

Abstract
A combination of genetic and environmental factors can cause autoimmune disease in animals. SKG mice, which are genetically prone to develop autoimmune arthritis, fail to develop the disease under a microbially clean condition, despite active thymic production of arthritogenic autoimmune T cells and their persistence in the periphery. However, in the clean environment, a single intraperitoneal injection of zymosan, a crude fungal β-glucan, or purified β-glucans such as curdlan and laminarin can trigger severe chronic arthritis in SKG mice, but only transient arthritis in normal mice. Blockade of Dectin-1, a major β-glucan receptor, can prevent SKG arthritis triggered by β-glucans, which strongly activate dendritic cells in vitro in a Dectin-1–dependent but Toll-like receptor-independent manner. Furthermore, antibiotic treatment against fungi can prevent SKG arthritis in an arthritis-prone microbial environment. Multiple injections of polyinosinic-polycytidylic acid double-stranded RNA also elicit mild arthritis in SKG mice. Thus, specific microbes, including fungi and viruses, may evoke autoimmune arthritis such as rheumatoid arthritis by stimulating innate immunity in individuals who harbor potentially arthritogenic autoimmune T cells as a result of genetic anomalies or variations.
Both genetic and environmental factors contribute to the development of common autoimmune diseases, such as rheumatoid arthritis (RA) and type 1 diabetes (1, 2). However, it remains obscure how each factor is arranged into a causal chain of pathogenesis. Given that T cells are the main mediators of many autoimmune diseases, one of the key issues for elucidating the mechanism of autoimmune disease would be to understand how genetic and environmental factors affect the control of the generation, activation, and expansion of pathogenic self-reactive T cells.
RA is a chronic systemic inflammatory disease that primarily affects the synovial membranes of multiple joints (1). Both genetic and environmental factors are involved in the pathogenesis (1, 3, 4). For example, MHC and non-MHC genes play significant roles in determining the genetic susceptibility to RA (1, 4, 5). Various infectious agents, including viruses and bacteria, have also been suspected to be causative agents of RA, although epidemiological evidence has been elusive (4, 6, 7). Because T cells, especially CD4+ T cells, play crucial roles at least in the initial phase of RA, a key question to understand the etiology of RA would be how genetic and environmental factors contribute to the generation and activation of arthritogenic T cells. In this paper, we have addressed this issue by using a newly established animal model of RA.
SKG mice spontaneously develop T cell–mediated chronic autoimmune arthritis as a consequence of a mutation of the gene encoding an SH2 domain of ZAP-70, a key signal transduction molecule in T cells (8). This mutation impairs positive and negative selection of T cells in the thymus, leading to thymic production of arthritogenic autoimmune CD4+ T cells. The mice succumb to symmetrical joint swelling beginning in small joints of the digits and progressing to larger joints, accompanying severe synovitis with formation of pannus invading and eroding adjacent cartilage and subchondral bone. They develop rheumatoid factor and other autoantibodies in the circulation and extra-articular lesions, such as interstitial pneumonitis, vasculitides, and subcutaneous necrobiotic nodules, not unlike rheumatoid nodules. Genetic deficiency of IL-6, IL-1, or TNF-α inhibits the development of SKG arthritis (9), similar to the effects of anticytokine therapy in human RA (1). These clinical and immunopathological characteristics of SKG arthritis make the strain a suitable model of human RA.
We show in this paper that SKG mice fail to develop arthritis in a microbially clean environment, despite their thymic production of arthritogenic autoimmune T cells that persist in the periphery. However, under this arthritis-resistant condition, zymosan, a crude yeast cell wall extract, can provoke severe arthritis in SKG mice, and glucose polymer β-1, 3-D-glucans (β-glucans), which are the main constituents of zymosan, are responsible for the arthritogenic effect. Blockade of Dectin-1, a major β-glucan receptor, is able to prevent SKG arthritis triggered by β-glucans. Furthermore, antibiotic treatment of fungi can prevent SKG arthritis in an arthritis-prone microbial environment. Polyinosinic-polycytidylic acid (poly[I:C]), a double-stranded RNA, also showed a mild arthritogenic effect in SKG mice. Thus, certain microbes, such as fungi and viruses, may activate arthritogenic T cells through stimulating innate immunity, thereby evoking chronic autoimmune arthritis in the individuals who are genetically prone to produce arthritogenic autoimmune T cells that persist in the periphery.
The present results illustrate how a specific combination of genetic and environmental factors in the generation and activation of autoimmune T cells leads to the development of a particular autoimmune disease, and will help design the measures to prevent autoimmune disease in genetically susceptible individuals by controlling autoimmune-eliciting environmental agents, or blocking their effects on the immune system at the molecular level.
Results
Environmental stimuli trigger chronic arthritis in SKG mice
SKG mice failed to develop chronic arthritis in our strictly controlled specific pathogen-free (SPF) environment, although all SKG mice developed the disease by 6 mo under our conventional microbial condition (non-SPF; Fig. 1 A and reference 8). To examine whether vertical or horizontal environmental agents are responsible for eliciting arthritis in SKG mice under the non-SPF condition, 7-d-old SKG mice born in the SPF environment were foster nursed to BALB/c mice in the non-SPF environment or 8-wk-old nonarthritic SPF SKG mice were transferred to the non-SPF facility. Both groups developed arthritis as severe as non-SPF SKG mice (Fig. 1 A).
Figure 1.

Chemical or biological compounds trigger arthritis in SKG mice. (A) Arthritis scores of 6-mo-old SKG mice maintained in the indicated environment. SPF-born nonarthritic SKG mice were foster nursed to non-SPF BALB/c mice at 7 d of age or transferred to the non-SPF environment at 8 wk of age. (B) 5 × 107 splenocytes, 108 thymocytes, or 5 × 106 BM cells prepared from each nonarthritic SPF SKG mouse were adoptively transferred to individual 6-wk-old nude or SCID mice. BM cells were T cell depleted by treatment with anti–Thy-1, anti-CD4, and anti-CD8 antibodies and rabbit complement before transfer. Arthritis scores 6 mo after transfer are shown. (C) Arthritis scores of SPF SKG mice 5 mo after administration of indicated compounds. SKG mice between 8 and 12 wk of age maintained in the SPF environment were treated as follows: i.p. injection of 2 mg zymosan, 1 μg LPS, 200 μg ConA, 500 μl pristane, 1 mg CP, or 150 μl PBS; i.v. administered with 400 ng PTX on days 0 and 2; i.p. 150 μg poly(I:C) or 150 μl PBS three times per week for 17 wk, 10 nmol CpG or non-CpG once per week for 4 wk; and i.v. with 0.3 mg agonistic anti-CD40 or control rat IgG on days 0, 7, and 14.
However, in the strict SPF condition, transfer of splenocytes or thymocytes from SPF-born nonarthritic SKG mice to BALB/c athymic nude mice produced severe arthritis (Fig. 1 B). Transfer of T cell–depleted BM cells from nonarthritic SPF SKG mice also produced severe arthritis in SCID mice, indicating that SKG BM cells differentiated through the SCID thymus to arthritogenic T cells and became activated in the periphery without exposure to a putative arthritogenic environmental agent (Fig. 1 B).
Together, SKG mice fail to develop arthritis in our microbially clean environment despite continuous thymic production of arthritogenic T cells and their persistence in the periphery. Certain environmental agents that act on SKG mice horizontally can trigger arthritis. Furthermore, antigen-nonspecific proliferative stimulation, as in the case of presumable homeostatic T cell proliferation upon transfer to the T cell–deficient environment of nude mice, suffices to activate dormant arthritogenic T cells to cause arthritis.
