

Abstract
Protein phosphorylation initiates signal transduction that triggers lymphocyte activation. However, other posttranslational modifications may contribute to this process. Here, we show that CD28 engagement induced protein arginine methyltransferase activity and methylation on arginine of several proteins, including Vav1. Methylation of Vav1 and IL-2 production were reduced by inhibiting S-adenosyl-L-homocysteine hydrolase, an enzyme that regulates cellular transmethylation. Methylated Vav1 was induced in human and mouse T cells and selectively localized in the nucleus, which suggested that this form marks a nuclear function of Vav1. Our findings uncover a signaling pathway that is controlled by CD28 that is likely to be important for T cell activation.
TCR stimulation alone is insufficient to drive full T cell expansion and differentiation; this limit is overcome by CD28 coengagement that favors a sequential activation of thousands of genes (1). CD28 shares some signaling effectors with the TCR (e.g., Akt, Vav1, Tec/ITK) and amplifies calcium increase, activation of small GTPases, and cytoskeletal changes, and thus, lowers the activation threshold (1). However, the vast array of immediate and late biologic responses that are induced by CD28 costimulation suggest that it controls as yet unknown pathways.
Studies of receptor-mediated lymphocyte activation revealed mechanisms based on protein and lipid phosphorylation that promote gene activation (2). This view may be incomplete because other posttranslational modifications regulate protein functions and gene expression. For instance, methylation of proteins on arginine (R) by S-adenosylmethionine (AdoMet)-dependent protein arginine methyltransferase (PRMT) may modify protein–protein and protein–RNA interactions during chromatin remodeling, transcription, RNA processing, nuclear import/export, and signal transduction (3, 4). Mass spectrometry (5, 6), protein microarrays (7), and anti-R-methylated antibodies (6) are expanding the list of PRMTs targets rapidly, and indicate that R-methylation is more widespread than believed previously. There are two classes of PRMTs: type I PRMTs (PRMT 1–4, 6) produce asymmetric dimethyl-R, whereas type II PRMTs (PRMTs 5, 7) form symmetric dimethyl-R (4). They differ in target sequences and protein domains that are likely to confer substrate specificity and activity regulation (3). Here, we report the first evidence that an increase of PRMT activity and R-methylation of several proteins occur during T cell activation. These events are controlled largely by CD28, and as such, uncover a novel property of the “second signal” that is required for T cell activation.
RESULTS AND DISCUSSION
CD28-induced an increase of R-methylation and protein arginine methyltransferase activity
To test whether T cell activation induced R-methylation of cellular proteins we used an antimethyl-R antibody (α-Dma). α-Dma reacted in immunoblot (Fig. 1 A, left) and immunoprecipitation (Fig. 1 A, middle) with an Arg-Gly-Gly (RGG)-containing sequence (P3) of Sam68 (8) after in vitro methylation by glutathione S-transferase (GST)-PRMT1, assessed by radioactive labeling (Fig. 1 A, right). α-Dma recognized a few proteins in unstimulated primary T cells by immunoblotting (Fig. 1 B). Their detection increased at 5 min after superantigen (sAg) and B7 costimulation, gradually rising up to 30 min and several other proteins (20–120 kD) became visible only after 10–30 min. α-DMA reactivity decreased toward proteins of stimulated T cells that were pretreated with the methyltransferases (MTases) inhibitor, methylthioadenosine (MTA; Fig. 1 B). These data are reminiscent of nerve growth factor–treated PC12 cells, where initial R-methylation was detected at similar times and increased during hours after stimulation (9). The kinetics of protein R-methylation is different from tyrosine phosphorylation that is induced only a few seconds after stimulation (2), and declines after ∼15–20 min. R-methylation was augmented by B7 stimulation alone and was increased slightly by sAg.
Figure 1.
