

Abstract
Pulmonary fibrosis is the consequence of a variety of diseases with no satisfying treatment option. Therapy-induced fibrosis also limits the efficacy of chemotherapy and radiotherapy in numerous cancers. Here, we studied the potential of platelet-derived growth factor (PDGF) receptor tyrosine kinase inhibitors (RTKIs) to attenuate radiation-induced pulmonary fibrosis. Thoraces of C57BL/6 mice were irradiated (20 Gy), and mice were treated with three distinct PDGF RTKIs (SU9518, SU11657, or Imatinib). Irradiation was found to induce severe lung fibrosis resulting in dramatically reduced mouse survival. Treatment with PDGF RTKIs markedly attenuated the development of pulmonary fibrosis in excellent correlation with clinical, histological, and computed tomography results. Importantly, RTKIs also prolonged the life span of irradiated mice. We found that radiation up-regulated expression of PDGF (A–D) isoforms leading to phosphorylation of PDGF receptor, which was strongly inhibited by RTKIs. Our findings suggest a pivotal role of PDGF signaling in the pathogenesis of pulmonary fibrosis and indicate that inhibition of fibrogenesis, rather than inflammation, is critical to antifibrotic treatment. This study points the way to a potential new approach for treating idiopathic or therapy-related forms of lung fibrosis.
Pulmonary fibrosis comprises a group of interstitial disorders of the lung parenchyma that develop as a consequence of multiple causes, including radiotherapy and chemotherapy for lung neoplasms (1, 2). The pathophysiologic events induced by radiation have striking similarities to those that occur after other types of lung injury, such as surgery, chemotherapy, and idiopathic pulmonary fibrosis (IPF; reference 3). IPF is the most common form of lung fibrosis with a prevalence of 16–18 cases per 100,000 (4, 5). Clinically, IPF is characterized by interstitial infiltrates, progressive dyspnea, and worsening of pulmonary function that may lead to death from respiratory failure (1, 6, 7). Despite the medical need, there has been remarkably little progress in the development of effective therapeutic strategies (1, 6–9).
It has been proposed that fibrogenesis is not a unique pathologic process but rather, is due to an excess of the same biologic events involved in normal tissue repair (10). Persistent and exaggerated wound healing ultimately leads to an excess of fibroblast replication and matrix deposition (1, 6–9). Evidence for the importance of platelet-derived growth factor (PDGF) signaling in the fibrotic process is provided by reports showing that a number of fibrogenic mediators such as TGF-β, IL-1, TNF-α, bFGF, and thrombin exhibit PDGF-dependent profibrotic activities (6, 11–14). From the involvement of PDGF in the fibrotic process, it has been suggested that the PDGF–PDGFR system might be a promising target for treating fibrotic disease (15). The PDGF family consists of a family of disulfide-bonded homodimers or heterodimers of four possible subunits (PDGF-A, PDGF-B, PDGF-C, and PDGF-D) that act on cells by binding to homodimers or heterodimers of the two PDGF receptor proteins (PDGFR-α and PDGFR-β) and activating their receptor tyrosine kinase activity (for a review see reference 16). An ideal animal model for IPF does not exist, but bleomycin- and radiation-induced lung fibrosis models have been used to study lung fibrosis (3, 8, 17–24).
In this study, we investigate the role of the PDGF–PDGFR system in the development of lung fibrosis in a radiation-induced fibrosis model using C57BL/6 mice. To evaluate a potential antifibrotic therapy, we also tested the hypothesis whether treatment with PDGF receptor tyrosine kinase inhibitors (RTKIs) might be an effective strategy to attenuate fibrogenesis, even if administered after radiation injury. We used three different RTKIs (SU9518, SU11657, and Imatinib/Gleevec) that overlap in their ability to inhibit both PDGFR-α and PDGFR-β. The encouraging results from this study provide a rationale for clinical trials based on available PDGF RTKIs in patients with pulmonary fibrosis.
Results
PDGF signaling in vitro
Because radiation causes tumor cells to express pro-angiogenic factors (25) and is clinically known to cause fibrosis, we asked if radiation might also induce profibrotic factors and which cells other than macrophages (26–28) might contribute to radiation-induced fibrosis. To this end, we used a coculture model of human fibroblasts in conjunction with either human endothelial cells (human umbilical vein endothelial cells [HUVECs] and human lung microvascular endothelial cells [HLMVECs]) or a human lung cancer cell line (A549). Selective radiation of either endothelial cells or A549 cells in the coculture immediately before adding transwells with the fibroblasts induced fibroblast proliferation by 2.4 ± 0.5- and 1.7 ± 0.3-fold, respectively. If SU9518 was added in the fibroblast compartment, radiation-induced fibroblast proliferation was inhibited by 65 ± 8% (A549) and 46 ± 7% (HUVEC), respectively (Fig. 1 A). Similar data were obtained using irradiated HLMVEC instead of HUVEC for paracrine stimulation (not depicted).
Figure 1.

Radiation-induced paracrine activation of fibroblasts in a coculture system. (A) Fibroblast proliferation induced by exposure to coculture medium (Control) or by prior 10 Gy irradiation of HUVECs or A549 cells in the absence (RT) or presence of SU9518 (SU9518+RT) in the fibroblast medium. Mean ± SD. *, P < 0.05. (B) Phosphorylation status (anti-phosphorylated tyrosine antibody, anti-pY) of PDGFRβ in quiescent fibroblasts, fibroblasts exposed to medium from 10 Gy irradiated endothelial cells (6 and 72 h after radiation, RT), or with additional exposure to PDGF RTKI, SU9518 (RT+SU). Equal loading of lanes was demonstrated with anti-PDGFRβ. (C) Real-time quantitative RT-PCR of PDGF-A, PDGF-B, PDGF-C, and PDGF-D isoforms at 6, 12, 24, 48, and 72 h after 10 Gy irradiation of HLMVECs and A549 cells. Data are means ± SD from at least three independent measurements and show relative expression levels compared with the nonirradiated control cells at each time point.
Immunoprecipitation (IP) western analysis of the fibroblast cells revealed that irradiation of the endothelial cells or A549 cells induced phosphorylation of PDGFR in fibroblasts at 6 and 72 h after irradiation, confirming the role of PDGF signaling in intercell communication. This paracrine activation of PDGFR was inhibited by the addition of SU9518 to the fibroblast compartment (Fig. 1 B). To confirm that endothelial and A549 cells have up-regulated PDGF after radiation and to determine which isoforms are released, we measured the expression levels of all four PDGF isoforms (A–D) using real-time quantitative RT-PCR (Fig. 1 C). All PDGF isoforms were significantly up-regulated after 10 Gy radiation in HLMVEC (P < 0.01) and A549 cells (P < 0.02), which persisted up to 72 h after irradiation. Although the PDGF-B (greater than fivefold), PDGF-C (greater than sixfold), and PDGF-D (greater than fourfold) were the predominant radiation-induced PDGF isoforms in HLMVEC, PDGF-A (greater than twofold) and PDGF-B (greater than fivefold) were more strongly up-regulated in irradiated A549 cells. Because fibroblast activation is thought to be a critical event in fibrogenesis, these in vitro results suggested that inhibition of PDGF signaling might be a way to attenuate this process.
Radiation-induced lung fibrosis
Exposure of normal lung tissue to irradiation has two well-recognized adverse effects: acute/subacute pneumonitis and fibrosis as a long-term sequela (2, 3, 19, 22–24, 29). To translate the coculture results in vivo, we conducted an experiment with a total of 312 C57BL/6 mice randomized into 8 groups of 30–56 animals. Six groups were irradiated with either 20 Gy or 40 Gy and were administered the vehicle or the PDGFR kinase inhibitor, SU9518, either 1 d before or 3 d after irradiation. In both arms, SU9518 was administered s.c. twice weekly throughout the observation period (up to 26 wk after irradiation). The two nonirradiated groups received either vehicle only or SU9518. To test the pharmacokinetics of SU9518, plasma levels were determined after a single s.c. dose (100 mg/kg) was administered to C57BL/6 mice. The plasma levels of SU9518 rose to a peak within 2 h of administration and decayed slowly over the next several days with a half-life of ∼73.6 h (Fig. 2 A). This was in agreement with our previous results showing that SU9518 is a potent inhibitor of both PDGFRα and PDGFRβ tyrosine kinases, and that it had a long duration of plasma exposure when administered s.c. to rats (30). To confirm the presence of an effective therapeutic dose using twice weekly administration, SU9518 plasma concentrations were measured every 4 wk (3 d after the last s.c. injection) during the entire therapy period. The SU9518 plasma concentration was remarkably constant with values varying between 710 ± 45 and 777 ± 160.03 ng/ml.
