell clustering (Fig. 4 B and Fig. 5F). Recent in vivo, real-time visualization techniques have provided new insights into DC–T cell interaction, showing that there are short, weak, and multiple interactions over approximately the first 8 h (23). It is timely to reevaluate the signals that induce DCs to form stable and specific clusters with T cells. We consider that pDCs deliver these signals to LNDCs and that CD2 plays a role in this process because LNDCs express some CD2 ligands (24). The formation of CD2-mediated LNDC–pDC conjugates (Fig. 2 H) is linked with the APC function of LNDCs for priming CTLs (Fig. 6 D). In this respect, in CD2-deficient mice, the induction of anti–lymphocytic choriomeningitis virus CTLs has been reported to require high doses of antigen (25, 26). It is possible that the extent of CD2's in vivo effect varies, depending on the type and dose of viral antigens. However, CD2 has also been reported to be involved in T cell responses against rare and cross-presented lymphocytic choriomeningitis virus antigens (25). In a cutaneous HSV-1 infection model, it is now clear that HSV-1 antigens are cross-presented to T cells (6). Thus, we consider that CD2 plays a significant role in cross-priming anti-HSV CTLs.
The role of CD40L on pDCs is of interest for licensing LNDCs to prime CTL immunity. Previous studies have suggested that CD40L on activated CD4+ T cells generally provides this signal to DCs (2, 27). However, in a primary immune response, CD4+ T cells first need to be activated by antigen-primed mDCs (5, 14), and it is not clear how mDCs initially receive these signals before activated CD4+ T cells appear. We have shown that LN-recruited pDCs rapidly express cell surface CD40L (Fig. 1 C and Fig. 2 G). Although the CD40L+ pDCs were only 6.3% of the total LN pDCs, this was relatively high when compared with the CD40L+CD4+ T cells, which constitute only 0.28% of LN T cells (unpublished data) on day 2 of infection. We believe that the activated pDCs recruited to LNs rapidly provide the initial CD40L signals to LNDCs, to promote their ability to present antigen to both CD4+ and CD8+ T cells. This interaction of CD40L and CD40 on DCs involves cell–cell contact and leads to the translocation of lipid rafts, aggregation of MHC class II and CD80 to enhance antigen presentation (28), activation of TRAF6 to prime antigen-specific CD4+ T cells (29), up-regulation of transporter associated with antigen processing to augment antigen processing (30), and the promotion of cross-presentation on MHC class I to CD8+ T cells (31).
Recent experiments have revealed that pDCs are potent producers of cytokines and chemokines in inflammation (7). IFN-α, in particular, plays an important role by not only preventing viral replication and spread, but also by stimulating DCs and NK cells. In DC-mediated viral immunity, IFN-α has been shown to promote the cross-priming of CTLs (32) and activate uninfected, bystander DCs (21), depending on the dose and timing. In contrast, in our culture conditions, impaired LNDCs failed to completely mature to induce CTLs, even in the presence of IFN-α (Fig. 6 A). However, DCs used in previous studies were “conventional” DCs, which are distinct from the virally impaired DCs used in this study (Fig. 6). We propose that pDCs help LNDCs in at least two steps: first, pDCs contact impaired LNDCs through CD2–CD2L interactions, and engagement of the CD40L–CD40 interaction sets the LNDCs into a recovered condition; and second, recovered LNDCs can now prime T cells using the appropriate signals, such as IFN-α. Inhibiting IFN-α reduced the ability of LNDCs to induce CTLs only after the LNDCs received help from pDCs (Fig. 6 D), indicating that IFN-α can stimulate only recovered/conventional LNDCs to prime CTLs, which is consistent with previous studies (21, 32). When we tested the in vivo effect of IFN-α by injecting locally into anti-Ly6G/C–treated mice at the time of infection, there was a slight recovery of CTL numbers (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20041961/DC1) with undefined mechanism. However, recovered LNDCs need additional IFN-α signals to prime T cells at the second stage (Fig. 6 D). Because pDCs were not completely depleted by anti-Ly6G/C Ab, it is plausible to speculate that the residual activity of remaining pDCs might have been compensated by exogenous IFN-α at the second stage.
