

Abstract
Rhinoviruses are the major trigger of acute asthma exacerbations and asthmatic subjects are more susceptible to these infections. To investigate the underlying mechanisms of this increased susceptibility, we examined virus replication and innate responses to rhinovirus (RV)-16 infection of primary bronchial epithelial cells from asthmatic and healthy control subjects.
Viral RNA expression and late virus release into supernatant was increased 50- and 7-fold, respectively in asthmatic cells compared with healthy controls. Virus infection induced late cell lysis in asthmatic cells but not in normal cells. Examination of the early cellular response to infection revealed impairment of virus induced caspase 3/7 activity and of apoptotic responses in the asthmatic cultures. Inhibition of apoptosis in normal cultures resulted in enhanced viral yield, comparable to that seen in infected asthmatic cultures. Examination of early innate immune responses revealed profound impairment of virus-induced interferon-β mRNA expression in asthmatic cultures and they produced >2.5 times less interferon-β protein. In infected asthmatic cells, exogenous interferon-β induced apoptosis and reduced virus replication, demonstrating a causal link between deficient interferon-β, impaired apoptosis and increased virus replication. These data suggest a novel use for type I interferons in the treatment or prevention of virus-induced asthma exacerbations.
Viral respiratory tract infections are responsible for up to 85% of asthma exacerbations (1, 2), with the most severe requiring hospitalization (3). These infections can trigger severe asthma exacerbations even when there is good asthma control by compliant patients taking optimal doses of inhaled corticosteroids (ICSs; 4, 5).
The most common pathogens associated with asthma exacerbations are rhinoviruses (RVs), which lead to increased lower airway inflammation (6) and increased bronchial responsiveness (7). Subjects with asthma have increased susceptibility to RV infection with more severe lower respiratory tract symptoms and reductions in lung function than normal subjects similarly infected (8). Although RV infects bronchial epithelial cells (BECs; 9) and has been detected in the lower airways (9, 10), the reasons why the asthmatic lower respiratory tract is more susceptible to the effects of infection with RV are unknown.
Rapid induction of apoptosis in virus-infected host cells is a critical component of innate antiviral responses (11), as early apoptosis prevents establishment of viral replication and promotes phagocytosis of infected cells. Type I IFNs are also an important component of the innate immune response, having a direct antiviral effect on infected and neighboring cells, while promoting acquired antiviral immune responses (12). Recently they have been linked to apoptotic responses to virus infections in antiviral defense (11). Thus, type 1 IFNs play critical roles in regulating apoptosis, as well as innate and acquired immune responses in antiviral defense. Therefore we hypothesized that asthmatic BECs have abnormal innate responses to virus infection characterized by impaired type 1 IFN production and impaired virus-induced apoptosis resulting in increased virus replication.
To address this hypothesis we investigated virus replication, type 1 IFN production and apoptotic responses in primary BECs from asthmatic and normal subjects. We initially studied cells from asthmatic subjects treated with ICSs, as the great majority of asthmatics are on regular prophylactic therapy with these agents. However, to determine whether ICSs therapy influenced our observations, we also studied cells from milder asthmatics completely naive to ICSs therapy.
Results
ICAM-1 expression and induction with infection
Primary BECs from 14 subjects with moderately severe asthma treated with ICSs and 10 normal healthy controls were studied (Table I). As the cellular receptor for RV-16 is ICAM-1 (13), we first determined whether surface ICAM-1 expression differed between asthmatic and normal BECs. Before infection ICAM-1 expression was not significantly different between subject groups (Fig. 1 A). By 24 h after infection, ICAM-1 surface protein expression was significantly induced in a virus-specific manner, consistent with previous studies using primary BECs (14; Fig. 1 B), however, levels were significantly greater in the normal cultures compared with expression in the asthmatic cultures (Fig. 1 B). RV-16 infection also provoked a vigorous inflammatory response in BECs from both asthmatic and healthy controls. There was a significant induction of RANTES and IL-6 protein release at 48 h after infection (Fig. 1, C and D). There were no significant differences between the two groups, suggesting comparable levels of proinflammatory responses in cultures from either group. UV-inactivated RV did not trigger a proinflammatory response.
Table I.
Subject characteristics
| Asthma (ICSs treated) | Asthma (ICSs naive) | Healthy controls | p-value | |
|---|---|---|---|---|
| Number | 14 | 10 | 10 | NA |
| Sex (percent male) | 69% | 60% | 60% | P = 0.8 |
| Mean age (range) | 32 (21–58) | 32 (12.6) | 29 (24–38) | P = 0.4 |
| Mean FEV1% predicted (SD) | 77.3a (15.5) | 103.6 (9.8) | 110.3 (13.6) | P < 0.001 |
| Mean dose of ICSs, BDP μg/day (SD) | 490 (260) | 0 | 0 | NA |
FEV1% predicted refers to the forced expiratory volume in 1 s expressed as a percentage of the predicted value.
ICSs refers to ICSs. Dose is expressed in dose of beclomethasone dipropionate (BDP) in μg per day where 1 μg BDP = 1 μg Budesonide or 0.5 μg Fluticasone.
aSignificantly lower than healthy controls, P < 0.001.
Figure 1.

ICAM-1 expression of normal and asthmatic BECs before and after RV infection. (A and B) ICAM-1 expression was measured by flow cytometry immediately before RV-16 infection (A) or 24 h after infection (B). Data are expressed as mean fluorescence intensity (MFI). Before infection, asthmatic cells had a tendency to a lower median MFI 31 IQR (12, 80) compared with healthy control cells 67 (34, 83) but this was not significant (P = 0.4; A). ICAM-1 was significantly induced in both asthmatic and healthy control cells by 24 h (B), asthmatic cells had an MFI of 54.6 (27.6, 145.2) healthy control cells 110.4 (65, 195.3; P = 0.3). In these and all later box whisker plots, the line inside the box represents the median, upper box border represents 75th quartile, lower 25th quartile, whiskers are 5th and 95th centiles, dots represent outliers. (C and D) IL-6 and RANTES production was measured 48 h after infection in the supernatant of cells by ELISA. In C there was virus-specific release of IL-6 from RV infected in asthmatic cells with a median fold increase from baseline of 14 (IQR 5.2, 19.8) and in healthy control cells of 4 (4, 22.4). This was significantly greater than in cells treated with medium alone or UV-inactivated RV (P < 0.01), but there was no difference between the groups. In D there was virus-specific release of RANTES from RV infected in asthmatic cells with a median fold increase from baseline of 66 (IQR 33.2, 125.8) and in healthy control cells of 82.3 (25.2, 215.9). This was significantly greater than in cells treated with medium alone or UV-inactivated RV (P < 0.01), but there was no difference between the groups. *, Significantly different from cells treated with medium alone and UV-inactivated RV-16, P < 0.05. Shaded bars, asthma; open bars, healthy controls.
Viral replication after epithelial cell infection and epithelial cell lysis
We next investigated whether primary BECs from asthmatic subjects were more susceptible to increased viral replication by determining levels of RV-16 vRNA expression 8 h after infection. We observed significantly increased vRNA expression (by >50-fold) in primary BECs from asthmatic compared with normal subjects (Fig. 2 A). This was accompanied by a progressive increase in cell lysis in virus infected BECs from asthmatic subjects, reaching a significant 3.4-fold increase above baseline at 48 h. In contrast, there was no significant induction of cell lysis in cells from normal subjects either at 24 or 48 h after infection (Fig. 2 B), indicating that the innate responses of these cells were sufficiently robust to protect them from substantial lytic cell death. The increase in lactate dehydrogenase (LDH) activity was significantly greater in asthmatic cells compared with the normal cells at 48 h (Fig. 2 C) and was specific to viral replication as there was no significant increase in LDH activity in asthmatic cells treated with medium alone or with UV inactivated RV-16 48 h after infection (Fig. 2 C).