Triggering of arthritis in SPF SKG mice by chemical or biological compounds
Next, we attempted to determine whether any chemical or biological compounds known to affect T cells, B cells, or APCs can trigger arthritis in SPF SKG mice (Fig. 1 C). A single i.p. injection of zymosan elicited the most severe arthritis at the highest incidence. Injection of poly(I:C), three times per week for 17 wk, also triggered mild arthritis in the majority of treated mice (10, 11). Cyclophosphamide (CP) at 1 mg/mouse, the dose causing temporary lymphopenia and used to elicit diabetes in NOD mice (12), produced mild arthritis at a significantly high incidence compared with PBS-treated controls. Administration of CpG or agonistic anti-CD40 mAb, at the doses eliciting autoimmunity in other systems, failed to trigger arthritis (13–15). A single injection of ConA, pristane, and LPS and two i.v. injections of pertussis toxin (PTX), at a dose reported to have biological effects, elicited mild arthritis at a low incidence (14, 16–18). SKG mice were sensitive to high doses of LPS (e.g., 1 mg); many of the treated mice died immediately after treatment and those that survived did not develop arthritis (unpublished data). Thus, zymosan, poly(I:C), and CP can effectively provoke arthritis in SPF SKG mice.
Possible roles of fungi in triggering arthritis in SKG mice
The strong arthritogenic effect of zymosan in SKG mice prompted us to examine possible fungal infection in non-SPF SKG mice. Specific detection of fungal 18S-ribosomal RNA gene (18SrDNA) revealed fungal infection in the lungs and some esophagi, but not in the livers or ankles, of 12-wk-old non-SPF SKG mice (Fig. 2 A and reference 19). Real-time quantitative PCR analysis revealed that the lungs from non-SPF mice had a significantly higher concentration of fungi than those in the SPF condition (Fig. 2 B). Non-SPF SKG mice harbored larger amounts of fungi than BALB/c (Fig. 2 B), indicating that SKG mice are more susceptible to infection than BALB/c mice, presumably due to the dysfunction of SKG T cells bearing the ZAP-70skg mutation (8). DNA sequencing of PCR products for 18SrDNA or internal transcriber spacer 1 (ITS1) identified five species of fungi in the lungs of non-SPF SKG mice (19, 20): Pneumocystis murina, Cladosporium cladosporioides, Graphium rubrum, Alternaria alternata, and Aspergillus nidulans (see Table S1, available at http://www.jem.org/cgi/content/full/jem.20041758/DC1). Methenamine silver staining of the lungs indeed revealed argyrophilic cysts in non-SPF SKG mice, corresponding to be P. murina infection (Fig. 2, C and D), but not in non-SPF BALB/c mice (Fig. 2, E and F). Inflammation was minimal surrounding the fungi, and filamentous fungi were histologically undetectable in non-SPF SKG lungs (Fig. 2, C and D).
Figure 2.
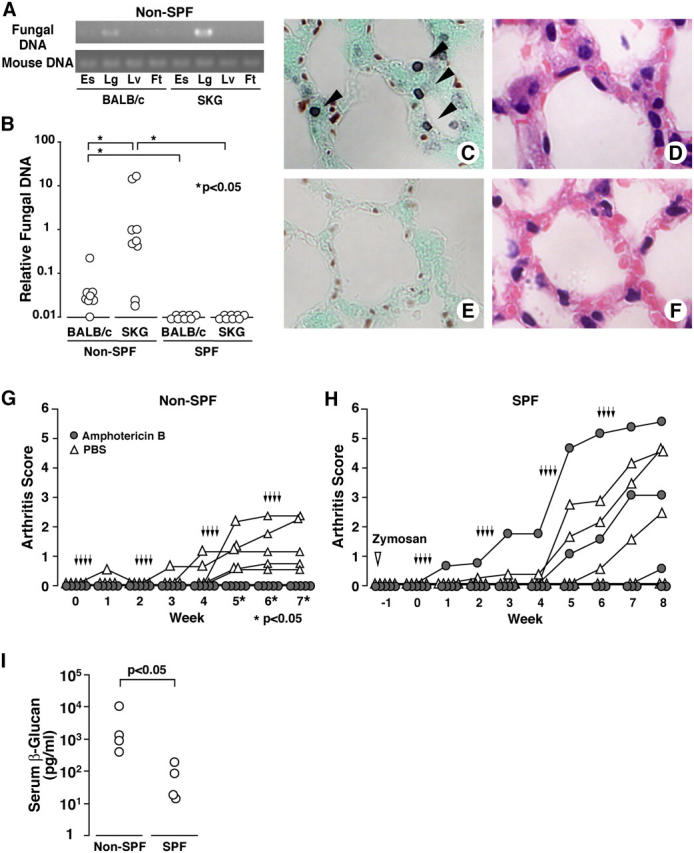
Silent fungal infections under non-SPF trigger arthritis in SKG mice. (A) PCRs of fungal 18SrDNA or mouse Rag 1 gene from esophagus (Es), lung (Lg), liver (Lv), or foot including ankle joint (Ft) of BALB/c or SKG mice in non-SPF. (B) Lungs of BALB/c or SKG mice in non-SPF or SPF were assessed for fungi by real-time quantitative PCR. (C–F) Methenamine silver (C and E) or hematoxylin and eosin (D and F) stainings of lungs from SKG mice (C and D) or BALB/c (E and F) of non-SPF. Arrows indicate argyrophilic fungal cysts. (G) Male SKG mice of 10 wk of age in the non-SPF facility were treated with four cycles of four daily i.p. injections of 75 μg amphotericin B (arrows) at 10-d intervals. (H) SKG mice in the SPF facility received the same protocol of antifungal treatment 10 d after i.p. administration of 2 mg zymosan (open inverted triangle). (I) Concentration of serum β-glucan in non-SPF or SPF 12-wk-old SKG mice.
To determine the possible contribution of fungi to triggering arthritis in SKG mice, non-SPF SKG mice were treated with amphotericin B, which has a broad antifungal property and reportedly reduces the viability of Pneumocystis carinii (21). The treatment prevented SKG arthritis in the non-SPF condition (Fig. 2 G), whereas it did not alter the course of zymosan-triggered SKG arthritis in the SPF condition (Fig. 2 H), indicating that the arthritis-preventive effect of amphotericin B is due to its antifungal activity, not to its possible immunosuppressive side effects. Furthermore, β-glucan, a major component of fungi, was detected in the sera of SKG mice under the non-SPF condition (3,175 ± 4,021 pg/ml), compared with under the SPF condition (63 ± 72 pg/ml; Fig. 2 I). These results collectively indicate that fungi can be a key agent to trigger arthritis in SKG mice, at least in our conventional non-SPF condition.
Purified β-glucans are strong inducers of arthritis in SKG mice
Among the constituents of zymosan, such as β-glucans, mannan, chitin, and proteins, β-glucan is known to be associated with fungal pathogenicity (22). To assess whether β-glucan is responsible for triggering arthritis in SKG mice, we administered the purified β-glucans, curdlan, or laminarin to SKG or BALB/c mice (Fig. 3). Curdlan is a linear β-1,3-glucan of ∼500-mers derived from the bacterium Alcaligenes faecalis; laminarin consists of short, soluble β-1,3- and some β-1,6-glucans (<10,000 MW) derived from seaweed Laminaria digitata. Administration of 3 mg curdlan or 30 mg laminarin triggered severe chronic arthritis in all the treated SKG mice and produced transient arthritis in 50 and <10%, respectively, of BALB/c mice (Fig. 3, A and B). SKG mice treated with 2 mg zymosan showed a similar severity and time course of disease progression (Fig. 2 H and Fig. 6 A). Histology of swollen joints in curdlan- or laminarin-treated mice, whether SKG or BALB/c, exhibited severe synovitis accompanying massive subsynovial infiltration of neutrophils, lymphocytes, macrophages, and plasma cells and villous proliferation of synoviocytes. However, synovitis in BALB/c mice was transient and reverted to normal histology in 4 mo (Fig. 3, C–H).