Figure 2.
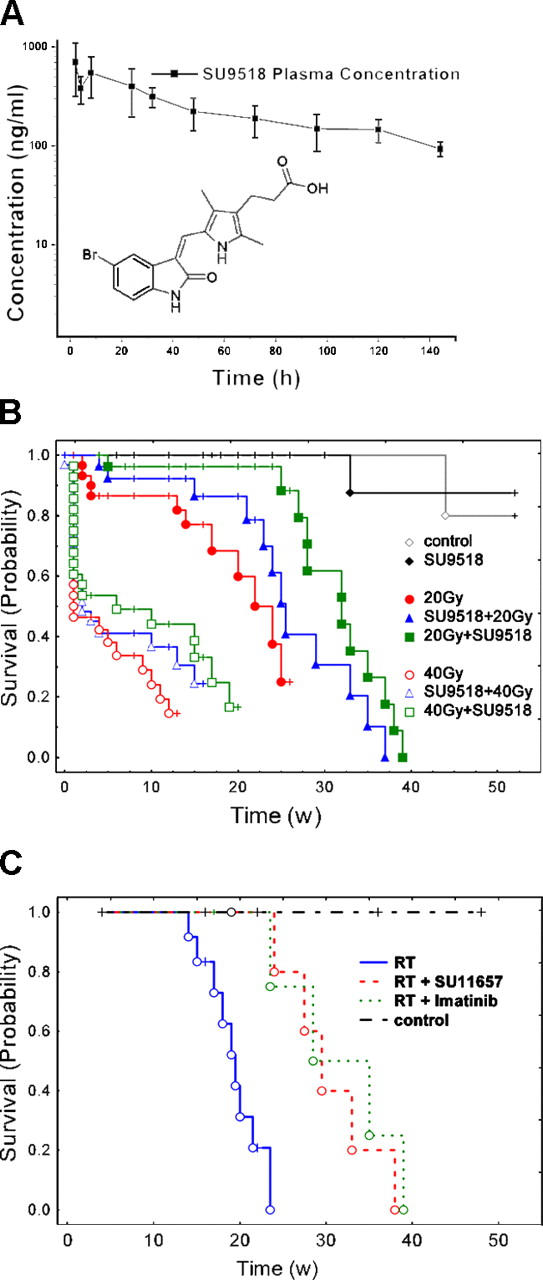
Inhibition of PDGF signaling prolongs survival of irradiated mice. (A) Pharmacokinetic analysis of plasma SU9518 (structure shown as inset) concentration after a single dose of 100 mg/kg administered s.c. to C57BL/6 mice. Tmax ≤ 2 h; Cmax = 706 ng/ml; T1/2 = 73.6 h. (B) Kaplan-Meier analysis of mouse survival after thoracic irradiation and SU9518 treatment. Death was considered complete (cause specific due to radiation) in all cases except those of planned euthanasia for histological assessment, which were considered as censored. Radiation (20 or 40 Gy) reduced survival (P < 0.001 vs. the control [100 μl carboxymethylcellulose twice weekly]; log-rank). SU9518 had no influence on survival of unirradiated mice (P > 0.95 vs. the control). The difference between prevention (SU9518 started 1 d before irradiation) and intervention (SU9518 started 3 d after irradiation) was significant for the 20 Gy group but not for the 40 Gy group (P = 0.035/20 Gy; P = 0.41/40 Gy). SU9518 prevention in both the 20 and 40 Gy group tended to increase survival versus radiation alone, but was not statistically significant (P = 0.15/20 Gy; P = 0.22/40 Gy). If SU9518 administration was started 3 d after 20 Gy irradiation (intervention, 20Gy+SU9518, 40Gy+SU9518), mouse survival increased markedly versus radiation alone (P = 0.006/20 Gy; P = 0.04/40 Gy). (C) Kaplan-Meier analysis of mouse survival from independent confirmation experiments using two other PDGF RTKIs (Imatinib and SU11657) with administration starting 3 d after thoracic irradiation (20 Gy, intervention). Both Imatinib and SU11657 significantly increased survival of irradiated mice (P < 0.01).
Antifibrotic therapy by PDGF inhibition prolongs mouse survival
We found that a single dose of thoracic irradiation (20 Gy) led to lung fibrosis and reduced animal survival, with a median survival of 22 wk versus untreated control mice that stayed alive for >1 yr (P < 0.0001). Challenging mice with the very high single radiation dose of 40 Gy resulted in a dramatic reduction of the median survival down to ∼1 wk after irradiation (Fig. 2 B). SU9518 treatment prolonged median survival by 10 wk (20 Gy: from 22 to 32 wk, P < 0.006) and 5 wk (40 Gy: from 1 to 6 wk, P < 0.04) if administration started 3 d after radiation. If SU9518 administration started before radiation injury, life span was also prolonged but was not statistically significant (20 Gy: P = 0.15; 40 Gy: P = 0.22).
SU9518 treatment also attenuated radiation-related clinical adverse effects such as weight loss (P < 0.02, at all time points after week 14). Similar beneficial effects of SU9518 treatment were observed in other clinical parameters that were monitored weekly during the entire observation period, including animal behavior (worse after irradiation, improved by SU9518), tachypnea, and heart rate (both higher after irradiation, reduced by SU9518).
Because the fibrosis process appeared to be more relevant to survival of mice irradiated with 20 Gy and less important for the animals treated with the very high dose of 40 Gy, we focused on 20 Gy to illustrate the radiological and pathological changes in subsequent experiments.
Confirmation using PDGFR inhibitors SU11657 and Imatinib
The above in vitro and in vivo data suggested that the effects resulted from inhibition of PDGFR in proliferating fibroblasts. However, because kinase inhibitors never exhibit exquisite selectivity, the possibility that the effects might be mediated by inhibition of other kinases cannot be completely excluded. Therefore, key experiments were repeated with Imatinib (Gleevec) and SU11657, two other PDGF RTKIs that are expected to have different kinase selectivity profiles. Because of the poor pharmacokinetic properties of Gleevec in mice (31) and the long duration of dosing, we chose oral administration with the compound formulated in the animal diet. As with the most effective regimen using SU9518 from the experiments described above, anti-PDGF treatment started 3 d after 20 Gy thoracic irradiation. Fig. 2 C shows that both SU11657 and Imatinib treatment significantly prolonged the survival of irradiated mice (P < 0.01). The median survival increased in this independent experiment markedly from 19 to ∼30 wk. We did not observe a statistically significant difference in animal survival between Imatinib and SU11657 therapy (P > 0.5). Furthermore, as with the SU9518 treatment reported above, SU11657 and Imatinib improved all clinical parameters (weight, heart rate, and breathing frequency) in 20 Gy irradiated mice that were monitored weekly over the 26-wk observation period.