Figure 2.

RV-16 replication and release from normal and asthmatic BECs. (A) RV-16 vRNA production was measured by qPCR after 8 h of infection. Median (IQR) production (×106) from asthmatic cells was significantly increased at 2.1 (0.16, 9.7) compared with 0.04 (0.009, 0.06) from healthy controls (P = 0.007). (B and C) Late cell lysis as a consequence of RV-16 infection was determined by analysis of LDH activity in culture supernatants; values represent the fold induction from baseline. Data points represent the mean and the SD. In asthmatic cells LDH activity progressively increased over time and was significantly increased from baseline at both 24 h (P = 0.01) and 48 h (P = 0.003) whereas in healthy control cells there was no significant increase even at 48 h (P = 0.2; B). By 48 h, the mean LDH activity from asthmatic cells was significantly greater than in normal cells, at 3.4 (0.1)-fold increased over baseline compared with only a 1.34 (0.1)-fold increase in the healthy control cells (P = 0.0001; C). Induction of LDH activity was shown to be virus replication dependent in that no significant change in LDH activity was seen in cells treated with medium alone or UV-inactivated RV (C). (D) RV-16 release into the supernatant of infected cells was determined by calculating the TCID50 × 104/ml by titration assay in Ohio HeLa cells. The supernatant from 14 ICSs requiring asthmatics and 10 healthy controls were examined on individual titration plates. Data points represent the mean and the standard deviation. By 48 h significantly more RV was detected from asthmatic cells with a mean TCID50 × 104/ml of 3.99 (0.8), compared with 0.54 (0.12) in healthy control cells (P = 0.001). *, Significantly different from cells treated with UV inactivated RV or medium alone. **, Results from asthmatic cells and healthy controls significantly different (P < 0.01). +, Significantly increased above baseline (P < 0.001). Shaded bars, asthma; open bars, healthy controls.
Having observed that RV infection of asthmatic primary BECs resulted in increased vRNA production and cell lysis, we examined whether increased intact virus particles were released into the supernatants. Recovery of viable RV was significantly increased at 24 h and, by 48 h the virus release was more than sevenfold greater in asthmatic compared with normal primary BECs (Fig. 2 D). These results confirmed a link between early vRNA production, later cell lysis and virus release and all were significantly increased in asthmatic primary BECs. The normal cells were protected against lytic virus replication, with 50 times lower levels of vRNA production, no significant cell lysis and release of almost eight times less new virus progeny. These data suggested that postbinding events early in the replication cycle were profoundly influencing early viral RNA production, later cell lysis and virus release from infected cells.
Bronchial epithelial cell viability and induction of caspases after RV infection
As apoptosis is a natural defense that protects against virus replication, our findings of increased replication in asthmatic cells led us to investigate whether early apoptotic responses were different between subject groups. There was a significant reduction in viable annexin-V (AxV−)/7-amino-actinomycin (7AAD−) cell numbers 8 h after RV-16 infection of normal BECs, such that only 63% of cells were viable and available for virus replication at this time point. This was not observed in cells treated with medium alone or UV-inactivated RV-16 (Fig. 3 A). Similarly, infection of asthmatic BECs with RV-16 also significantly reduced numbers of viable cells 8 h after infection and this was also dependent on virus replication (Fig. 3 A). However the reduction in viable cells was of a significantly lesser magnitude with 80% of cells remaining viable and available for virus replication (Fig. 3 A). By analyzing changes in frequencies of apoptotic cells and necrotic cells over the first 8 h after RV-16 infection, we found that there was a significant increase in apoptotic normal BECs, which was not observed in cells treated with medium alone or UV-inactivated RV-16 confirming that induction of apoptosis was directly related to virus replication (Fig. 3 B). Infection of asthmatic BECs with RV-16 also significantly increased numbers of apoptotic cells at 8 h after infection (Fig. 3 B), however, the increased apoptotic cells (therefore not available for virus replication) in asthmatic BECs (1.4-fold increase from baseline), was significantly less than that observed in healthy control BECs (2.2-fold; Fig. 3 B), confirming impairment of apoptotic responses in asthmatic cells. There were no significant differences between subject groups in the number of necrotic cells at 8 h after virus infection (unpublished data), confirming that alterations in early cell viability were a result of increased apoptosis, not early necrosis.
Figure 3.
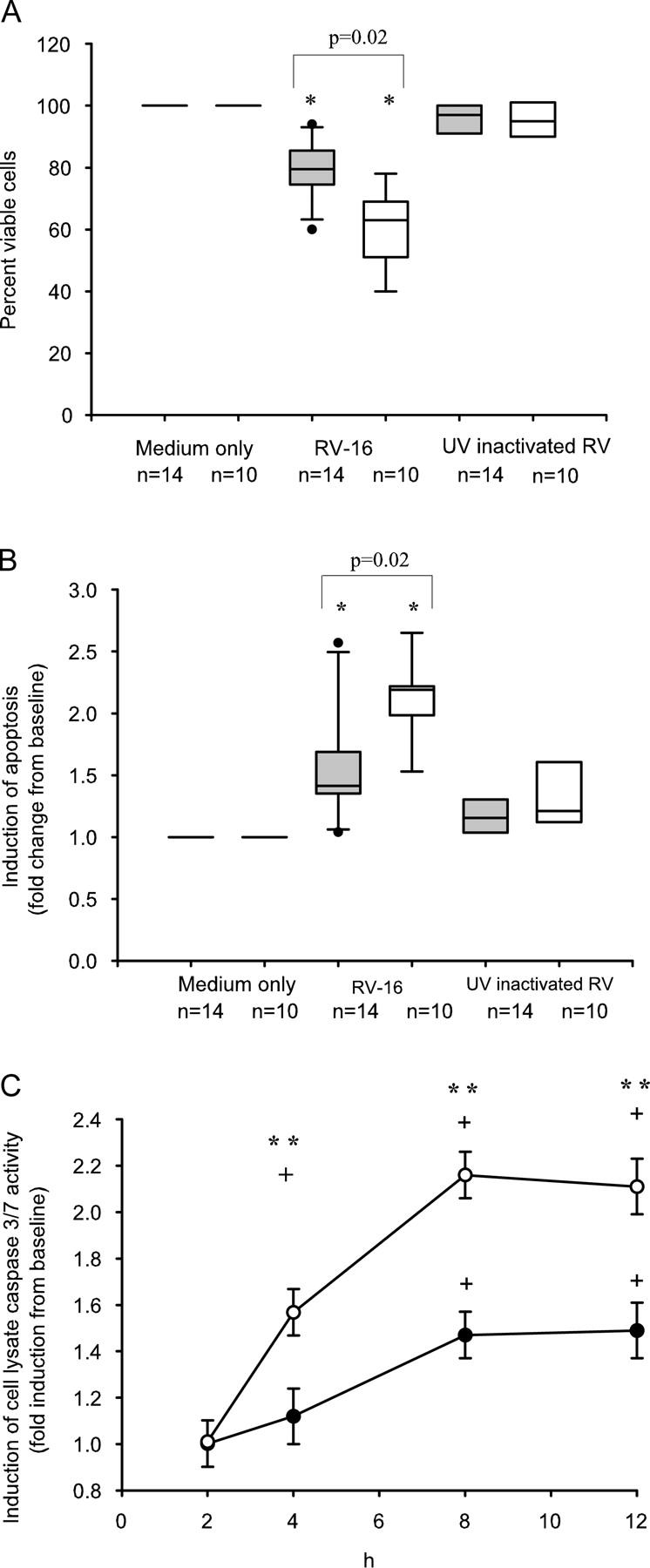