Figure 3.
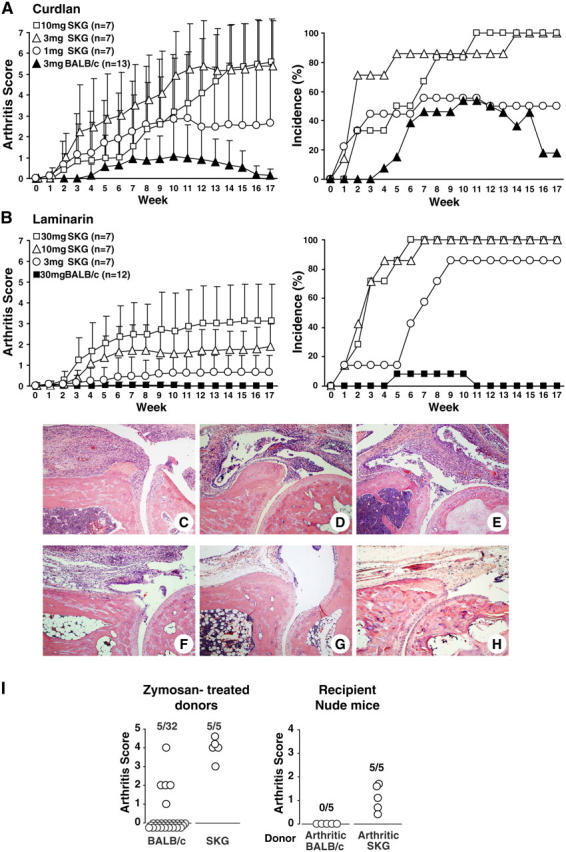
Purified β-glucans trigger arthritis in SKG mice. (A and B) Arthritis score and incidence of SKG mice or BALB/c in the SPF environment after a single i.p. injection of indicated amounts of (A) curdlan or (B) laminarin. Vertical bars represent the means ± SD. Arthritis scores are significantly different (P < 0.05) between SKG and BALB/c mice treated with 3 mg curdlan during 2–17 wk; between SKG mice treated with 3 and 10 mg curdlan during 4–7 wk; between SKG and BALB/c mice treated with 30 mg laminarin during 3–17 wk; and between SKG mice treated with 3 and 30 mg laminarin during 4–17 wk. (C–H) Hematoxylin and eosin staining of ankle joints. Arthritic joints of SPF SKG mice 20 wk after treatment with 2 mg zymosan (C), 30 mg laminarin (D), or 3 mg curdlan (E). (F and G) An arthritic (F, 8 wk after treatment) or cured (G, 20 wk) ankle joint from BALB/c mice with transient arthritis triggered by 3 mg curdlan. (H) An ankle joint of 40-wk-old nonarthritic SKG mice in SPF. (I) Arthritis scores of BALB/c or SKG donor mice 9 wk after administration of 2 mg zymosan. Nude mice were transferred with 5 × 107 splenocytes from arthritic BALB/c or SKG mice and assessed for arthritis 12 wk after transfer.
Figure 6.
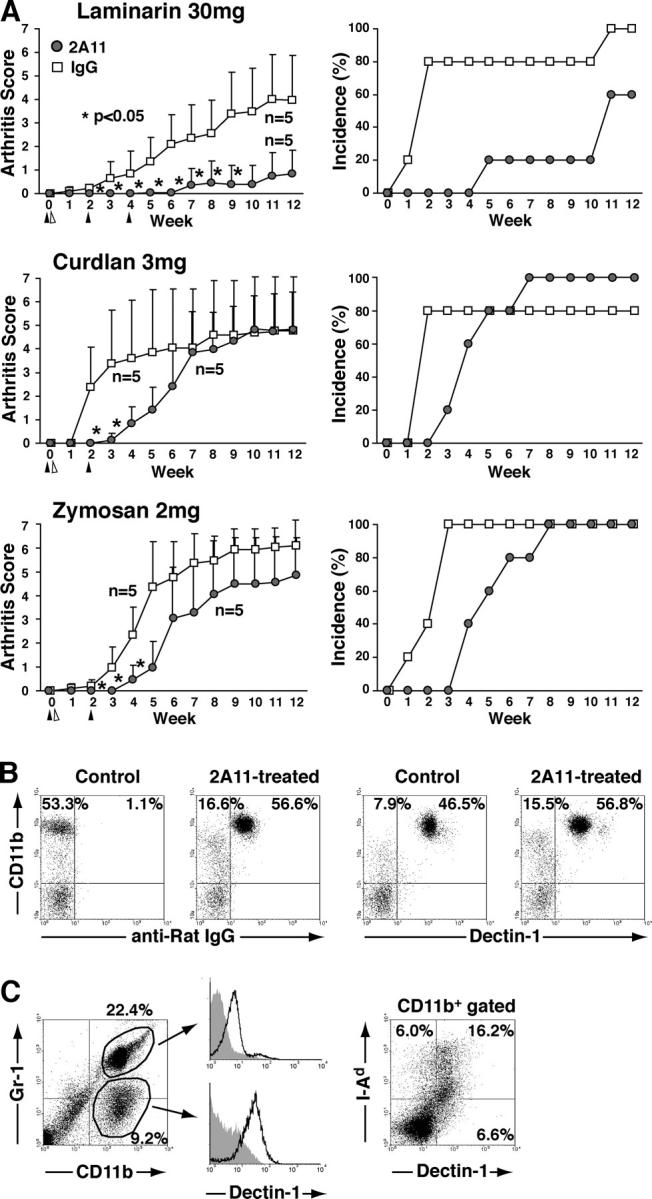
Dectin-1 as a major receptor for β-glucans in triggering SKG arthritis by β-glucans. (A) 0.5 mg 2A11 anti–Dectin-1 mAb or rat IgG were administered i.v. into SPF SKG mice two or three times, 2 wk apart (closed triangle), before or after i.p. injection of indicated amounts of laminarin, curdlan, or zymosan (open triangle). Vertical bars represent the means ± SD. Asterisks indicate significant differences (P < 0.05) at the indicated time points. (B) SKG mice were treated i.v. with 0.5 mg 2A11 or rat IgG, and the expression of CD11b and surface-bound rat IgG or Dectin-1 on peripheral blood was analyzed 1 wk later. (C) The synovium from arthritic SKG mice treated with 30 mg laminarin was digested for 1 h; resulting single cell suspensions were analyzed for the expression of indicated surface markers. Shaded histograms indicate staining controls. A representative result of three independent experiments is shown in B and C.
To determine whether β-glucan–induced arthritis in SKG or BALB/c mice was a T cell–mediated autoimmune disease, we transferred splenocytes from arthritic SKG or BALB/c mice to BALB/c nude mice (Fig. 3 I). Transfer of splenocytes from arthritic SKG produced arthritis in all the recipients in 3 mo, whereas transfer from BALB/c mice did not. Thus, β-glucans can evoke T cell–mediated autoimmune arthritis in SKG mice, but only transient synovial inflammation in BALB/c mice; the latter is apparently not mediated by autoimmune T cells.