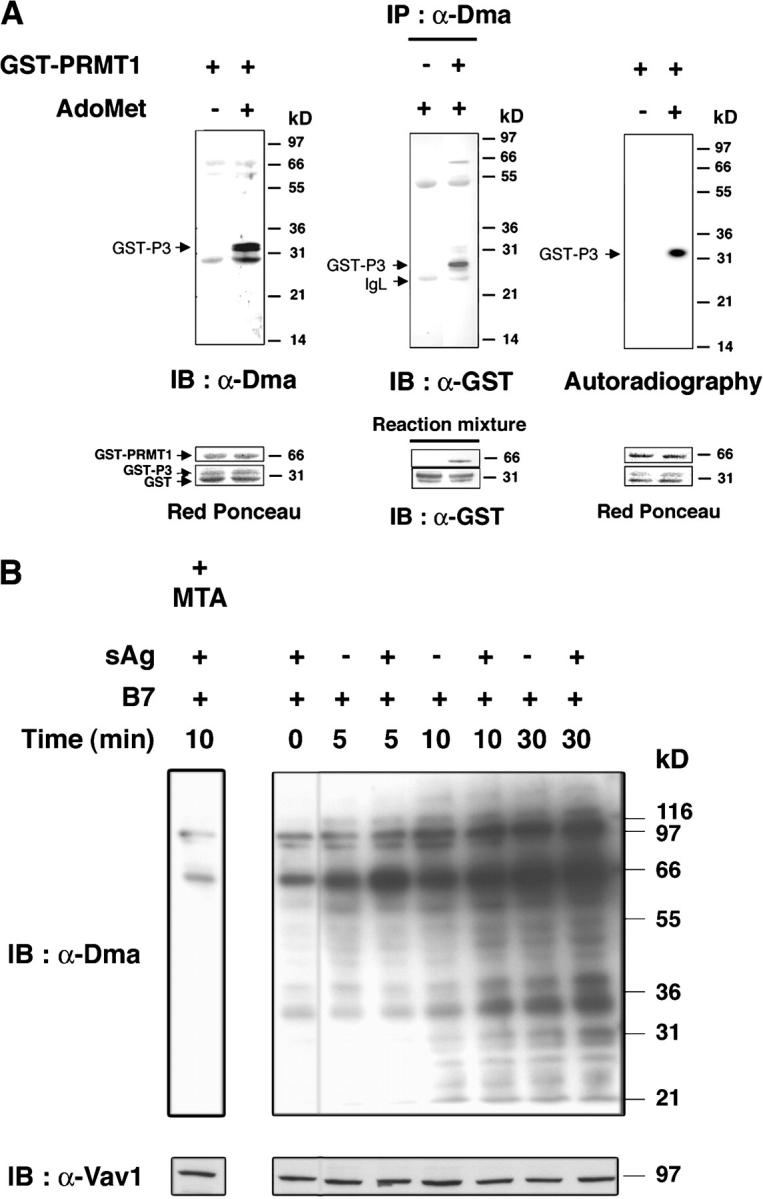

T cell activation increases protein R-methylation and PRMT activity. (A) GST-P3 was reacted with or without AdoMet and GST-PRMT1 (left), or with or without GST-PRMT1 and AdoMet (middle; see Materials and methods) and immunoblotted with α-Dma antibody (left), or immunoprecipitated by α-Dma antibody and then immunoblotted with α-GST antibody (middle). PRMT1-dependent 3H incorporation in P3 in the presence of [3H]-AdoMet (right). Incubation, SDS-PAGE, and blotting were as in the left panel. The apparent discrepancy in migration of GST-P3 between the immunoprecipitate and reaction mixture (middle) was due to the use of NuPAGE Bis-Tris/MOPS and Tris-glycine SDS-gels, respectively. No 3H was detected in GST. Control for reaction mixture is below each experiment. (B) 106 T cells stimulated with 2.5 × 105 5–3.1-B7 cells prepulsed or not with a sAg cocktail (1 μg/ml) for the indicated times as described in Materials and methods. Lysates were immunoblotted with α-Dma (top). Equal loading was controlled by α-Vav1 (bottom). MTA was 0.3 mM for 1 h before stimulation. Shown is one representative of two experiments. (C) PRMT activity was assessed as in A (left) on a lysate of 2 × 106 T cells stimulated as in B with 5–3.1-B7 cells prepulsed (white bars) or not (black bars) with sAg. Substrate content (GST-P3 arrow, bottom) was controlled by α-GST, quantified to normalize radioactivity content (cpm/mm2) associated GST-P3. (D) PRMT activity was detected as in C in lysates of 1.5 × 106 CD28WT (black bars) or CD28Del30 (white bars) cells stimulated 0 or 10 min with 5 × 105 5–3.1-B7 cells. Substrate content was quantified as in C. Data are representative of two experiments.