Differences in cell viability and apoptotic response after RV-16 infection in asthmatic and normal BECs. (A) Viable (AxV−/7AAD−) cell number was determined 8 h after infection and expressed as percent viability compared with cells treated with medium alone. Infection with RV-16 led to a significant reduction in median (IQR) cell viability in both asthmatic and control cells compared with both medium alone (P = 0.03 and 0.02, respectively) and UV-inactivated RV-16 (P = 0.02 and 0.001, respectively). In asthmatic cells there was significantly better viability, median 80% (74, 86), compared with healthy controls 63% (51, 69; P = 0.02). (B) Apoptotic (AxV+/7AAD−) cells were also analyzed 8 h after RV-16 infection. Data are expressed as median (IQR) fold change in apoptosis from baseline. There was a significant and virus-specific increase in cell apoptosis in response to infection in cells from both groups, however, this response was significantly impaired in asthmatic cells with a fold increase of only 1.4 (1.3, 1.7), compared with 2.2 (2.1, 2.3) in healthy controls (P = 0.02). (C) The time course for activation of caspase 3/7 by RV-16 was determined in cells from 10 ICSs requiring asthmatics and 10 healthy controls, all conditions were done in quadruplicate, values represent the fold induction from baseline. There was significant mean (SD) induction of active caspase 3/7 in response to infection in normal cells at 4, 8, and 12 h, induction reached a plateau at 8 h. In asthmatic cells the induction was later, being significantly increased above baseline only at 8 and 12 h and was of significantly reduced magnitude at each time point compared with normal cells. At 8 h there was a significantly impaired induction of active caspase 3/7 in asthmatic cells (mean [SD] = 1.47 [0.13]) compared with healthy controls (mean [SD] =2.16 [0.34]; P = 0.004). *, Significantly different from cells treated with UV-inactivated RV-16 and medium only (P < 0.01). **, Asthmatic cells significantly different from healthy controls (P < 0.05). +, Significantly different from baseline (P < 0.05). Shaded bars, asthma; open bars, healthy controls.
To investigate whether RV-induced apoptosis involved the activation of caspase 3/7, we next determined levels of activated caspase 3/7. There was significant induction of caspase 3/7 activity in healthy control cells at 4 h, peaking at 8 h and still elevated 12 h after infection. Induction of caspase 3/7 activity in normal cells was significantly greater than that seen in asthmatic cells (Fig. 3 C). These data demonstrate that RV-induced apoptosis occurs via the caspase 3/7 pathway and that activation of this pathway is defective in asthmatic subjects.
To confirm whether increased virus production by asthmatic BECs is a direct consequence of an impaired apoptotic response, we then treated cells with the caspase 3 inhibitor ZVD-fmk, before and after infection with RV-16. Caspase 3 inhibition impaired RV-induced apoptosis in the normal cultures but had no significant effect on asthmatic cells compared with infection alone (Fig. 4 A). Treatment of the normal BECs with ZVD-fmk also had a direct impact on RV-16 production with a significant >4.5-fold increase in RV titer at 48 h compared with those infected with RV-16 alone (Fig. 4 B). Consistent with the fact that there was no significant inhibition of apoptosis in asthmatic cells treated with ZVD-fmk, we observed no significant increase in virus title (Fig. 4 B). These data provide direct evidence that inhibition of early apoptotic responses to viral infection is associated with increased later virus release and that inhibition of apoptosis in normal cells results in a phenotype typical of asthmatic cells (that is impaired apoptosis and increased virus replication). Notably, inhibition of apoptosis in normal cells was sufficient to increase virus replication to levels similar to those observed in the asthmatic cells.
Figure 4.
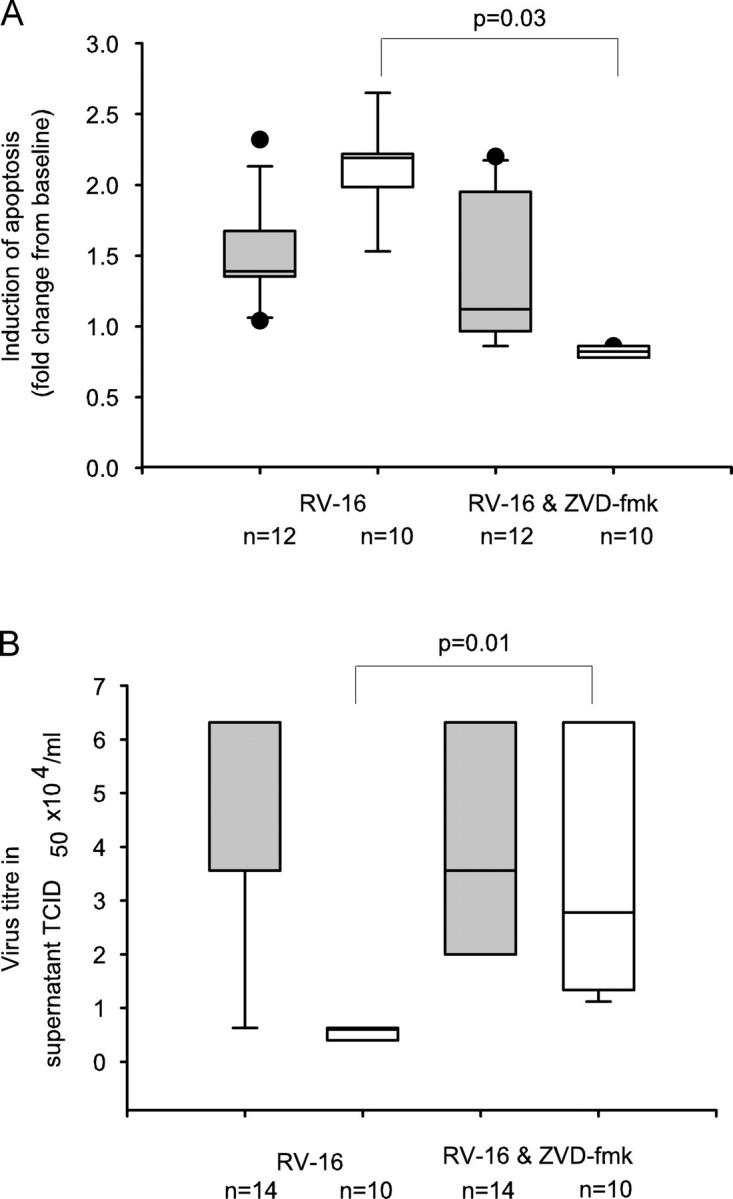
Inhibition of caspase activity inhibits apoptosis and increases RV-16 replication. (A) The effect of inhibition of caspase-3 using the inhibitor, ZVD-fmk, was measured by flow cytometry. Cells were treated with RV-16 alone or with ZVD-fmk, before and after infection with RV-16. Results are expressed as the fold change in apoptotic cells compared with cells treated with medium alone. In asthmatic cells were there was a median (IQR) induction of apoptosis above baseline of 1.4 (1.3, 1.7) with RV-16 alone; pretreatment of cells with the ZVD-fmk, had little effect on apoptosis (median (IQR) = 1.12 (1.01, 1.8); (P = 0.4). However, in healthy controls cells, RV-16 infection resulted in a median (IQR) fold induction of apoptosis above baseline of 2.2 (2.1, 2.3) and this was abolished by pretreatment with ZVD-fmk (median [IQR] 0.82 [0.76, 0.86; P = 0.03]). (B) The effect of caspase-3 inhibition on RV-16 production was measured by HeLa titration assay on the BEC supernatant removed after 48 h of infection. There was no difference seen in the TCID50 × 104/ml in the supernatant removed from asthmatic cells infected with RV-16 (median [IQR] = 3.56 [3.50–3.62]) compared with infected cells treated with ZVD-fmk (median [IQR] = 3.5 [3.45–3.62]; P = 0.94). However, for healthy control BECs, the TCID50 × 104/ml increased greater than fourfold, from a median (IQR) value of 0.6 (0.4, 0.63) with infection alone to 2.78 (0.63, 6.32; P = 0.01) in the presence of RV-16 and ZVD-fmk. Shaded bars, asthma; open bars, healthy controls.
