

Abstract
Humoral immune responses are thought to be enhanced by complement-mediated recruitment of the CD21–CD19–CD81 coreceptor complex into the B cell antigen receptor (BCR) complex, which lowers the threshold of B cell activation and increases the survival and proliferative capacity of responding B cells. To investigate the role of the CD21–CD35 complement receptors in the generation of B cell memory, we analyzed the response against viral particles derived from the bacteriophage Qβ in mice deficient in CD21–CD35 (Cr2−/−). Despite highly efficient induction of early antibody responses and germinal center (GC) reactions to immunization with Qβ, Cr2−/− mice exhibited impaired antibody persistence paralleled by a strongly reduced development of bone marrow plasma cells. Surprisingly, antigen-specific memory B cells were essentially normal in these mice. In the absence of CD21-mediated costimulation, Qβ-specific post-GC B cells failed to induce the transcriptional regulators Blimp-1 and XBP-1 driving plasma cell differentiation, and the antiapoptotic protein Bcl-2, which resulted in failure to generate the precursor population of long-lived plasma cells residing in the bone marrow. These results suggest that complement receptors maintain antibody responses by delivery of differentiation and survival signals to precursors of bone marrow plasma cells.
Protective immunological memory against reinfection with most viruses largely depends on the induction of long-lasting antibody responses. This concept provides the basis of all successful vaccines used to date (1). B cell memory is characterized by increased frequencies of long-lived memory B cells and elevated levels of specific antibodies (2). Both memory B cells and BM antibody-secreting cells (ASCs), which sustain long-term antibody production (3, 4), are thought to originate in germinal centers (GCs; 5). However, the mechanisms underlying recruitment of GC B cells into the memory B cell or BM plasma cell compartment remain ill defined. Selective accumulation of high affinity ASCs in the BM has suggested that high antigen affinity of the B cell Ag receptor (BCR) favors differentiation of GC B cells into plasma cells (6, 7). Although a minimal threshold of signal strength is required for differentiation into a long-lived plasma cell, selection into the memory B cell population appears to be less stringent (6, 7). Additional signals have been reported to drive these two pathways; for instance CD40L, IL-4, or ligation of CD27 direct differentiation of GC B cells toward a memory phenotype (8–10) whereas commitment to a plasma cell fate is promoted by IL-10 and requires IL-6 (9, 11, 12). Signals determining plasma cell fate decision are dependent on the induction of the transcription factors Blimp-1 and XBP-1 for formation of Ig-producing cells (13, 14). Together these regulators drive terminal differentiation of B cells into ASCs, by promoting a plasma cell phenotype and extinguishing gene expression programs involved in proliferation and GC function (15).
Survival of B cells in GCs during the antigen-driven selection process leading to high-affinity memory B cells and plasma cells is dependent on signaling through the CD21–CD19 complex (16). The interaction of CD21 with complement-coated antigen appears to provide a selective advantage to GC B cells. Two additional mechanisms have been proposed by which CD21–CD35 enhances humoral immunity (17–19). First, recruitment of the CD21–CD19–CD81 complex into the BCR complex lowers the threshold of B cell activation. Second, complement receptors CD21–CD35 enhance trapping of antigen on follicular dendritic cells (FDCs) thereby driving the GC reaction and maintaining B cell memory. Insight into the role of complement receptors in humoral responses has been gained through the study of mice with a genetically disrupted Cr2 locus, deficient for the expression of CD21 (complement receptor 2) and CD35 (complement receptor 1). These mice have been reported to have impaired antibody responses and defective GC formation in response to T cell–dependent and –independent antigens (20–22). However, antibody responses were affected to a varying degree dependent on the nature and amount of antigen used in these studies. The role of CD21–CD35 in the generation of immunological memory also remains controversial. Although Cr2−/− mice infected with vesicular stomatitis virus maintained memory antibody titers comparably to controls (23), accelerated loss of serum antibody was reported in responses to the hapten (4-hydroxy-3-nitrophenyl)acetyl (NP; 24). Furthermore, expression of CD21–CD35 was essential for generation of memory B cells to carrier-coupled NP in the absence but not in the presence of adjuvants (25).
To dissect the role of complement receptors in the induction of immunological B cell memory to a highly repetitive antigen capable of efficient cross-linking of surface Ig on B cells, virus-like particles from the RNA phage Qβ were used as a model antigen. Qβ capsids form icosahedral particles of ∼30 nm diam (26) with a highly ordered repetitive structure, which makes them potent B cell immunogens in the absence of adjuvant (27, 28). Therefore, Qβ particles exhibit the geometry and size of a prototype virus without displaying potentially complicating factors such as viral replication. Immunization with Qβ induces an early, T cell–independent IgM response, followed by a persistent and slowly declining T cell–dependent IgG response (29). Qβ particles efficiently induce GC formation, with antigen-specific GC B cells peaking around day 12 and being still detectable at late stages after immunization (29). Immunization of Cr2−/− mice with Qβ showed that short-term primary responses, induction of GCs and memory B cell formation were independent of complement receptors. In contrast, maintenance of long-lasting antibody titers by BM plasma cells required CD21–CD35. CD21 promoted differentiation of a plasma cell precursor population expressing the plasma cell–specific transcription factors Blimp-1 and XBP-1 as well as the antiapoptotic protein Bcl-2. These results suggest that engagement of complement receptors on B cells by complement-coated antigen is critical for generation of long-lived plasma cells in the BM responsible for maintenance of memory antibody titers.
Results
Maintenance of antibody titers is impaired in Cr2−/− mice
Immunization with a single dose of virus-like particles derived from the bacteriophage Qβ elicits strong, long-lasting IgG responses (29). To assess the role of complement receptors in the induction and maintenance of antibody responses to this antigen, Cr2−/− and WT mice were immunized i.v. with 10 μg Qβ and antibody titers were measured at several time points after immunization. Anti-Qβ IgG antibody levels were similar in Cr2−/− mice and WT mice early after immunization (Fig. 1 A). However, whereas in WT mice serum anti-Qβ IgG antibodies increased and reached a peak around day 21, antibody production was not sustained in Cr2−/− mice. After the third week after immunization, antibody titers in Cr2−/− mice were substantially reduced compared with WT littermates and exhibited nearly a 12-fold reduction 9 wk after immunization. Thus, Cr2−/− mice generate normal short-term anti-Qβ responses but fail to maintain antibody titers in the later phase of the response.
Figure 1.