PRMT activity can change in cells that are stimulated by growth/differentiation factor (9). To determine whether this was the case during T cell activation, we measured P3 methylation in lysates of T cells that were stimulated by B7 alone, or together with sAg. With this assay (Fig. 1 C), we consistently detected basal PRMT activity that increased after 5–10 min of B7 stimulation, and was augmented weakly by sAg. Longer kinetics showed a small increase at 30 min (unpublished data). To assess the role of CD28 signaling in this event directly, we used Jurkat cells which expressed CD28 wild-type (CD28WT), or a tail deletion mutant (Del30) unable to costimulate (10). Fig. 1 D demonstrates that only intact CD28 induces an increase in PRMT activity, whereas no change over background was seen in CD28Del30 cells. Although R-methylation of P3 may be due to PRMT1, we cannot exclude the implication of other PRMTs because RGGs can be methylated by other PRMTs (7). How PRMT activity is regulated by extracellular stimuli is unknown, but PRMTs possess interaction domains and potential posttranslational modification sites that may serve this function (3). These data provide evidence for the first time that CD28, and to a lesser extent the TCR, connects to a pathway that regulates cellular R-methylation.
Induction of Vav1 R-methylation by CD28 engagement
We then asked whether known effectors of CD28 signaling (1) were PRMT targets. Thus, detergent lysates of CD28WT, CD28Del30, and CD28Neg (lacking CD28) cells were stimulated with 5–3.1-B7 and immunoprecipitated with α-Dma, followed by immunoblot for these effectors. α-Dma immunoprecipitated an ∼95-kD protein that reacted with α-Vav1 antibodies at ∼30 min after stimulation in CD28WT cells, but not in CD28Del30 or CD28Neg cells (Fig. 2 A). The 95-kD protein recognized by α-Dma co-migrated with Vav1 (Fig. 2 B) and was not seen in α-Dma immunoprecipitates from B7-stimulated Vav1-deficient Jurkat (Fig. 2 C). In contrast, Akt, Lck, and Grb2 did not seem to react with α-Dma (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050176/DC1). Because of closely migrating IgH and L with Lck, Akt, and Grb-2 (Fig. S1), these experiments were repeated by probing the immunoblots with horseradish peroxidase-ProtA and by direct identification of the proteins that were immunoprecipitated (Fig. 2 B); it confirmed that Akt, Lck, and Grb2 did not react with α-Dma in unstimulated or B7-stimulated cells. These data suggested that Vav1 was a PRMT target. α-CD28 antibody cross-linking of CD28WT cells, but not CD28Del30 cells, resulted in Vav1 detection by α-Dma at 40 min (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20050176/DC1), and supported a direct role for CD28 signal in this event. In contrast, Vav1 was detected faintly at the same time point (or before) upon TCR cross-linking with α-CD3 antibody (Fig. S2 B). Vav1 signal detection was >10-fold less than that of α-CD28 stimulation (Fig. S2, B and C). The higher capacity of CD28 over TCR signal to induce detection of Vav1 by α-Dma was assessed further in a more physiologic setting using Staphylococcus aureus enterotoxin B (SEB)-loaded APC stimulation of CD28WT and CD28Del30 cells (10). Thus, TCR and CD28 costimulation of CD28WT cells with SEB-loaded 5–3.1-B7 cells (Fig. 2 D), but not of CD28Del30 cells, induced Vav1 recognition by α-Dma at 40 min. Vav1 was detected in CD28Del30 cells only at a 20-fold higher SEB concentration (unpublished data). TCR and CD28 coengagement induced stronger Vav1 detection than CD28 alone (unpublished data). B7-induced Vav1 detection by α-Dma was confirmed in primary T cells (Fig. 2 E), and was inhibited by MTA pretreatment (Fig. 2 E); this is consistent with it being methylation dependent. Inhibition also was observed after T cell exposure to the Src-protein tyrosine kinase inhibitor, PP2 (Fig. 2 F), which suggested an upstream control by tyrosine kinases.
Figure 2.

