

Abstract
The membrane phospholipid phosphatidylinositol 4, 5-bisphosphate [PI(4,5)P2] is a critical signal transducer in eukaryotic cells. However, the physiological roles of the type I phosphatidylinositol phosphate kinases (PIPKIs) that synthesize PI(4,5)P2 are largely unknown. Here, we show that the α isozyme of PIPKI (PIPKIα) negatively regulates mast cell functions and anaphylactic responses. In vitro, PIPKIα-deficient mast cells exhibited increased degranulation and cytokine production after Fcɛ receptor-I cross-linking. In vivo, PIPKIα−/− mice displayed enhanced passive cutaneous and systemic anaphylaxis. Filamentous actin was diminished in PIPKIα−/− mast cells, and enhanced degranulation observed in the absence of PIPKIα was also seen in wild-type mast cells treated with latrunculin, a pharmacological inhibitor of actin polymerization. Moreover, the association of FcɛRI with lipid rafts and FcɛRI-mediated activation of signaling proteins was augmented in PIPKIα−/− mast cells. Thus, PIPKIα is a negative regulator of FcɛRI-mediated cellular responses and anaphylaxis, which functions by controlling the actin cytoskeleton and dynamics of FcɛRI signaling. Our results indicate that the different PIPKI isoforms might be functionally specialized.
Engagement of mast cell FcɛRI by IgE, followed by the aggregation of multiple IgE-bearing FcɛRI molecules by polyvalent antigen, leads to cellular activation and the initiation of allergic reactions (1–3). Activated mast cells release preformed granule-associated chemical mediators, transcribe multiple cytokine genes, and secrete newly synthesized arachidonic acid metabolites and various proteins that trigger allergic inflammation. At the molecular level, the cross-linking of mast cell FcɛRI molecules initiates various signaling cascades that lead to the generation of second messengers, protein phosphorylation, and protein–protein and lipid–protein interactions (4). Among the most important intracellular signaling molecules activated after FcɛRI cross-linking are the phospholipid-metabolizing enzymes, which include phospholipase C (PLC; reference 5), src homology 2-containing inositol 5-phosphatase 1 (6), and the phosphoinositide 3-kinases (PI3Ks; references 7, 8).
Phosphorylated derivatives of phosphatidylinositol, collectively referred to as phosphoinositides, make up a minor proportion of membrane phospholipids, but are important intermediates in eukaryotic cellular responses (9, 10). Two major second messengers, phosphatidylinositol 3, 4, 5-trisphosphate (PIP3) and inositol 1,4,5-trisphosphate (IP3), are produced by PI3Ks and PLC, respectively. PIP3 and IP3 are required for intracellular signal transduction pathways activated by most cell surface receptors, including cytokine/growth factor receptors, the T cell receptor, the B cell receptor, and receptors for Ig. There is increasing evidence that the metabolism of PIP3 and IP3 has physiological and pathophysiological significance for immune system regulation (for reviews see references 11–15).
Phosphatidylinositol 4, 5-bisphosphate [PI(4,5)P2] is a substrate shared by PI3K and PLC. Phosphorylation of PI(4,5)P2 by PI3K generates PIP3, which controls the intracellular localization and activity of several proteins containing pleckstrin homology domains (16). Conversely, hydrolysis of PI(4,5)P2 by PLC produces IP3 and DAG, second messengers necessary for the entry of calcium into cells and protein kinase C activation, respectively (13). In addition to its well-established role as a precursor of second messengers, a more direct function for PI(4,5)P2 in the control of diverse cellular processes has been demonstrated recently (17–20). Based on biochemical assays and studies of permeable cell preparations, PI(4,5)P2 appears to be particularly important for the regulation of cytoskeletal reorganization and intracellular membrane transport (9, 17, 18, 21–23).
The phosphatidylinositol phosphate kinases (PIPKs) are enzymes important for PI(4,5)P2 synthesis. There are two types of PIPKs: type I (PIPKI) and type II (PIPKII). Three genes encode the α,β and γ PIPKI isoforms, and the γ gene further generates three splice variants (24–28); in addition, three genes encode PIPKII (29). PI(4,5)P2 can be synthesized either by phosphorylation of PI(4)P by PIPKI, or by phosphorylation of PI(5)P by PIPKII. In mammalian cells, PI(4)P is at least fifty times more abundant than PI(5)P. Moreover, PIPKI (but not PIPKII) can induce dramatic changes in the actin cytoskeleton. Thus, it is generally accepted that the majority of PI(4,5)P2 molecules in a mammalian cell are derived from PIPKI-mediated phosphorylation of PI(4)P. Given the broad range of potential functions of PI(4,5)P2, and the fact that the steady-state level of PI(4,5)P2 is much higher than that of other lipid second messengers, it is likely that some kind of functional compartmentalization of PI(4,5)P2 production exists inside the cell (17, 30). Such compartmentalization could be achieved by the interplay of multiple PIPKI molecules.
In this work, we genetically inactivated the α isozyme of PIPKI (PIPKIα) in mice. PIPKIα deficiency leads to enhanced degranulation and cytokine production in response to FcɛRI stimulation in cultured mast cells. Moreover, anaphylactic responses are enhanced in mice deficient for PIPKIα. From a mechanistic standpoint, our results indicate that PIPKIα is crucial for the integrity of the actin cytoskeleton and FcɛRI translocation to lipid rafts. Thus, our data are the first genetic evidence for a nonredundant role of PIPKIα in vivo: the restraint of allergic reactions.
Results
Generation of PIPKIα knockout mice and bone marrow–derived mast cells
Murine embryonic stem cells heterozygous for a deletion mutation of the PIPKIα gene were generated by replacing 1.7 kb of the PIPKIα gene (including the region encoding the NH2-terminal amino acids 68–106, indispensable for kinase activity) with a PGK-Neo cassette (Fig. 1 A). Southern blot analysis using a short arm flanking probe confirmed disruption of the gene (Fig. 1 B). No random integrations of the PGK-Neo cassette were detected (unpublished data). These cells were used to derive homozygous PIPKIα−/− mice, which were born at the expected Mendelian ratio, were healthy and fertile, and displayed no histological abnormalities up to 12 mo of age. There were no overt differences from the WT in lymphocyte numbers or in subpopulations present in the thymus, lymph node, spleen, and bone marrow (unpublished data).
Figure 1.

Gene targeting of murine PIPKIα and characterization of PIPKIα2/− BMMCs. (A) Partial restriction map of the genomic PIPKIα sequence and construction of the targeting vector bearing the neomycin resistance (Neo r) gene. Exon 3, which encodes a portion of the kinase core domain, was replaced with a PGK-Neo cassette. The PIPKIα flanking probe used for Southern blotting and expected fragment sizes after digestion of WT and mutant genomic DNA are indicated. H, Hind III; E, EcoRV; S, SmaI; DT-A, diphtheria toxin A subunit. (B) Southern blot of genomic DNA from wild type (+/+), PIPKIα+/− (+/−), and PIPKIα−/− (−/−) E14 embryonic stem cells hybridized to the probe indicated in A. (C) Western blot of PIPKI isozyme expression in BMMCs using antibodies specifically recognizing the indicated proteins. (D) Flow cytometric analysis of the normal surface expression of c-Kit (left) and FcɛRI (right) on PIPKIα−/− BMMCs. (E) Equivalent cumulative cell numbers of PIPKIα+/+ (open circles) and PIPKIα−/− BMMCs (closed circles, dotted line) in cultures maintained for the indicated number of days. For all figures, results shown are representative of at least three independent experiments using three pairs of simultaneously established PIPKIα+/+ and PIPKIα−/− BMMCs.