In vivo detection of PDGF signaling
PDGF-B, PDGFR-β, and activated PDGFR-β (phosphorylated) were analyzed in mouse lung specimens using immunohistochemistry and IP Western blotting (IP-western). We found that radiation-induced persistent expression of PDGF-B and phosphorylation of the PDGFR (Fig. 3 A) in lungs for most of the time points analyzed during the 26-wk observation period. This radiation-induced PDGFR phosphorylation was reduced by RTKI treatment. The activation of PDGFRβ was confirmed by IP-western, which demonstrated radiation-induced phosphorylation of PDGFR. IP-western also demonstrated that radiation-induced phosphorylation of PDGFR could be inhibited by SU9518 at various time points e.g., at 2, 4, and 20 wk after radiation (Fig. 3 B). This attenuation of PDGFR phosphorylation was seen in the group where SU9518 treatment started before irradiation (prevention, SU9518+RT) as well as in the group where SU9518 treatment started after irradiation (intervention, RT+SU9518). In addition, real-time quantitative RT-PCR revealed significant up-regulation of all four PDGF isoforms in irradiated lung specimen at 2 and 4 wk after irradiation (P < 0.01) as well as for PDGF-A, PDGF-B, and PDGF-C isoforms at 20 wk (P < 0.001). The PDGF-B and PDGF-C RNA levels were elevated stronger at the early time points (2 and 4 wk), whereas PDGF-A was the predominant isoform at 20 wk. Interestingly, the PDGF-A, PDGF-B, and PDGF-D isoforms appeared to be slightly (between 1- and 1.2-fold) elevated after the PDGF RTK inhibitor SU9518 (P < 0.05). A similar up-regulation of ligands has been shown for other RTKIs in human trials using VEGFR TKIs. In contrast, the fibrosis-related expression of the PDGF-A, PDGF-B, and PDGFC isoforms was significantly impaired at 20 wk after irradiation in SU9518-treated mice (P < 0.02). In accordance with the late onset of fibrosis development that is attenuated by PDGF RTKI, PDGF expression is not markedly affected by SU9518 at earlier time points. Again, these data underscore that the inhibition of fibrosis is the principal target of PDGF RTKI.
Figure 3.
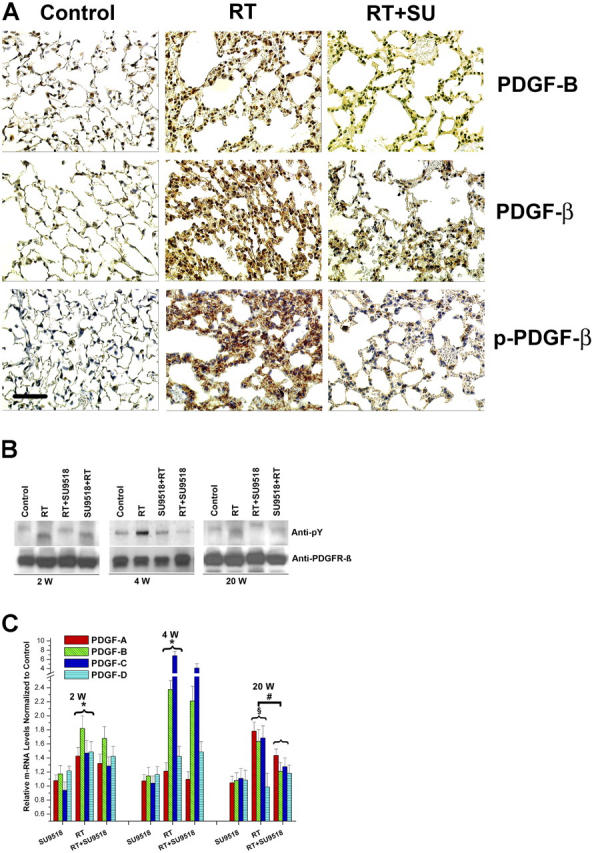
Constitutive activation of PDGF signaling in mouse lungs. (A) Immunohistochemical analysis demonstrates strong induction of PDGF-B and PDGFR-β expression in lung sections 4 wk after 20 Gy total thoracic irradiation (RT). Phosphorylation of PDGFR-β (p-PDGFR-β) was also enhanced in irradiated (RT) versus control mice. Treatment with SU9518 (RT+SU) significantly reduced phosphorylation of PDGFR-β. Bar, 100 μm. (B) 2, 4, and 20 wk after 20 Gy irradiation (RT) and administration of SU9518 (SU+RT and RT+SU), mice were killed and the lungs were harvested for protein isolation. The amount of phosphorylated PDGFR-β (anti-pY) was determined by immunoblotting. Equal loading of lanes was demonstrated with anti-PDGFRβ. (C) Real-time quantitative RT-PCR of PDGF-A, PDGF-B, PDGF-C, and PDGF-D isoforms detected in RNA from mouse lung after SU9518 treatment, 20 Gy thoracic irradiation (RT), and SU9518 administered after lung radiation (RT+SU9518). In irradiated lung specimen, significant up-regulation of all four PDGF isoforms are shown at 2 and 4 wk after irradiation (*, P < 0.01) as well as for PDGF-A, PDGF-B, and PDGF–C isoforms at 20 wk (§, P < 0.001). The expression of the PDGF-A, PDGF-B, and PDGF-C isoforms is significantly impaired at 20 wk after irradiation in SU9518-treated mice (#, P < 0.02), whereas the PDGF expression at earlier time points is not markedly affected by PDGF RTK. This data underscore that the inhibition of fibrosis is the principal target for PDGF RTKIs. Bars are means ± SD from three independent measurements and show relative expression levels compared with the nonirradiated lung tissue at 2, 4, and 20 wk after radiation.
High resolution computed tomography (CT) of mouse lungs
To obtain an independent qualitative and quantitative measure of lung fibrosis that could be repeated in the same animal over time, we used CT. After week 16, typical CT features of lung fibrosis were observed in the 20 Gy group, including irregular septal thickening, patchy peripheral reticular abnormalities with intralobular linear opacities, and subpleural honeycombing (Fig. 4 A). The extent of fibrotic disease progression in CT correlated well with histology and clinical impairment. SU9518 treatment markedly reduced the radiological/morphological signs of fibrosis in both the prevention and intervention groups (P < 0.01). In addition to morphologic assessment, CT enabled quantitation of fibrosis by assessment of lung density (quantified in Hounsfield units [HU]). Lung density dramatically increased during weeks 12–24 after radiotherapy (Fig. 4 B). SU9518 in both the intervention and prevention arms strongly inhibited this increase by ∼50% (Fig. 4, C and D; P < 0.001). The intervention arm appeared to be slightly more effective than the prevention arm after week 20 (Fig. 4 B; P < 0.05). In the independent confirmation experiments using Imatinib and SU11657 treatments, lung densities were also dramatically reduced in irradiated mice by ∼50% by both compounds (Fig. 4 E; P < 0.05).
Figure 4.

High resolution CT as a noninvasive tool for qualitative and quantitative longitudinal monitoring of pulmonary fibrosis progression in mice. Treatment with SU9518 started either 1 d before 20 Gy irradiation (SU9518+RT) or 3 d after irradiation (RT+SU9518). (A) Representative CT scans showing progression of pulmonary fibrosis in mice after 20 Gy whole thorax irradiation (RT) and treatment with SU9518 treatment starting 3 d after RT. Fibrosis (black arrows) is characterized by diffuse bilateral areas of “ground-glass” attenuation and intralobular reticular opacities. (B, C, and D) Quantitative lung density values derived from CT scans. The same 10 randomly chosen mice in each treatment group were examined in a longitudinal study by CT every 2 wk. Five regions of interest were randomly selected in the lungs, and the lung density (in HU) was determined for each region of interest. The time course in B shows that intervention and prevention schedule were both effective in attenuating radiation-induced lung density, and SU9518 intervention (RT+SU9518) was slightly more effective than SU9518 prevention after week 20 (P < 0.05). Bars are mean ± SE. *, P < 0.001 versus the control; #, P < 0.01 versus RT (B and D). (E) Independent confirmatory experiment using Imatinib and SU11657. Inhibitor administration started on day 3 after 20 Gy thoracic irradiation (20 Gy). Both compounds are very effective in attenuating radiation-induced enhancement of lung density. Bars show lung density derived from CT scans (mean ± SE). *, P < 0.01 versus control; #, P < 0.01 versus RT.
Histological assessment of lung fibrosis
To better understand the pathogenesis of the radiation-induced lung fibrosis process and to evaluate the modulation by PDGF RTKI, mice were selected for analysis of leukocyte infiltration, edema, and collagen deposition with associated thickening of the alveolar septum.