Our observations demonstrate a novel role for LN-recruited pDCs as “immune networkers” in antiviral CTL immunity, in addition to their known function as a source of IFN-α. Although distinct subsets of DCs can induce the corresponding effector T cells ex vivo (2, 5, 6), in vivo LNDC–T cell networks are more interactive, both temporally and spatially. The creation of appropriate cytokine fields and the mediation of cell communication by pDC-derived cytokines, and chemokines are essential to antiviral host defense. Viral escape creates a major problem in immunotherapy, and some viruses evade immune surveillance even in hosts vaccinated with antigen-primed DCs or CTLs generated ex vivo (4, 24). DC dysfunction is also seen in cancer patients and is a major factor impairing the generation of anti-tumor CTLs (4, 24). Our study suggests the potent ability of pDCs to improve the activity and communication of impaired DC networks and may provide a novel strategy for the development of DC-based vaccination, to improve CTL induction against persistent viral infection, such as hepatitis and AIDS, and also in cancer.
Materials and Methods
Mice
Specific pathogen-free mice were used in all experiments. Female C57BL/6 mice (8–9-wk old) were obtained from CLEA Japan Inc. CXCR3-deficient mice were generated as previously reported (33) and backcrossed into C57BL/6 mice for eight generations. B6 background CD40L-deficient mice (provided by R. Abe, Tokyo University of Science, Tokyo, Japan) were bred as previously reported (34). All animal experiments complied with the guidelines set by the University of Tokyo Graduate School of Medicine.
HSV-1 and P. acnes infection
The KOS strain of HSV-1 (provided by T. Kaburaki, University of Tokyo, Tokyo, Japan) was propagated and titered using Vero cells grown in MEM with 10% FCS. Mice were infected with 5 × 104 PFU in the right hind foot, and the right PLNs were removed for examination at the times indicated in the figures. Virus titers were determined on day 7 in standard serial dilution plaque assays. Mice were infected with heat-killed P. acnes (1 mg/100 μl PBS; 11828; American Type Culture Collection) via tail vein injections (9–11, 14). In some experiments, mice were injected with BrdU (500 μg/100 μl PBS; Sigma-Aldrich) 1 h before death (9, 14).
Cell sorting and culture and flow cytometry
DC and T cell populations were isolated using a cell sorter (EPICS ELITE ESP; Beckman Coulter) or a MACS system (Miltenyi Biotech) as previously described (9–11, 14). For cell sorting, MHCII-, CD19-, CD3-, and DX5-depleted blood cells or CD19-, CD3-, and DX5-depleted LN cells were stained with anti-CD11c–FITC mAb and anti-B220–PE mAb (BD Biosciences). Analysis of the sorted populations showed purities >95%. For DC phenotyping, blood and LN cells were stained with biotinylated mAbs against CD2, CD40, CD40L, CD86 (BD PharMingen), and Cy-Chrome–conjugated streptavidin, followed by incubation with anti-CD11c–FITC mAb and anti-B220–PE mAb.
Purified B220+CD11c+ cells (105 cells/200-μl well) were incubated for 16 h in round bottom 96-well culture plates in RPMI 1640 with 20% FCS, 100 U/ml penicillin G, and 100 μ/ml streptomycin in the presence or absence of irradiated HSV (30 PFU/well). IFN-α (PBL-Biomedical) in the culture supernatants was assayed using an ELISA (10).
pDC depletion and reconstitution experiments
To inhibit pDC precursor recruitment to LNs, mice were injected i.v. with 100 μg anti-CXCL9 (R&D Systems) plus E-selectin (10) or control (goat IgG; Sigma-Aldrich) Abs immediately before HSV infection. To deplete the pDC fractions, mice were injected i.p. with 200 μg anti-Ly6G/C mAb (clone RB6-8C5; BD Biosciences), 100 μg of anti–PDCA-1 mAb (clone JF05-1C2.4.1, functional grade; Miltenyi Biotec), or control Ab (rat IgG; Sigma-Aldrich) 1.5 d before and 0.5 d after infection (Fig. 3 and Fig. S1 C).