It has been demonstrated that the PI3Ks and PLCγ, which utilize PI(4,5)P2 as a substrate to generate either PIP3 or IP3, respectively, play critical roles in the development and effector functions of mast cells (31–34). In addition, PI(4,5)P2 modulates the organization of the actin cytoskeleton, another cellular process that has been implicated in the degranulation response (35, 36). To investigate the physiological role of PIPKIα in mast cells, we established IL-3–dependent bone marrow–derived mast cell (BMMC) lines from PIPKIα+/+ and PIPKIα−/− littermates. BMMCs from PIPKIα−/− mice lacked PIPKIα protein, but showed normal expression of two other PIPKIs, PIPKIβ and PIPKIγ (Fig. 1 C). Both PIPKIα+/+ and PIPKIα−/− BMMCs expressed similar levels of c-Kit and FcɛRI, markers that are characteristic of mature BMMCs (Fig. 1 D), and PIPKIα−/− BMMCs proliferated at the WT rate (Fig. 1 E). Thus, PIPKIα expression appears to be dispensable for the cytokine-dependent emergence and differentiation of BMMCs.
Enhanced degranulation, cytokine gene expression, and FcɛRI signaling in PIPKIα−/− BMMCs
To determine whether PIPKIα was required for mast cell functions, we investigated mast cell granule release by measuring the extracellular activity of β-hexosaminidase, a marker enzyme for histamine-containing granules. The total activity of β-hexosaminidase per cell did not differ between PIPKIα+/+ and PIPKIα−/− BMMCs, and treatment with IgE alone did not induce granule release from BMMCs of either genotype. However, FcɛRI-evoked degranulation was increased in PIPKIα-deficient mast cells compared with WT controls (Fig. 2 A). From a total of >15 experiments using five pairs of BMMC lines from littermate mice, we calculated that the degranulation at 5 min after 10 ng/ml DNP stimulation was 56.7% ± 6.2 for PIPKIα−/− BMMCs compared with 23.2% ± 3.6 for WT BMMCs. This difference was statistically significant as determined by the Mann-Whitney's U test (P = 0.00078) and suggested that PIPKIα is involved in the control of FcɛRI signaling.
Figure 2.
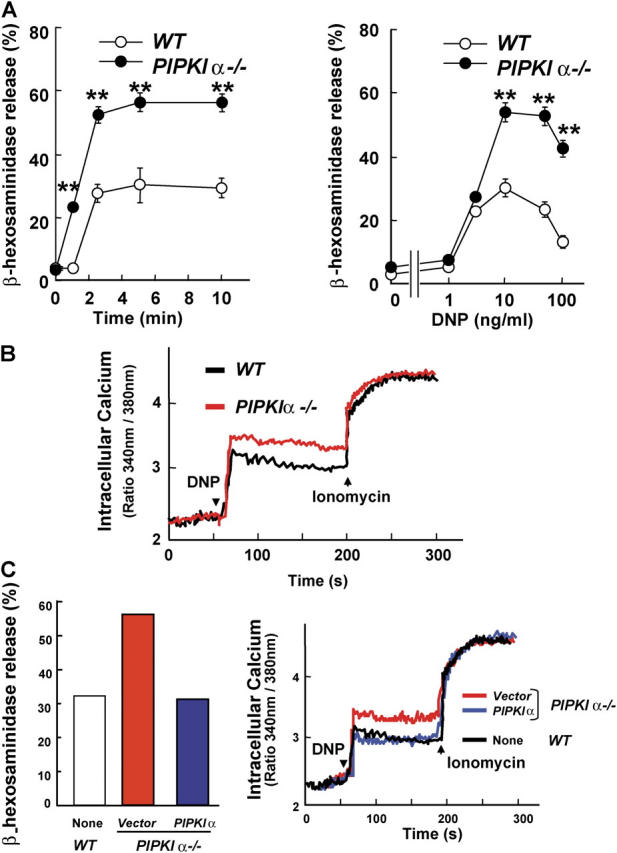
Enhanced FcɛRI-mediated degranulation and calcium mobilization in PIPKIα2/− mast cells. (A) Increased β-hexosaminidase release. PIPKIα+/+ (WT; open circles) and PIPKIα−/− (closed circles) BMMCs were preloaded with mouse anti-DNP IgE and stimulated with 50 ng/ml−1 DNP-HSA for the indicated times (left) or stimulated for 10 min with the indicated concentrations of DNP-HSA (right). The percentage of total cellular β-hexosaminidase that was released was taken as degranulation. Data shown are the mean ± SD of triplicate samples. **, P < 0.01 for PIPKIα−/− cells compared with PIPKIα+/+ cells, as determined by Student's t test. (B) Increased calcium mobilization. IgE-sensitized, Fura-2-loaded BMMCs were stimulated with 50 ng/ml−1 DNP-HSA and Ca2+ flux was monitored by spectrofluorimetry. (C) Restoration of normal degranulation and Ca2+ flux after introduction of PIPKIα cDNA. Degranulation (left) and Ca2+ flux (right) were measured as in A and B, respectively, in WT BMMCs or PIPKIα−/− BMMCs reconstituted with either empty vector (red) or PIPKIα cDNA (blue).
It is well known that a transient increase in intracellular calcium is essential for degranulation (34, 37). We found that the amplitude of Ca2+ elevation was increased in PIPKIα−/− BMMCs compared with that in WT cells (Fig. 2 B), consistent with the enhanced degranulation observed in the mutant cells. To confirm that the enhanced degranulation truly resulted from the loss of PIPKIα expression, we reestablished PIPKIα expression in PIPKIα−/− BMMCs using retroviral infection. Degranulation and Ca2+ mobilization were restored to normal by infection of PIPKIα−/− BMMCs with a retrovirus containing PIPKIα, but not by infection with a control virus (Fig. 2 C). Together, these results demonstrate that a lack of PIPKIα expression engenders increased antigen-induced degranulation mediated by high affinity FcɛRI.
Cytokines produced by mast cells are critical mediators in allergy and inflammation. Therefore, we investigated whether PIPKIα played a role in mast cell cytokine production. Quantitative RT-PCR was performed to determine the induction of various cytokine mRNA transcripts in WT and PIPKIα−/− BMMCs. FcɛRI engagement induced higher levels of all cytokine mRNAs examined in PIPKIα−/− BMMCs compared with WT cells (Fig. 3 A). IL-2 and IL-3 were the molecules most markedly affected; relative quantitation revealed >50-fold increases in these mRNAs in PIPKIα−/− cells that had been stimulated for 1 h (Fig. 3 A and not depicted).
Figure 3.

Augmented cytokine gene expression and FcɛRI signaling in PIPKIα2/− BMMCs. (A) Increased cytokine mRNA expression. PIPKIα+/+ and PIPKIα−/− BMMCs were sensitized with IgE and stimulated with DNP (50 ng/ml−1) for the indicated times. Induction of mRNA expression for the indicated cytokines was detected by RT-PCR. (B) Enhanced signaling molecule phosphorylation after FcɛRI cross-linking. BMMCs were sensitized with IgE and stimulated with DNP for the indicated times. For both panels, 30 μg of cell lysates were subjected to successive rounds of immunoblotting using phospho-specific and total antibodies recognizing the indicated proteins. Syk phosphorylation was monitored by immunoprecipitation of Syk followed by immunoblotting using antiphosphotyrosine antibody. One trial representative of a minimum of three experiments is shown in each case. White lines indicate that intervening lanes have been spliced out. (C) Band intensities in B were quantified using Dolphin-1 software, and relative phosphorylation levels were normalized to protein levels as described in Materials and methods. Values shown are the fold increase in the phosphorylated form of each molecule in PIPKIα−/− BMMCs compared with the value in WT cells activated for 2 min (except for SAPK, which was 10 min). (D) Normal signaling molecule phosphorylation after IL-3 or SCF stimulation. WT and PIPKIα−/− BMMCs were stimulated with either 30 ng/ml−1 IL-3 or 30 ng/ml−1 SCF and the phosphorylation of ERK and p38 was assessed as in B.