α-Dma detects Vav1 upon CD28 costimulation. (A) CD28WT, CD28Del30, and CD28Neg cells (1.5 × 107) were stimulated with 0.5 × 107 5–3.1-B7 (+), or 5–3.1 (−) cells for the indicated times. Lysates were immunoprecipitated with α-Dma and immunoblotted with α-Vav1. Immunoblot for comparable protein content in lysates is shown (bottom). This is one representative of five independent experiments. (B) CD28WT cells were stimulated as in A for the indicated times. Lysates were immunoprecipitated with α-Dma in parallel with α−Vav1, α-Lck, α-Grb-2, and α-Akt antibodies. Immunoblots were with the indicated antibodies. (C) Vav1-deficient Jurkat cells (JVav) and Jurkat cells were stimulated with 5–3.1 or 5–3.1-B7 cells for the indicated times and lysates were treated as in A. Controls for Vav1 were done by α-Vav1 immunoblot (bottom). (D) CD28WT and CD28Del30 cell lines (1.5 × 107) were stimulated with 0.5 μg/ml SEB prepulsed 5–3.1-B7 cells (0.5 × 107) for the indicated times. Lysates were treated as in A and immunoblotted with α-Vav1. (bottom) Comparable Vav1 content in lysates. One experiment is shown of two giving similar results. (E) 2 × 107 cultured T cells were pretreated for 1 h with 0.3 mM MTA before stimulation with 0.5 × 107 5–3.1-B7 cells. Cell lysates were reacted to α-Dma as in A, followed by α-Vav1 immunoblotting. Similar results were obtained in two experiments. (F) CD28WT cells treated with or without PP2 (10 μM) were stimulated for 3 or 45 min and processed as in A. 10 μM of PP2 strongly inhibited B7-induced Vav1 tyrosine phosphorylation (not depicted).
Our data indicated that Vav1 is methylated directly on arginine or that it is bound to an R-methylated protein. To distinguish between these possibilities, we used a mouse T cell hybridoma (T 8.1) that overexpresses myc-tagged Vav1. α-Dma immunoprecipitated Vav1 in these cells stimulated by B7 (Fig. 3 A) with a kinetics similar to human T cells (Fig. 2). Conversely, Vav1 immunoprecipitated with α-myc antibody from B7-stimulated cells was detected by α-Dma immunoblot (Fig. 3 B), which indicated that Vav1 was methylated on arginine. Signals detected in unstimulated cells of human or mouse origin with α-Vav1, α-myc, or α-Dma were not always observed. This might be due to stimulation by receptors other than CD28 or the metabolic status of the cells.
Figure 3.

B7 induces R-methylation of Vav1. (A) 1.5 × 107 T 8.1-Vav1 cells were stimulated with 0.5 × 107 DAP3 (−), or DAP3-B7 (+) cells for the indicated times. Lysates that were immunoprecipitated with α-Dma were immunoblotted with α-myc antibody. (B) Same experiment as in A, but lysates were immunoprecipitated with α-myc and immunoblotted with α-Dma. Controls for Vav1 amounts in α-myc immunoprecipitates are shown (bottom). Comparable data were obtained in three other experiments.
Vav1 contains no RGG sequence, a PRMT1 target (7), but it bears five Arg-Gly sites that also can serve as PRMT1 substrates (8). However, PRMT1-targeted sequences that are divergent from these have been found (11). Moreover, PRMT4 (or CARM1) target sequences do not show a clear consensus (7). Whereas, for other PRMTs, no substrate sequence has been defined (12, 13). Recent data indicate that PRMTs 1–6 can be expressed in T cells (14). The loose prediction of the R-methylation site on Vav1 will require mass spectrometry and genetic studies for its unambiguous identification. Together, these data provide strong evidence for CD28-induced Vav1 R-methylation prevailing over the TCR signal in triggering this event, similar to global R-methylation and an increase in PRMT activity.
Vav1 R-methylation and IL-2 production depend on S-adenosyl-L-homocysteine hydrolase activity
Methylation reactions generate S-adenosyl-homocysteine, a potent feedback inhibitor of all MTases that is eliminated rapidly by S-adenosyl-L-homocysteine hydrolase (SAHase; reference 15). Pharmacologic SAHase blockers strongly inhibit T cell responses (16). To begin investigating the relationship between CD28-induced R-methylation and T cell activation, CD28WT cells (Fig. 4 A) were treated with MDL28, 842 (MDL), a potent irreversible inhibitor of SAHase, and stimulated with sAg-pulsed B7-bearing cells. Fig. 4 A shows that Vav1 R-methylation decreased with increasing doses of MDL. Similar data were obtained with primary T cells (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050176/DC1) and agreed with inhibition of PRMTs by S-adenosyl-homocysteine accumulation at MDL concentrations and pretreatment conditions (1 h) that did not cause cell death (not depicted). MDL inhibits IL-2 production in normal T cells (16), but it was unclear whether this effect depended on a blockade of early or late biochemical changes. Therefore, we measured the effect of MDL on IL-2 production in CD28WT cells instead of primary T cells, because in the former it only takes 2 h to detect IL-2 after sAg/B7 costimulation (unpublished data). This allowed us to determine whether MDL inhibition on IL-2 expression was due to interference with early biochemical events, including R-methylation. Fig. 4 B shows that MDL decreased IL-2 production at 4 h (and 2 h, not depicted) after stimulation, within the same range of concentrations (1–5 μM) that inhibited Vav1 methylation. MDL did not reduce tyrosine phosphorylation that was induced by TCR or CD28 signals (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20050176/DC1). This suggested that PRMTs do not control early tyrosine phosphorylation or that MDL caused no toxic affect on it. No significant change in SAHase activity upon CD28 stimulation or of protein expression was detected (unpublished data). Although other MTases—in addition to PRMTs—may be inhibited by MDL, these data suggest that CD28-induced R-methylation may be one important event for IL-2 production.