For short-term reconstitution assays, 2 × 106 blood pDC precursors, collected on day 2 from HSV-infected WT or CXCR3−/− mice or from P. acnes–primed mice, were injected i.v. on day 1.5 into anti-Ly6G/C Ab–treated mice infected with HSV. pDC-reconstituted mice were killed 0.5, 1.5, and 5.5 d later (Fig. 5 A). Before cell transfer, 90–92% pure MACS-sorted pDCs were labeled with 2.5 μM CFSE (Molecular Probes) for 10 min at 37°C or with 5 μM 5-(and-6)-([(4-chloromethyl)benzoyl]amino) tetramethylrhodamine (CMTMR; Molecular Probes) for 15 min at 37°C (9, 10).
Antibodies and immunohistochemistry
Immunohistochemical staining was performed as previously reported (9, 14). The following anti–mouse mAbs were used: CD2 (clone RM2-5), CD3ɛ (145-2C11), CD4 (RM4-5), CD8α (53-6.7), CD11c (HL3), CD19 (1D3), CD40 (HM40-3), CD45R/B220 (RA3-6B2), CD86 (GL1), CD154/CD40L (MR1) and Pan–NK cell (DX5; all from BD Biosciences), DEC-205 (BMA), and CD11c (N418; Serotec). Rabbit pAb to CCL21 was made in house (9). As secondary Abs, we used Alexa 488–labeled anti–rat Ig, Alexa 564–labeled anti–hamster, rabbit, and rat Igs, Alexa 647–labeled anti–rat Ig, and streptoavidin labeled with Alexa 564 or 647 (Molecular Probes). For control Abs, we used rat IgG, rabbit IgG, and hamster IgG (Sigma-Aldrich).
ELISPOT assay
Nitrocellulose 96-well plates (Millipore) were coated with 2 μg/ml of purified anti–IFN-γ mAb (clone R4-6A2; BD Biosciences) overnight at 4°C and washed five times in RPMI 1640 with 10% FCS. For CTL precursor frequency assays (Fig. 3, B, F, and J), 92–96% pure MACS-isolated CD8+ T cells obtained from HSV-infected PLN on day 7 were plated at 106 cells/well in the presence or absence of irradiated HSV (30 PFU/well). For ex vivo APC assays (Fig. 4 A and Fig. 5 E), MACS-isolated CD4+ or CD8+ T cells obtained from HSV-infected PLN on day 2 were plated at 106 cells/well without restimulation by exogenous antigens. The indicated numbers of sorted DC populations (mDCs, CD8α+ DCs, and pDCs) from HSV-infected PLN on day 2 were separately added, in triplicate, to the wells in 100-μl aliquots. After overnight incubation at 37°C, the plates were washed eight times with 0.05% Tween–PBS and incubated with 2 μg/ml of biotinylated anti–IFN-γ mAb (clone XMG1.2; BD Biosciences) for 2 h, followed by streptavidin–horseradish peroxidase conjugate (Zymed Laboratories) for 2 h and aminoethyl carbazole substrate (Vector Laboratories). The red spots were counted using a dissecting microscope (SZX9; Olympus). In CTL precursor frequency assays, no spots were detected when CD8+ T cells were cultured in the absence of HSV.
Cluster formation assay
An ex vivo cluster formation assay was performed as described previously (35), with slight modifications. For DC–T cell clusters (Fig. 4 B and Fig. 5 F), 105 MACS-separated B220− LNDCs from the indicated mice and 106 MACS-separated CD3+ T cells from the PLNs of HSV-infected mice at day 2 were co-cultured for 2 h in 500 μl of medium, rotating at 40 rpm at 37°C. For LNDC–pDC conjugates (Fig. 2, G and H), 5 × 104 MACS-separated B220− LNDCs and 5 × 104 pDCs from the PLNs of uninfected or HSV-infected mice at day 2 were incubated for 2 h in 500 μl of medium, rotating at 40 rpm at 37°C with anti-CD2, anti-CD2 plus CXCL10, or 100 μg/ml of control Abs. Cluster formation was estimated by directly counting 1,000 free and clustered LNDCs on cytosmear preparations. In DC–T cell clusters, an LNDC cluster was judged as three or more T cells. In DC–DC conjugates, an LNDC conjugate was judged as containing one or more B220+ pDCs after immunostaining for B220. In some experiments, MACS-sorted LNDCs or pDCs were prelabeled with 10 or 2.5 μm CFSE, respectively, to determine the distribution of CD40L (Fig. 2 G). Clustered LNDCs were expressed as a percentage of the total number of cells counted.