β-Glucans activate BM-DCs in a Dectin-1–dependent manner
Although zymosan is known to activate BM-DCs through Toll-like receptor (TLR)2 and 6 as well as β-glucan receptors, it is not clear yet whether purified β-glucans have a similar effect (23, 24). Adding curdlan or laminarin to the culture of immature DCs that were induced in vitro from BM cells revealed that these β-glucans were able to activate immature DCs as indicated by up-regulation of CD86 in a dose-dependent fashion (Fig. 4 A). Laminarin, curdlan, and zymosan also up-regulated the expression of CD40, CD80, and class II MHC (I-Ad) on DCs (Fig. 4 B). To assess the possibility that DC activation was induced by contaminated LPS or by direct interaction between β-glucan and TLRs, we cultured immature BM-DCs prepared from TLR4-, MyD88-, or TLR2-deficient mice with purified β-glucans and assessed CD86 expression (Fig. 4 C). Laminarin or curdlan equally enhanced the expression of CD86 on DCs prepared from either these knockout or wild type mice, whereas LPS and Pam3CSK4, a synthetic ligand of TLR2, was ineffective on TLR4-deficient DCs and TLR2- or MyD88-deficient DCs, respectively (25–27). This indicates that DC activation by β-glucans is not due to direct stimulation of TLR2 or MyD88-dependent pathway or to stimulation of TLR4 by possible LPS contaminant in the β-glucan preparation.
Figure 4.

Purified β-glucans activate BM-DCs. (A) The expression of CD86 on CD11c+ BM-DCs cultured with laminarin, curdlan, or zymosan at indicated concentrations. (B) The expression of indicated surface markers on CD11c+ BM-DCs cultured with 1 μg/ml LPS, 10 mg/ml laminarin, 100 μg/ml curdlan, or 100 μg/ml zymosan. Percentages of positive cells are also shown. (C) The expression of CD86 on TLR4-, TLR2-, or MyD88-deficient or wild-type CD11c+ BM-DCs cultured with 10 mg/ml laminarin (LAM), 100 μg/ml curdlan (CDL), 10 μg/ml LPS, or 10 μg/ml Pam3CSK4. (D) BM-DCs were incubated with or without 100 μg/ml of laminarin for 1 h before the culturing with 10 μg/ml of curdlan or zymosan (ZYM), and assessed for CD86 expression. (E) The expression of Dectin-1 on unstimulated DCs or DCs activated by 1 μg/ml LPS, 10 mg/ml laminarin, 100 μg/ml curdlan, or 100 μg/ml zymosan. (F) BM-DCs were incubated with 10 μg/ml of 2A11 anti–Dectin-1 mAb or isotype-control IgG for 1 h before culture with indicated β-glucans or cultured with β-glucans alone. The expression of CD86 on CD11c+ BM-DCs 24 h after the stimulation is shown. Shaded histograms indicate unstimulated DCs in A, C, D, and F or staining controls in B and E. Ordinate indicates cell number. A representative result of three independent experiments is shown in A–F.
Laminarin is known to competitively inhibit the binding of other β-glucans to their receptors (28–31). Indeed, a nonstimulatory dose (100 μg/ml) of laminarin blocked the activation of BM-DCs by stimulatory doses (10 μg/ml) of curdlan (Fig. 4 D), indicating that these two β-glucans share the same receptor other than the TLR. In contrast, this dose of laminarin failed to block the activation of BM-DCs by 10 μg/ml zymosan, which can also interact with TLR2 and TLR6 (Fig. 4 D and reference 24).
Among β-glucan receptors, Dectin-1 is known to play a major role in mediating many biological responses to β-glucans (32, 33). Staining with 2A11 anti–Dectin-1 mAb revealed that Dectin-1 was highly expressed on DCs, especially on immature DCs, and activation slightly decreased the expression (Fig. 4 E). When 2A11 was added before culturing DCs with curdlan or zymosan, DC activation with curdlan was blocked in a dose-dependent manner, but the activation by zymosan was not (Fig. 4 F and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20041758/DC1).
Although Dectin-1 stimulation is known to enhance TLR-stimulated cytokine formation, it is unclear whether Dectin-1 stimulation alone can provoke cytokine formation (32, 33). In Fig. 5 A, purified β-glucans induced TNF-α production by DCs as shown previously with a macrophage cell line (34). 2A11 completely blocked this curdlan-stimulated TNF-α production, whereas it did not affect zymosan-stimulated production (Fig. 5 B). Furthermore, MyD88 deficiency only slightly reduced curdlan-stimulated TNF-α production, in contrast with significant reduction in zymosan-stimulated production (Fig. 5 C). These results collectively indicate that Dectin-1 stimulation alone, without TLR engagement, can induce TNF-α production, although zymosan appears to induce the production through both Dectin-1 and TLR (32, 33).
Figure 5.
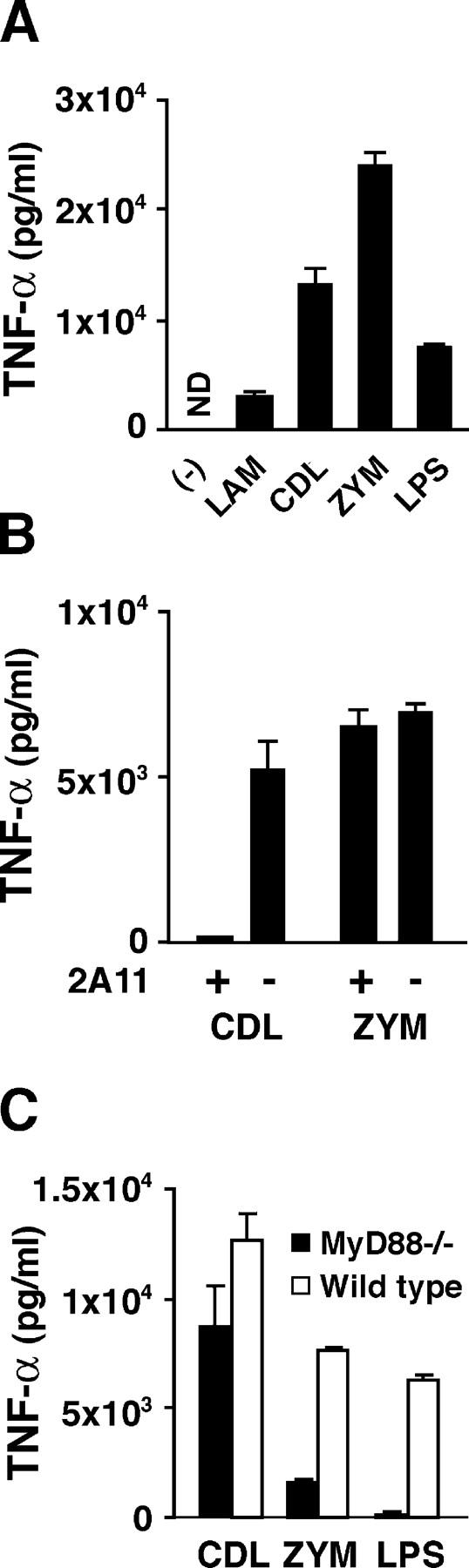
Production of TNF-α by β-glucan–stimulated DCs. (A) BM-DCs were incubated with 10 mg/ml laminarin, 100 μg/ml curdlan, 100 μg/ml zymosan, or 1 μg/ml LPS from day 5 for 24 h and the amount of TNF-α was assessed by ELISA. (B) TNF-α production by BM-DCs that were incubated with 10 μg/ml of 2A11 mAb or control IgG2b for 1 h before culture with 100 μg/ml curdlan or 100 μg/ml zymosan. (C) TNF-α production by MyD88-deficient or wild-type BM-DCs that were cultured with 100 μg/ml curdlan, 100 μg/ml zymosan, or 10 μg/ml LPS. Vertical bars represent the means ± SD of triplicates.