Release of interferon-β from BECs after infection
Because IFN-α/β has recently been shown to induce apoptotic responses in antiviral immunity (11), and because IFN-β is known to be secreted first and to strongly induce the IFN-α subfamily (15), we investigated whether impaired production of IFN-β was the underlying mechanism regulating the abnormal antiviral response in asthmatic cells. RV infection induced a robust almost fourfold increase in IFN-β mRNA expression in healthy control BECs 8 h after RV-16 infection, significantly greater than both medium and UV-inactivated virus controls, confirming the induction to be live-virus specific (Fig. 5 A). In marked contrast, a similar increase in IFN-β mRNA expression in response to RV infection at 8 h was absent in asthmatic cells (Fig. 5 A). Impaired induction of IFN-β mRNA expression by RV-infected asthmatic BECs was also reflected by impaired protein production with IFN-β protein levels being more than twofold higher in supernatants from healthy control cells compared with those from asthmatic cells (Fig. 5 B). Confirming the relationships between IFN-β production and apoptosis, and between apoptosis and suppression of virus replication, there was a positive correlation between induction of apoptosis at 8 h and induction of IFN-β mRNA at 8 h, r = 0.43 (P = 0.04) and an even stronger negative correlation between IFN-β release and virus release at 48h (TCID50 × 104/ml), r = −0.79 (P = 0.01). These data confirmed that IFN-β induction by RV infection of primary BECs from asthmatic subjects is profoundly impaired and this impairment is related to impaired apoptosis and increased viral replication.
Figure 5.

Impaired IFN-β production in asthma and its role in restoring apoptotic and antiviral response in asthmatic cells. (A) Induction of IFN-β mRNA was measured by qPCR after 8 h of RV-16 infection. There was no significant induction of IFN-β mRNA from asthmatic cells 8 h after infection with RV-16, median (IQR) fold induction from baseline control of 0.3 (0.3, 0.8), which was not significantly different from cells treated with medium alone or UV inactivated RV-16. In contrast there was a 3.6 (3.4, 3.6)-fold increase in IFN-β mRNA expression in cells from healthy controls (P = 0.004). (B) Release of IFN-β into culture supernatants 48 h after infection was measured by ELISA. For asthmatic BECs, median (IQR) IFN-β levels were significantly reduced at 721 (464, 1,290) pg/ml, compared with 1,854 pg/ml (758, 3,766; P = 0.03) in healthy controls. In both groups there was a significant increase above cells treated with medium alone and UV-inactivated RV-16 (unpublished data). (C) The ability of IFN-β to restore induction of apoptosis in RV-16–infected asthmatic cells was measured by FACS analysis as described in the legend to Fig. 3. Asthmatic cells were either pretreated with IFN-β (100 IU) for 24 h or simultaneously exposed to RV-16 and IFN-β. To mimic the presence of viral RNA, cells were also exposed to poly(I)/poly(C) a synthetic double-stranded RNA oligonucleotide, instead of RV-16. Results are expressed as the median (IQR) fold increase in apoptotic cells from baseline at 8 h after infection. There was no significant increase in apoptosis in cells exposed to either IFN-β 1.2 (1.1, 1.8; P = 0.3), RV-16 1.7 (1.3, 1.9; P = 0.2) or poly(I)/poly(C) 1.9 (1.7, 3.5; P = 0.08) alone. In cells simultaneously treated with RV-16 and IFN-β there was a tendency to increased apoptosis 3.8 (1.7, 5.0; P = 0.11) whereas in those pretreated with IFN-β for 24 h and then infected there was a significant induction of apoptosis 5.6 (3.9, 5.7; P = 0.02, compared with virus alone). In cells exposed to poly(I)/poly(C) alone there was a similar trend toward an increase in apoptosis 1.9 (1.7, 3.5; P = 0.08) which was significantly enhanced by simultaneous treatment with IFN-β 5.1 (3.9, 5.6; P = 0.01) and further enhanced by 24 h pretreatment with IFN-β 9.3 (6.6, 9.3; P = 0.001), compared with poly(I)/poly(C) alone. (D) The effect of IFN-β on virus release from asthmatic cells was measured by HeLa titration assay of asthmatic BEC culture supernatants removed 48 h after infection. Cells were either pretreated with IFN-β (100 IU) for 12 h and then exposed to RV-16 or were treated with IFN-β immediately after infection. There was a significant reduction in virus release in cells treated with IFN-β after infection median TCID50 × 104/ml 2.78 (2, 3.56; P = 0.04) and a further reduction in cells pretreated with IFN-β 1.12 (0.28, 1.34) compared with cells infected with RV-16 alone 3.56 (3.5–3.62; P = 0.012). *, Significantly different from medium alone. Shaded bars, asthma; open bars, healthy controls.
To examine whether these findings might be related to differences in ICAM-1 expression levels, or were confined to a major group RV, experiments were performed using BECs from eight asthmatic subjects and eight healthy controls, which were infected with the minor group RV, RV-1B. In this case, we also found evidence of early apoptosis in BECs from healthy controls with a 2.3-fold increase (interquartile range [IQR] 1.98, 2.43) at 8 h after infection, but this was again impaired in BECs from asthmatic subjects where the fold increase was only 1.32 (IQR 1.15, 1.4; P = 0.03). In addition, healthy control BECs demonstrated a vigorous IFN-β response at 48 h with median levels of 1,799 pg/ml (IQR 696, 3,402) compared with BECs from asthmatic donors, which released 453 pg/ml (IQR 254, 886; P = 0.03).