Cytokine gene expression is regulated by several signaling cascades, including those governed by mitogen-activated protein kinases (38). We stimulated PIPKIα+/+ and PIPKIα−/− BMMCs with IgE plus antigen and monitored the phosphorylation (activation) of various signaling proteins. FcɛRI-triggered phosphorylation of ERK1/ERK2 (T202/Y204), SAPK (T183/Y185), p38 (T180/Y182), PLCγ-1 (Y783), PKB (Ser473), and Syk was increased in PIPKIα−/− BMMCs compared with WT BMMCs (Fig. 3, B and C), indicating that multiple signaling cascades are activated in the absence of PIPKIα. Of interest, normal phosphorylation of ERK and p38 occurred in PIPKIα−/− BMMCs stimulated with either IL-3 or SCF (Fig. 3 D), and the basal phosphorylation of mitogen-activated protein kinases was not increased in PIPKIα−/− BMMCs (Fig. 3, B and D). Therefore, an absence of PIPKIα does not lead to hyperphosphorylation of these signaling molecules per se. Rather, the hyperphosphorylation of signaling molecules in PIPKIα−/− BMMCs depends on FcɛRI stimulation.
Increased severity of local and systemic anaphylactic reactions in PIPKIα-deficient mice
Anaphylaxis is an extreme form of allergic reaction triggered by allergen-induced cross-linking of allergen-specific IgE present on the surface of mast cells. Mast cell degranulation is induced, which leads to the release of copious amounts of vasoactive amines and inflammatory mediators. To determine whether PIPKIα functions as a negative regulator of mast cell activation in vivo, we examined IgE-dependent anaphylactic reactions in PIPKIα−/− mice. Antigen challenge of mice that had been sensitized previously with monoclonal DNP-specific IgE antibody demonstrated that PIPKIα−/− mice exhibited a greater degree of systemic anaphylaxis than WT mice, as assessed by core temperature changes (Fig. 4 A). An increase in passive cutaneous anaphylaxis was also observed in the mutants (Fig. 4 B). Mast cell numbers were identical in the ear skin of WT (12 ± 0.9 per field; n = 8) and PIPKIα−/− (12 ± 1.0 per field; n = 8) mice, so that the augmented anaphylaxis in PIPKIα−/− mice was most likely due to mast cell hyperreactivity to antigens. These data imply that a major physiological role of PIPKIα is to prevent inappropriate mast cell degranulation and cytokine production and, thus, anaphylaxis.
Figure 4.

Enhanced anaphylactic responses in PIPKIα−/− mice. (A) Systemic anaphylaxis. WT and PIPKIα−/− mice (n = 7 mice per genotype) received 5 μg anti-DNP IgE i.v., followed by stimulation with 1 mg DNP-HSA per mouse. The systemic anaphylactic response was monitored by measuring rectal temperature at the indicated times after antigen injection. (B) Passive cutaneous anaphylaxis. WT and PIPKIα−/− mice (n = 9 per genotype) received 100 μg anti-TNP IgE i.v. After 24 h, mice were epicutaneously challenged with 10 μl 1% picryl chloride on the right ears, and with 10 μl 1% oxazolone on the left ears. Net ear swelling (thickness of the right ear minus that of the left ear) was measured with a caliper at the indicated times. Data are expressed as mean ± SD. *, P < 0.05 and **, P < 0.01 for PIPKIα−/− mice compared with PIPKIα+/+ mice as determined by Student's unpaired t test.
Decreased PI(4,5)P2 levels and atypical actin cytoskeleton in PIPKIα-deficient mast cells
In view of the in vitro evidence that PIPKIα produces PI(4,5)P2, the precursor of IP3 and PIP3, the finding of increased mast cell activation and allergic reactions in the absence of PIPKIα was an unexpected result. Therefore, we determined the relative contribution of PIPKIα to overall PI(4,5)P2 production. The intracellular PI(4,5)P2 content was assessed in BMMCs that had been metabolically labeled with [3H]inositol for 48 h to achieve a stable equilibrium. Lipid extraction followed by HPLC analysis revealed a statistically significant decrease in PI(4,5)P2 in BMMCs lacking PIPKIα (Fig. 5 A). A concomitant increase in PI(4)P, the substrate of PIPKIα, was observed in PIPKIα−/− BMMCs. These results provide the first genetic evidence that PIPKIα acts as a functional PI(4)P 5-kinase in vivo, and are in agreement with a recent paper describing a partial decrease in PI(4,5)P2 levels in HeLa cells treated with small interference RNA for PIPKIα (39). We conclude that the loss of PIPKIα can be compensated for by other PIPKs in the context of bulk PI(4,5)P2 synthesis, but that a specific PI(4,5)P2 pool may exist that is exclusively maintained by PIPKIα.
Figure 5.
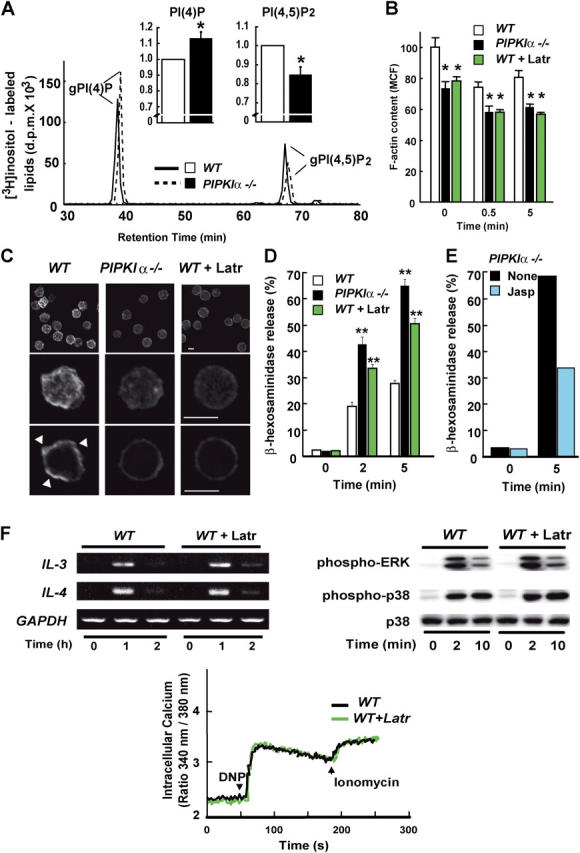
PI(4,5)P2 levels and actin cytoskeleton in PIPKIα−/− BMMCs. (A) Altered phospholipids. HPLC analysis of phospholipids prepared from WT and PIPKIα−/− BMMCs that were metabolically labeled with [3H]inositol for 48 h. The chromatographic tracings shown are one result representative of five independent trials. The decrease in PI(4,5)P2 (0.84-fold) and increase in PI(4)P (1.13-fold) in PIPKIα−/− BMMCs were statistically significant (insets). *, P < 0.05 for PIPKIα−/− cells compared with untreated WT cells. (B) Decreased F-actin content as determined by flow cytometry. IgE-sensitized PIPKIα−/− BMMCs and IgE-sensitized WT BMMCs, which were either left untreated or pretreated with 0.5 μM latrunculin (Latr) for 15 min, were stimulated with 50 ng/ml−1 DNP for the indicated times. F-actin was stained with Alexa 488–labeled phalloidin and analyzed by flow cytometry. The mean channel fluorescence (MCF) in untreated WT BMMCs was arbitrarily assigned a value of 100. Data shown are the mean percentage of the control value ± SD of triplicate samples. *, P < 0.05 for PIPKIα−/− cells or latrunculin-treated WT cells compared with untreated WT cells at the indicated times. (C) Decreased F-actin content as determined by confocal fluorescence microscopy. F-actin in the cells examined in B was visualized using Alexa 488–labeled phalloidin. Confocal images were collected every 1 μm, and summation images (top and middle rows) or single images from the center of representative cells (bottom row) are presented. Bar, 10 μm. The WT cells exhibited a more jagged circumferential F-actin structure compared with PIPKIα−/− cells (arrowheads). (D) Increased degranulation induced by latrunculin. IgE-sensitized PIPKIα−/− BMMCs, and IgE-sensitized WT BMMCs that were either left untreated or pretreated with latrunculin (Latr) for 15 min were stimulated with 50 ng/ml−1 DNP for the indicated times. Degranulation was measured as in Fig. 2 A. **, P < 0.01 for latrunculin-treated WT cells or PIPKIα−/− cells compared with untreated WT cells at 5 min after cross-linking (n = 4). (E) Suppression of degranulation induced by jasplakinolide. IgE-sensitized PIPKIα−/− BMMCs that were either left untreated or pretreated with 1μM jasplakinolide (Jasp) for 15 min were stimulated with DNP for 5 min. Degranulation was assessed as for in D. (F) Normal cytokine mRNA expression, signaling molecule phosphorylation, and Ca2+ mobilization in the presence of latrunculin. IgE-sensitized WT BMMCs that were either left untreated or pretreated with latrunculin for 15 min were stimulated with DNP for the indicated times. Cytokine gene expression, protein phosphorylation, and Ca2+ mobilization were determined as in Fig. 3, A and B, and Fig. 2 B, respectively.