We observed a biphasic radiation response, initially consisting of acute and subacute pneumonitis (Fig. 5 and Figs. S1–S3, available at http://www.jem.org/cgi/content/full/jem.20041393/DC1), which was followed by the onset of fibrogenesis. The characteristic histologic findings in the pneumonitis phase of the radiation response were prominent inflammatory cell infiltrates in the alveoli and lung interstitium with simultaneous interstitial edema (Fig. 5, C and E). Both parameters exhibited similar kinetics in the acute phase, reaching their maximum ∼72 h after radiation injury. After the acute radiation response, both leukocyte count and septal edema spontaneously subsided within a few days. SU9518 administration starting 1 d before irradiation (prevention, but then given continuously for the observation period) reduced the edema by ∼40 ± 10% at 24 h (P < 0.02). When SU9518 administration started 3 d after radiation (intervention, but then given continuously for the observation period) a significant reduction of edema (by 50 ± 15%) was first detected at 1 wk after radiation (P < 0.02). Interestingly, SU9518 treatment did not significantly decrease this first, radiation-induced acute leukocyte peak (P > 0.2).
Figure 5.
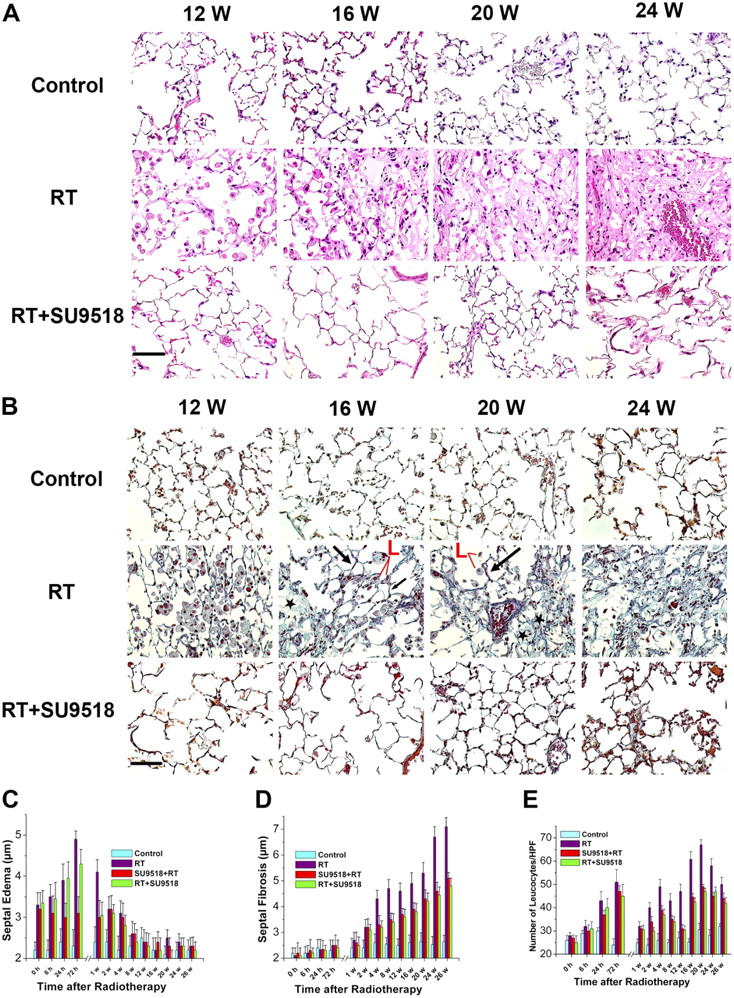
Histological assessment of lung fibrosis. (A) Photomicrographs of hematoxylin and eosin–stained lung tissue sections from control mice, irradiated mice (20 Gy, RT), and mice treated with SU9518 after thoracic irradiation (intervention, RT+SU9518) at 12, 16, 20, and 24 wk after radiation. Bar, 100 μm. (B) Masson's trichrome staining of lung sections 12, 16, 20, and 24 wk after irradiation without (RT) or with administration of SU9518 (RT+SU9518). Examples of focal fibrotic lesions (*), areas of “normal” lung (black arrows), and inflammatory cell infiltration (L, red) are marked. Scale bar, 100 μm. (C) Time course of septal edema development in irradiated mice (RT) with maximum edema at 72 h after radiation (P < 0.05 RT vs. control for each time from 0 h to 4 wk). The preventive or interventive administration of SU9518 (SU9518+RT, RT+SU9518) had significant suppressive effects on edema development, except at 72 h after radiation (P < 0.05 vs. RT). (D) Septal fibrosis develops progressively several weeks after radiation (P < 0.05 RT vs. control for each time point after week 2). Both the SU9518 intervention (RT+SU9518) and prevention (SU9518+RT) treatment significantly reduced the progression of septal fibrosis, as measured by the extent of alveolar wall thickness, at all time points after week 2 (P < 0.05 vs. RT for each time point). However, septal fibrosis was not completely inhibited by the compound (P < 0.05 vs. the control after week 2). (E) Quantitative analysis of leukocyte numbers at various times after irradiation (20 Gy, RT) reveals two peaks. SU9518 before (SU9518+RT) or after (RT+SU9518) irradiation did not significantly attenuate the early leukocyte peak (P > 0.2). Both the preventive (SU9518+RT) and interventional (RT+SU9518) administration of SU9518 significantly inhibited the fibrosis-associated second inflammatory response during weeks 2–26 (P < 0.05 vs. RT for each time point). However, the second fibrosis-associated inflammation was not completely inhibited by SU9518 (P < 0.05 vs. the control after week 2). Bars are mean ± SD.
In irradiated mice, the later fibrogenesis phase was accompanied by a strong second onset of leukocyte infiltration that began several weeks after irradiation and reached a peak at ∼20 wk after irradiation. Masson's trichrome staining of irradiated lungs further showed the development of fibrosis by progressive collagen deposition after week 12. This fibrogenesis phase was characterized by development of typical fibroblast foci, with abnormal wound healing/repair leading to replication of mesenchymal cells, as characterized by fibroblast/myofibroblast migration and proliferation and exuberant deposition of extracellular matrix in irradiated lungs. SU9518 treatment markedly reduced collagen deposition, alveolar wall thickness (P < 0.05), and all other histologically visible signs of fibrosis (Fig. 5, B and D).
At later time points (>20 wk), the fibroblast foci evolved and coalesced into more widespread fibrosis with remodeling of the lung architecture (Fig. 5, A and B). Thus, in the irradiated lungs, the second onset of progressive fibrogenesis-related infiltration of leukocytes persisted until the morphologically described fibrosis process was completed (after week 26). SU9518 intervention and prevention significantly inhibited this fibrosis-associated second inflammatory response, as demonstrated by reduced leukocyte counts from weeks 2–26 (P < 0.05 for each time point).
Discussion
Here, we show the pivotal role of PDGF signaling in the pathogenesis of radiation-induced pulmonary fibrosis. Importantly, our findings suggest that direct inhibition of fibrogenesis by PDGF RTKIs is an effective strategy to attenuate lung fibrosis. We found that three distinct inhibitors of PDGF signaling (Imatinib/Gleevec, SU9518, and SU11657) markedly attenuated pulmonary fibrosis in a mouse model of radiation-induced lung fibrosis (C57/Bl6).
We could demonstrate that treatment of irradiated mice with PDGF RTKIs markedly attenuates the development of fibroblast foci, the hallmark of pulmonary fibrosis, and the subsequent remodeling of the lung architecture. The morphological results were in agreement with qualitative and quantitative high resolution CT scans of mouse lungs, demonstrating that PDGFR inhibition could dramatically attenuate the typical radiological features of lung fibrosis induced by thoracic irradiation.
Interestingly, with respect to the time course of fibrosis development, we found that PDGF RTKIs were more effective when administered after thoracic irradiation in an intervention schedule than when giving them before irradiation in a prevention schedule. No antifibrotic therapy has been clearly shown to prolong survival in patients with lung fibrosis (1, 6, 7, 9). Therefore, we conclude from our data that demonstrate a significant survival benefit and reduced clinical morbidity in PDGF RTKI–treated mice that a rationale exists for clinical testing of this class of compounds in patients developing pulmonary fibrosis.