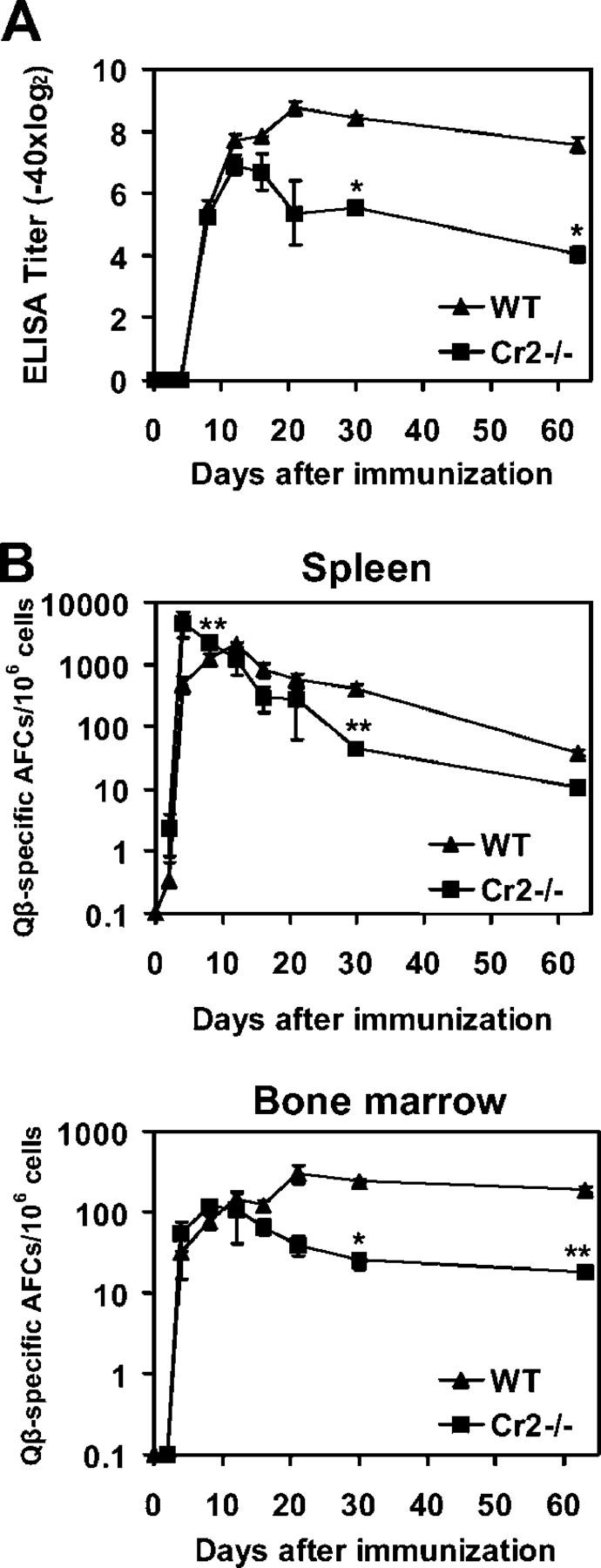
Maintenance of anti-Qβ antibody titers and generation of BM ASCs are impaired in Cr2−/− mice. (A) C57BL/6 and Cr2−/− mice were immunized i.v. with 10 μg Qβ and Qβ-specific serum IgG titers were determined by ELISA. (B) Frequencies of Qβ-specific IgG ASCs in spleen and BM of Cr2−/− and WT mice were determined by ELISPOT assay. Values are given as the mean ± SEM, with significant differences between means indicated by asterisks (*, P < 0.01; **, P < 0.05). The analysis was repeated twice on days 8, 21, and 100 with a similar result.
BM plasma cells but not memory B cells are reduced in Cr2−/− mice at late stages after immunization
Elevated levels of IgG antibodies are thought to be maintained by long-lived plasma cells residing in the BM (30) as well as by memory B cells continually differentiating into ASCs through activation by persisting antigen (31). We therefore followed the kinetics of ASCs and memory B cells in Cr2−/− and WT mice (Fig. 1 B). As expected from serum antibody levels, up to 12 d after immunization numbers of Qβ-specific ASCs were similar or slightly increased in spleen and BM of Cr2−/− mice compared with WT controls. At later stages after immunization, the number of cells secreting anti-Qβ IgG antibodies, especially those in the BM, were reduced in Cr2−/− mice and the frequency of Qβ-specific cells in the BM plasma cell pool was more than 10-fold lower in Cr2−/− mice than in WT controls 9 wk after immunization (Fig. 1 B). Therefore, generation of presumably short-lived plasma cells in the spleen was normal in Cr2−/− mice but the formation of long-lived BM plasma cells was strongly reduced.
To quantify Qβ-specific memory B cells we used an antigen-specific B cell detection system relying on detection of bound Qβ to specific isotype-switched B cells by flow cytometry (Fig. 2 A; 29). Activated and isotype-switched B lymphocytes, defined as (IgM; IgD; CD4; CD8; CD11b; Gr-1; YO-PRO-1)−B220+, were gated and analyzed for Qβ-binding on several days after immunization. Qβ-specific B cells were increased in Cr2−/− mice early in the immune response, but antigen-specific memory B cells reached similar frequencies in Cr2−/− and WT mice 9 wk after immunization (Fig. 2 B). Therefore, generation and persistence of Qβ-specific memory B cells was normal in Cr2−/− mice. Frequencies of Qβ-specific B cells in LNs reflected those found in the spleen, while no Qβ-binding memory B cells could be detected in the BM (unpublished data). To confirm that the Qβ-binding (IgM; IgD; CD4; CD8; CD11b; Gr-1; YO-PRO-1)−B220+ cells identified at late stages after immunization were memory B cells, we adoptively transferred sorted cells into irradiated recipients and assessed whether they mounted an anamnestic response. 6 d after adoptive transfer and immunization, relative antibody titers (titer per transferred cells) were significantly higher in recipient mice that had received Qβ-binding IgMlowIgDlow B cells than in mice transferred with naive IgM+IgD+ B cells or total isotype-switched B cells (Fig. 2 C). Thus, cells identified as (IgM; IgD; CD4; CD8; CD11b; Gr-1; YO-PRO-1)−B220+ binding Qβ are bona fide memory cells.
Figure 2.

Induction of memory B cells is normal in Cr2−/− mice. (A) Representative staining of splenocytes from naive and immunized mice to identify Qβ-specific (IgM; IgD; CD4; CD8; CD11b; Gr-1; YO-PRO-1)−B220+ cells. PNA binding on Qβ-specific isotype-switched B cells is shown. (B) Frequency of Qβ-specific isotype-switched B cells in spleens of Cr2−/− and WT mice. The analysis was repeated twice on days 8, 21, and 100 with a similar result. (C) Anti-Qβ antibody levels induced in irradiated WT mice 6 d after immunization and adoptive transfer of Qβ-binding (IgM; IgD; CD4; CD8; CD11b; Gr-1; YO-PRO-1)−B220+ B cells, total (IgM; IgD; CD4; CD8; CD11b; Gr-1; YO-PRO-1)−B220+ B cells, or B220+IgM+IgD+ B cells. Results are expressed as serum IgG ELISA titers per transferred cells. (D) Qβ-specific serum IgG levels in Cr2−/− and WT mice 6 mo after primary immunization and 6 d after secondary challenge with Qβ. (E) B cell responses induced in Cr2−/− and WT mice by injection of 25 μg AP205. Frequencies of AP205-specific IgG ASCs in spleen and BM and of AP205-specific memory B cells in spleen were determined 9 wk after immunization. AP205-specific serum IgG was measured 13 wk after primary immunization and 6 d after antigen recall. All data represent the mean ± SEM, mean values statistically different from WT levels are indicated by asterisks (*, P < 0.01; **, P < 0.05).