CTL assay
To investigate DC-mediated CTL activity (Fig. 6, A–C), 106 PLN CD8+ T cells, obtained from HSV-infected mice on day 2, were co-cultured with 105 mitomycin C–treated d2 DCs of the indicated subsets for 48 h without any restimulation in vitro. Four sets of culture experiments were performed using (a) LNDCs from anti-Ly6G/C Ab–treated mice (impaired LNDC) supplemented with or without 1,000 U/ml of recombinant IFN-α (Hycult Biotechnology); (b) impaired LNDCs plus pDCs at ratios of 1:1, 3:1, and 10:1; (c) LNDCs from control Ab–treated mice; and (d) pDCs from control Ab–treated mice. To exclude the T cell–derived factors, a two-step co-culture system was also set up (Fig. 6 D). Equal numbers (105) of impaired LNDCs and pDCs were co-cultured for 16 h, supplemented with a neutralizing anti–IFN-α (Hycult Biotechnology), anti-CD2, anti-CD40L mAb, or 100 μg/ml of control Abs. After washing, these DCs were co-cultured for a further 48h with 106 PLN CD8+ T cells obtained from HSV-infected mice on day 2. Cytotoxicity was measured in standard 4-h LDH release assays using a Cytotoxicity Detection Kit (Boehringer) as described previously (11, 36). Specific cytotoxicity was determined using HSV-infected splenocytes as target cells and calculated relative to LDH release in medium alone. In some experiments (Fig. 6 E), we used pDCs from HSV-infected, CD40L-deficient mice. Because CD40L−/− mice show reduced number of LN cells as reported (37), we pooled right inguinal, popliteal, and lumbar LNs to collect a sufficient number of cells. To investigate whether CTLs were generated in vivo (Figs. 3, C, G, and K and Fig. 5G), 106 PLN CD8+ T cells obtained from HSV-infected mice on day 7 (and day 0 for negative control) were tested without adding DCs.
Online supplemental material
Fig. S1 provides data for the effect of anti-Ly6G/C Ab (A and B) and anti–PDCA-1 (C) on the percentage of PLN pDCs. Fig. S2 shows the in vivo effect of recombinant IFN-α on anti-HSV CTL generation in anti-Ly6G/C Ab–treated mice. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20041961/DC1.
Acknowledgments
We are grateful to Dr. R. Abe for providing CD40L−/− mice and to Dr. T. Kaburaki for providing KOS-HSV.
This work was supported in part by Solution Oriented Research for Science and Technology, the Japan Science and Technology Agency, and a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
The authors have no conflicting financial interests.
Abbreviations used: Ab, antibody; BrdU, 5′-bromo- 2′-deoxyuridine; CFSE, carboxyfluorescein diacetate succinimidyl ester; HEV, high endothelial venule; mDC, myeloid DC; pDC, plasmacytoid DC; PDCA, pDC antigen; PLN, popliteal LN; rpm, revolutions per minute.