To gain insights into the functional characteristics of the PI(4,5)P2 compartment controlled by PIPKIα, we analyzed the production of the PI(4,5)P2–derived second messenger IP3. At 1 min after FcɛRI stimulation, IP3 production was enhanced in PIPKIα−/− BMMCs (2.8 ± 0.37 pmol/106) compared with WT cells (2.1 ± 0.17 pmol/106; P = 0.13; n = 8). In addition, the observation that PKB activation was increased in PIPKIα−/− BMMCs (Fig. 3 B) suggests that PIP3 formation is also enhanced in the absence of PIPKIα. Thus, somewhat surprisingly, the production of the second messengers IP3 and PIP3 is increased, rather than decreased, in the absence of PIPKIα.
In sharp contrast, the absence of PIPKIα had a clearly suppressive effect on the filamentous actin cytoskeleton (F-actin) in BMMCs. F-actin in PIPKIα−/− BMMCs was consistently decreased to 70–80% of the WT level (Fig. 5 B). We used confocal fluorescence microscopy to examine the morphology and distribution of F-actin in PIPKIα−/− BMMCs. A dramatic reduction in F-actin staining was found in PIPKIα−/− BMMCs in comparison with WT cells (Fig. 5 C). Notably, the pattern of F-actin distribution and the cell shape of PIPKIα−/− BMMCs were similar to those of WT BMMCs treated with latrunculin, a potent inhibitor of actin polymerization (40). Cortical actin filaments were decreased in both cases. Together, these results indicate that PIPKIα is dispensable for IP3 and PIP3 production, but is a critical regulator of actin cytoskeletal reorganization.
A role for F-actin in the down-regulation of mast cell degranulation, but not cytokine expression
The peripheral actin cytoskeleton is essential for many biological processes involving changes to the plasma membrane architecture (41, 42). In the rat basophilic leukemia (RBL) cell line, it has been demonstrated that FcɛRI-triggered actin polymerization plays a negative regulatory role in degranulation, cytokine production, and Ca2+ elevation as well as in FcɛRI signaling (35, 43). To test whether F-actin was involved in BMMC responses, we first examined the effect of latrunculin on degranulation. WT BMMCs pretreated with latrunculin showed enhanced degranulation in response to FcɛRI engagement (Fig. 5 D). Moreover, jasplakinolide, a cell-permeable stabilizer of actin filaments, suppressed degranulation in BMMCs upon FcɛRI cross-linking (Fig. 5 E). Latrunculin and jasplakinolide also had parallel effects on BMMC degranulation induced by ionomycin (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20041891/DC1). These results suggest that polymerized actin can restrain degranulation, and that the augmented degranulation observed in PIPKIα−/− BMMCs can be attributed to their decreased polymerized actin content.
Next, we examined the possible involvement of the actin cytoskeleton in FcɛRI-mediated cytokine production and protein phosphorylation. Contrary to its striking effect on degranulation, treatment of WT BMMCs with latrunculin before FcɛRI cross-linking did not enhance cytokine induction or phosphorylation of signaling proteins, and did not lead to elevated intracellular calcium (Fig. 5 F). These observations may account for the fact that latrunculin only partially mimics the effect of PIPKIα disruption on BMMC degranulation. Our findings concur with a previous paper that demonstrated normal activation of ERK and p38 despite increased actin polymerization in BMMCs lacking Wiskott-Aldrich syndrome protein-interacting protein (44). Thus, contrary to the situation in RBL cells, alterations to the actin cytoskeleton in BMMCs do not necessarily result in anomalies to all FcɛRI-evoked cellular responses. Rather, our data show that PIPKIα-mediated suppression of FcɛRI-mediated cellular responses in mast cells appears to operate via at least two different mechanisms, only one of which depends on regulation of actin polymerization.
Increased localization of FcɛRI in lipid rafts in PIPKIα−/− BMMCs
Although most FcɛRI-mediated responses were potentiated in PIPKIα-deficient BMMCs, those induced by IL-3 and SCF treatment were similar in magnitude to those of WT BMMCs. Therefore, we investigated whether an absence of PIPKIα could lead to a change in the dynamics of FcɛRI distribution on the plasma membrane. Lipid rafts are defined as plasma membrane microdomains enriched with glycosphingolipids and cholesterol. These structures are now generally recognized as “signaling platforms” (45, 46). It has been shown that, after cross-linking, FcɛRI molecules are recruited to the rafts where they undergo tyrosine-phosphorylation as the initiation step of several downstream signaling cascades. Indeed, disruption of lipid rafts with methyl-β-cyclodextrin inhibited FcɛRI signaling in BMMCs, as exemplified by a decrease in ERK and p38 phosphorylation (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20041891/DC1).
Due to their low density, lipid rafts can be separated from nonraft plasma membrane components by ultracentrifugation of detergent-lysed cells in sucrose density gradients. As shown in Fig. 6 A, engagement of FcɛRI induced translocation of at least the FcɛRIγ chain component of FcɛRI to the lipid raft fractions (fractions 3 and 4) of both WT and PIPKIα−/− BMMCs. Importantly, the presence of FcɛRIγ in the raft fractions was significantly enhanced in PIPKIα−/− BMMCs even before aggregation. Immunoblotting for the lipid raft marker linker for activation of T cells (LAT) demonstrated that the partitioning of LAT between the lipid rafts and nonlipid raft regions of the membrane was not altered in the absence of PIPKIα (Fig. 6 B). It should also be noted that the amounts of FcɛRIγ in the lipid rafts were not affected by latrunculin, suggesting that the actin cytoskeleton does not play a major role in regulating the redistribution of FcɛRI. These results suggest that PIPKIα suppresses the interaction between FcɛRI and the lipid rafts independently of actin cytoskeletal organization. Such a role for PIPKIα could explain its apparent ability to impede the propagation of FcɛRI-mediated signaling cascades leading to BMMC cytokine production and degranulation. The precise mechanism by which PIPKIα regulates FcɛRI localization is under investigation.
Figure 6.

Regulation of FcɛRI localization to lipid rafts by PIPKIα. (A) Enhanced localization of FcɛRIγ to lipid rafts. BMMCs sensitized with anti-DNP IgE were either left untreated or treated with latrunculin for 15 min. Cells were incubated with or without DNP (stimulation) for 2 min, lysed in 0.5% Triton X-100 buffer, and subjected to sucrose gradient ultracentrifugation to purify lipid rafts. (left) Fractions were separated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted (IB) with anti-FcɛRI γ-chain antibody. Fractions 3 and 4 contain the lipid rafts. (right) The distribution of FcɛRIγ in fractions 3 and 4 was quantitated by densitometric analysis of the immunoblot. (B) Normal distribution of LAT. Fractions from A were immunoblotted with anti-LAT antibody as for in A (right) and LAT distribution was densitometrically quantitated as in A (left).
Discussion
Mast cells play a key role in allergic reactions due to their ability to synthesize and release proinflammatory mediators and cytokines (1–3). Upon exposure to allergens, specific IgE bound to FcɛRI on mast cells becomes cross-linked and intracellular signals are transduced that lead to cellular activation. These intracellular signals are tightly regulated, as spurious signals could result in unwanted, and possibly deleterious, responses. Although recent work has identified many of the proteins that positively regulate FcɛRI signaling, little is known about the negative regulators of these signaling cascades. In this study, we have identified a physiological role for PIPKIα as a negative regulator of FcɛRI-mediated mast cell functions. BMMCs from PIPKIα-deficient mice exhibit enhanced degranulation and cytokine gene expression. As a result, loss of PIPKIα culminates in aggravated systemic and local passive anaphylaxis in vivo.