The effect of interferon-β on epithelial cell apoptosis in response to RV infection and treatment with poly(I)/poly(C)
To confirm that the impairment of IFN-β production in asthmatic cells was functionally related to impaired apoptotic responses, we tested the ability of exogenous IFN-β to induce apoptosis in RV-16–infected asthmatic BECs alone. To mimic the effects of IFN-β during both initial infection and the secondary wave of infection consequent upon new virus released from neighboring infected cells, we studied cells exposed to exogenous IFN-β just after infection or both before and after infection respectively. In both cases (Fig. 5 C) treatment of cells with IFN-β during infection caused a doubling in the number of apoptotic cells compared with treatment with IFN-β or virus alone, neither of which caused significant increases in the frequency of apoptotic cells. IFN-β was also capable of inducing apoptosis in the presence of poly(I)/poly(C) (Fig. 5 C), confirming that signals involving recognition of double stranded RNA are sufficient for commitment to apoptosis in response to IFN-β.
The effect of exogenous interferon-β on recovery of RV from asthmatic BECs
Finally, we investigated whether reconstitution of type 1 IFN responses in asthmatic cells with exogenous IFN-β was able to overcome the increased RV replication observed in asthmatic primary BECs. In line with its ability to induce apoptosis of virally infected asthmatic BEC, IFN-β caused a significant reduction in RV-16 infectious virus release into supernatants of asthmatic BECs (Fig. 5 D). The protection afforded was greater when IFN-β was present both before and after infection indicating that type 1 IFN production during the initial phase of infection was likely critical in prevention of the secondary wave of infection consequent upon new virus released from neighboring infected cells.
The effect of corticosteroids on virus replication, cell viability, and interferon-β release
To determine whether the deficient innate antiviral responses observed in the cells from subjects with moderately severe asthma (who were all treated with ICSs) was related either to asthma severity or to corticosteroid exposure, we next studied a group of mild asthmatics that had never used inhaled or parenteral corticosteroids (ICSs naive, Table I). In addition we treated cells from ICSs naive asthmatic cells with dexamethasone at doses of 10 and 100 nM for 24 h before infection with RV-16 and investigated their response in terms of interferon-β and apoptotic responses to infection and virus replication.
The release of IFN-β after RV-16 infection was just as profoundly impaired in primary BECs from ICSs-naive asthma as it was in cells from ICSs-treated asthma (Fig. 6 A). In vitro pretreatment of cells from healthy controls and ICSs naive subjects with dexamethasone (10 or 100 nM) had no significant effect on RV-16–induced IFN-β (Fig. 6 A). Similarly, RV-16 induction of apoptosis of BECs from ICSs naive asthmatics was also just as profoundly impaired when compared with cells from healthy controls and again treatment with dexamethasone had no significant effect in or ICSs-naive asthma (Fig. 6 B). In terms of virus replication, cells from ICSs naive mild asthmatics were intermediately susceptible to RV replication with virus titers exactly half way between those observed in cells from ICSs requiring moderate asthmatics and cells from normal controls (Fig. 6 C), again there was no significant effect of pretreatment of cells from healthy controls and ICSs naive asthmatics with dexamethasone (Fig. 6 C).
Figure 6.
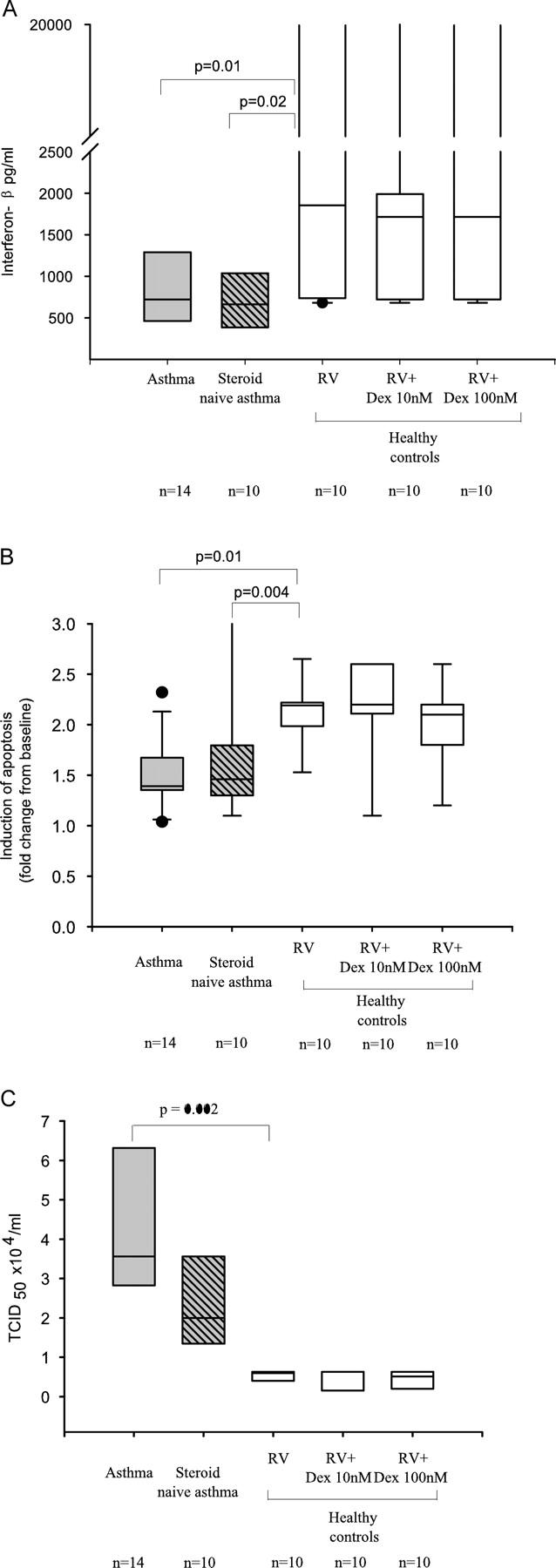
The effect of corticosteroids on interferon-β release apoptotic responses and virus replication. The results from asthmatic cells (those requiring ICSs) and healthy controls were compared with asthmatic subjects who had never used corticosteroids (ICSs naive). Cells from healthy controls and ICSs naive asthmatics were treated with RV-16 and pretreated for 24 h with dexamethasone (Dex) at 10 and 100 nM and then infected with RV-16. only the results from healthy control cells are displayed in the figures. (A) Release of IFN-β into culture supernatants 48 h after infection was measured by ELISA. As noted in asthmatic BECs, median (IQR) IFN-β levels were significantly reduced at 721 (464, 1,290) pg/ml, compared with 1,854 pg/ml (758, 3,766; P = 0.01) in healthy controls. This was also seen in ICSs naive asthmatic cells 666 (387, 1,039) compared with healthy controls (P = 0.02). The addition of Dex did significantly alter these levels. In healthy control cells; 10 nM 1717 (720, 1,990) compared with infected control cells (P = 0.55) or 100 nM 990 (721, 2,656). This was also seen in ICSs-naive asthma; 10 nM 772 (581, 1,220) compared with ICSs-naive asthma cells infected alone (P = 0.7), though at 100 nM; 449 (389, 609) there appeared to be a reduction, this did not reach statistical significance (P = 0.2). (B) Apoptotic (AxV+/7AAD−) cells were also analyzed 8 h after RV-16 infection. Data are expressed as median (IQR) fold change in apoptosis from baseline. Healthy controls cells had significantly more virus-specific apoptosis 2.19 (1.99, 2.2), compared with asthmatic cells (ICSs requiring) 1.39 (1.35, 1.67; P = 0.01) and ICSs naive asthmatic cells 1.36 (1.2, 1.5; P = 0.004). The addition of Dex to healthy control cells had no significant impact on apoptosis compared with cells not pretreated, at either 10 nM 2.16 (1.86, 2.6; P = 0.96) or 100 nM 1.87 (1.65, 2.14; P = 0.09). This was also seen in ICSs-naive asthma cells at 10nM 1.26 (1.2, 1.8) compared with ICSs naive cells infected alone (P = 0.97) and 100 nM 1.16 (1.1, 1.4; P = 0.08). (C) RV-16 release into the supernatant of infected cells was determined by calculating the TCID50 × 104/ml by titration assay in Ohio HeLa cells. By 48 h significantly more RV was detected from asthmatic cells with a mean TCID50 × 104/ml of 3.99 (0.8), compared with 0.54 (0.12) in healthy control cells (P = 0.002). There was a trend to higher titer in ICSs naive asthmatics 2.2 (1.2), but this was not significantly different from either ICSs requiring asthmatics or healthy controls. The addition of dexamethasone 10 nM in control cells 0.46 (0.3) and ICSs-naive asthma cells 2.1 (1.3) or 100 nM in control cells 0.42 (0.3) and ICSs-naive asthma 2.1 (1.3) had no significant effect. Shaded bars, asthma (ICSs treated); hatched bars, asthma (ICSs naive); open bars, healthy controls.