In contrast with TNF-α production, the amount of IL-12 produced by laminarin- or curdlan-stimulated DCs was low (30 pg/ml) or undetectable, respectively, compared with ∼200 pg/ml of IL-12 produced by zymosan-stimulated DCs (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20041758/DC1). This indicates that stimulation of DCs through TLRs, rather than Dectin-1, is crucial for IL-12 production (32).
Together, purified β-glucans can induce maturation of DCs (Fig. 4) and production of cytokines (Fig. 5) mainly through Dectin-1, but not through TLRs or other β-glucan receptors such as CR3, scavenger receptors, or lactosylceramide.
Blockade of Dectin-1 inhibits β-glucan–induced arthritis in SKG mice
To determine whether the in vivo arthritogenic effect of curdlan, laminarin, and zymosan is via Dectin-1, we treated SKG mice with 2A11 before and after injection of these compounds. The treatment significantly delayed the onset of arthritis in SKG mice treated with either compound, and significantly reduced both the incidence and severity of arthritis in laminarin-treated SKG mice (Fig. 6 A). The administered antibody bound to Dectin-1–expressing cells but did not deplete them, indicating that the mAb blocked Dectin-1 in vivo (Fig. 6 B). Analysis of collagen-digested synovial cell suspensions from laminarin-treated arthritic SKG mice revealed several types of cells in the inflamed synovium (Fig. 6 C); i.e., ∼10% of cells were Gr-1low CD11b+ monocytes/macrophages, which were Dectin-1high; ∼20% were Gr-1+ CD11b+ granulocytes. More than half of Dectin-1–expressing CD11b+ cells were class II MHC+, indicating that the arthritic synovium of SKG mice contains numerous Dectin-1+ macrophages. These results, together with the development of transient arthritis in normal BALB/c mice by i.p. injection of β-glucan (Fig. 3, A and B), indicate that circulating β-glucans can activate Dectin-1–expressing synovial cells, including synovial macrophages/DCs and granulocytes, and that blockade of Dectin-1 on the synovial cells can inhibit the activation of synovial cells to mediate synovitis and also to activate arthritogenic T cells.
Discussion
The main findings in this paper are that microbial compounds, especially β-glucans, can evoke T cell–mediated autoimmune arthritis in SKG mice, which are genetically prone to produce arthritogenic self-reactive T cells, but fail to develop the disease in a clean environment. This indicates that a specific combination of a genetic T cell abnormality and exposure to particular microbes capable of strongly activating innate immunity is able to produce overt T cell–mediated autoimmune disease, but either alone is not.
SKG mice spontaneously suffer from severe arthritis in a conventional environment, but not under SPF conditions. Transfer of nonarthritic SPF SKG mice to a non-SPF environment after weaning elicited arthritis. However, under a strict SPF condition, simply transferring peripheral T cells or thymocytes from nonarthritic SKG mice to syngeneic nude mice effectively produced arthritis. When taken together, these results indicate the following: (a) the thymi of SKG mice are continuously producing arthritogenic T cells, which persist in the periphery but are unable to mediate arthritis unless exogenously stimulated; (b) environmental agents, including microbes, that horizontally affect the immune system may trigger T cell–mediated chronic arthritis in SKG mice; and, importantly, (c) antigen-nonspecific general activation of T cells, like homeostatic proliferation as seen upon transfer of T cells to T cell–deficient nude mice, may suffice to activate dormant arthritogenic T cells to mediate arthritis.
Among various chemical or biological compounds tested in the present work, the most potently arthritogenic in SKG mice were β-glucans. Dectin-1, a receptor mediating main biological response to β-glucans in vitro and in vivo, is expressed on cells of the monocyte/macrophage and neutrophil lineages, and at lower levels on splenic DCs and a subpopulation of splenic T cells (35). It has been shown that β-glucans, zymosan in particular, induce the production of reactive oxygen, nitrogen intermediates, and proinflammatory cytokines by macrophages and neutrophils and enhance phagocytosis by macrophages (22, 36). We have shown in this paper that purified β-glucans can stimulate BM-DCs to maturate and form cytokines including TNF-α, and that this DC activation was dependent on Dectin-1 but not TLRs. Furthermore, blockade of Dectin-1 significantly reduced the incidence and severity of arthritis. Although expression of Dectin-1 on T cells suggests the possibility that β-glucans may also directly stimulate arthritogenic T cells, this is unlikely as the major mechanism of β-glucan–induced SKG arthritis because the expression of Dectin-1 on splenic T cells was confined to a minor subset of CD8+ GR-1+ T cells in SKG and BALB/c mice (35) and because β-glucans exhibited no direct cell-proliferative or cytokine-inducing effects on T cells in vitro (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20041758/DC1). Together, it is likely in SKG mice that administered β-glucans, or their degraded products with lower molecular weights, activate Dectin-1–expressing APCs, including DCs/macrophages in the synovium, enhancing their presentation of joint self-antigens to arthritogenic T cells. The strong arthritogenic activity of β-glucans could also be attributed in part to their persistence in the circulation. Fungal β-glucans produced at the infection sites, such as lung, may circulate to the joints and elicit arthritis by activating synovial cells (37, 38). However, it is of note that in contrast with effective triggering of arthritis by parenteral exposure to β-glucans, oral administration of a large amount of zymosan for 3 wk failed to elicit arthritis in SKG mice, indicating that orally taken β-glucans may be little absorbed in the digestive system (unpublished data).
Administration of β-glucans elicited not only chronic arthritis in SKG mice but also transient and reversible arthritis in BALB/c mice. The latter can also be produced in normal animals by direct injection of zymosan into the joint (39). However, the β-glucan–induced arthritis in BALB/c mice was not transferable to nude mice by splenic T cells. These findings collectively indicate that β-glucans can directly activate synovial macrophages/DCs and cause synovial inflammation presumably because synovial cells are sensitive to various chemical or biological stimuli and intrinsically capable of producing proinflammatory cytokines upon stimulation (8, 9, 40, 41). However, this synovial inflammation is not sufficient to evoke chronic T cell–mediated autoimmune arthritis unless the mice harbor arthritogenic T cells sufficient in number or TCR specificity, or both, to mediate chronic synovitis. Supporting this role of environmental compounds to trigger T cell–mediated autoimmune disease, several papers have shown that inoculation of CpG, heat-shock proteins, or agonistic anti-CD40 monoclonal antibody initiated overt autoimmune disease such as diabetes and encephalomyelitis in mice expressing transgenic TCRs specific for self- or surrogate self-antigens expressed in the target organs/tissues (14, 15, 42). On the other hand, there are reports that administration of activated DCs pulsed with organ specific self-peptides can only produce transient or local autoimmunity in normal animals; for example, local administration of activated DCs loaded with type II collagen peptides elicited only localized arthritis at the site of injection (43, 44). Thus, our results together with these findings indicate that activation of APCs by environmental agents may provoke transient and self-limiting or chronic and progressive autoimmune disease depending on the quality and quantity of autoimmune T cells present in the host at the time of exposure to the agents. Among various types of human arthritides of unknown etiology, some, especially chronic ones like RA, may be caused by a combination of exposure to APC-activating environmental agents and the production/persistence of arthritogenic T cells; others, especially infectious ones, may be due to direct effects of environmental agents on the synovium, which is highly sensitive to various stimuli and, upon activation, capable of secreting proinflammatory cytokines and chemical mediators (1). Our results also imply that one-time “hit-and-run” exposure to an arthritogenic environmental compound may suffice to trigger chronic arthritis if arthritogenic autoimmune T cells are already present in the immune system.