PIPKIs are lipid kinases that are critical for intracellular signaling due to their production of the versatile phospholipid PI(4,5)P2. PIPKIs and PI(4,5)P2 have been implicated in the regulation of the actin cytoskeleton, vesicular trafficking, cell migration, adhesion, phagocytosis, and apoptosis (9, 17, 18, 21, 22). Three genes encoding PIPKIs have been identified that show considerable homology to each other, but not to other lipid kinases. However, it has been difficult to clarify whether the multiple PIPKI isozymes have overlapping or redundant functions. The genetic examination of PIPKIα function presented in this work clearly demonstrates an essential function for a PIPKI isozyme as a modifier of FcɛRI-mediated mast cell activation, and suggests that each PIPKI isozyme may play a unique physiological role. Consistent with this concept is the recent paper by Di Paolo et al., which asserts that disruption of the PIPKIγ isoform in mice leads to early postnatal lethality and synaptic defects (47). However, there must also be an overlap of PIPKI isozyme functions because PIPKIα-deficient mice are viable and fertile and display no overt histological abnormalities despite the wide tissue distribution of PIPKIα expression.
It has long been hypothesized that cortical actin filaments act as a barrier to prevent secretory granules from accessing the plasma membrane (48). This inference has been supported mainly by pharmacological evidence that inhibitors of actin polymerization potentiate degranulation. In vitro, latrunculin reportedly enhances FcɛRI-mediated degranulation in both mouse BMMCs and RBL cells, although FcɛRI stimulation results in a decrease in F-actin content in the former (44) and an increase in the latter (35). Thus, a major outstanding question in this field has been whether this actin-based regulation of degranulation operates physiologically. Our work shows that PIPKIα regulates actin reorganization in mast cells in a manner that is functionally important for degranulation, supporting a physiologically relevant role for the control of degranulation via the actin cytoskeleton. Moreover, our results provide insight into a potential molecular mechanism that can maintain sufficient F-actin in mast cells to suppress inappropriate degranulation. This mechanism may define the threshold for the occurrence of allergic reactions in vivo.
IP3 production and PIP3-dependent activation of PKB are enhanced in PIPKIα−/− BMMCs, suggesting that PIPKIα is dispensable for supplying PI(4,5)P2 for the generation of second messengers. It has recently become clear that intact PI(4,5)P2 (but not the products of its metabolism) can act directly as a signaling lipid (17–20). This function of PI(4,5)P2 is mediated by actin regulatory proteins and components of the exocytosis/endocytosis machinery that possess PI(4,5)P2 binding domains (9, 17, 18, 21–23). Given the essential role of PI(4,5)P2 in actin reorganization, our data indicate that the pool of PI(4,5)P2 produced specifically by PIPKIα is responsible for the maintenance of the actin cytoskeleton in mast cells. We propose that PI(4,5)P2 synthesis by PIPKIα must take place in a defined membrane compartments because, despite its striking effect on F-actin content, the overall PI(4,5)P2 level was only partially reduced in BMMCs lacking PIPKIα (Fig. 5). However, the putative specialized membrane compartments in which PIPKIα-mediated PI(4,5)P2 synthesis occurs remain to be characterized. It should also be noted that our findings do not contradict previous studies that have unequivocally demonstrated that PI(4,5)P2 interacts with and activates several proteins needed for the docking and fusion of secretory granules (49, 50). Our work suggests that, although PIPKs other than PIPKIα can supply sufficient PI(4,5)P2 to allow membrane fusion, PIPKIα has an exclusive role in modulating the actin cytoskeleton and mast cell degranulation.
The mechanism underlying the augmented FcɛRI signaling response and increased cytokine gene expression caused by PIPKIα deficiency appears to be distinct from that underlying the enhanced degranulation because these phenotypes could not be induced by latrunculin-mediated reduction of F-actin. As PIPKIα-deficient BMMCs responded normally to IL-3 and SCF, whereas virtually all responses mediated by FcɛRI were enhanced, we speculate that PIPKIα acts at the level of FcɛRI activation as well as at the level of actin cytoskeleton reorganization. Recent studies have revealed that LAT in lipid rafts plays a central role in FcɛRI signaling in mast cells (51). Aggregation of FcɛRI induces the inclusion of this receptor and Syk into lipid rafts. Subsequently, Syk phosphorylates LAT to create protein-binding sites that facilitate the assembly of a macromolecular complex of signaling proteins that include Grb2, Gads, SLP76, Vav, and PLCγ. Thus, the FcɛRI signaling pathway becomes broadly divergent after the stage of lipid raft recruitment (36, 52, 53). In our work, we showed that the association of the FcɛRIγ subunit with lipid rafts was increased in PIPKIα−/− BMMCs. Does the distribution of FcɛRIγ fairly represent the distribution of FcɛRI? Although the FcɛRIγ chain associates with a variety of receptors, including FcγR (2), BMMCs do not degranulate in response to IgG (unpublished data). Furthermore, other receptors in which FcɛRIγ participates, such as FcαR, are not expressed on BMMCs (2, 54). Therefore, we believe that the distribution of FcɛRIγ truly represents the localization of functional FcɛRI on the mast cell surface. To our knowledge, our investigation of PIPKIα provides the first identification of a molecule that modulates the localization of FcɛRI to lipid rafts. Thus, loss of this regulatory function may account for the broad impact of PIPKIα deficiency on the multiple cellular responses elicited by FcɛRI engagement (Fig. 7). It should be noted that some FcɛRI-mediated responses are unaltered in the absence of LAT (51), indicating that signaling pathways independent of lipid rafts exist. The role of PIPKIα in such pathways remains to be defined.
Figure 7.

A model for the role of PIPKIα in mast cell activation and anaphylactic responses mediated by FcɛRI. PIPKIα acts both to control FcɛRI signaling after cross-linking and to promote actin polymerization, thereby inhibiting both degranulation and cytokine production. Thus, anaphylaxis is prevented.
Our work provides clues to the mechanisms involved in FcɛRI dynamics in the plasma membrane. Because phosphoinositides (including PI(4,5)P2) are reportedly enriched in lipid rafts (55), it will be interesting to analyze FcɛRI distribution in mutant cells deficient for other phosphoinositide-metabolizing enzymes such as PIPKIβ and PIPKIγ. These types of studies may further establish that the PI(4,5)P2 synthesized by each PIPK isozyme is functionally compartmentalized. More importantly, such investigations may clarify the precise mechanisms regulating FcɛRI dynamics in the plasma membrane that influence the outcome of allergen challenge in vivo.
Our findings may have applications in the clinical arena. FcɛRI engagement is known to trigger allergic reactions, and an association between human FcɛRIβ chain polymorphisms and atopic phenotypes has been reported previously (56). FcɛRI engagement has also been linked to the pathogenesis of parasitic diseases and autoimmune disorders (2). These associations arise because FcɛRI is found on monocytes, eosinophils, platelets, Langerhans cells, and dendritic cells as well as on mast cells. Thus, the functional specialization reported here for a PIPKI isoform in the molecular attenuation of FcɛRI signaling may represent a distinct mechanism underlying a subset of allergic hypersensitivities, parasite susceptibilities or autoimmune diseases. Such specialization among PIPKs could provide clinical researchers with novel therapeutic targets for these disorders.
Materials and Methods
Generation of PIPKIα-deficient mice
Mice deficient for PIPKIα were generated using homologous recombination in embryonic stem cells as described previously (57). For further experimental details, see Supplemental Materials and methods (available at http://www.jem.org/cgi/content/full/jem.20041891/DC1). All experimental protocols were reviewed and approved by the Akita University Institutional Committee for Animal Studies.