DISCUSSION
Using in vitro–cultured primary BECs from asthmatic and normal volunteers, we have demonstrated that asthmatic cells have an abnormal innate response to infection by RV-16, resulting in increased virus replication and cell lysis compared with cells from healthy normal controls. Asthmatic cells produced markedly increased levels of viral RNA, released almost eight times as many new virus progeny and infection led to progressive cell lysis. A similar response was also observed in asthmatic BECs exposed to the minor group RV, HRV-1B, which gains entry into the cell via the very low density lipoprotein receptor (16). This suggests that the observed response of asthmatic epithelial cells is a function of altered intracellular signaling rather than differences in ICAM expression and viral entry into the cells.
To explore the mechanisms behind this increased susceptibility of asthmatic epithelial cells to viral replication and cytolysis, we studied innate antiviral defences in the cells from both subject groups. Asthmatic cells were shown to be resistant to early apoptosis after infection with RV-16 and to have a profoundly deficient type I interferon response that was related to increased virus replication. These findings are in contrast with previous studies in which we demonstrated that asthmatic BECs are more susceptible to oxidant induced apoptosis (17). Thus, even though other stimuli can activate readily the apoptotic mechanism in asthmatic epithelial cells, it is not triggered in response to virus infection. The early apoptotic response in virally infected normal cells was shown to be a key protective mechanism since inhibition of apoptosis in healthy control cells led to enhanced virus release, comparable to levels observed in asthmatic cells. A link between this and the actions of IFN-β was demonstrated by the ability of exogenous IFN-β to induce apoptosis in asthmatic cells infected with RV-16 and to reduce virus production to levels similar to those observed in normal cells. We could find no evidence of infection with mycoplasma or other adventitious agents that might provide an explanation for the low level of IFN-β production by the asthmatic cell cultures. Furthermore, based on the ability of exogenous IFN-β to induce apoptosis in the asthmatic epithelial cells infected with RV or dsRNA, it seemed unlikely that there was deactivation of downstream signaling pathways as a consequence of chronic infection. A similar abnormal apoptotic and deficient interferon-β response was seen in cells derived from ICSs naive asthmatics and in vitro pretreatment of steroid naive cells with dexamethasone had no significant effect upon IFN-β production, apoptosis, or virus replication. Epidemiological evidence demonstrates that asthmatic subjects develop more severe lower respiratory symptoms and reductions in lung function when infected with RV (8), however the mechanisms behind this increased susceptibility to RV infection were unknown. The data presented in this report indicate that impaired type 1 interferon production is likely to be an important mechanism.
It has been demonstrated previously that antiviral pathways are redundant, and that loss of one pathway does not lead to increased susceptibility to virus infection. Although we observed that production of IFNβ from RV-16–infected asthmatic epithelial cells was lower than normal, their proinflammatory responses to viral infection appeared to be intact, as levels IL-6 and RANTES were similarly elevated in normal and asthmatic epithelial cell cultures. These findings are similar to other studies in which we compared cytokine production from normal and asthmatic BECs and failed to see differences in IL-8 and GM-CSF production in response to TNFα, IL-4, IL-13, or house dust mite allergen, but found lower TGFα production from asthmatic BECs under the same conditions (18). Given that some of the antiviral responses remain intact in asthmatic BECs, it seems unlikely that all antiviral pathways are defective, rather that the lesion is selective for the pathway(s) that trigger expression of IFN-β. This abnormality is most likely to result in only a subtle impairment of the immune response, making it more likely that an inflammatory response will need to occur in the airways to eliminate the infection.
The main role of type 1 IFNs, however, is in innate immunity, where they are critical regulators of a wide array of innate protective responses. Interferon-β is induced first, it then induces interferon-α. Both induce a wide variety of antiviral proteins; including RNases that digest viral double-stranded RNA to limit viral replication. Interferon-α/β also induce the antiviral protein kinase (PKR), which limits viral replication and induces apoptosis (19). Type I interferons have also been shown to induce apoptosis via activation of the tumor suppressor gene p53 in response to vesicular stomatitis virus infection in mice, further enhancing antiviral activity (11) and demonstrating that early apoptosis is a key protective innate immune antiviral response. We have confirmed this important role for type I interferons and apoptosis in relation to RV infection in human cells. The release of type I interferons from infected cells not only reduces virus spread from an infected cell to neighboring cells but also causes surrounding cells to become primed to an antiviral state with early apoptosis on exposure to virus. Infected apoptotic cells would then be removed by phagocytosis, virus replication minimized, and this would limit the magnitude of inflammatory responses to infection.
Antiinflammatory treatment with ICSs improves airway inflammation and symptoms associated with stable atopic asthma, but does not influence airway inflammation associated with acute exacerbations precipitated by RV infection (20) nor does it effectively prevent virus-induced exacerbations (21). In addition, ICSs are recommended for maintenance therapy for all forms of asthma except mild intermittent asthma (22). Although it is possible that long-term treatment of asthmatics with ICSs may have influenced the phenotype of the cultured cells, it is unlikely that the observed effects are a consequence of residual corticosteroid in the cultures as cells were investigated at passage two after repeated changes of media and the observed proinflammatory responses to RV-16 infection were normal. Nonetheless, to investigate both of these possibilities, we examined responses to infection in cells from a group of ICSs naive asthmatics and studied the effect of in vitro treatment of these cells with dexamethasone. These cells were intermediately susceptible to virus replication, and they still demonstrated profoundly impaired apoptotic and type 1 interferon responses. In vitro treatment with dexamethasone at high doses was unable to induce the impaired innate responses seen in the cells from moderately severe asthmatics requiring ICSs. For these reasons the in vitro data appear entirely consistent with clinical observations already made (20). The similar response seen in ICSs naive cells from mild asthma confirms that the deficient innate responses and increased virus replication are not likely due to corticosteroid therapy.