In addition to β-glucans, other chemical or biological compounds could trigger arthritis in SKG mice at various degrees. For example, repeated administration of poly(I:C) produced mild arthritis in all the treated mice, presumably by stimulating the production of type I interferons and activating synovial macrophages/DCs via TLR3 (45). Although a single dose of LPS evoked arthritis in some SKG mice, it remains to be determined whether multiple injections can trigger the disease at a higher incidence. Mild arthritis was also induced by PTX, which causes nonspecific proliferation of T cells via activation of APCs with its catalytic ADP-ribosyltransferase activity (46), or by cyclophosphamide, which produces transient lymphocytopenia and reduction of CD25+CD4+ natural regulatory T cells (12, 47, 48). The ZAP-70skg mutation itself affects the function of natural CD25+CD4+ regulatory T cells in the control of arthritogenic T cells (unpublished data). Thus, not only fungi but also viruses or bacteria may be potentially able to trigger arthritis in SKG mice by activating innate immunity, or by peripheral control of arthritogenic T cells. Furthermore, it is likely that SKG mice are more susceptible to such arthritogenic microbial infections than normal mice because the ZAP-70skg mutation not only alters thymic T cell selection and the resulting T cell repertoire, but also reduces the responsiveness of peripheral T cells to microbes (8). It remains to be investigated whether other biological, chemical, or physical insults that activate innate immunity or alter peripheral control of T cell activation/expansion can also trigger arthritis in SKG mice.
In conclusion, environmental agents, such as fungi and viruses, may evoke autoimmune arthritis similar to RA in genetically susceptible individuals who already harbor arthritogenic T cells through stimulating APCs in an antigen nonspecific manner and thereby activating preexisting arthritogenic T cells. Similar combinations of genetic and environmental factors may cause other autoimmune diseases as well.
Materials and Methods
Mice.
SKG or BALB/c mice (Clea Japan) were maintained in our animal facility under non-SPF conditions or strictly controlled SPF facility. TLR2, TLR4, or MyD88-deficient mice were backcrossed to C57BL6 more than eight times and maintained in SPF conditions (25, 27). All experiments were conducted according to the institutional guidelines for animal welfare.
Clinical assessment of SKG arthritis.
Joint swelling was monitored by inspection and scored as follows: 0, no joint swelling; 0.1, swelling of one finger joint; 0.5, mild swelling of wrist or ankle; and 1.0, severe swelling of wrist or ankle. Scores for all fingers and toes, wrists, and ankles were totaled for each mouse. Female mice were used unless otherwise stated.
Reagents.
Cyclophosphamide, ConA, pristane, poly(I:C), PTX, LPS, zymosan A, laminarin, and rat IgG were purchased from Sigma-Aldrich; curdlan was obtained from Wako; anti-CD4 (H129.19), anti-CD8a (53–6.7), anti-CD11b (M1/70), anti-CD40 (3/23), anti-CD11c (HL3), anti-CD80 (16-10A1), anti-CD86 (GL-1), and anti-IAd (AMS-32.1) were obtained from BD Biosciences; isotype-control rat IgG2b (KLH/Gb-1-2) was obtained from eBioscience; and FITC-F(ab′)2 mouse anti–rat IgG was obtained from Jackson ImmunoResearch Laboratories. Oligodeoxynucleotide (ODN) 1668 CpG, 5′-TCCATGACGTTCCTGATGCT-3′, and ODN-AP1 non-CpG, 5-GCTTGATGACTCAGCCGGAA-3′ were synthesized by Hokkaido System Science (13). Anti–Dectin-1 (2A11) (31) or agonistic anti-CD40 (FGK 45) (49) have been reported previously. Curdlan or laminarin were suspended or dissolved in PBS at 30 or 100 mg/ml, respectively, before i.p. injection. Curdlan for culture was dissolved in 0.15 N NaOH at 10 mg/ml as described previously (34).
PCR.
Tissues were lysed in 50 mM Tris-HCl, pH 7.5, 20 mM EDTA, 1% SDS, 100 mM NaCl, and 400 μg/ml proteinase K (Nacalai Tesque) and DNA was extracted by phenol-chloroform. For fungal 18S rDNA and ITS1, the reaction mixture (25 μl) contained 0.2 mM dNTP, 0.8 μM forward and reverse primer, and 1.0 U Ex-Taq DNA polymerase (Takara) in supplier's buffer. PCRs were performed on a PTC-200 Programmable Thermal Control (MJ Research Inc.). PCRs consisted of a 5-min at 94°C denaturation step followed by 40 cycles of 30 s at 94°C, 30 s at 60°C, and 30 s at 72°C. For the murine Rag1 gene, PCRs were performed as described before except that denaturing temperature was 95°C for 9 min, annealing temperature was 56°C, and the number of cycles was 35 with Taq Gold (Applied Biosystems). The primer sequences were as follows: fungal 18S rDNA (19), B2F, 5′-ACTTTCGATGGTAGGATAG-3′ and B4R, 5′-TGATCGTCTTCGATCCCCTA-3′; fungal ITS1 (20), 18SF1, 5′-AGGTTTCCGTAGGTGAACCT-3′ and 58SR1, 5′-TTCGCTGCGTTCTTCATCGA-3′; and murine Rag1, Rag1F 5′-CTTCGGAATGCCGAGAAAGT-3′ and Rag1R, 5′-TGTGAAGGGACCATTCAGGT-3′. 18S rDNA or Rag1 were quantified by real-time PCR using the ABI/PRISM 7700 sequence detection system (Applied Biosystems). For 18SrDNA PCRs contained 0.3 μM primers, SYBR green PCR Kit (QIAGEN) and MgCl2 to 3.5 mM, and consisted of a 15-min at 95°C denaturation step followed by 47 cycles of 15 s at 94°C, a 30-s annealing step at 53°C, a 30-s extension step at 72°C, and a 15-s detection step at 76°C. For Rag1 detection, the annealing step was 54°C and the detection was merged with extension at 72°C for 30 s with default MgCl2 concentration.
PCR products of 18S rDNA or ITS1 were electrophoresed in agarose 1.5% gel, visualized by staining with ethidium bromide, cut out, and purified with a gel extraction kit (QIAGEN). Initial PCR products were directly sequenced with ABI PRISM 3100 Genetic Analyzer (Applied Biosystems) according to manufacturer's manual. When the result shows a mixture of different sequences, the PCR product was subcloned with pGEMeasy (Promega) and sequenced. Obtained sequence was analyzed with BLAST in the National Center for Biotechnology Information.
Measurement of serum β-glucan.
Mixture of serum and double volume of 0.32 M perchloric acid was incubated at 37°C for 20 min and centrifuged at 1,000 g for 15 min; supernatant was mixed with the same volume of 0.18 N NaOH and further diluted with 0.01 N NaOH (50). The concentration of β-glucan was measured by BGSTAR KIT (Wako), a limulus test specific for β-glucan, according to manufacturer's instruction (37).
Histology.
Lungs or ankle joints were fixed in buffered 10% formalin, and paraffin-embedded sections were stained with methenamine silver or hematoxylin and eosin.
DC cultures.