Retroviral gene transfer
BMMC reconstitution assays using the retroviral vector pBabe-puro were performed as described previously (58). In brief, Myc epitope-tagged full-length PIPKIα cDNA (59) was cloned into the SalI site of pBabe-puro. This vector was transfected into a phoenix-E packaging cell line. PIPKIα−/− BMMCs were cultured in the supernatant for 1 d and selected in 2.5 mg/ml−1 puromycin for 3 wk before being used for experiments.
Degranulation and calcium mobilization
2 × 106 cells ml−1 BMMCs were sensitized with 0.2 μg/ml−1 anti-DNP IgE (SPE-7) for 15 h. The cells were washed, resuspended in OPTI-MEM (GIBCO BRL) containing 0.1% BSA, and challenged with DNP-HSA (Sigma-Aldrich). The percentage of total cellular β-hexosaminidase that was released was taken as degranulation as described previously (60). For calcium mobilization, the sensitized BMMCs were incubated at room temperature for 45 min with 2 μM Fura-2-AM (Molecular Probes) in Tyrode's buffer (10 mM Hepes, pH 7.4, 112 mM NaCl, 2.7 mM KCl, 0.4 mM NaH2PO4, 1.6 mM CaCl2, 1 mM MgCl2, 2 mM glucose, and 1% BSA). FcɛRI-mediated calcium mobilization was measured every 0.5 s using a spectrophotometer (Shimadzu) set for dual excitation at 340 and 380 nm and emission at 510 nm.
Immunoblotting, immunoprecipitation, and in vitro PI3K assay
BMMCs were sensitized as described before. After washing and resuspension in OPTI-MEM containing 0.1% BSA, the cells (5–10 × 106) were stimulated with 50 ng ml−1 DNP-HSA. Reactions were terminated by the addition of ice-cold PBS and cell lysates were prepared in RIPA buffer (1% Triton X-100, 10 mM Tris/HCl, pH 7.4, 150 mM NaCl, 30 mM Na4P2O7, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail from Roche Molecular Chemicals). Immunoprecipitation and immunoblotting were performed as described previously (61). The relative phosphorylation of each signaling protein was normalized to its protein level in each sample (for details, see Supplemental Materials and methods). Immune complex lipid kinase assays were performed as described previously (57).
Passive systemic and cutaneous anaphylaxis
For passive systemic anaphylaxis, mice (10–15 wk old) were sensitized for 24 h by intravenous injection of 5 μg anti-DNP IgE (SPE-7). The mice were subsequently challenged with an intravenous injection of 100 μg DNP-HSA. Body temperature was monitored using a rectal probe (Shibaura) starting from the time of antigen injection. For passive cutaneous anaphylaxis, mice (10–15 wk old) were injected intravenously with 100 μg anti-TNP IgE (62). After 24 h, ear-swelling responses were elicited by painting 10 μl 1% picryl chloride (Nakalai Tesque) in acetone on the right ears of each animal, and 10 μl 1% 4-ethoxymethylene-2-phenyl-2-oxazoline-5-one (oxazolone; Sigma-Aldrich) in acetone on the left ear. Ear thickness was measured as described previously (62).
Cellular PI(4,5)P2 measurement and actin cytoskeleton assessments
BMMCs were labeled for 48 h with 10 μCi ml−1 [3H]-myo-inositol (Amersham Biosciences) in inositol-free DMEM containing dialyzed 10% heat-inactivated FCS. Labeling was quenched and lipids were extracted as described (57). Dried lipids were deacylated and analyzed by HPLC according to Serunian et al. using a Partisphere SAX column (Whatman; reference 63). Radioactivity was assayed in 0.5-ml fractions using a liquid scintillation counter. To compare data among experiments, the raw radioactive counts determined for PI(4)P and PI(4,5)P2 were normalized to the raw radioactive counts for total phosphoinositides. The normalized amounts of PI(4)P or PI(4,5)P2 present in PIPKIα+/+ BMMCs in each experiment were assigned a value of 1, and the relative amounts of PI(4)P or PI(4,5)P2 in PIPKIα−/− BMMCs were calculated. Significance was assessed with Student's t test. p-values <0.05 were considered significant. Flow cytometry and fluorescence microscopy were used to analyze the status of the actin cytoskeleton (for details, see Supplemental Materials and methods).
RT-PCR
107 BMMCs sensitized with anti-DNP IgE were stimulated with 50 ng ml−1 DNP-HSA. Total RNA was prepared using TRIzol reagent (GIBCO BRL), and first-strand cDNA was synthesized using 5 μg total RNA with M-MLV Reverse Transcriptase (Toyobo). Specific PCR primers and amplification conditions for cytokine gene expression are described in Supplemental Materials and methods.
Lipid raft preparation
Preparation of lipid rafts was performed as described previously with some modifications (64). In brief, 2 × 107 BMMCs sensitized with anti-DNP IgE were incubated for 2 min at 37°C with or without DNP. Cells were washed and pellets were lysed in 1 ml MBS buffer (0.5% Triton X-100, 25 mM MES, pH 6.5, 150 mM NaCl, 1 mM Na3VO4, and protease inhibitor cocktail) and incubated for 30 min on ice. Subsequent steps were performed at 4°C. Lysates were mixed with 0.5 ml 85% sucrose in MBS, transferred to 5PA centrifuge tubes (Hitachi), and overlaid with 2.4 ml 35% sucrose followed by 1.5 ml 5% sucrose. After centrifugation for 18 h at 200,000 g in a Hitachi P55ST2 rotor at 4°C, 10 0.5-ml fractions were collected starting at the top of the gradient. For analysis of protein composition, aliquots were mixed directly with 4× SDS-PAGE sample buffer and analyzed by Western blotting. Distribution of FcɛRI and LAT in lipid rafts was determined by densitometric analysis of immunoblots using Dolphin-1D software (Kurabo).
Online supplemental material
Primers used for cytokine gene expression and procedures for the generation of PIPKIα-deficient mice and the analysis of FcɛRI surface expression, actin cytoskeleton assessments, and immunoblotting are available in Supplemental Materials and methods. Fig. S1 shows the effects of latrunculin and jasplakinolide on ionomycin-induced degranulation in WT and PIPKIα−/− BMMCs. Fig. S2 shows methyl-β-cyclodextrin inhibition of FcɛRI-mediated activation of p38 and ERK. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20041891/DC1.
Acknowledgments
We thank Dr. U. Blank for providing anti-Syk antibody, Dr. J. Rivera for providing anti-FcɛRIβ antibody, and Dr. T. Kitamura for the retroviral vectors. We thank Drs. H. Nishina, T. Katada, M. Ui, M. Mori, Y. Hachiya, K. Yamashita, K. Nagata, O. Kaminuma, S. Miyatake, N. Yajima, Y. Horie, K. Hamada, M. Yamada, Y. Sato, Y. Tsuya, and T. Nakano for valuable comments and advice.
Part of this work was supported by research grants from the Ministry of Education, Science, Sports and Culture; the Japan Society for the Promotion of Science (to J. Sasaki, T. Sasaki, A. Suzuki, and Y. Kanaho); Japan Science and Technology; a Grant-in-Aid for Cancer Research from the Japanese Ministry of Health, Labor and Welfare (to T. Sasaki); and Yamanouchi Foundation for Research on Metabolic Disorders and Intelligent Cosmos Academic Foundation (to J. Sasaki).
The authors have no conflicting financial interests.
Abbreviations used: BMMC, bone marrow–derived mast cell; IP3, inositol 1,4,5-trisphosphate; LAT, linker for activation of T cells; PI3K, phosphoinositide 3-kinase; PIP3, phosphatidylinositol 3, 4, 5-trisphosphate; PIPKI, type I phosphatidylinositol phosphate kinase; PIPKIα, α isozyme of PIPKI; PLC, phospholipase C; RBL, rat basophilic leukemia.