Peripheral blood monocytes taken from asthmatic subjects and cultured with RV have a reduced interferon-γ response, suggesting asthmatics may have a deficient Th1-acquired immune response to RV infection, though the mechanism for this is unclear (23). Because type 1 interferons (IFN-α/β) are known to promote antiviral acquired immune responses (12), we suggest that deficient innate epithelial IFN-α/β response may be one mechanisms leading to a deficient Th1-acquired immune response. In vivo studies investigating both innate and acquired immune responses will be required to confirm whether this is true.
Our results offer a logical explanation for asthmatic subjects having lower respiratory tract symptoms and reductions in lung function of greater severity and duration as a consequence of RV infection (8). Although infection of BECs is limited in nonasthmatic subjects by an innate antiviral response and induction of apoptosis in infected cells, a deficiency of IFN-β in asthma facilitates virus replication and cytolysis with increased infection of neighboring cells. Exaggerated inflammatory responses are likely to result in asthmatics in vivo, consequent upon the increased replication and the cytolytic effects of the virus infection. Crucially, this defect can be restored in vitro by provision of exogenous IFN-β that restores apoptotic responses and limits virus replication to levels observed in normal cells. Thus, we propose that IFN-β may have therapeutic utility in preventing or treating virus-induced exacerbations of asthma.
MATERIALS and METHODS
All subjects were nonsmokers, with no exacerbations or respiratory tract infections in the preceding 4 wk. Allergy skin tests used a panel of common aeroallergens and were considered positive if the wheal response was >3 mm than the negative control. Lung function was assessed by spirometry and bronchial hyperresponsiveness by histamine challenge. Asthma was diagnosed in atopic individuals with a consistent history and evidence of bronchial hyperresponsiveness (defined by a PC20 histamine <8 mg/ml) and was categorized in accordance with the GINA guidelines (24). Subjects with asthma were divided into those with moderate disease, who had stable symptoms and used ICSs or had mild intermittent symptoms requiring the use of bronchodilators but who had never used inhaled or parenteral corticosteroids (Table I). Healthy controls had no previous history of lung disease, normal lung function, no evidence of bronchial hyperresponsiveness, and were nonatopic. The study was approved by the Southampton University Hospital Ethics Committee. All subjects gave written informed consent.
Bronchial epithelial cell tissue culture
Primary BECs were grown from bronchial brushings (>95% epithelial cells), which were obtained by fiber-optic bronchoscopy in accordance with standard guidelines (25); there was no significant difference in the proportion of columnar and basal cells isolated from normal or asthmatic donors. Cell culture and characterization was performed as described previously (17, 26). The cultured cells were all cytokeratin positive and exhibited a basal cell phenotype, as evidenced by the expression of cytokeratin 13, irrespective of the type of donor of the original brushings. Primary cultures were established by seeding freshly brushed BECs into hormonally supplemented bronchial epithelial growth medium (Clonetics) containing 50 U/ml penicillin and 50 μg/ml streptomycin. At passage two, cells were seeded onto 12-well trays and cultured until 80% confluent (17) before exposure to RV-16; where indicated, the caspase 3 inhibitor, ZVD-fmk (120 μM; Calbiochem), dexamethasone (10 and 100 nM; Sigma-Aldrich), or human IFN-β (100 IU; Sigma-Aldrich) or their vehicles were also added to the cells.
Generation and titration of RV
RV-16 and RV-1B stocks were generated and titrated from infected cultures of Ohio HeLa cells as described previously (14). Cells were infected at a multiplicity of infection of 2. Confirmation of infection and quantification of viral production was assessed by HeLa titration assay (14) and reverse transcription quantitative polymerase chain reaction (RT-qPCR), as described below. As negative controls, cells were treated with medium alone and UV inactivated RV-16 (14).
Assessment of cell viability
Viability and apoptosis were assessed by flow cytometry using AxV conjugated to PE and the vital dye, 7AAD, as described previously (27). The active forms of caspase 3/7 were detected using the Apo-One Homogenous Caspase 3/7 assay (Promega). Caspase activity was corrected for cell mass determined using methylene blue uptake. Cell lysis was measured as LDH release into the culture supernatant using pyruvate as a substrate (Sigma-Aldrich).
RT-qPCR and ELISA
RT-qPCR analysis of IFN-β mRNA and RV-16 viral RNA (vRNA) gene expression was performed on DNase treated RNA extracted from BECs using TRIzol (Life Technologies). Total RNA (1 μg) was reverse transcribed using avian myeloblastosis virus transcriptase (Promega) and random hexamers for IFN-β mRNA and 18S rRNA analysis or oligo (dT)15 for RV-16 vRNA. Real-time detection used an iCyclerIQ detection system using a PCR protocol as follows: 42 cycles at 95°C for 15 s, 60°C for 1 min and 72°C for 15 s. IFN-β signals were normalized to 18S rRNA and relative quantification performed using the ΔΔCT method. Comparisons were made 8 h after infection. Quantification of RV-16 was achieved using a TAQman assay located in the 5′ UTR in conjunction with the standard curve method. The standard curve was constructed using 10-fold serial dilutions of RV-16 5′ NTR cDNA cloned into PCR 2.1 TOPO (Invitrogen). Relative values for RV detection were calculated by normalizing to the starting cell number. Probe: FAM/TAMRA 6-FAMTGAGTCCTCCGGCCCCTGAATG, forward primer (RVTM-1) 5′-GTGAAGAGCCSCRTGTGCT-3′, reverse primer (RVTM-2) 5′-GCTSCAGGG-TTAAGGTTAGCC-3′.
ICAM-1 was measured by flow cytometry using a monoclonal antibody to ICAM-1 (anti–human CD54; eBioscience) and a FITC-labeled secondary antibody (DakoCytomation).
IFN-β (Biosource International), IL-6, and RANTES (Biosource International) release were measured by ELISA according to the manufacturer's instructions. The sensitivities were <250 pg/ml for IFN-β, <3 pg/ml for IL-6, and 2 pg/ml for RANTES.
Statistical analysis
When data were normally distributed the mean and SD have been used, differences between groups have been analyzed using Student's t test, when not normally distributed data were analyzed using nonparametric equivalents and summarized using the median and IQR, multiple comparisons were first analyzed by the Kruskal Wallis test and then by individual testing if significant. Correlations were analyzed by Spearman's test. A p-value of <0.05 was considered significant.
Acknowledgments
The authors would like to acknowledge the technical help given toward this project by Mrs. G. Sanderson, Dr. A.L. Andrews, and Dr. L.M. Hamilton.
Dr. P.A.B. Wark was funded by the National Health and Medical Research Council (Australia), Neil Hamilton Fairley fellowship. The work was also funded by the British Medical Association, HC Roscoe fellowship and Asthma UK, and by British Lung Foundation/Severin Wunderman Family Foundation Lung Research Programme grant number P00/2.
The authors have no conflicting financial interests.
Abbreviations used: AxV, annexin-V; BEC, bronchial epithelial cell; ICS, inhaled corticosteroid; IQR, interquartile range; LDH, lactate dehydrogenase; MFI, mean fluorescence intensity; RV, rhinovirus.
P.A.B. Wark and S.L. Johnston contributed equally to this work.