References
- 1.Vossen, M.T., E.M. Westerhout, C. Soderberg-Naucler, and E.J. Wiertz. 2002. Viral immune evasion: a masterpiece of evolution. Immunogenetics. 54:527–542. [DOI] [PubMed] [Google Scholar]
- 2.Carbone, F.R., and W.R. Heath. 2003. The role of dendritic cell subsets in immunity to viruses. Curr. Opin. Immunol. 15:416–420. [DOI] [PubMed] [Google Scholar]
- 3.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 4.Steinman, R.M., and M. Pope. 2002. Exploiting dendritic cells to improve vaccine efficacy. J. Clin. Invest. 109:1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao, X., E. Deak, K. Soderberg, M. Linehan, D. Spezzano, J. Zhu, D.M. Knipe, and A. Iwasaki. 2003. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus–2. J. Exp. Med. 197:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allan, R.S., C.M. Smith, G.T. Belz, A.L. van Lint, L.M. Wakim, W.R. Heath, and F.R. Carbone. 2003. Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science. 301:1925–1928. [DOI] [PubMed] [Google Scholar]
- 7.Colonna, M., G. Trinchieri, and Y.J. Liu. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5:1219–1226. [DOI] [PubMed] [Google Scholar]
- 8.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoneyama, H., K. Matsuno, Y. Zhang, M. Murai, M. Itakura, S. Ishikawa, G. Hasegawa, M. Naito, H. Asakura, and K. Matsushima. 2000. Regulation by chemokines of circulating dendritic cell precursors and the formation of portal tract–associated lymphoid tissue in a granulomatous liver disease. J. Exp. Med. 193:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoneyama, H., K. Matsuno, Y. Zhang, T. Nishiwaki, M. Kitabatake, S. Ueha, S. Narumi, S. Morikawa, T. Ezaki, B. Lu, et al. 2004. Evidence for recruitment of plasmacytoid dendritic cell precursors to inflamed lymph nodes through high endothelial venules. Int. Immunol. 16:915–928. [DOI] [PubMed] [Google Scholar]
- 11.Zhang, Y., H. Yoneyama, Y. Wang, S. Ishikawa, S. Hashimoto, J.L. Gao, P. Murphy, and K. Matsushima. 2004. Mobilization of dendritic cell precursors into the circulation by administration of MIP-1alpha in mice. J. Natl. Cancer Inst. 96:201–209. [DOI] [PubMed] [Google Scholar]
- 12.Janatpour, M.J., S. Hudak, M. Sathe, J.D. Sedgwick, and L.M. McEvoy. 2001. Tumor necrosis factor–dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. J. Exp. Med. 194:1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Fontecha, A., L.L. Thomsen, S. Brett, C. Gerard, M. Lipp, A. Lanzavecchia, and F. Sallusto. 2004. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 5:1260–1265. [DOI] [PubMed] [Google Scholar]
- 14.Yoneyama, H., S. Narumi, Y. Zhang, M. Murai, M. Baggiolini, A. Lanzavecchia, T. Ichida, H. Asakura, and K. Matsushima. 2002. Pivotal role of dendritic cell–derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J. Exp. Med. 195:1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 16.Krug, A., A.R. French, W. Barchet, J.A. Fischer, A. Dzionek, J.T. Pingel, M.M. Orihuela, S. Akira, W.M. Yokoyama, and M. Colonna. 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 21:107–119. [DOI] [PubMed] [Google Scholar]
- 17.Dalod, M., T. Hamilton, R. Salomon, T.P. Salazar-Mather, S.C. Henry, J.D. Hamilton, and C.A. Biron. 2003. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon α/β. J. Exp. Med. 197:885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krug, A., R. Veeraswamy, A. Pekosz, O. Kanagawa, E.R. Unanue, M. Colonna, and M. Cella. 2003. Interferon-producing cells fail to induce proliferation of naive T cells but can promote expansion and T helper 1 differentiation of antigen-experienced unpolarized T cells. J. Exp. Med. 197:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruse, M., O. Rosorius, F. Kratzer, G. Stelz, C. Kuhnt, G. Schuler, J. Hauber, and A. Steinkasserer. 2000. Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J. Virol. 74:7127–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollara, G., K. Speidel, L. Samady, M. Rajpopat, Y. McGrath, J. Ledermann, R.S. Coffin, D.R. Katz, and B. Chain. 2003. Herpes simplex virus infection of dendritic cells: balance among activation, inhibition, and immunity. J. Infect. Dis. 187:165–178. [DOI] [PubMed] [Google Scholar]