J. Sasaki and T. Sasaki contributed equally to this work.
References
- 1.Metcalfe, D.D., D. Baram, and Y.A. Mekori. 1997. Mast cells. Physiol. Rev. 77:1033–1079. [DOI] [PubMed] [Google Scholar]
- 2.Kinet, J.P. 1999. The high-affinity IgE receptor (FcɛRI): from physiology to pathology. Annu. Rev. Immunol. 17:931–972. [DOI] [PubMed] [Google Scholar]
- 3.Galli, S.J. 2000. Mast cells and basophils. Curr. Opin. Hematol. 7:32–39. [DOI] [PubMed] [Google Scholar]
- 4.Turner, H., and J.P. Kinet. 1999. Signalling through the high-affinity IgE receptor FcɛRI. Nature. 402:B24–B30. [DOI] [PubMed] [Google Scholar]
- 5.Zhang, J., E.H. Berenstein, R.L. Evans, and R.P. Siraganian. 1996. Transfection of Syk protein tyrosine kinase reconstitutes high affinity IgE receptor-mediated degranulation in a Syk-negative variant of rat basophilic leukemia RBL-2H3 cells. J. Exp. Med. 184:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osborne, M.A., G. Zenner, M. Lubinus, X. Zhang, Z. Songyang, L.C. Cantley, P. Majerus, P. Burn, and J.P. Kochan. 1996. The inositol 5′-phosphatase SHIP binds to immunoreceptor signaling motifs and responds to high affinity IgE receptor aggregation. J. Biol. Chem. 271:29271–29278. [DOI] [PubMed] [Google Scholar]
- 7.Barker, S.A., K.K. Caldwell, A. Hall, A.M. Martinez, J.R. Pfeiffer, J.M. Oliver, and B.S. Wilson. 1995. Wortmannin blocks lipid and protein kinase activities associated with PI 3-kinase and inhibits a subset of responses induced by FceR1 cross-linking. Mol. Biol. Cell. 6:1145–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu, H., K. Saito, L.D. Klaman, J. Shen, T. Fleming, Y. Wang, J.C. Pratt, G. Lin, B. Lim, J.P. Kinet, and B.G. Neel. 2001. Essential role for Gab2 in the allergic response. Nature. 412:186–190. [DOI] [PubMed] [Google Scholar]
- 9.Takenawa, T., and T. Itoh. 2001. Phosphoinositides, key molecules for regulation of actin cytoskeletal organization and membrane traffic from the plasma membrane. Biochim. Biophys. Acta. 1533:190–206. [DOI] [PubMed] [Google Scholar]
- 10.Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science. 296:1655–1657. [DOI] [PubMed] [Google Scholar]
- 11.Katso, R., K. Okkenhaug, K. Ahmadi, S. White, J. Timms, and M.D. Waterfield. 2001. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 17:615–675. [DOI] [PubMed] [Google Scholar]
- 12.Vanhaesebroeck, B., S.J. Leevers, K. Ahmadi, J. Timms, R. Katso, P.C. Driscoll, R. Woscholski, P.J. Parker, and M.D. Waterfield. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70:535–602. [DOI] [PubMed] [Google Scholar]
- 13.Rebecchi, M.J., and S.N. Pentyala. 2000. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80:1291–1335. [DOI] [PubMed] [Google Scholar]
- 14.Lewis, R.S. 2001. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 19:497–521. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki, T., A. Suzuki, J. Sasaki, and J.M. Penninger. 2002. Phosphoinositide 3-kinases in immunity: lessons from knockout mice. J. Biochem. (Tokyo). 131:495–501. [DOI] [PubMed] [Google Scholar]
- 16.Lemmon, M.A., and K.M. Ferguson. 2000. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 350:1–18. [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh, T., and T. Takenawa. 2002. Phosphoinositide-binding domains: functional units for temporal and spatial regulation of intracellular signalling. Cell. Signal. 14:733–743. [DOI] [PubMed] [Google Scholar]
- 18.Czeck, M.P. 2000. PIP2 and PIP3: complex roles at the cell surface. Cell. 100:603–606. [DOI] [PubMed] [Google Scholar]
- 19.Cremona, O., G. Di Paolo, M. Wenk, A. Luthi, W. Kim, K. Takei, L. Daniell, Y. Nemoto, S. Shears, R. Flavell, et al. 1999. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 15:179–188. [DOI] [PubMed] [Google Scholar]
- 20.Caroni, P. 2001. New EMBO members' review: actin cytoskeleton regulation through modulation of PI(4,5)P(2) rafts. EMBO J. 20:4332–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botelho, R.J., H. Tapper, W. Furuya, D. Mojdami, and S. Grinstein. 2002. Fc gamma R-mediated phagocytosis stimulates localized pinocytosis in human neutrophils. J. Immunol. 169:4423–4429. [DOI] [PubMed] [Google Scholar]
- 22.Mejillano, M., M. Yamamoto, R. AL, H. Sun, X. Wang, and H. Yin. 2001. Regulation of apoptosis by phosphatidylinositol 4,5-bisphosphate inhibition of caspases, and caspase inactivation of phosphatidylinositol phosphate 5-kinases. J. Biol. Chem. 276:1865-1872. [DOI] [PubMed] [Google Scholar]
- 23.Janmey, P., W. Xian, and L. Lanagan. 1999. Controlling cytoskeleton structure by phosphoinositide-protein interactions: phosphoinositide binding protein domains and effects of lipid packing. Chem. Physiol. Lipids. 101:93–107. [DOI] [PubMed] [Google Scholar]
- 24.Loijens, J.C., and R.A. Anderson. 1996. Type I phosphatidylinositol-4-phosphate 5-kinases are distinct members of this novel lipid kinase family. J. Biol. Chem. 271:32937–32943. [DOI] [PubMed] [Google Scholar]
- 25.Ishihara, H., Y. Shibasaki, N. Kizuki, H. Katagiri, Y. Yazaki, T. Asano, and Y. Oka. 1996. Cloning of cDNAs encoding two isoforms of 68-kDa type I phosphatidylinositol-4-phosphate 5-kinase. J. Biol. Chem. 271:23611–23614. [DOI] [PubMed] [Google Scholar]
- 26.Di Paolo, G., L. Pellegrini, K. Letinic, G. Cestra, R. Zoncu, S. Voronov, S. Chang, J. Guo, M.R. Wenk, and P. De Camilli. 2002. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 γ by the FERM domain of talin. Nature. 420:85–89. [DOI] [PubMed] [Google Scholar]
- 27.Ling, K., R.L. Doughman, A.J. Firestone, M.W. Bunce, and R.A. Anderson. 2002. Type I γ phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 420:89–93. [DOI] [PubMed] [Google Scholar]
- 28.Giudici, M.L., P.C. Emson, and R.F. Irvine. 2004. A novel neuronal-specific splice variant of Type I phosphatidylinositol 4-phosphate 5-kinase isoform γ. Biochem. J. 379:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rameh, L.E., K.F. Tolias, B.C. Duckworth, and L.C. Cantley. 1997. A new pathway for synthesis of phosphatidylinositol-4,5-bis-phosphate. Nature. 390:192–196. [DOI] [PubMed] [Google Scholar]
- 30.Hinchlife, K. 2000. Intracellular signalling: is PIP2 a messenger too? Curr. Biol. 10:104–105. [DOI] [PubMed] [Google Scholar]
- 31.Liu, Q., T. Sasaki, I. Kozieradzki, A. Wakeham, A. Itie, D.J. Dumont, and J.M. Penninger. 1999. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 13:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukao, T., T. Yamada, M. Tanabe, Y. Terauchi, T. Ota, T. Takayama, T. Asano, T. Takeuchi, T. Kadowaki, J. Hata Ji, and S. Koyasu. 2002. Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat. Immunol. 3:295–304. [DOI] [PubMed] [Google Scholar]
- 33.Laffargue, M., R. Calvez, P. Finan, A. Trifilieff, M. Barbier, F. Altruda, E. Hirsch, and M.P. Wymann. 2002. Phosphoinositide 3-kinase γ is an essential amplifier of mast cell function. Immunity. 16:441–451. [DOI] [PubMed] [Google Scholar]