BALB/c BM-DCs were induced by culturing BM cells with 10 ng/ml GM-CSF (PeproTech) and 120 μ/ml polymyxin B as described previously (51). LPS, Pam3CSK4 (Calbiochem), curdlan, zymosan, or laminarin were added at the indicated concentration on day 5 and the expression of surface markers or the concentration of cytokines in the supernatant was analyzed on day 6 using a FACSCalibur (BD Biosciences) or ELISA (eBioscience), respectively.
Preparation of synovial cells.
Synovial tissues from ankle joints were digested with 400 Mandl U/ml of Liberase Brendzyme II (Roche) in RPMI 1640 medium for 1 h at 37°C; digested cells were filtrated through a nylon mesh to prepare single cell suspensions.
Statistics.
Mann-Whitney U test was used for statistical analyses.
Online supplemental material.
Table S1 shows the incidence of fungal infection in non-SPF SKG mice. PCR products of fungal 18S rDNA or ITS1 in their lung DNA were sequenced to determine the infecting fungal species. Fig. S1 shows that 2A11 anti–Dectin-1 mAb blocked in a dose-dependent fashion the enhancement of CD86 expression in BM-DCs by curdlan. Fig. S2 shows the production of IL-12, assessed by ELISA, from BM-DCs stimulated with laminarin, curdlan, zymosan, or LPS. Fig. S3 shows the influence of β-glucans on the proliferation of BALB/c CD4+ T cells stimulated by soluble anti-CD28 and plate-bound anti-CD3. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20041758/DC1.
Acknowledgments
We thank Z. Fehervari for discussion, H. Hata for providing aged SKG mice, and T. Matsushita and K. Kogishi for preparing histology.
This work was supported by Grants-in-Aid from the Ministry of Education, Sports and Culture, the Ministry of Human Welfare of Japan, and the Japan Science and Technology Agency.
The authors have no conflicting financial interests.
Abbreviations used: CP, cyclophosphamide; ITS1, internal transcriber spacer 1; poly(I:C), polyinosinic-polycytidylic acid; PTX, pertussis toxin; RA, rheumatoid arthritis; SPF, specific pathogen-free; TLR, Toll-like receptor.
H. Yoshitomi and N. Sakaguchi contributed equally to this work.
T. Tagami's present address is Ajinomoto Co., Inc., Kawasaki 210-8681, Japan.
T. Sakihama's present address is Laboratory for Systems Biology and Medicine, Research Center for Advanced Science and Technology, University of Tokyo, Tokyo 153-8904, Japan.
References
- 1.Firestein, G.S. 2003. Evolving concepts of rheumatoid arthritis. Nature. 423:356–361. [DOI] [PubMed] [Google Scholar]
- 2.Andre, I., A. Gonzalez, B. Wang, J. Katz, C. Benoist, and D. Mathis. 1996. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc. Natl. Acad. Sci. USA. 93:2260–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klareskog, L., J. Lorentzen, L. Padyukov, and L. Alfredsson. 2002. Genes and environment in arthritis: can RA be prevented? Arthritis Res. 4:S31–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silman, A.J., and J.E. Pearson. 2002. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 4:S265–S272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckner, J.H., and G.T. Nepom. 2002. Genetics of rheumatoid arthritis: is there a scientific explanation for the human leukocyte antigen association? Curr. Opin. Rheumatol. 14:254–259. [DOI] [PubMed] [Google Scholar]
- 6.Krause, A., T. Kamradt, and G.R. Burmester. 1996. Potential infectious agents in the induction of arthritides. Curr. Opin. Rheumatol. 8:203–209. [DOI] [PubMed] [Google Scholar]
- 7.Murai, C., Y. Munakata, Y. Takahashi, T. Ishii, S. Shibata, T. Muryoi, T. Funato, M. Nakamura, K. Sugamura, and T. Sasaki. 1999. Rheumatoid arthritis after human parvovirus B19 infection. Ann. Rheum. Dis. 58:130–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakaguchi, N., T. Takahashi, H. Hata, T. Nomura, T. Tagami, S. Yamazaki, T. Sakihama, T. Matsutani, I. Negishi, S. Nakatsuru, and S. Sakaguchi. 2003. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 426:454–460. [DOI] [PubMed] [Google Scholar]
- 9.Hata, H., N. Sakaguchi, H. Yoshitomi, Y. Iwakura, K. Sekikawa, Y. Azuma, C. Kanai, E. Moriizumi, T. Nomura, T. Nakamura, and S. Sakaguchi. 2004. Distinct contribution of IL-6, TNF-α, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J. Clin. Invest. 114:582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas, V.A., B.A. Woda, E.S. Handler, D.L. Greiner, J.P. Mordes, and A.A. Rossini. 1991. Altered expression of diabetes in BB/Wor rats by exposure to viral pathogens. Diabetes. 40:255–258. [DOI] [PubMed] [Google Scholar]
- 11.Moriyama, H., L. Wen, N. Abiru, E. Liu, L. Yu, D. Miao, R. Gianani, F.S. Wong, and G.S. Eisenbarth. 2002. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc. Natl. Acad. Sci. USA. 99:5539–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasunami, R., and J.F. Bach. 1988. Anti-suppressor effect of cyclophosphamide on the development of spontaneous diabetes in NOD mice. Eur. J. Immunol. 18:481–484. [DOI] [PubMed] [Google Scholar]
- 13.Sparwasser, T., L. Hultner, E.S. Koch, A. Luz, G.B. Lipford, and H. Wagner. 1999. Immunostimulatory CpG-oligodeoxynucleotides cause extramedullary murine hemopoiesis. J. Immunol. 162:2368–2374. [PubMed] [Google Scholar]
- 14.Waldner, H., M. Collins, and V.K. Kuchroo. 2004. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J. Clin. Invest. 113:990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garza, K.M., S.M. Chan, R. Suri, L.T. Nguyen, B. Odermatt, S.P. Schoenberger, and P.S. Ohashi. 2000. Role of antigen-presenting cells in mediating tolerance and autoimmunity. J. Exp. Med. 191:2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tiegs, G., J. Hentschel, and A. Wendel. 1992. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 90:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potter, M., and J.S. Wax. 1981. Genetics of susceptibility to pristane-induced plasmacytomas in BALB/cAn: reduced susceptibility in BALB/cJ with a brief description of pristane-induced arthritis. J. Immunol. 127:1591–1595. [PubMed] [Google Scholar]
- 18.Munoz, J.J., M.G. Peacock, and W.J. Hadlow. 1987. Anaphylaxis or so-called encephalopathy in mice sensitized to an antigen with the aid of pertussigen (pertussis toxin). Infect. Immun. 55:1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makimura, K., S.Y. Murayama, and H. Yamaguchi. 1994. Detection of a wide range of medically important fungi by the polymerase chain reaction. J. Med. Microbiol. 40:358–364. [DOI] [PubMed] [Google Scholar]
- 20.Makimura, K., T. Mochizuki, A. Hasegawa, K. Uchida, H. Saito, and H. Yamaguchi. 1998. Phylogenetic classification of Trichophyton mentagrophytes complex strains based on DNA sequences of nuclear ribosomal internal transcribed spacer 1 regions. J. Clin. Microbiol. 36:2629–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneshiro, E.S., M.S. Collins, and M.T. Cushion. 2000. Inhibitors of sterol biosynthesis and amphotericin B reduce the viability of pneumocystis carinii f. sp. carinii. Antimicrob. Agents Chemother. 44:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown, G.D., and S. Gordon. 2003. Fungal beta-glucans and mammalian immunity. Immunity. 19:311–315. [DOI] [PubMed] [Google Scholar]