On a cellular level, our coculture data suggest that paracrine PDGF secretion is an important source of radiation-induced fibroblast proliferation. Likewise, we found that radiation induced persistent exaggerated release of all four PDGF (A–D) isoforms with consequent activation of PDGFR in fibroblasts both in vitro and in vivo in mice lungs, thus indicating an important role for PDGF signaling in radiation-induced fibrogenesis and pulmonary fibrosis.
The high antifibrotic efficacy of Imatinib, SU11657, and SU9518 in our mouse study might be explained in part by the fact that all RTKIs are potent inhibitors of both PDGFR-α and PDGFR-β isoforms. Thus, it is conceivable that PDGF signaling was effectively attenuated through all stages of the disease, regardless of preferential expression of a specific PDGF isoform at a given time. The compound SU9518 exhibits great potency with high selectivity for inhibition of PDGF-induced proliferation relative to FGF- and EGF-induced proliferation (30). The IC50 values for SU9518 inhibition of PDGF-, FGF-, and EGF-induced BrdU incorporation using mouse fibroblasts were 0.053, 4.40, and 9.63 μMol, respectively. Further, SU9518 exhibited no inhibition of isolated EGFR kinase using a biochemical assay (30). However, considering the large number of human kinases (32), the possibility that the effects might be mediated by inhibition of other kinases cannot be completely excluded. This issue would be applicable to most available small molecule RTKIs, because current kinase inhibitors never exhibit exquisite selectivity (33–36). Although their effects are not fully elucidated, some RTKIs have entered clinical applications. For example, Imatinib is successfully used for (a) the treatment of chronic myelogenous leukemia with the primary rationale inhibition of Bcr/Abl kinase; (b) gastrointestinal stromal tumors (inhibition of c-Kit); and (c) malignant brain tumors, lung cancer, and other solid tumors (inhibition of PDGFR; references 34 and 37).
To strengthen our concept of PDGF involvement in fibrosis, we present data using two other small molecule RTKIs (besides SU9518) that are considered PDGFR inhibitors with different kinase selectivity profiles: Imatinib (Gleevec) with high activity against three kinases, Bcr/Abl, c-Kit, and PDGFR-α and PDGFR-β (31, 34, 37, 38), and SU11657, a multi-targeted inhibitor of class III/V receptor tyrosine kinases with selectivity similar to SU11248 (inhibits PDGFR-α and PDGFR-β, VEGFR-2, flt3, and c-kit), which is currently investigated in phase I/II clinical cancer trials (34, 39). Thus, the significant overlap among the three PDGFR inhibitors used is PDGFR inhibition. All three inhibitors were found to reduce lung fibrosis after radiation injury and prolonged animal survival. Together with our data on radiation-induced PDGF expression and phosphorylation of PDGFR in vitro and in vivo, we suggest that the inhibition of the PDGFR signaling is the key mechanism behind our functional findings.
In the past, an important assumption was that fibrosis might be avoided if the inflammatory cascade was interrupted before irreversible tissue injury occurred (6, 7). This led to initial enthusiasm for corticosteroid and cytotoxic therapy for IPF. However, it is now accepted that current antiinflammatory therapies, even in combination with potent immunosuppressives, fail to improve the disease outcome (1, 6–9). Therefore, acute inflammation is not the only critical step in development of the fibrotic response (7, 8, 40, 41) and a new hypothesis seems in order (Fig. 6). We found that although the acute inflammatory response induced by radiation injury was not completely abrogated, inhibition of PDGF signaling attenuated the onset and development of lung fibrosis. The fibrotic tissue alteration was accompanied by a second chronic inflammatory response that was dramatically attenuated by PDGF RTKIs. Accordingly, the acute radiation-induced expression of PDGF isoforms was mildly affected by the PDGF RTK, whereas the fibrosis-related PDGF expression at later time points (week 20) was significantly attenuated. Thus, the potent direct inhibition of fibrogenesis consisting of stromal cell migration, proliferation, and extracellular matrix deposition seems to be the principal target of PDGF RTKIs.
Figure 6.

Model of fibrosis development integrating our findings. The data suggest that a fibrosis-generating stimulus such as ionizing radiation initiates an inflammatory response consisting of leukocyte infiltration and the release of proinflammatory/profibrotic cytokines such as PDGF. This acute tissue response initiates a constitutive chronic stimulation and activation of the fibroblasts by PDGF, with subsequent proliferation and extracellular matrix deposition. PDGF RTKIs inhibits fibrogenesis induced by the release of PDGF from injured endothelial cells, epithelial cells, and infiltrated macrophages.
The potential of PDGF inhibition for the treatment of lung fibrosis is supported by previous data implicating the PDGF–PDGFR system in IPF-, asbestos-, bleomycin-, and radiation-induced lung fibrosis (20, 27, 42–46) as well as fibrosis in other organs such as the kidneys, liver, skin, and heart (47–50). Administration of the PDGF RTK AG1296, a tyrophostin analogue, has been shown to reduce pulmonary fibrosis in a rat model of metal-induced lung injury (35). Imatinib has been shown to ameliorate chronic allograft nephropathy in rats (36), to attenuate bone marrow fibrosis in patients with chronic myelogenous leukemia (38), and very recently to inhibit TGF-β–dependent bleomycin-mediated fibrosis (51).
An advantage of our radiation-induced lung fibrosis model might be that the development of lung fibrosis occurs relatively slowly up to 26 wk after irradiation, thus resembling the time course in humans. In other lung fibrosis models, the fibrotic end-stage is typically reached by 4 wk or earlier after the instillation of, e.g., bleomycin or vanadium pentoxid (35, 51). In patients with advanced lung cancer, radiotherapy is considered the mainstay of therapy. However, therapy-related normal tissue radiosensitivity that leads to pulmonary fibrosis limits the delivery of effective radiation doses for a curative treatment of lung cancer patients. Thus, the ability to reduce normal tissue complication susceptibility could allow oncologists to increase the radiation dose to be given, which in turn could enhance cancer cure rates. On the other hand, because PDGF RTKI are also being investigated as cancer drugs (25, 31), a twofold rationale for their use in radiation oncology might unfold.
Taken together, we demonstrate that direct intervention in fibrogenesis using PDGF RTKIs might be a useful approach to the therapy of radiation-induced lung fibrosis. However, it is unlikely that single pathway inhibition can completely prevent lung fibrosis in humans, considering the intricate genetic networking (52, 53). Nevertheless the PDGF RTKIs that are undergoing clinical studies or are approved for certain oncological indications (e.g., SU11248 or Gleevec [STI571/Imatinib]; reference 34) may have appropriate potency, selectivity, and safety profiles for the treatment of fibrosis-related diseases. This could be of benefit for patients with idiopathic forms of fibrosis and also for lung cancer patients to enhance the therapeutic efficacy by simultaneously reducing the radiation-induced undesired side effects.
Materials and Methods
Reagents and cell culture
Primary isolated human dermal fibroblasts, HUVECs (Promocell), HLMVECs (Clonetics/ Cambrex Bio Science), and human non-small cell lung cancer cell line A549 (Tumorbank DKFZ) were cultured as described previously (25, 54).
An in vitro coculture system was used as described previously with minor modifications (25, 54). In brief, endothelial and A549 cells were grown in 24-well plates and irradiated with 10 Gy. Separately, collagen I–coated transwell inserts (Becton Dickinson) were plated with 20,000 fibroblasts and preincubated with 5 μM SU9518 for 1 h. After irradiation of A549 cells and endothelial cells, the transwells containing the fibroblasts were transferred into the 24-well plates and incubated for 72 h before counting the fibroblasts.