- 34.Wen, R., S.T. Jou, Y. Chen, A. Hoffmeyer, and D. Wang. 2002. Phospholipase C γ 2 is essential for specific functions of FceR and Fc gamma R. J. Immunol. 169:6743–6752. [DOI] [PubMed] [Google Scholar]
- 35.Frigeri, L., and J. Apgar. 1999. The role of actin microfilaments in the down-regulation of the degranulation response in RBL-2H3 mast cells. J. Immunol. 162:2243–2250. [PubMed] [Google Scholar]
- 36.Holowka, D., E.D. Sheets, and B. Baird. 2000. Interactions between Fc(epsilon)RI and lipid raft components are regulated by the actin cytoskeleton. J. Cell Sci. 113:1009–1119. [DOI] [PubMed] [Google Scholar]
- 37.Baram, D., Y. Mekori, and R. Sagi-Eisenberg. 2001. Synaptotagmin regulates mast cell functions. Immunol. Rev. 179:25–34. [DOI] [PubMed] [Google Scholar]
- 38.Hata, D., Y. Kawakami, N. Inagaki, C.S. Lantz, T. Kitamura, W.N. Khan, M. Maeda-Yamamoto, T. Miura, W. Han, S.E. Hartman, et al. 1998. Involvement of Bruton's tyrosine kinase in FcɛRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 187:1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pardon, D., Y.J. Wang, M. Yamamoto, H. Yin, and G. Roth. 2003. Phosphatidylinositol phosphate 5-kinase Iβ recruits AP-2 to the plasma membrane and regulates rates of constitutive endocytosis. J. Cell Biol. 162:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morton, W., K. Ayscough, and P. McLaughlin. 2000. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat. Cell Biol. 2:376–378. [DOI] [PubMed] [Google Scholar]
- 41.Oliver, J.M. 1978. Cell biology of leukocyte abnormalities–membrane and cytoskeletal function in normal and defective cells. Am. J. Pathol. 93:221–270. [PMC free article] [PubMed] [Google Scholar]
- 42.Pollard, T.D., and G.G. Borisy. 2003. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 112:453–465. [DOI] [PubMed] [Google Scholar]
- 43.Narasimhan, V., D. Holowka, and B. Baird. 1990. Microfilaments regulate the rate of exocytosis in rat basophilic leukemia cells. Biochem. Biophys. Res. Commun. 171:222–229. [DOI] [PubMed] [Google Scholar]
- 44.Kettner, A., L. Kumar, I.M. Anton, Y. Sasahara, M. de la Fuente, V.I. Pivniouk, H. Falet, J.H. Hartwig, and R.S. Geha. 2004. WIP regulates signaling via the high affinity receptor for immunoglobulin E in mast cells. J. Exp. Med. 199:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Draber, P., and L. Draberova. 2002. Lipid rafts in mast cell signaling. Mol. Immunol. 38:1247–1252. [DOI] [PubMed] [Google Scholar]
- 46.Pike, L.J. 2004. Lipid rafts: heterogeneity on the high seas. Biochem. J. 378:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Paolo, G., H.S. Moskowitz, K. Gipson, M.R. Wenk, S. Voronov, M. Obayashi, R. Flavell, R.M. Fitzsimonds, T.A. Ryan, and P. De Camilli. 2004. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 431:415–422. [DOI] [PubMed] [Google Scholar]
- 48.Burgoyne, R.D., and T.R. Cheek. 1985. Reorganisation of peripheral actin filaments as a preclude to expcytosis. Biosci. Rep. 7:281–288. [DOI] [PubMed] [Google Scholar]
- 49.Hay, J.C., P.L. Fisette, G.H. Jenkins, K. Fukami, T. Takenawa, R.A. Anderson, and T.F. Martin. 1995. ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature. 374:173–177. [DOI] [PubMed] [Google Scholar]
- 50.Tucker, W.C., J.M. Edwardson, J. Bai, H.J. Kim, T.F. Martin, and E.R. Chapman. 2003. Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J. Cell Biol. 162:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saitoh, S., R. Arudchandran, T.S. Manetz, W. Zhang, C.L. Sommers, P.E. Love, J. Rivera, and L.E. Samelson. 2000. LAT is essential for FcɛRI-mediated mast cell activation. Immunity. 12:525–535. [DOI] [PubMed] [Google Scholar]
- 52.Rivera, J., J. Cordero, Y. Furumoto, C. Luciano-Montalvo, C. Gonzalez-Espinosa, M. Kovarova, S. Odom, and V. Parravicini. 2002. Macromolecular protein signaling complexes and mast cell responses: a view of the organization of IgE-dependent mast cell signaling. Mol. Immunol. 38:1253–1258. [DOI] [PubMed] [Google Scholar]
- 53.Wilson, B.S., J.R. Pfeiffer, and J.M. Oliver. 2002. FcɛRI signaling observed from the inside of the mast cell membrane. Mol. Immunol. 38:1259–1268. [DOI] [PubMed] [Google Scholar]
- 54.Lobell, R.B., K.F. Austen, and H.R. Katz. 1994. FcgR-mediated endocytosis and expression of cell surface FcγRIIb1 and FcγRIIb2 by mouse bone marrow culture-derived progenitor mast cells. J. Immunol. 152:811–818. [PubMed] [Google Scholar]
- 55.Hope, H.R., and L.J. Pike. 1996. Phosphoinositides and phosphoinositide-utilizing enzymes in detergent-insoluble lipid domains. Mol. Biol. Cell. 7:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shirakawa, T., A. Li, M. Dubowitz, J.W. Dekker, A.E. Shaw, J.A. Faux, C. Ra, W.O. Cookson, and J.M. Hopkin. 1994. Association between atopy and variants of the beta subunit of the high-affinity immunoglobulin E receptor. Nat. Genet. 7:125–129. [DOI] [PubMed] [Google Scholar]
- 57.Sasaki, T., J. Irie-Sasaki, R.G. Jones, A.J. Oliveira-dos-Santos, W.L. Stanford, B. Bolon, A. Wakeham, A. Itie, D. Bouchard, I. Kozieradzki, et al. 2000. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science. 287:1040–1046. [DOI] [PubMed] [Google Scholar]
- 58.Sasaki, T., T. Wada, H. Kishimoto, J. Irie-Sasaki, G. Matsumoto, T. Goto, Z. Yao, A. Wakeham, T.W. Mak, A. Suzuki, et al. 2001. The stress kinase mitogen-activated protein kinase kinase (MKK)7 is a negative regulator of antigen receptor and growth factor receptor-induced proliferation in hematopoietic cells. J. Exp. Med. 194:757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Honda, A., M. Nogami, T. Yokozeki, M. Yamazaki, H. Nakamura, H. Watanabe, K. Kawamoto, K. Nakayama, A.J. Morris, M.A. Frohman, and Y. Kanaho. 1999. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 99:521–532. [DOI] [PubMed] [Google Scholar]
- 60.Manetz, T.S., C. Gonzalez-Espinosa, R. Arudchandran, S. Xirasagar, V. Tybulewicz, and J. Rivera. 2001. Vav1 regulates phospholipase C γ activation and calcium responses in mast cells. Mol. Cell. Biol. 21:3763–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Irie-Sasaki, J., T. Sasaki, W. Matsumoto, A. Opavsky, M. Cheng, G. Welstead, E. Griffiths, C. Krawczyk, C.D. Richardson, K. Aitken, et al. 2001. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 409:349–354. [DOI] [PubMed] [Google Scholar]
- 62.Naito, K., M. Hirama, C. Okumura, and C. Ra. 1995. Soluble form of the human high-affinity receptor for IgE inhibits recurrent allergic reaction in a novel mouse model of type I allergy. Eur. J. Immunol. 25:1631–1637. [DOI] [PubMed] [Google Scholar]
- 63.Serunian, L.A., K.R. Auger, and L.C. Cantley. 1991. Identification and quantification of polyphosphoinositides produced in response to platelet-derived growth factor stimulation. Methods Enzymol. 198:78–87. [DOI] [PubMed] [Google Scholar]
- 64.Saito, K., K.F. Tolias, A. Saci, H.B. Koon, L.A. Humphries, A. Scharenberg, D.J. Rawlings, J.P. Kinet, and C.L. Carpenter. 2003. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity. 19:669–678. [DOI] [PubMed] [Google Scholar]