Because normal frequencies of Qβ-specific memory B cells were observed in Cr2−/− mice, we tested their ability to mount an efficient recall response late after immunization. Cr2−/− and WT mice were primed and challenged 6 mo later with Qβ; the increase in antibody titer was analyzed 6 d after secondary immunization. Anti-Qβ titers increased substantially in both strains, more than 70-fold in Cr2−/− mice and nearly 50-fold in WT mice (Fig. 2 D), indicating that recall responses were normal in Cr2−/− mice.
Similar results were obtained for a second virus-like particle, derived from the bacteriophage AP205. Cr2−/− mice immunized with AP205 exhibited reduced maintenance of antibody titers and generation of BM plasma cells compared with controls 9 and 13 wk after immunization (Fig. 2 E), despite normal induction of early antibody responses (unpublished data). In contrast, as observed for Qβ, frequencies of AP205-specific memory B cells and the capacity to mount efficient recall responses to AP205 particles was comparable in Cr2−/− and WT mice (Fig. 2 E).
Thus, in the absence of complement receptors normal antigen-specific memory B cells were induced by immunization with virus-like particles, but complement receptors were required for generation and/or maintenance of long-lived BM plasma cells.
GCs are efficiently induced in Cr2−/− mice by immunization with Qβ
As long-lived plasma cells, along with memory B cells are generated in the GC reaction (5) we assessed the induction of Qβ-specific GC B cells in Cr2−/− and WT mice. For this purpose, isotype-switched Qβ-specific B cells were analyzed for binding to the GC marker peanut agglutinin (PNA; Fig. 2 A) and the frequency of PNAhigh Qβ-specific B cells in spleens of Cr2−/− and WT mice was determined at several time points after immunization (Fig. 3 A). In both groups of mice, high frequencies of PNAhigh Qβ-specific B cells were observed (Fig. 3 A) and similar peak frequencies of specific GC B cells were reached. Surprisingly, Qβ-specific GC B cells were induced earlier in Cr2−/− mice than in WT mice, but reduced numbers of GC cells were observed at later stages after immunization. The frequency of Qβ-specific PNAlow B cells was comparable in the spleen of Cr2−/− and WT mice at all time points analyzed (unpublished data).
Figure 3.
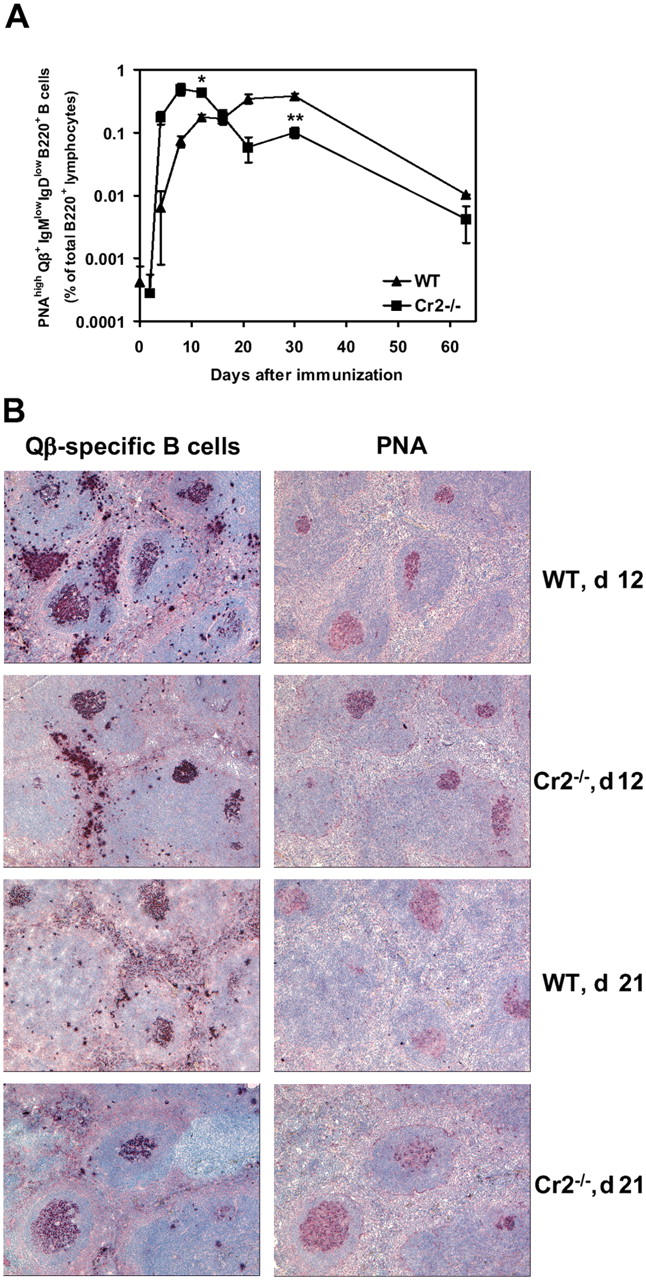
Immunization with Qβ induces efficient GC formation in Cr2−/− mice. (A) Frequency of Qβ-specific PNAhigh B cells in spleens of Cr2−/− and WT mice. Isotype-switched Qβ-binding PNAhigh B cells were identified as shown in Fig. 2 A. Data are expressed as the mean ± SEM, mean values statistically different from WT levels are indicated by asterisks (*, P < 0.01; **, P < 0.05). (B) Immunohistochemical detection of Qβ-specific B cells and PNA-binding cells in serial sections of the spleen of Cr2−/− and WT mice 12 and 21 d after immunization. Original magnification: ×62.5 (day 12); ×75 (day 21).
These results were confirmed by immunohistochemistry (Fig. 3 B). Staining of serial spleen sections for Qβ-specific B cells (29) and PNA-binding revealed no significant difference in the number and size of Qβ-specific GCs generated in Cr2−/− and WT mice 12 and 21 d after immunization. Thus, Cr2−/− mice exhibited no obvious deficiency in size or architecture of GCs induced by immunization with Qβ particles.
Blimp-1, XBP-1, and Bcl-2 fail to be induced in isotype-switched PNAlow B cells specific for Qβ in Cr2−/− mice
The induction of normal numbers of GC B cells in Cr2−/− mice by immunization with Qβ suggested a role for complement receptors in the differentiation process after antigen-driven B cell expansion. Terminal differentiation of plasma cells has been shown to require the transcription factors Blimp-1 (13) and XBP-1 (14). We therefore analyzed the induction of these two regulators of plasmacytic differentiation in B220highIgMlowIgDlow Qβ-binding PNAhigh GC B cells and in B220highIgMlowIgDlow Qβ-specific B cells with a PNAlow phenotype. Antigen-specific PNAhigh and PNAlow B220+ cells were gated as shown in Fig. 2 A and purified by FACS from spleens of Cr2−/− and WT mice 12 d after injection of Qβ. Blimp-1 and XBP-1 mRNA levels were determined by quantitative RT-PCR. As apparent in Fig. 4 A, Blimp-1 and XBP-1 mRNA was up-regulated 12–13-fold in WT mice in the PNAlow but not in the PNAhigh Qβ-specific B cell population. In contrast, in Cr2−/− mice significant levels of Blimp-1 and XBP-1 failed to be induced in PNAlow Qβ-specific B cells. Expression of Blimp-1 and XBP-1 in antigen-specific GC B cells from WT mice was comparable to background levels found in purified T cells (unpublished data). The spliced form of XBP-1, which has been reported to appear late in plasma cell differentiation and to be associated with increased Ig synthesis (32), could not be detected in any of the samples (unpublished data).