Figure 4.

MDL 28, 842 inhibits Vav1 R-methylation and IL-2 production. (A) 1.5 × 107 CD28WT cells were pretreated for 1 h with 0.1 and 1 μM MDL and stimulated for 40 min with 0.5 × 107 SEB-pulsed 5–3.1-B7 cells, as in Fig. 2 B. Lysates were processed, immunoprecipitated with α-Dma, and Vav1 was detected as in Fig. 2 B. Numbers indicate relative amounts of methylated Vav1 signal after normalization for protein (bottom). Similar results were obtained in two experiments. (B) 5 × 105 CD28 WT cells (triplicates) were treated for 1 h with 0.1, 1, and 5 μM of MDL. After washing, they were stimulated for 3.5 h with 105 5–3.1-B7 cells pulsed with SEB (0.5 μg/ml). Brefeldin A (10 μg/ml) was added after 1 h of incubation. Cells were treated as described in Materials and methods for IL-2 FACS analysis. Similar results were obtained in four experiments.
Methylated Vav1 segregates within the nuclear fraction
Although the CD28-driven increase in PRMT activity and global R-methylation began within 5 min after stimulation, Vav1 methylation was detected later (20–30 min; Figs. 1 and 2). Recent data showed that FcɛR stimulation induces a nuclear localization sequence (NLS)-dependent Vav1 nuclear accumulation within ∼20 min (17). Because R-methylation may be required for nuclear residency (3, 8), we wondered whether R-methylated Vav1 localized in the nucleus. Cytoplasmic and nuclear fractions of CD28WT cells that were stimulated with 5–3.1-B7 cells were immunoprecipitated with α-Vav1, followed by α-Dma immunoblotting. Fig. 5 A demonstrates that R-methylated Vav1, detected at 30–40 min after B7 stimulation, localized selectively in the nuclear fraction, whereas most of the Vav1 immunoprecipitated from the cytosol did not react with α-Dma. Loading equal amounts of proteins from nuclear and cytoplasm extracts (Fig. 5 B) further confirmed the presence of Vav1 in the nuclear fraction after stimulation. Controls for cytoplasmic (ZAP-70) and mitochondrial (Hsp60) markers indicated minimal contamination of cytoplasmic content in the nuclear fraction; Ku70 protein, which is expressed in both, indicated that the nuclear fraction was obtained (Fig. 5 B), as confirmed in other experiments using CDK7 (not depicted). These data, reproduced several times in Jurkat cells, were confirmed in T8.1–Vav-1 hybridoma (unpublished data). Vav1 R-methylation seemed to be delayed slightly with respect to Vav1 detection in the nuclear fraction (Fig. 5 A and not depicted); this suggested that Vav1 migrates into the nucleus where it becomes R-methylated. Therefore, although increases in PRMT activity may coincide with R-methylation of some substrates (Fig. 1, B and C), this modification may require appropriate subcellular localization for others (e.g., Vav1; reference 3). It is of interest that Itk also was R-methylated after CD28 costimulation with kinetics similar to Vav1 (unpublished data).
Figure 5.

R-methylated Vav1 localizes in the nuclear fraction. (A) CD28WT cells (107) stimulated with 0.3 × 107 5–3.1-B7 cells for the indicated times were processed to obtain cytosol (C) or nuclear (N) proteins. α-Vav1 immunoprecipitates from each fraction were immunoblotted with α-Dma (top). Vav1 content was controlled after stripping and reprobing with α-Vav1 (bottom). (B) 20 μg of C and N protein were loaded on SDS-PAGE and immunoblotted with indicated antibodies.