- 23.Edwards, A.D., S.P. Manickasingham, R. Sporri, S.S. Diebold, O. Schulz, A. Sher, T. Kaisho, S. Akira, and C. Reis e Sousa. 2002. Microbial recognition via Toll-like receptor-dependent and -independent pathways determines the cytokine response of murine dendritic cell subsets to CD40 triggering. J. Immunol. 169:3652–3660. [DOI] [PubMed] [Google Scholar]
- 24.Ozinsky, A., D.M. Underhill, J.D. Fontenot, A.M. Hajjar, K.D. Smith, C.B. Wilson, L. Schroeder, and A. Aderem. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. USA. 97:13766–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 11:115–122. [DOI] [PubMed] [Google Scholar]
- 26.Aliprantis, A.O., R.B. Yang, M.R. Mark, S. Suggett, B. Devaux, J.D. Radolf, G.R. Klimpel, P. Godowski, and A. Zychlinsky. 1999. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 285:736–739. [DOI] [PubMed] [Google Scholar]
- 27.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 11:443–451. [DOI] [PubMed] [Google Scholar]
- 28.Czop, J.K., and K.F. Austen. 1985. A beta-glucan inhibitable receptor on human monocytes: its identity with the phagocytic receptor for particulate activators of the alternative complement pathway. J. Immunol. 134:2588–2593. [PubMed] [Google Scholar]
- 29.Giaimis, J., Y. Lombard, P. Fonteneau, C.D. Muller, R. Levy, M. Makaya-Kumba, J. Lazdins, and P. Poindron. 1993. Both mannose and beta-glucan receptors are involved in phagocytosis of unopsonized, heat-killed Saccharomyces cerevisiae by murine macrophages. J. Leukoc. Biol. 54:564–571. [DOI] [PubMed] [Google Scholar]
- 30.Thornton, B.P., V. Vetvicka, M. Pitman, R.C. Goldman, and G.D. Ross. 1996. Analysis of the sugar specificity and molecular location of the beta-glucan-binding lectin site of complement receptor type 3 (CD11b/CD18). J. Immunol. 156:1235–1246. [PubMed] [Google Scholar]
- 31.Brown, G.D., P.R. Taylor, D.M. Reid, J.A. Willment, D.L. Williams, L. Martinez-Pomares, S.Y. Wong, and S. Gordon. 2002. Dectin-1 is a major β-glucan receptor on macrophages. J. Exp. Med. 196:407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gantner, B.N., R.M. Simmons, S.J. Canavera, S. Akira, and D.M. Underhill. 2003. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 197:1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown, G.D., J. Herre, D.L. Williams, J.A. Willment, A.S. Marshall, and S. Gordon. 2003. Dectin-1 mediates the biological effects of β-glucans. J. Exp. Med. 197:1119–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kataoka, K., T. Muta, S. Yamazaki, and K. Takeshige. 2002. Activation of macrophages by linear (1right-arrow3)-beta-D-glucans. Impliations for the recognition of fungi by innate immunity. J. Biol. Chem. 277:36825–36831. [DOI] [PubMed] [Google Scholar]
- 35.Taylor, P.R., G.D. Brown, D.M. Reid, J.A. Willment, L. Martinez-Pomares, S. Gordon, and S.Y. Wong. 2002. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J. Immunol. 169:3876–3882. [DOI] [PubMed] [Google Scholar]
- 36.Underhill, D.M., A. Ozinsky, A.M. Hajjar, A. Stevens, C.B. Wilson, M. Bassetti, and A. Aderem. 1999. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 401:811–815. [DOI] [PubMed] [Google Scholar]
- 37.Obayashi, T., M. Yoshida, T. Mori, H. Goto, A. Yasuoka, H. Iwasaki, H. Teshima, S. Kohno, A. Horiuchi, A. Ito, et al. 1995. Plasma (1→3)-beta-D-glucan measurement in diagnosis of invasive deep mycosis and fungal febrile episodes. Lancet. 345:17–20. [DOI] [PubMed] [Google Scholar]
- 38.Yasuoka, A., N. Tachikawa, K. Shimada, S. Kimura, and S. Oka. 1996. (1→3) beta-D-glucan as a quantitative serological marker for Pneumocystis carinii pneumonia. Clin. Diagn. Lab. Immunol. 3:197–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keystone, E.C., H.U. Schorlemmer, C. Pope, and A.C. Allison. 1977. Zymosan-induced arthritis: a model of chronic proliferative arthritis following activation of the alternative pathway of complement. Arthritis Rheum. 20:1396–1401. [DOI] [PubMed] [Google Scholar]
- 40.Keffer, J., L. Probert, H. Cazlaris, S. Georgopoulos, E. Kaslaris, D. Kioussis, and G. Kollias. 1991. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10:4025–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kyburz, D., J. Rethage, R. Seibl, R. Lauener, R.E. Gay, D.A. Carson, and S. Gay. 2003. Bacterial peptidoglycans but not CpG oligodeoxynucleotides activate synovial fibroblasts by toll-like receptor signaling. Arthritis Rheum. 48:642–650. [DOI] [PubMed] [Google Scholar]
- 42.Millar, D.G., K.M. Garza, B. Odermatt, A.R. Elford, N. Ono, Z. Li, and P.S. Ohashi. 2003. Hsp70 promotes antigen-presenting cell function and converts T-cell tolerance to autoimmunity in vivo. Nat. Med. 9:1469–1476. [DOI] [PubMed] [Google Scholar]
- 43.Eriksson, U., R. Ricci, L. Hunziker, M.O. Kurrer, G.Y. Oudit, T.H. Watts, I. Sonderegger, K. Bachmaier, M. Kopf, and J.M. Penninger. 2003. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 9:1484–1490. [DOI] [PubMed] [Google Scholar]
- 44.Leung, B.P., M. Conacher, D. Hunter, I.B. McInnes, F.Y. Liew, and J.M. Brewer. 2002. A novel dendritic cell-induced model of erosive inflammatory arthritis: distinct roles for dendritic cells in T cell activation and induction of local inflammation. J. Immunol. 169:7071–7077. [DOI] [PubMed] [Google Scholar]
- 45.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 46.Wakatsuki, A., P. Borrow, K. Rigley, and P.C. Beverley. 2003. Cell-surface bound pertussis toxin induces polyclonal T cell responses with high levels of interferon-gamma in the absence of interleukin-12. Eur. J. Immunol. 33:1859–1868. [DOI] [PubMed] [Google Scholar]
- 47.Sakaguchi, S. 2004. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22:531–562. [DOI] [PubMed] [Google Scholar]
- 48.Ghiringhelli, F., N. Larmonier, E. Schmitt, A. Parcellier, D. Cathelin, C. Garrido, B. Chauffert, E. Solary, B. Bonnotte, and F. Martin. 2004. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 34:336–344. [DOI] [PubMed] [Google Scholar]
- 49.Rolink, A., F. Melchers, and J. Andersson. 1996. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 5:319–330. [DOI] [PubMed] [Google Scholar]
- 50.Obayashi, T. 1984. Addition of perchloric acid to blood samples for colorimetric limulus test using chromogenic substrate: comparison with conventional procedures and clinical applications. J. Lab. Clin. Med. 104:321–330. [PubMed] [Google Scholar]
- 51.Fehervari, Z., and S. Sakaguchi. 2004. Control of Foxp3+ CD25+ CD4+ regulatory cell activation and function by dendritic cells. Int. Immunol. 16:1769–1780. [DOI] [PubMed] [Google Scholar]