References
- 1.Johnston, S.L., P.K. Pattemore, G. Sanderson, S. Smith, F. Lampe, L. Josephs, P. Symington, S. O'Toole, S.H. Myint, D.A.J. Tyrell, and S.T. Holgate. 1995. Community study of the role of viral infections in exacerbations of asthma in 9–11-year old children. BMJ. 310:1225–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nicholson, K.G., J. Kent, and D.C. Ireland. 1993. Respiratory viruses and exacerbations of asthma in adults. BMJ. 307:982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnston, S.L., P.K. Pattemore, G. Sanderson, S. Smith, M.J. Campbell, L.K. Josephs, A. Cunningham, B.S. Robinson, S.H. Myint, M.E. Ward, et al. 1996. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am. J. Respir. Crit. Care Med. 154:654–660. [DOI] [PubMed] [Google Scholar]
- 4.Reddel, H., S. Ware, G. Marks, C. Salome, C. Jenkins, and A. Woolcock. 1999. Differences between asthma exacerbations and poor asthma control. Lancet. 353:364–369. [DOI] [PubMed] [Google Scholar]
- 5.Doull, I.J.M., F.C. Lampe, S. Smith, J. Schreiber, N.J. Freezer, and S.T. Holgate. 1997. Effect of inhaled corticosteroids on episodes of wheezing associated with viral infection in school age children: randomised double blind placebo controlled trial. BMJ. 315:858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraenkel, D.J., P. Bardin, G. Sanderson, S.T. Holgate, and S.L. Johnston. 1995. Lower airway inflammation during rhinovirus colds in normal and in asthmatic subjects. Am. J. Respir. Crit. Care Med. 151:879–886. [DOI] [PubMed] [Google Scholar]
- 7.Grunberg, K., H.H. Smits, M.C. Timmers, E.P. de Klerk, R.J. Dolhain, E.C. Dick, P.S. Hiemstra, and P.J. Sterk. 1997. Experimental rhinovirus 16 infection: effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am. J. Respir. Crit. Care Med. 156:609–616. [DOI] [PubMed] [Google Scholar]
- 8.Corne, J.M., C. Marshall, S. Smith, J. Schreiber, G. Sanderson, S.T. Holgate, and S.L. Johnston. 2002. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet. 359:831–834. [DOI] [PubMed] [Google Scholar]
- 9.Gern, J.E., D.M. Galagan, N.N. Jarjour, E.C. Dick, and W.W. Busse. 1997. Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am. J. Respir. Crit. Care Med. 155:1159–1161. [DOI] [PubMed] [Google Scholar]
- 10.Papadopoulos, N.G., P.J. Bates, P.G. Bardin, A. Papi, S.H. Leir, D.J. Fraenkel, J. Meyer, P.M. Lackie, G. Sanderson, S.T. Holgate, and S.L. Johnston. 2000. Rhinoviruses infect the lower airways. J. Infect. Dis. 181:1875–1884. [DOI] [PubMed] [Google Scholar]
- 11.Takaoka, A., S. Hayakawa, Y. Hideyuki, D. Stoiber, H. Negishi, H. Kikuchi, S. Shigeru, S. Imai, T. Shibue, K. Honda, and T. Taniguchi. 2003. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature. 424:516–523. [DOI] [PubMed] [Google Scholar]
- 12.Parronchi, P., M. De Carli, R. Manetti, C. Simonelli, S. Sampognaro, M. Piccinni, D. Macchia, E. Maggi, G. Del Prete, and S. Romagnani. 1992. IL-4 and IFN (alpha and gamma) exert opposite regulatory effects on the development of cytolytic potential by Th1 or Th2 human T cell clones. J. Immunol. 149:2977–2983. [PubMed] [Google Scholar]
- 13.Staunton, D.E., V.J. Merluzzi, R. Rothlein, R. Barton, S.D. Marlin, and T.A. Springer. 1989. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 56:849–853. [DOI] [PubMed] [Google Scholar]
- 14.Papi, A., and S.L. Johnston. 1999. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-B-mediated transcription. J. Biol. Chem. 274:9707–9720. [DOI] [PubMed] [Google Scholar]
- 15.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya, M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 13:539–548. [DOI] [PubMed] [Google Scholar]
- 16.Vlasak, M., S. Blomqvist, T. Hovi, E. Hewat, and D. Blaas. 2003. Sequence and structure of human rhinoviruses reveal the basis of receptor discrimination. J. Virol. 77:6923–6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bucchieri, F., J. Lordon, A. Richter, D. Buchanan, R. Djukanovic, S.T. Holgate, and D.E. Davies. 2001. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am. J. Respir. Cell Mol. Biol. 27:179–185. [DOI] [PubMed] [Google Scholar]
- 18.Lordan, J.L., F. Bucchieri, A. Richter, A. Konstantinidis, J.W. Holloway, M. Thornber, S.M. Puddicombe, D. Buchanan, S.J. Wilson, R. Djukanovic, et al. 2002. Cooperative effects of Th2 cytokines and allergen on normal and asthmatic bronchial epithelial cells. J. Immunol. 169:407–414. [DOI] [PubMed] [Google Scholar]
- 19.Balachandran, S., C.N. Kim, W.C. Yeh, T.W. Mak, K. Bhalla, and G.N. Barber. 1998. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signalling. EMBO J. 23:6888–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunberg, K., R.F. Sharon, J.K. Sont, J.C. In't Veen, W.A. Van Schadewijk, E.P. De Klerk, C.R. Dick, J.H. Van Krieken, and P.J. Sterk. 2001. Rhinovirus-induced airway inflammation in asthma. Effect of treatment with inhaled corticosteroids before and during experimental infection. Am. J. Respir. Crit. Care Med. 164:1816–1822. [DOI] [PubMed] [Google Scholar]
- 21.Doull, I.J.M., F.C. Lampe, S. Smith, J. Schreiber, N.J. Freezer, and S.T. Holgate. 1997. Effect of inhaled corticosteroids on episodes of wheezing associated with viral infection in school age children: randomised double blind placebo controlled trial. BMJ. 315:858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.British Thoracic Society, S.I.G.N.S. 2003. British guideline on the management of asthma. Thorax. 58 (Suppl. 1): i1–i94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks, G.D., K.A. Buchta, C.A. Swenson, J.E. Gern, and W. Busse. 2003. Rhinovirus induced interferon-γ and airway responsiveness in asthma. Am. J. Respir. Crit. Care Med. 168:1091–1094. [DOI] [PubMed] [Google Scholar]
- 24.National Heart, Lung and Blood Institute. 1995. Global Strategy for Asthma Management and Prevention. 96–369.
- 25.Hurd, S.Z. 1991. Workshop summary and guidelines: investigative use of bronchoscopy. J. Allergy Clin. Immunol. 88:808–814. [DOI] [PubMed] [Google Scholar]
- 26.Lordan, J.L., F. Bucchieri, A. Richter, A. Konstantinidis, J.W. Holloway, M. Thornber, S.M. Puddicombe, D. Buchanan, S.J. Wilson, R. Djukanovic, et al. 2002. Cooperative effects of Th2 cytokines and allergen on normal and asthmatic bronchial epithelial cells. J. Immunol. 169:407–414. [DOI] [PubMed] [Google Scholar]
- 27.Puddicombe, S.M., C. Torres-Lozano, A. Richter, F. Bucchieri, J. Lordan, P.H. Howarth, B. Vrugt, R. Albers, R. Djukanovic, S.T. Holgate, et al. 2003. Increased expression of p21waf cyclin-dependent kinase inhibitor in asthmatic bronchial epithelium. Am. J. Respir. Cell Mol. Biol. 28:61–68. [DOI] [PubMed] [Google Scholar]