Vav1 regulates cytoskeleton remodeling, calcium increase, and Erk activation that were detected seconds/minutes after TCR/CD28 coligation (1). In contrast, methylated Vav1 accumulated in the nucleus ∼30 min after stimulation, which suggests that this modification marks another function of Vav1. Nuclear Vav1 seems to associate with NFAT- and NF-κB–containing transcriptional complexes (17), and coactivators can be regulated by R-methylation (14, 18). Future studies should elucidate whether Vav1 R-methylation is required for nuclear retention or modulation of transcriptional activities. Understanding the impact of Vav1 R-methylation on T cell activation should await mutation analysis of the target residue(s). However, a few considerations suggest it to have biologic relevance. Densitometry analysis (Figs. 2 B, 5 A, and not depicted) revealed that R-methylated Vav1 represents 1–3% of total Vav1, a figure that is similar, for example, to the levels of TCR-bound phosphorylated ZAP-70 (19) that is sufficient to trigger T cell activation. Moreover, the conservation of Vav1 R-methylation in human and mouse demonstrated here suggests an important function. Because six ATP molecules are required for each methylation to occur (20), its functional benefit ought to overbalance this high energetic cost for it to be conserved in evolution.
The role of R-methylation and PRMTs in the regulation of the immune system is beginning to emerge. CARM1 plays a role in T cell development (21), and PRMT1-mediated R-methylation of the nuclear cofactor Nip45 modulates cytokine gene expression (14). Our study should raise interest in further understanding the significance of R-methylation in lymphocyte activation, as we show that this modification is induced rapidly upon costimulation and gradually increases thereafter. Dimethylated-R is a stable protein modification and no R-dimethylase has been identified to date (22); this suggests that R-methylation encompasses a signaling time-scale, which is distinct from protein phosphorylation. R-methylation accumulation during activation may stabilize gene expression programs or impart long-term “transcriptional memory” (22) in the differentiating T cell. One possibility is that CD28-induced R-methylation provides a more complete explanation for why costimulation elicits a remarkable series of robust biologic effects (1).
MATERIALS AND METHODS
Cells.
CD28WT, CD28Del30, CD28Neg, 5–3.1, and 5–3.1-B7 cells have been described previously (10). The mouse T cell hybridoma T8.1-Vav1 that overexpresses myc-tagged human Vav1 was from F. Michel (Pasteur Institute, Paris, France). The Vav1-deficient Jurkat cell line JVav (23) was from R.T. Abraham (The Burnham Institute, La Jolla, California). DAP.3 and DAP.3-B7 cell lines were obtained from R. Germain (National Institutes of Health, Bethesda, Maryland). PBMCs stimulated by PHA (2 μg/ml) and IL-2 (50 ng/ml; Chiron Corp.) were maintained for 9–11 d before use.
Antibodies, chemicals, and plasmids.
α-PhosphoTyr (4G10, UBI); α-CD28 (CD28.2, Coulter-Beckman); α-KU-70, α-Lck, α-Grb-2, and α-Akt (Santa Cruz Biotechnology Inc.), α-Itk (2F12, UBI); α-ZAP-70 (Transduction Laboratories), α-Vav1 (Vav30 obtained from J. Griffin, Dana Farber Cancer Institute, Boston, MA); α-myc (clone 9E10, Roche); α-hIL-2 (BD Biosciences); goat-α–mouse IgG1 antibody (SBA); α-mono-dimethyl-arginine (α-Dma; Abcam); α-GST and α-HSP-60 (Sigma-Aldrich); Staphylococcus enterotoxins (SE)A, B, E and C3 (Toxin Technologies). Z-4′,5′-didehydro-5′-deoxy-5′-fluoroadenosine MDL 28,842 was obtained from G. Zenke (Novartis, Basel, Switzerland). MTA and PP2 were from Sigma-Aldrich and Calbiochem, respectively. The plasmids, pGEX-GST-P3 (obtained from S. Richard, McGill University, Montreal, Quebec, Canada) and pGEX-GST (GE Healthcare), were used to obtain GST and GST-P3 by affinity chromatography according to the manufacturer's instructions.
Cell stimulation and immunoprecipitation.