Experimental protocol and animal model
All animal procedures were approved by institutional and governmental authorities. Fibrosis-prone mice (8-wk-old female C57BL/6J mice with an approximate body weight of 20 g; Charles River Laboratories) were used. For thoracic irradiation, mice were anesthetized by intraperitoneal application of 0.2 mg/kg Domitor (Pfizer) and 100 mg/kg ketamin 10% (Parke-Davis). Cobalt-60 γ radiation (Siemens Gammatron S) was administered as single dose (20 or 40 Gy as indicated) to the entire thorax (0.441 Gy/min; source surface distance of 0.7 m) using one standing field anterior-posterior. Other organs, above and beyond the thorax, were shielded. Animals were supplied with diet and water ad libitum. The first set of animal experiments was performed to analyze the effects of the PDGF RTKI SU9518 in radiation-induced lung fibrosis. The second set of experiments was performed to independently confirm the results using Imatinib and SU11657, two other PDGFR RTKIs with otherwise different kinase inhibitory spectrum.
Treatment with RTKI
PDGF RTKIs were synthesized at SUGEN Inc. SU9518 was administered s.c. at 100 mg/kg (in 0.5% carboxymethylcellulose sodium USP, 0.9% sodium chloride, 0.4% polysorbate 80, 0.9% benzyl alcohol, and 4 ml/kg deionized water) twice weekly until the end of the observation period. To achieve a clinically relevant dose (31), Imatinib and SU11657 were formulated in standard mouse chow at 0.5 (Imatinib) and 0.0125 mg/g (SU11657), respectively. SU9518 is a selective and potent inhibitor of cellular PDGFR-α, PDGFR-β, and PDGF receptor–induced cell proliferation and has demonstrated activity in preventing arterial stenosis in a rat model (30). Imatinib (Gleevec) has high activity against three kinases: Bcr/Abl, c-Kit, and PDGFR-α and PDGFR-β (31, 34, 37, 38). SU11657 has been shown to inhibit VEGFR-1, VEGFR-2, and PDGFR-α and PDGFR-β (34, 39). Thus, among known kinases tested, Imatinib/Gleevec, SU11657, and SU9518 overlap in their inhibition of PDGFR-α and PDGFR-β.
Real-time quantitative RT-PCR
Expression levels of RNA transcripts were quantitated by real-time PCR. Total RNA from A549 cells, HLMVECs, and frozen lung tissues (mouse) were isolated using RNeasy Kit (QIAGEN). RNA quality was ensured by lab-on chip technology according to the manufacturer's instructions (2100 bio-analyzer in combination with the RNA 6000 Lab Chip kit; Agilent Technologies) and spectrophotometric analysis. The RNA only was used with a 28S/18S ribosomal RNA ratio of 2.0 (±0.3). SYBR Green assay and the ABI Prism 7900HT Sequence Detection System (Applied Biosystems) were used for the real-time quantitation of the RNA. A consensus sequence, derived from Locus Link/refseq database (NCBI, http://www.ncbi.nlm.nih.gov/LocusLink; accession nos: mPDGF-A: NM_008808; mPDGF-B: NM_011057; mPDGF-C: NM_019971; mPDGF-D: NM_027924; hPDGF-A: NM_002607; hPDGF-B: NM_002608; hPDGF-C: NM_016205; and hPDGF-D: NM_025208), was used to design the forward and reverse primers to the sequences using ABI's Primer Express software (Cytomyx). The primers were as follows: mPDGF-A: forward 5′-GGCTGGCTCGAAGTCAGATC-3′ and reverse 5′-CCTCAGCCCCTACGGAGTCT-3′; mPDGF-B: forward 5′-AAGCTCGGGTGACCATTCG-3′ and reverse 5′-ACTTTCGGTGCTTGCCTTTG-3′; mPDGF-C: forward 5′-CTCGGGCTGAGTCCAACCT-3′ and reverse 5′-TTGCACTCCGTTCTGTTCCTT-3′; mPDGF-D: forward 5′-CCAAGGAACCTGCTTCTGACA-3′ and reverse 5′-TCCGAATTGATGGTCAAAGGA-3′; hPDGF-A: forward 5′-GTGGCCAAGGTGGAATACGT-3′ and reverse 5′-CGCACTCCAAATGCTCCTCTA-3′; hPDGF-B: forward 5′-AGATCGAGATTGTGCGGAAGA-3′ and reverse 5′-GCTGCCACTGTCTCACACTTG-3′; hPDGF-C: forward 5′-GGACTCAGGCGGAATCCAA-3′ and reverse 5′-CTGAGGATCTTGTACTCCGTTCTG-3′; and hPDGF-D: forward 5′-CAATGATGATGCCAAGCGTTA-3′ and reverse 5′-TGGCCAACTTCAGCTCTTCTC-3′. After RNA isolation, contaminating genomic DNA was removed by DNase 1 treatment (DNA-free; Ambion). First strand cDNA was reverse transcribed from total RNA using the cDNA Archive Kit (MultiScribe Reverse Transcriptase; Applied Biosystems) and stored at −20°C until use. Complementary DNAs were mixed with SYBR Green PCR master mix (Applied Biosystems) and primers, and real-time PCR was performed. In addition to profiling all samples for the target sequence, samples were profiled for 18S (ribosomal RNA) expression as endogenous control. For each single well amplification reaction, a threshold cycle (CT) was observed in the exponential phase of amplification, and the quantitation of relative expression levels was achieved using standard curves for both the target and endogenous controls. All assays were performed in triplicate.
Pharmacokinetic analysis
Pharmacokinetic analysis was performed as described previously (30). In brief, blood samples from mice were obtained and RTKIs were extracted from the plasma. HPLC/mass spectrometric analysis was performed.
Lung histology and immunohistochemistry
Histological analysis from mice tissues was performed systematically at early and later time points after radiation until 26 wk as described previously (55). In brief, lungs were fixed by intratracheal instillation of 4% formalin followed by overnight fixation, embedding in paraffin, sectioning at 5 μm, and staining with hematoxylin and eosin or Masson's trichrome. The total count of leukocytes, intraalveolar edema, and septal thickness were determined by morphometric evaluation (Q 600 Quantimet; Leica).
Immunohistochemistry was performed for PDGFB, PDGFRβ, and phopho-PDGFR. In brief, lung sections were deparaffinized and stained with anti–PDGF-B (no. sc-7878; Santa Cruz Biotechnology, Inc.), anti–PDGFR-β (no. sc-432; Santa Cruz Biotechnology, Inc.), or anti–phospho-PDGFR (nos. 3161 and 3391; Cell Signaling Technologies) primary antibodies. Biotin-conjugated goat anti–rabbit IgG second antibody (BA-1000; Vector Laboratories) was used and the sections were detected with DAB+ reagent (no. K3468; DakoCytomation) after incubation with Elite ABC reagent (Vector Laboratories) according to the manufacturer's instructions. Control sections were routinely analyzed in other organs including the kidney, skin, muscle, and spleen to investigate potential side effects of RTKIs.
High resolution CT
To obtain an independent qualitative and quantitative measure for lung fibrosis in the mice, we used high resolution CT. CT is the radiological method of choice for monitoring fibrosis in patients (1, 5, 7). This radiological method allows noninvasive and repeated measurements in the same mice in a longitudinal study (55). CT was performed in 10 randomly selected mice from each group every second week during the entire observation period. CT images were captured on a Somatom Plus 4 Volume Zoom multi-slice CT scanner (Siemens). 120 kV with 100 mAS were applied. 0.5-mm thin slices with 0.5-mm inter-slice distance spanned the complete mouse chest (a total acquisition time of 0.5 s). Multiplanar reconstructions were performed for semiquantitative analysis. HU of section slides from the upper and lower lung region were determined. Eight regions of interest were defined in the following areas: the right upper anterior and posterior regions, the left upper anterior and posterior regions, the right lower anterior and posterior regions, and the left lower anterior and posterior regions. Total arithmetic means + SD of the HU were calculated. For the Imatinib/Gleevec and SU11657 confirmation study, a different CT scanner was used (Aquilion 32; Toshiba).
PDGFR phosphorylation in lungs
After protein isolation and homogenization of the frozen mouse lung, the protein content was determined, and equal amounts of protein were used in each IP. The lysate was incubated with anti-PDGFRβ antibody (no. 06-498; Upstate Biotechnology) and protein A agarose beads overnight at 4°C. The beads were collected by centrifugation and washed. The protein on the beads was dissolved in SDS-PAGE buffer, fractionated on a 12% gel, and transferred to a nitrocellulose membrane. After blocking, the blot was probed for phosphotyrosine with PY99 antibody (no. sc-7020; Santa Cruz Biotechnology, Inc.) and visualized with ECL reagents (Pierce Chemical Co.). The blots were then stripped and reprobed for total PDGFRβ with anti-PDGFRβ antibody (no. sc-432; Santa Cruz Biotechnology).