Figure 4.

Qβ-specific isotype-switched PNAlow B cells from Cr2−/− mice exhibit reduced levels of Blimp-1, XBP-1, and Bcl-2 mRNA. Qβ-binding isotype-switched PNAhigh and PNAlow B cells, identified as shown in Fig. 2 A, were purified by FACS from three to four pooled spleens 12 d after immunization. Blimp-1, XBP-1, and Bcl-2 mRNA levels were determined by quantitative RT-PCR. (A) Expression of Blimp-1 and XBP-1 in purified cells from Cr2−/− and WT mice. (B) Blimp-1 and XBP-1 expression in Qβ-specific B cells from chimeric mice having Cr2−/− B cells and WT FDCs and from control chimeras. 5 × 107 splenocytes from Cr2−/− or C57BL/6 mice were adoptively transferred into sublethally irradiated C57BL/6-CD45.1 recipients and mRNA levels were determined in CD45.1− B cells from immunized recipient mice. (C) Bcl-2 mRNA levels in sorted cells from Cr2−/− and WT mice. Expression levels are depicted in relation to β-actin expression. Quantitative RT-PCR of each sample was performed in triplicate. Results are represented as the mean ± SD. One of two similar experiments is shown.
To dissect the role of CD21–CD35 on B cells versus FDCs in the induction of Blimp-1 and XBP-1, we analyzed the expression of these transcription factors in Qβ-specific B cells from chimeric mice having normal FDCs but Cr2−/− B cells. For this purpose we transferred splenocytes derived from Cr2−/− or WT mice into sublethally irradiated C57BL/6-CD45.1 recipients. On days 8 and 12 after immunization anti-Qβ antibodies were present at comparable levels in both groups of chimeras, but not in irradiated control mice that had not received any B cells (unpublished data). As observed for Cr2−/− mice, sorted Qβ-specific isotype-switched CD45.1−B220+PNAlow B cells from chimeric mice having Cr2−/− B cells and WT FDCs displayed strongly reduced Blimp-1 and XBP-1 mRNA levels when compared with cells from control chimeras (Fig. 4 B). Hence, in the absence of stimulation through complement receptors, Qβ-specific isotype-switched PNAlow B cells were unable to induce sufficient levels of the key transcription factors driving plasma cell differentiation. This indicates that the failure of Cr2−/− mice to generate long-lived plasma cells is a B cell–intrinsic defect and is not related to the absence of CD21–CD35 on FDCs.
The reduction of Blimp-1 and XBP-1 levels in PNAlow Qβ-specific B cells from Cr2−/− mice was concomitant to a reduced expression of the antiapoptotic protein Bcl-2 (Fig. 4 C). This observation is consistent with in vitro studies showing that recruitment of the B cell coreceptor during antigen-dependent B cell activation induced Bcl-2 expression (33). Thus, absence of survival mechanisms regulated by Bcl-2 may further explain the loss of BM plasma cells in Cr2−/− mice.
Qβ-specific isotype-switched B220highPNAlow cells are not secreting antibody
We next set out to characterize further the Qβ-specific isotype-switched B220highPNAlow B cell population that expressed the transcriptional regulators Blimp-1 and XBP-1 and the antiapoptotic protein Bcl-2 that was absent in Cr2−/− mice. As Blimp-1 and XBP-1 are expressed in plasma cells, we assessed whether Qβ-specific PNAlow B cells were secreting antibody and determined the phenotype of splenic plasma cells 12 d after immunization. For detection of Qβ-specific plasma cells, splenocytes were permeabilized and intracellular binding of fluorescently labeled Qβ particles to (CD4; CD8; CD11b)−B220high and B220low B cells was determined by flow cytometry. Surface staining was blocked by preincubation with unlabeled Qβ. As shown in Fig. 5 A, Qβ-specific plasma cells, expressing high levels of cytoplasmic antibodies, had exclusively a B220lowPNAlow phenotype. These bright intracellularly stained cells could only be detected after permeabilization, consistent with the fact that terminally differentiated plasma cells down-regulate surface Ig expression. Cells expressing Qβ-specific cytoplasmic antibodies were absent when B220high B cells were gated, therefore excluding that the Blimp-1– and XBP-1–expressing PNAlow B cells, which displayed a B220high phenotype, were terminally differentiated plasma cells. A population of B220lowPNAlow cells exhibiting cytoplasmic Igs specific for Qβ could also be detected in Cr2−/− mice. The frequency of these cells was comparable in the spleen of Cr2−/− and WT mice (Fig. 5 B). This is consistent with the normal ASC numbers detected on day 12 in Cr2−/− mice by ELISPOT assay, which were in the same range as those obtained by flow cytometry.
Figure 5.

B220highPNAlow B cells specific for Qβ are not secreting antibody. (A) Splenocytes from naive and immunized (day 12) WT mice were permeabilized and intracellular expression of Qβ-specific antibodies was detected with Alexa 647-labeled Qβ. (CD4; CD8; CD11b)−B220high and B220low cells were gated and analyzed for PNA binding. Mean percentages of Qβ-specific plasma cells are indicated. Surface staining was blocked by preincubation with unlabeled Qβ; the specificity of the staining was controlled with Alexa 647-conjugated AP205. (B) Frequency of Qβ-specific B220lowPNAlow plasma cells in spleen of Cr2−/− and WT mice on day 12 after immunization. Values are given as the mean ± SEM.
These results suggest that the Qβ-binding isotype-switched B220highPNAlow population that expresses the transcription factors Blimp-1 and XBP-1 and fails to develop in Cr2−/− mice, are antigen-experienced B cells that have left GCs and are committed to a plasma cell fate, but have not yet fully differentiated into ASCs.
Qβ-specific GC-derived BM plasma cell precursors are absent in Cr2−/− mice
A population of B cells representing an intermediary stage before terminal plasma cell differentiation has been described recently (34). These post-GC B cells, which are direct precursors to plasma cells, were identified in the BM and display a phenotype intermediate between splenic B cells and terminally differentiated plasma cells. Such plasma cell precursors were shown to retain expression of the BCR, B220, and MHCII, albeit lower levels than splenic B cells, and to express the plasma cell marker CD138 as well as receptors capable of interacting with BM stroma, such as VLA-4, LFA-1, and CD44 (34). To confirm the identity of Qβ-specific PNAlow Blimp-1–XBP-1–expressing B cells as precursors of plasma cells, the expression of these surface markers identifying post-GC plasma cell precursors was determined on isotype-switched PNAlowB220high B cells binding Qβ 12 d after immunization. As shown in Fig. 6 A, CD138 was induced on a proportion of Qβ-binding PNAlow B cells but not on specific GC B cells. The integrins VLA-4 and LFA-1 as well as CD44 were also up-regulated on Qβ-specific PNAlow B cells compared with the PNAhigh B cell population. Therefore, cell surface markers, which are known to be up-regulated in the differentiation process of post-GC B cells to BM plasma cells, were induced in isotype-switched Qβ-specific PNAlow B cells. The presence of cells with this plasma cell precursor phenotype was also determined in immunized Cr2−/− mice (Fig. 6 B). Consistent with the fact that Blimp-1–XBP-1–positive isotype-switched B cells binding Qβ were absent in these mice, a population of cells with up-regulated CD138, VLA-4, LFA-1, and CD44 expression failed to be induced. These results indicate that generation of post-GC precursors to plasma cells requires the interaction of complement-coated antigen with its receptors.