CD28WT, CD28Del30, and CD28Neg were stimulated with 5–3.1 and 5–3.1-B7 cells with or without SEB as described previously (10). PHA-blasts, IL-2–starved for 2 d, were stimulated with 5–3.1-B7 cells pulsed with a cocktail of SEA, SEB, SEE, and SEC3 (at 1 μg/ml each). The T8.1-Vav1 cell line was stimulated with DAP.3 or DAP.3-B7 as described (24). In some experiments, time 0 was the mix of separate B7-expressing cells and T cells lysates. For IL-2 detection, cells pooled from triplicate wells were fixed with 4% paraformaldehyde and permeabilized with 0.5% saponin in PBS/1% BSA. α-huIL-2-PE (BD Biosciences; 1 μg/ml) was added for 30 min, and cells were analyzed by FACS to detect the percentage of positive cells. Permeabilization and specificity controls were with α-Bcl-2-PE and isotype-matched irrelevant IgG1-PE (BD Biosciences), respectively. 0% inhibition was equivalent to the percentage of positive cells in the absence of MDL. Immunoprecipitation, immunoblots, and chemiluminescence detection and quantification were as described previously (10).
Subcellular fractionation.
Stimulated cells were treated for nuclear and cytoplasmic extraction using NE-PER kit according to the manufacturer's instructions (Pierce Chemical Co.). Proteins were quantified by the Bradford method (Bio-Rad Laboratories) and subjected to immunoprecipitation after detergent solubilization. The same concentrations of nuclear and cytoplasmic proteins were analyzed on SDS-PAGE to control for cross-contamination.
In vitro methylation and protein arginine methyltransferase activity.
GST-PRMT1 (2 μg) was reacted for 2 h at 37°C with 5 μg of GST or GST-P3 in 0.5 μM S-adenosyl-L-[methyl-3H]methionine in 50 μl 25 mM Tris-HCl, pH 7.4. P3 cold methylation was done with 50 μM AdoMet. The reaction was stopped at 100°C for 5 min in SDS-PAGE sample buffer. PRMT activity was adapted from Cimato et al. (9). Stimulated T cells were subjected to Dounce homogenization in 50 mM Tris, pH 7.5, with protease inhibitors on ice. Lysates were incubated with GST or GST-P3–coated glutathione beads together with 2 μ Ci [H3]-AdoMet for 1.5 h. After washing, P3-associated radioactivity (cpm/mm2) was detected after blotting, quantified on a β-Imager 2000 (Biospace), and normalized to protein content.
Online supplemental material.
Fig. S1 shows that Lck, Grb-2, and Akt are not immunoprecipitated by α-Dma after CD28 engagement. Fig. S2 shows that anti-CD28 triggering induces recognition of Vav1 by α-Dma antibody. Fig. S3 shows that MDL 28, 842 inhibits Vav1 R-methylation in normal T cells. Fig. S4 shows that MDL 28, 842 does not interfere with activation-induced tyrosine phosphorylation. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050176/DC1.
Acknowledgments
We thank A. Weiss for reagents and F. Michel, V. Di Bartolo, and G. Langsley for reading the manuscript.
This work was supported by the Institut Pasteur and the Centre National de la Recherche Scientifique. F. Blanchet received fellowships from the Ministere de la Recherche et de L'enseignement superieur and the Association pour la Recherche sur le Cancer. M.S. Hershfield was supported by U.S. National Institutes of Health grant DK20902.
The authors have no conflicting financial interests.