Statistics
Mouse survival curves after thoracic irradiation and RTKI treatments were calculated with the Kaplan-Meier method and compared using the log-rank test. Other quantitative data are given as mean values ± SD or as indicated. For analysis of differences between the groups, ANOVA followed by the appropriate post hoc test for individual comparisons between the groups was performed. All tests were two-tailed. P < 0.05 was considered statistically significant.
Online supplemental material
Additional data on longitudinal histological and CT assessments are available at http://www.jem.org/cgi/content/full/jem.20041393/DC1.
Acknowledgments
We would like to thank Thuy Trinh, Ingrid Moll, Heike Zieher, Klaus J. Weber, Peter Peschke, Michael Wannenmacher, and Jürgen Debus from the University of Heidelberg and German Cancer Research Center (DKFZ), Heidelberg. We thank also Wilhelm Ansorge, Russ Hodge, and Jonathon Blake of the EMBL, Heidelberg. We further thank Tinya J. Abrams, Anthony R. Howlett, Emile Plise, Cesar Medina, Asaad Nematalla, Anand Sistla, and P. Cho Tang of SUGEN, Inc., and Elaine Krul of Pharmacia Corporation.
This study was supported by the Research Program of the University of Heidelberg Medical School.
The authors have no conflicting financial interests.
Abbreviations used: CT, computed tomography; HLMVEC, human lung microvascular endothelial cell; HU, hounsfield units; HUVEC, human umbilical vein endothelial cell; IP, immunoprecipitation; IPF, idiopathic pulmonary fibrosis; IP-western, IP Western blotting; PDGF, platelet-derived growth factor; RT, radiation; RTKI, receptor tyrosine kinase inhibitor.
References
- 1.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. 2002. Am. J. Respir. Crit Care Med. 165:277–304. [DOI] [PubMed]
- 2.Movsas, B., T.A. Raffin, A.H. Epstein, and C.J. Link Jr. 1997. Pulmonary radiation injury. Chest. 111:1061–1076. [DOI] [PubMed] [Google Scholar]
- 3.Rubin, P., C.J. Johnston, J.P. Williams, S. McDonald, and J.N. Finkelstein. 1995. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int. J. Radiat. Oncol. Biol. Phys. 33:99–109. [DOI] [PubMed] [Google Scholar]
- 4.Coultas, D.B., R.E. Zumwalt, W.C. Black, and R.E. Sobonya. 1994. The epidemiology of interstitial lung diseases. Am. J. Respir. Crit. Care Med. 150:967–972. [DOI] [PubMed] [Google Scholar]
- 5.Hodgson, U., T. Laitinen, and P. Tukiainen. 2002. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax. 57:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen, J.T., and M.A. Spiteri. 2002. Growth factors in idiopathic pulmonary fibrosis: relative roles. Respir. Res. 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gross, T.J., and G.W. Hunninghake. 2001. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 345:517–525. [DOI] [PubMed] [Google Scholar]
- 8.Kamp, D.W. 2003. Idiopathic pulmonary fibrosis: the inflammation hypothesis revisited. Chest. 124:1187–1190. [DOI] [PubMed] [Google Scholar]
- 9.Mason, R.J., M.I. Schwarz, G.W. Hunninghake, and R.A. Musson. 1999. NHLBI Workshop Summary. Pharmacological therapy for idiopathic pulmonary fibrosis. Past, present, and future. Am. J. Respir. Crit. Care Med. 160:1771–1777. [DOI] [PubMed] [Google Scholar]
- 10.Border, W.A., and N.A. Noble. 1994. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 331:1286–1292. [DOI] [PubMed] [Google Scholar]
- 11.Battegay, E.J., E.W. Raines, R.A. Seifert, D.F. Bowen-Pope, and R. Ross. 1990. TGF-beta induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell. 63:515–524. [DOI] [PubMed] [Google Scholar]
- 12.Battegay, E.J., E.W. Raines, T. Colbert, and R. Ross. 1995. TNF-alpha stimulation of fibroblast proliferation. Dependence on platelet-derived growth factor (PDGF) secretion and alteration of PDGF receptor expression. J. Immunol. 154:6040–6047. [PubMed] [Google Scholar]
- 13.Raines, E.W., S.K. Dower, and R. Ross. 1989. Interleukin-1 mitogenic activity for fibroblasts and smooth muscle cells is due to PDGF-AA. Science. 243:393–396. [DOI] [PubMed] [Google Scholar]
- 14.Kolb, M., P.J. Margetts, D.C. Anthony, F. Pitossi, and J. Gauldie. 2001. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest. 107:1529–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonner, J.C. 2004. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 15:255–273. [DOI] [PubMed] [Google Scholar]
- 16.Ostman, A., and C.H. Heldin. 2001. Involvement of platelet-derived growth factor in disease: development of specific antagonists. Adv. Cancer Res. 80:1–38. [DOI] [PubMed] [Google Scholar]
- 17.Gurujeyalakshmi, G., M.A. Hollinger, and S.N. Giri. 1996. Inhibitory effect of interferon gamma, interleukin-1, interleukin-6 and platelet-derived growth factor-A mRNA expression in bleomycin-mouse model of lung fibrosis. Res. Commun. Pharmacol. Toxicol. 1:1–15. [Google Scholar]
- 18.Gurujeyalakshmi, G., M.A. Hollinger, and S.N. Giri. 1999. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am. J. Physiol. 276:L311–L318. [DOI] [PubMed] [Google Scholar]
- 19.Hallahan, D.E., L. Geng, and Y. Shyr. 2002. Effects of intercellular adhesion molecule 1 (ICAM-1) null mutation on radiation-induced pulmonary fibrosis and respiratory insufficiency in mice. J. Natl. Cancer Inst. 94:733–741. [DOI] [PubMed] [Google Scholar]
- 20.Maeda, A., K. Hiyama, H. Yamakido, S. Ishioka, and M. Yamakido. 1996. Increased expression of platelet-derived growth factor A and insulin-like growth factor-I in BAL cells during the development of bleomycin-induced pulmonary fibrosis in mice. Chest. 109:780–786. [DOI] [PubMed] [Google Scholar]
- 21.Nagase, T., N. Uozumi, S. Ishii, Y. Kita, H. Yamamoto, E. Ohga, Y. Ouchi, and T. Shimizu. 2002. A pivotal role of cytosolic phospholipase A(2) in bleomycin-induced pulmonary fibrosis. Nat. Med. 8:480–484. [DOI] [PubMed] [Google Scholar]
- 22.Sharplin, J., and A.J. Franko. 1989. A quantitative histological study of strain-dependent differences in the effects of irradiation on mouse lung during the intermediate and late phases. Radiat. Res. 119:15–31. [PubMed] [Google Scholar]
- 23.Sharplin, J., and A.J. Franko. 1989. A quantitative histological study of strain-dependent differences in the effects of irradiation on mouse lung during the early phase. Radiat. Res. 119:1–14. [PubMed] [Google Scholar]
- 24.Travis, E.L. 1980. The sequence of histological changes in mouse lungs after single doses of x-rays. Int. J. Radiat. Oncol. Biol. Phys. 6:345–347. [DOI] [PubMed] [Google Scholar]
- 25.Abdollahi, A., K.E. Lipson, X. Han, R. Krempien, T. Trinh, K.J. Weber, P. Hahnfeldt, L. Hlatky, J. Debus, A.R. Howlett, and P.E. Huber. 2003. SU5416 and SU6668 attenuate the angiogenic effects of radiation-induced tumor cell growth factor production and amplify the direct anti-endothelial action of radiation in vitro. Cancer Res. 63:3755–3763. [PubMed] [Google Scholar]