Figure 6.

Qβ-specific isotype-switched PNAlow B cells exhibiting a partial plasma cell phenotype are absent in Cr2−/− mice. (A) Expression of CD138, VLA-4, LFA-1, and CD44 on Qβ-specific PNAlow and PNAhigh B cells from WT mice. B220+ splenocytes were purified by magnetic cell sorting; IgMlowIgDlow B cells binding Qβ and low or high levels of PNA were gated and analyzed for expression of the indicated surface markers. One of three similar experiments is shown. (B) Comparison of CD138, VLA-4, LFA-1, and CD44 expression on Qβ-specific PNAlow and PNAhigh isotype-switched B220+ splenocytes from Cr2−/− and WT mice on day 12 after immunization. (C) Phenotype of Qβ-specific B cells in the blood of WT mice on day 12 after immunization. PNA-binding on Qβ-specific (IgM; IgD; CD4; CD8; CD11b; Gr-1; YO-PRO-1)−B220+ cells was determined. Expression of cytoplasmic Ig in Qβ-specific PNAlow B cells was assessed by analysis of binding of Alexa647-labeled Qβ to permeabilized (CD4; CD8; CD11b)−B220low cells.
In support of the hypothesis that the identified population represented plasma cell precursors destined to migrate to the BM, Qβ-specific B cells identified in the blood of immunized mice had a B220highPNAlow phenotype and did not express cytoplasmic Igs (Fig. 6 C). This suggests that Qβ-specific precursors of plasma cells homed to the BM before terminal differentiation, in accordance with the presence of a plasma cell precursor population in this organ (34). However, cells with surface Ig specific for Qβ could not be detected in the BM at any time point. This is an indication that upon arrival in the BM, precursors of plasma cells rapidly lose surface Ig expression and acquire cytoplasmic Ig expression as required for antibody secretion.
Preplasma memory B cells are formed normally in Cr2−/− mice
A population of memory B cells, which originate in GCs and can been distinguished from classical recirculating B220+ memory B cells on the basis of their B220− CD138− phenotype, has been described previously (35, 36). These cells have a greater propensity to form plasma cells than the B220+ memory B cell subset and rapidly differentiate into plasma cells after antigen recall (35). Nonsecreting B220−CD138− B cells have been referred to as preplasma memory B cells and their formation has been shown to require Blimp-1 expression (13). Because complement receptors played a role in the induction of Blimp-1, we analyzed the generation of preplasma memory B cells in Cr2−/− mice immunized with Qβ. As shown in Fig. 7 A, a population of Qβ-binding (IgD; CD4; CD8; YO-PRO-1)− cells with a B220−CD138− phenotype could be detected in the spleen of immunized Cr2−/− and WT mice. The frequency of these cells on days 12 and 21 was comparable in Cr2−/− and WT controls (Fig. 7 B). Thus, generation of preplasma memory B cells was not dependent on complement receptors, despite their formation reportedly requiring Blimp-1 expression. As already mentioned, cells with surface Ig receptors specific for Qβ could not be detected in the BM and therefore no Qβ-specific preplasma memory B cells could be identified in this organ.
Figure 7.
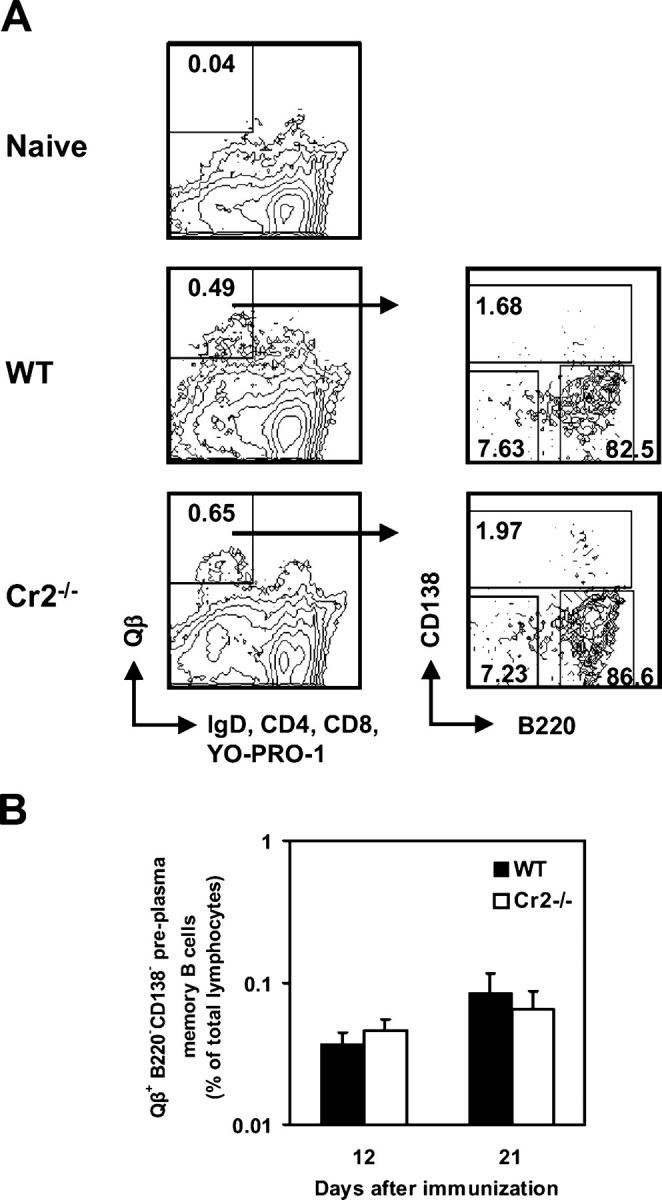
Preplasma memory B cells are induced normally in Cr2−/− mice. (A) Analysis of B220 and CD138 expression on Qβ-binding (IgD; CD4; CD8; YO-PRO-1)− splenocytes to identify B220−CD138− preplasma memory B cells. Mean percentages of B220-CD138−, B220+CD138−, and CD138+ cells in Cr2−/− and WT mice are indicated. (B) Frequency of Qβ-specific preplasma memory B cells in spleens of Cr2−/− and WT mice 12 and 21 d after immunization. Results are expressed as the mean ± SEM.