References
- 1.Acuto, O., and F. Michel. 2003. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat. Rev. Immunol. 3:939–951. [DOI] [PubMed] [Google Scholar]
- 2.Acuto, O., and D. Cantrell. 2000. T cell activation and the cytoskeleton. Annu. Rev. Immunol. 18:165–184. [DOI] [PubMed] [Google Scholar]
- 3.McBride, A.E., and P.A. Silver. 2001. State of the arg: protein methylation at arginine comes of age. Cell. 106:5–8. [DOI] [PubMed] [Google Scholar]
- 4.Boisvert, F.M., C.A. Chenard, and S. Richard. 2005. Protein interfaces in signaling regulated by arginine methylation. Sci. STKE. 2005:re2. [DOI] [PubMed]
- 5.Ong, S.E., G. Mittler, and M. Mann. 2004. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods. 1:116–126. [DOI] [PubMed] [Google Scholar]
- 6.Boisvert, F.M., J. Cote, M.C. Boulanger, and S. Richard. 2003. A proteomic analysis of arginine-methylated protein complexes. Mol. Cell. Proteomics. 2:1319–1330. [DOI] [PubMed] [Google Scholar]
- 7.Lee, J., and M.T. Bedford. 2002. PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep. 3:268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cote, J., F.M. Boisvert, M.C. Boulanger, M.T. Bedford, and S. Richard. 2003. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol. Biol. Cell. 14:274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cimato, T.R., J. Tang, Y. Xu, C. Guarnaccia, H.R. Herschman, S. Pongor, and J.M. Aletta. 2002. Nerve growth factor-mediated increases in protein methylation occur predominantly at type I arginine methylation sites and involve protein arginine methyltransferase 1. J. Neurosci. Res. 67:435–442. [DOI] [PubMed] [Google Scholar]
- 10.Michel, F., G. Attal-Bonnefoy, G. Mangino, S. Mise-Omata, and O. Acuto. 2001. CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity. 15:935–945. [DOI] [PubMed] [Google Scholar]
- 11.Smith, J.J., K.P. Rucknagel, A. Schierhorn, J. Tang, A. Nemeth, M. Linder, H.R. Herschman, and E. Wahle. 1999. Unusual sites of arginine methylation in Poly(A)-binding protein II and in vitro methylation by protein arginine methyltransferases PRMT1 and PRMT3. J. Biol. Chem. 274:13229–13234. [DOI] [PubMed] [Google Scholar]
- 12.Tang, J., J.D. Gary, S. Clarke, and H.R. Herschman. 1998. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem. 273:16935–16945. [DOI] [PubMed] [Google Scholar]
- 13.Frankel, A., N. Yadav, J. Lee, T.L. Branscombe, S. Clarke, and M.T. Bedford. 2002. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem. 277:3537–3543. [DOI] [PubMed] [Google Scholar]
- 14.Mowen, K.A., B.T. Schurter, J.W. Fathman, M. David, and L.H. Glimcher. 2004. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes. Mol. Cell. 15:559–571. [DOI] [PubMed] [Google Scholar]
- 15.Turner, M.A., X. Yang, D. Yin, K. Kuczera, R.T. Borchardt, and P.L. Howell. 2000. Structure and function of S-adenosylhomocysteine hydrolase. Cell Biochem. Biophys. 33:101–125. [DOI] [PubMed] [Google Scholar]
- 16.Wolos, J.A., K.A. Frondorf, and R.E. Esser. 1993. Immunosuppression mediated by an inhibitor of S-adenosyl-L-homocysteine hydrolase. Prevention and treatment of collagen-induced arthritis. J. Immunol. 151:526–534. [PubMed] [Google Scholar]
- 17.Houlard, M., R. Arudchandran, F. Regnier-Ricard, A. Germani, S. Gisselbrecht, U. Blank, J. Rivera, and N. Varin-Blank. 2002. Vav1 is a component of transcriptionally active complexes. J. Exp. Med. 195:1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chevillard-Briet, M., D. Trouche, and L. Vandel. 2002. Control of CBP co-activating activity by arginine methylation. EMBO J. 21:5457–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weil, R., J.F. Cloutier, M. Fournel, and A. Veillette. 1995. Regulation of Zap-70 by Src family tyrosine protein kinases in an antigen-specific T-cell line. J. Biol. Chem. 270:2791–2799. [DOI] [PubMed] [Google Scholar]
- 20.Gary, J.D., and S. Clarke. 1998. RNA and protein interactions modulated by protein arginine methylation. Prog. Nucleic Acid Res. Mol. Biol. 61:65–131. [DOI] [PubMed] [Google Scholar]
- 21.Kim, J., J. Lee, N. Yadav, Q. Wu, C. Carter, S. Richard, E. Richie, and M.T. Bedford. 2004. Loss of CARM1 results in hypomethylation of thymocyte cyclic AMP-regulated phosphoprotein and deregulated early T cell development. J. Biol. Chem. 279:25339–25344. [DOI] [PubMed] [Google Scholar]
- 22.Kubicek, S., and T. Jenuwein. 2004. A crack in histone lysine methylation. Cell. 119:903–906. [DOI] [PubMed] [Google Scholar]
- 23.Cao, Y., E.M. Janssen, A.W. Duncan, A. Altman, D.D. Billadeau, and R.T. Abraham. 2002. Pleiotropic defects in TCR signaling in a Vav-1-null Jurkat T-cell line. EMBO J. 21:4809–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michel, F., L. Grimaud, L. Tuosto, and O. Acuto. 1998. Fyn and ZAP-70 are required for Vav phosphorylation in T cells stimulated by antigen-presenting cells. J. Biol. Chem. 273:31932–31938. [DOI] [PubMed] [Google Scholar]