- 26.Kalluri, R., and E.G. Neilson. 2003. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112:1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinet, Y., W.N. Rom, G.R. Grotendorst, G.R. Martin, and R.G. Crystal. 1987. Exaggerated spontaneous release of platelet-derived growth factor by alveolar macrophages from patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 317:202–209. [DOI] [PubMed] [Google Scholar]
- 28.Oliver, J.A. 2002. Unexpected news in renal fibrosis. J. Clin. Invest. 110:1763–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Travis, E.L. 2001. Organizational response of normal tissues to irradiation. Semin. Radiat. Oncol. 11:184–196. [DOI] [PubMed] [Google Scholar]
- 30.Yamasaki, Y., K. Miyoshi, N. Oda, M. Watanabe, H. Miyake, J. Chan, X. Wang, L. Sun, C. Tang, G. McMahon, and K.E. Lipson. 2001. Weekly dosing with the platelet-derived growth factor receptor tyrosine kinase inhibitor SU9518 significantly inhibits arterial stenosis. Circ. Res. 88:630–636. [DOI] [PubMed] [Google Scholar]
- 31.Bergers, G., S. Song, N. Meyer-Morse, E. Bergsland, and D. Hanahan. 2003. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 111:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manning, G., D.B. Whyte, R. Martinez, T. Hunter, and S. Sudarsanam. 2002. The protein kinase complement of the human genome. Science. 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- 33.Bischof, M., A. Abdollahi, P. Gong, C. Stoffregen, K.E. Lipson, J.U. Debus, K.J. Weber, and P.E. Huber. 2004. Triple combination of irradiation, chemotherapy (pemetrexed), and VEGFR inhibition (SU5416) in human endothelial and tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 60:1220–1232. [DOI] [PubMed] [Google Scholar]
- 34.Pietras, K., T. Sjoblom, K. Rubin, C.H. Heldin, and A. Ostman. 2003. PDGF receptors as cancer drug targets. Cancer Cell. 3:439–443. [DOI] [PubMed] [Google Scholar]
- 35.Rice, A.B., C.R. Moomaw, D.L. Morgan, and J.C. Bonner. 1999. Specific inhibitors of platelet-derived growth factor or epidermal growth factor receptor tyrosine kinase reduce pulmonary fibrosis in rats. Am. J. Pathol. 155:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savikko, J., E. Taskinen, and E. Von Willebrand. 2003. Chronic allograft nephropathy is prevented by inhibition of platelet-derived growth factor receptor: tyrosine kinase inhibitors as a potential therapy. Transplantation. 75:1147–1153. [DOI] [PubMed] [Google Scholar]
- 37.Pietras, K. 2004. Increasing tumor uptake of anticancer drugs with imatinib. Semin. Oncol. 31:18–23. [DOI] [PubMed] [Google Scholar]
- 38.Hasselbalch, H.C., O.W. Bjerrum, B.A. Jensen, N.T. Clausen, P.B. Hansen, H. Birgens, M.H. Therkildsen, and E. Ralfkiaer. 2003. Imatinib mesylate in idiopathic and postpolycythemic myelofibrosis. Am. J. Hematol. 74:238–242. [DOI] [PubMed] [Google Scholar]
- 39.Cain, J.A., J.L. Grisolano, A.D. Laird, and M.H. Tomasson. 2004. Complete remission of TEL-PDGFRB-induced myeloproliferative disease in mice by receptor tyrosine kinase inhibitor SU11657. Blood. 104:561–564. [DOI] [PubMed] [Google Scholar]
- 40.Katzenstein, A.L., and J.L. Myers. 1998. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am. J. Respir. Crit. Care Med. 157:1301–1315. [DOI] [PubMed] [Google Scholar]
- 41.McBride, W.H. 1995. Cytokine cascades in late normal tissue radiation responses. Int. J. Radiat. Oncol. Biol. Phys. 33:233–234. [DOI] [PubMed] [Google Scholar]
- 42.Antoniades, H.N., M.A. Bravo, R.E. Avila, T. Galanopoulos, J. Neville-Golden, M. Maxwell, and M. Selman. 1990. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J. Clin. Invest. 86:1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoyle, G.W., J. Li, J.B. Finkelstein, T. Eisenberg, J.Y. Liu, J.A. Lasky, G. Athas, G.F. Morris, and A.R. Brody. 1999. Emphysematous lesions, inflammation, and fibrosis in the lungs of transgenic mice overexpressing platelet-derived growth factor. Am. J. Pathol. 154:1763–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu, J.Y., G.F. Morris, W.H. Lei, C.E. Hart, J.A. Lasky, and A.R. Brody. 1997. Rapid activation of PDGF-A and -B expression at sites of lung injury in asbestos-exposed rats. Am. J. Respir. Cell Mol. Biol. 17:129–140. [DOI] [PubMed] [Google Scholar]
- 45.Tada, H., F. Ogushi, K. Tani, Y. Nishioka, J.Y. Miyata, K. Sato, T. Asano, and S. Sone. 2003. Increased binding and chemotactic capacities of PDGF-BB on fibroblasts in radiation pneumonitis. Radiat. Res. 159:805–811. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida, M., J. Sakuma, S. Hayashi, K. Abe, I. Saito, S. Harada, M. Sakatani, S. Yamamoto, N. Matsumoto, and Y. Kaneda. 1995. A histologically distinctive interstitial pneumonia induced by overexpression of the interleukin 6, transforming growth factor beta 1, or platelet-derived growth factor B gene. Proc. Natl. Acad. Sci. USA. 92:9570–9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beljaars, L., B. Weert, A. Geerts, D.K. Meijer, and K. Poelstra. 2003. The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to fibroblast-like cells in fibrotic tissue. Biochem. Pharmacol. 66:1307–1317. [DOI] [PubMed] [Google Scholar]
- 48.Pinzani, M., S. Milani, H. Herbst, R. DeFranco, C. Grappone, A. Gentilini, A. Caligiuri, G. Pellegrini, D.V. Ngo, R.G. Romanelli, and P. Gentilini. 1996. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am. J. Pathol. 148:785–800. [PMC free article] [PubMed] [Google Scholar]
- 49.Ponten, A., X. Li, P. Thoren, K. Aase, T. Sjoblom, A. Ostman, and U. Eriksson. 2003. Transgenic overexpression of platelet-derived growth factor-C in the mouse heart induces cardiac fibrosis, hypertrophy, and dilated cardiomyopathy. Am. J. Pathol. 163:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamakage, A., K. Kikuchi, E.A. Smith, E.C. LeRoy, and M. Trojanowska. 1992. Selective upregulation of platelet-derived growth factor α receptors by transforming growth factor beta in scleroderma fibroblasts. J. Exp. Med. 175:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daniels, C.E., M.C. Wilkes, M. Edens, T.J. Kottom, S.J. Murphy, A.H. Limper, and E.B. Leof. 2004. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Invest. 114:1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdollahi, A., P. Hahnfeldt, C. Maercker, H.J. Grone, J. Debus, W. Ansorge, J. Folkman, L. Hlatky, and P.E. Huber. 2004. Endostatin's antiangiogenic signaling network. Mol. Cell. 13:649–663. [DOI] [PubMed] [Google Scholar]
- 53.Huber, P.E., K. Hauser, and A. Abdollahi. 2004. Genome wide expression profiling of angiogenic signaling and the Heisenberg uncertainty principle. Cell Cycle. 3:1348–1351. [DOI] [PubMed] [Google Scholar]
- 54.Abdollahi, A., K.E. Lipson, A. Sckell, H. Zieher, F. Klenke, D. Poerschke, A. Roth, X. Han, M. Krix, M. Bischof, et al. 2003. Combined therapy with direct and indirect angiogenesis inhibition results in enhanced antiangiogenic and antitumor effects. Cancer Res. 63:8890–8898. [PubMed] [Google Scholar]
- 55.Plathow, C., M. Li, P. Gong, H. Zieher, F. Kiessling, P. Peschke, H.U. Kauczor, A. Abdollahi, and P.E. Huber. 2004. Computed tomography monitoring of radiation-induced lung fibrosis in mice. Invest. Radiol. 39:600–609. [DOI] [PubMed] [Google Scholar]