Generation of isotype-switched Qβ-specific PNAlow B cells is GC dependent
The fact that short-term anti-Qβ antibody responses were normal in Cr2−/− mice suggested that complement receptors were required mainly for differentiation of GC-derived plasma cells. Consequently, if isotype-switched PNAlow B cells, which required complement receptors for induction of Blimp-1 and XBP-1, are post-GC plasma cell precursors, this population should not be induced in the absence of GCs. To address this question we analyzed the generation of isotype-switched B220+ PNAhigh and PNAlow B cells in TNFR1−/− mice, which lack mature FDC networks and do not form GCs (37). As expected, on day 12 after immunization, Qβ-specific PNAhigh GC B cells were absent in spleens of TNFR1−/− mice (Fig. 8 A). Qβ-specific B cells with a PNAlow phenotype were also more than 10-fold reduced, indicating that these cells are generated in GCs (Fig. 8 A). Further, lower frequencies of splenic B220lowPNAlow cells expressing cytoplasmic Igs specific for Qβ were detected in TNFR1−/− mice (Fig. 8 B), suggesting that a proportion of these antibody-producing cells derived from GC B cells. Cells with this phenotype were present in normal numbers in Cr2−/− mice (Fig. 5 B). Hence, some of the plasma cells in the spleen of Cr2−/− mice did arise from the GC reaction and are likely to represent splenic short-lived plasma cells.
Figure 8.
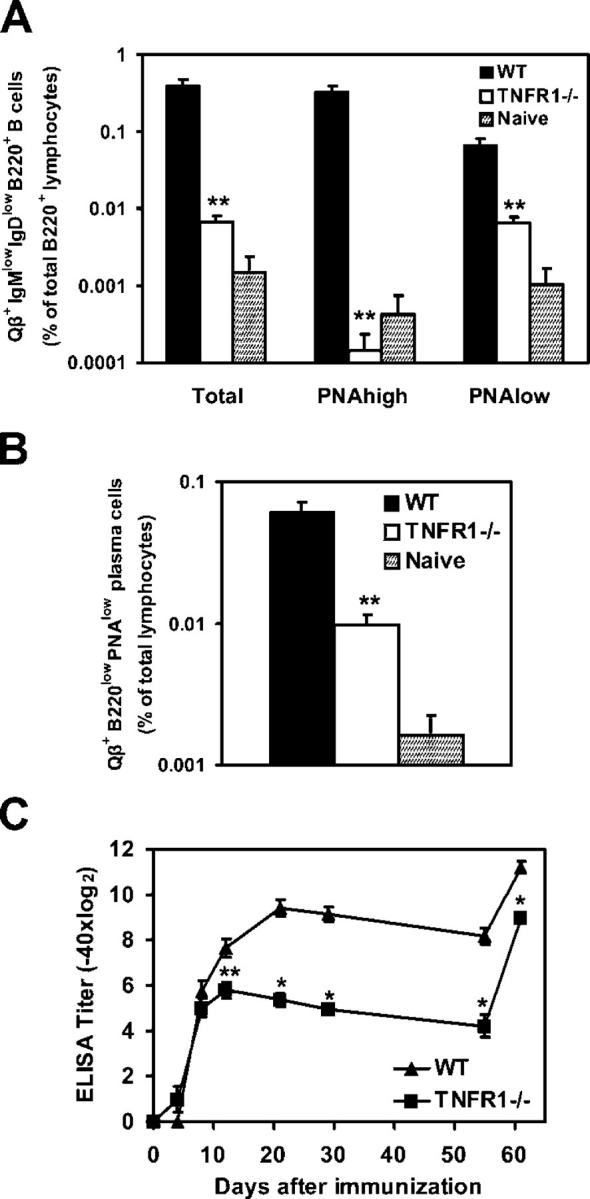
Isotype-switched Qβ-specific PNAlow B cells are reduced in the absence of GC formation. (A) Frequency of total, PNAhigh and PNAlow Qβ-binding isotype-switched B220+ cells in spleens of TNFR1−/− and WT mice 12 d after immunization. (B) Frequency of splenic B220low plasma cells with cytoplasmic Ig specific for Qβ in immunized TNFR1−/− and WT mice. (C) Levels of anti-Qβ serum IgG antibodies induced in TNFR1−/− and WT mice by immunization with Qβ. Mice were boosted on day 55 after primary immunization for analysis of recall responses. All data represent the mean ± SEM, mean values statistically different from WT levels are indicated by asterisks (*, P < 0.01; **, P < 0.05).
The lack of GC formation resulted in reduced anti-Qβ IgG antibody levels in TNFR1−/− mice compared with WT controls (Fig. 8 C). However, early antibody production was normal, similarly to what was observed in Cr2−/− mice. Not affected by the absence of GCs was also the ability of TNFR1−/− mice to mount efficient recall responses. These results are consistent with earlier studies showing normal induction of memory B cells and reduced persistence of antibody titers in TNFR1−/− mice after immunization with vesicular stomatitis virus (38).
The fact that in the absence of GCs the course of the anti-Qβ antibody response was remarkably similar to the response observed in Cr2−/− mice, suggests that Cr2−/− mice have a defect in the generation of GC-derived plasma cells. The reduction of Qβ-specific isotype-switched PNAlow B cells in the absence of GCs provides further evidence that this population, which requires complement receptors for up-regulation of Blimp-1 and XBP-1, originates in GCs.
Short-term antigen trapping is efficient but long-term antigen persistence is reduced in Cr2−/− mice
Complement receptors together with Fcγ receptors mediate antigen trapping on FDCs thereby sustaining humoral immunity. Therefore, we analyzed antigen retention in the spleen of Cr2−/− and WT mice 12 and 21 d after injection of 100 μg Qβ; at these time points, deposits of Qβ particles are found exclusively in B cell follicles (Fig. 9). Histological staining for Qβ antigen showed that antigen was efficiently trapped in the spleen of Cr2−/− mice at day 12 (Fig. 9); note that at this time point Blimp-1 and XBP-1 expression in the PNAlow B cell population was already dramatically different between Cr2−/− and WT mice. However, 3 wk after immunization, deposits of Qβ antigen were reduced in Cr2−/− mice compared with controls (Fig. 9). This suggests that binding of Ag-IgG complexes to Fcγ receptors was sufficient to ensure short-term antigen trapping but complement receptors were required for long-term antigen persistence. The reduced time span of Qβ trapping in Cr2−/− mice may be responsible for the faster decline of GC reactions in these mice.
Figure 9.
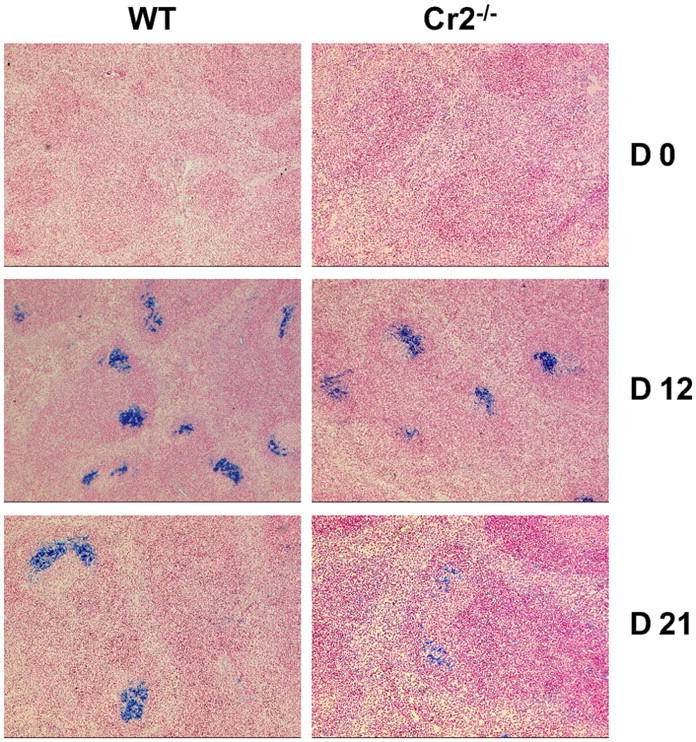
Short-term trapping of Qβ particles is efficient but long-term antigen retention is reduced in Cr2−/− mice. Histological staining of Qβ antigen on spleen sections from WT and Cr2−/− mice on days 0, 12, and 21 after immunization with 100 μg Qβ. Original magnification: ×50 (B6, day 0); ×75 (Cr2−/−, day 0); ×69 (day 12); ×82.5 (day 21).