- 21.Pollara, G., M. Jones, M.E. Handley, M. Rajpopat, A. Kwan, R.S. Coffin, G. Foster, B. Chain, and D.R. Katz. 2004. Herpes simplex virus type-1-induced activation of myeloid dendritic cells: the roles of virus cell interaction and paracrine type I IFN secretion. J. Immunol. 173:4108–4119. [DOI] [PubMed] [Google Scholar]
- 22.Salio, M., M. Cella, M. Suter, and A. Lanzavecchia. 1999. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 29:3245–3253. [DOI] [PubMed] [Google Scholar]
- 23.Mempel, T.R., S.E. Henrickson, and U.H. Von Andrian. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 427:154–159. [DOI] [PubMed] [Google Scholar]
- 24.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- 25.Bachmann, M.F., M. Barner, and M. Kopf. 1999. CD2 sets quantitative thresholds in T cell activation. J. Exp. Med. 190:1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans, C.F., G.F. Rall, N. Killeen, D. Littman, and M.B. Oldstone. 1993. CD2-deficient mice generate virus-specific cytotoxic T lymphocytes upon infection with lymphocytic choriomeningitis virus. J. Immunol. 151:6259–6264. [PubMed] [Google Scholar]
- 27.Behrens, G., M. Li, C.M. Smith, G.T. Belz, J. Mintern, F.R. Carbone, and W.R. Heath. 2004. Helper T cells, dendritic cells and CTL Immunity. Immunol. Cell Biol. 82:84–90. [DOI] [PubMed] [Google Scholar]
- 28.Clatza, A., L.C. Bonifaz, D.A. Vignali, and J. Moreno. 2003. CD40-induced aggregation of MHC class II and CD80 on the cell surface leads to an early enhancement in antigen presentation. J. Immunol. 171:6478–6487. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi, T., P.T. Walsh, M.C. Walsh, K.M. Speirs, E. Chiffoleau, C.G. King, W.W. Hancock, J.H. Caamano, C.A. Hunter, P. Scott, et al. 2003. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 19:353–363. [DOI] [PubMed] [Google Scholar]
- 30.Khanna, R., L. Cooper, N. Kienzle, D.J. Moss, S.R. Burrows, and K.K. Khanna. 1997. Engagement of CD40 antigen with soluble CD40 ligand up-regulates peptide transporter expression and restores endogenous processing function in Burkitt's lymphoma cells. J. Immunol. 159:5782–5785. [PubMed] [Google Scholar]
- 31.Delamarre, L., H. Holcombe, and I. Mellman. 2003. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J. Exp. Med. 198:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Bon, A., N. Etchart, C. Rossmann, M. Ashton, S. Hou, D. Gewert, P. Borrow, and D.F. Tough. 2003. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 4:1009–1015. [DOI] [PubMed] [Google Scholar]
- 33.Hancock, W.W., B. Lu, W. Gao, V. Csizmadia, K. Faia, J.A. King, S.T. Smiley, M. Ling, N.P. Gerard, and C. Gerard. 2000. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 192:1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Habiro, K., M. Kotani, K. Omoto, S. Kobayashi, K. Tanabe, H. Shimmura, K. Suzuki, T. Hayashi, H. Toma, and R. Abe. 2003. Mechanism of allorecognition and skin graft rejection in CD28 and CD40 ligand double-deficient mice. Transplantation. 76:854–858. [DOI] [PubMed] [Google Scholar]
- 35.Kushnir, N., L. Liu, and G.G. MacPherson. 1998. Dendritic cells and resting B cells form clusters in vitro and in vivo: T cell independence, partial LFA-1 dependence, and regulation by cross-linking surface molecules. J. Immunol. 160:1774–1781. [PubMed] [Google Scholar]
- 36.Murai, M., H. Yoneyama, T. Ezaki, M. Suematsu, Y. Terashima, A. Harada, H. Hamada, H. Asakura, H. Ishikawa, and K. Matsushima. 2003. Peyer's patch is the essential site in initiating murine acute and lethal graft-versus-host reaction. Nat. Immunol. 4:154–160. [DOI] [PubMed] [Google Scholar]
- 37.Renshaw, B.R., W.C. Fanslow III, R.J. Armitage, K.A. Campbell, D. Liggitt, B. Wright, B.L. Davison, and C.R. Maliszewski. 1994. Humoral immune responses in CD40 ligand–deficient mice. J. Exp. Med. 180:1889–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]