Discussion
Complement receptors provide an important link between innate and acquired immunity and their role in the induction of humoral immune responses has been clearly demonstrated (20, 22, 39). In this study, we dissected the role of CD21–CD35 complement receptors in the induction of different effectors of humoral memory. We showed that in response to repetitive antigens, such as virus-like particles derived from bacteriophages Qβ and AP205, generation of antigen-specific memory B cells was normal, but differentiation of GC B cells into long-lived plasma cells residing in the BM was substantially reduced. As a consequence, Cr2−/− mice were unable to maintain persistent antibody titers, a hallmark of protective, long-lasting humoral immunity. Considering that both memory B cells and BM plasma cells are end products of the GC reaction, it was surprising that only generation of BM plasma cells was affected by absence of complement receptors. Distinct differentiation pathways have been postulated for the selection of these two compartments in GCs. Whereas high affinity for antigen is required for commitment to a plasma cell fate, the memory B cell population appears to be more heterogeneous with respect to affinity and its survival in GCs relies on antigen-dependent signals preventing apoptosis (6, 7). Our results suggest that not only does coligation of the CD21–CD19–CD81 coreceptor and the BCR by complement-coated antigen decrease the affinity threshold needed for B cell activation (40), but it also increases the avidity of the interaction of B cells with antigen as required for promoting differentiation of GC B cells into BM plasma cells. In the absence of CD21, the threshold of signal strength required for differentiation into long-lived plasma cells may not be reached by most GC B cells, but signaling may still be sufficient for differentiation and survival of memory B cells. We therefore speculate that those few BM plasma cells that develop in Cr2−/− mice (Fig. 1 B) are clones with high affinity BCR, which compensates for the lack of CD21. This would be consistent with increased affinity maturation in Cr2−/− mice, which has been reported but is subject to debate (24, 25). Short-term antibody responses induced by immunization with Qβ were normal in Cr2−/− mice. This indicates that different mechanisms underlie regulation of short-lived, non-GC–derived plasma cells participating to the early phase of the antibody response and BM plasma cells responsible for long-term maintenance of memory titers.
The degree of antigen organization is crucial to the activation of B cells (41) as well as to the requirement for costimulatory molecules (42). Highly repetitive antigens such as viral particles are capable of efficient cross-linking of BCRs, which induces potent antibody responses even in the absence of T cells (43) or CD21–CD35 (28). Qβ capsids display a highly ordered structure comparable to that of viruses, conferring on them the ability to efficiently cross-link surface Ig on B cells. This may explain why antibody responses and GC reactions were efficiently induced by immunization with Qβ particles in Cr2−/− mice and is consistent with previous reports of normal antibody responses to vesicular stomatitis virus (23) and influenza virus (44) in these mice. In contrast, efficient antibody responses to other experimental antigens lacking the structural feature repetitiveness seem to be more dependent on CD21 (21, 22, 24). However, although antigen repetitiveness was able to compensate for the absence of CD21 to some extent, generation of persistent serum antibody was not achieved by immunization with Qβ. Similarly, an increase of antigen load or administration of antigen in inflammatory adjuvants has been shown to mitigate the defects in humoral responses of Cr2−/− mice (20, 24, 25); nevertheless antibody persistence was impaired even with optimal antigen doses in adjuvant (24).
Reduction of Qβ-specific BM plasma cells in Cr2−/− mice correlated with a failure to induce Blimp-1 and XBP-1 expression in post-GC B cells, from which long-lived BM ASCs are thought to arise (45). This observation suggests that complement receptors are essential for Blimp-1–XBP-1–mediated induction of plasmacytic differentiation in GCs. Blimp-1 has been described to be expressed in a small subset of GC B cells (46, 47). Owing to their partial plasma phenotype these Blimp-1+ GC cells were assumed to be committed to exit GCs and to differentiate into plasma cells. In contrast to previous reports we did not detect Blimp-1 expression in Qβ-specific GC B cells, but transcription of Blimp-1 was present in isotype-switched B cells with a PNAlow phenotype and surface Ig specific for Qβ. Consistent with the expression of transcriptional regulators driving plasma cell differentiation, these cells displayed a partial plasma cell phenotype, characterized by the up-regulation of CD138, VLA-4, LFA-1, and CD44. A corresponding population of Qβ-specific B cells with this phenotype was absent in Cr2−/− mice. This is in accordance with a failure of post-GC to induce Blimp-1 and XBP-1 in the absence of complement-mediated stimulation. Despite expression of Blimp-1 and XBP-1, Qβ-specific PNAlowB220high B cells did not stain for intracellular Ig, indicating that BM plasma cell precursors may leave the spleen before they secrete antibodies.
A clear role for complement receptors on FDCs in the maintenance of B cell memory has been demonstrated in chimeric mice with Cr2−/− FDC stroma and normal B cells (48). FDCs are thought to mediate long-term antigen retention, which may continually stimulate differentiation of memory B cell into ASCs (31, 49). The reduced persistence of Qβ particles on FDCs observed in Cr2−/− mice in this study, confirmed the role of complement receptors in long-term antigen retention. However, the presence of substantial Qβ depots in Cr2−/− mice on day 12 suggests that Ag–IgG complexes on FDCs were efficiently trapped through Fcγ receptors for short periods after immunization. Despite normal antigen trapping at this time point, Cr2−/− mice failed to induce Blimp-1, XBP-1, and Bcl-2 expression and to up-regulate surface molecules characteristic of plasma cell precursors. This indicates that reduced antigen retention was not responsible for the observed phenotype and is in agreement with previous studies reporting a direct role for CD21–CD35 on B cells for induction of long-lasting antibody responses (50, 51). Nevertheless, a lack of complement receptors on FDCs and consequent reduced long-term antigen trapping may contribute to the inability of Cr2−/− mice to maintain long-term ASCs and is compatible with our observation that GC reactions decayed more rapidly.
In conclusion, our results suggest that induction of long-lasting antibody production, which is mediated primarily by BM plasma cells, requires more than BCR signaling, even with antigens that are capable of efficient BCR cross-linking, such as viral particles. A complement-mediated signal, revealing the activation of the innate immune system, is essential. This allows focusing long-term antibody production on pathogens and keeps the induction of plasma cells capable of secreting specific IgG antibodies over extended periods of time under tight control of both the adaptive and the innate immune system.
Materials and Methods
Mice and antigens
C57BL/6 mice (Harlan), Cr2−/− mice (20), TNFR1−/− mice (52), and C57BL/6–CD45.1 mice were immunized i.v. with 10 or 100 μg Qβ or 25 μg AP205. Animal experiments were conducted in accordance with protocols approved by the Swiss Federal Veterinary Office.
Qβ capsids were expressed using the vector pQβ10 and purified as described previously (53). AP205 coat protein (54) was cloned into the pQb10 vector (26) and expressed and purified similarly as Qβ.
ELISA.
ELISAs were performed as described previously (29). Titers represent log2 dilutions of 40-fold prediluted sera at half maximal OD.
ELISPOT assay
Qβ/AP205-specific ASC frequencies were determined as described previously (55). In brief, 24-well plates were coated with 10 μg/ml Qβ or AP205. Spleen or BM cells were added in MEM containing 2% FCS and incubated for 5 h at 37°C. Cells were washed off and plates were incubated successively with goat anti–mouse IgG (EY Labs) and alkaline phosphatase-conjugated donkey anti–goat IgG antibodies (Jackson ImmunoResearch Laboratories) before development of alkaline phosphatase color reactions.
Flow cytometry
Detection of B cells expressing Qβ-specific surface Ig was performed by incubation with Qβ, followed by a polyclonal rabbit anti-Qβ serum (produced by RCC Ltd.) and Cy5-conjugated donkey anti–rabbit IgG serum (Jackson ImmunoResearch Laboratories). AP205-specific B cells were identified similarly, using a polyclonal rabbit anti-AP205 serum (generated in the laboratory of Dr. P. Pumpens, University of Latvia, Riga, Latvia).
Isotype-switched B cells were detected with a mixture of FITC-conjugated antibodies (anti-IgD, 11-26c.2a; goat anti-IgM serum; Jackson ImmunoResearch Laboratories; anti-CD4, GK1.5; anti-CD8, 53–6.7; anti-CD11b, M1/70; anti-Gr-1, RB6-8C5), and PE-TxR–conjugated anti-B220 (RA3-6B2). Biotinylated PNA (Vector Laboratories) and streptavidin-PE were used to assess PNA-binding. Preplasma memory B cells were detected with biotinylated anti-CD138 (281–2), streptavidin-Tricolor, PE-conjugated anti-B220 (RA3-6B2), and FITC-conjugated antibodies to IgD (11-26c.2a), CD4 (GK1.5), and CD8 (53–6.7). Dead cells were excluded by staining with 0.005 μg/ml YO-PRO-1 (Molecular Probes).
To characterize PNAlow Qβ-specific B cells, B220+ splenocytes, purified by magnetic cell sorting with B220 MicroBeads (Miltenyi Biotec), were stained with biotinylated antibodies (anti-CD138, 281–2; anti-CD11a, M17/4; anti-CD49d, R1-2; and anti-CD44, IM7) followed by streptavidin-Tricolor (Caltag) and PE-conjugated goat anti–mouse IgM F(ab′)2 (Southern Biotechnology Associates, Inc.), PE-conjugated rat anti–mouse IgD (11–26; eBioscience), FITC-conjugated PNA.
Qβ-specific plasma cells were detected by incubation with unlabeled Qβ, to block binding to surface IgG, and biotinylated PNA followed by streptavidin-PE, PE-TxR–conjugated anti-B220 (RA3-6B2), and FITC-conjugated antibodies to CD4 (GK1.5), CD8 (53–6.7), and CD11b (M1/70). After permeabilization, cells were incubated at room temperature with Qβ particles labeled with the fluorochrome Alexa 647, using the Alexa Fluor 647 Protein Labeling Kit (Molecular Probes).
Fc-receptors were blocked with anti–mouse CD16/32 (2.4G2). Antibodies were purchased from BD Biosciences unless otherwise specified.
Adoptive transfer experiments
5 × 107 splenocytes from naive Cr2−/− and C57BL/6 mice were transferred with 10 μg Qβ into sublethally irradiated (450 rads) C57BL/6-CD45.1–recipient mice. Irradiated control mice were given antigen but no cells.
For adoptive transfer of memory B cells, Qβ-binding IgMlowIgDlow and total IgMlowIgDlow splenocytes were purified by FACS from C57BL/6 mice immunized 6 wk previously. Control naive B cells (IgM+IgD+) were sorted from unimmunized mice. Single cell suspensions of 104 Qβ-binding IgMlowIgDlow, 105 IgMlowIgDlow, or 105 IgM+IgD+ B cells were injected together with 107 purified CD4+ cells into sublethally irradiated recipients, which were immunized with 10 μg Qβ.
Immunohistochemistry
Freshly removed organs were snap frozen in liquid nitrogen. Tissue sections of 5 μm thickness were cut in a cryostat and fixed with acetone. For detection of Qβ antigen, sections were incubated with rabbit anti-Qβ serum (RCC), followed by biotinylated sheep anti–rabbit Igs (The Binding Site) and alkaline phosphatase–labeled streptavidin (Roche). Alkaline phosphatase was visualized using the Vector Blue substrate (Vector Laboratories). Sections were counterstained with Vector Nuclear Fast Red (Vector Laboratories).
For detection of Qβ-specific B cells, spleen sections were incubated with Qβ and bound particles were detected with a polyclonal anti-Qβ serum as described previously (29). PNA-binding cells were stained with biotinylated PNA (Vector Laboratories) followed by avidin–biotin–peroxidase complexes (DAKO) before alkaline phosphatase was visualized.
Quantitative RT-PCR
5 × 104–105 specific B cells were sorted into TRI Reagent (Molecular Research Center) and total RNA was extracted according to the manufacturer's instructions. First strand cDNA was synthesized using random nonamer primers and SuperScript II reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed on an iCycler Thermal Cycler (Bio-Rad Laboratories) using the following primers for amplification (sense primer is given first): for Blimp-1 ATGGAGGACGCTGATATGAC and GATGCCTCGGCTTGAAC; for XBP-1 CGTAGACGTTTCCTGGCTATG and GGACCGGGTACCATGAG; for Bcl-2 TCGTGACTTCGCAGAGATG and AACTCAAAGAAGGCCACAATC; for β-actin TCACCATGGATGATGATATCGC and TGAAGGTCTCAAACATGATCTGG. Quantification of β-actin cDNA was performed for each sample to allow for normalization between samples. Dissociation curve analysis was performed to verify the presence of a single PCR product. Quantification of the transcripts was determined with the iCycler iQ Optical System Software (Bio-Rad Laboratories) using the comparative threshold cycle method.
Statistical analysis
Levels of statistical significance between means were determined using a Student's t test.
Acknowledgments
We thank M. Carroll for providing Cr2−/− mice, M. Bauer for cell sorting, A. ter Steege for help with immunohistochemistry, and E. Devevre, S. Muntwiler, A. Titz, and P. Sebbel for technical support.
The authors have no conflicting financial interests.
Abbreviations used: ASC, antibody-secreting cell; BCR, B cell Ag receptor; FDC, follicular dendritic cell; GC, germinal center; NP, (4-hydroxy-3-nitrophenyl)acetyl; PNA, peanut agglutinin.
References
- 1.Nossal, G.J. 1999. Vaccines. Fundamental Immunology. W.E. Paul, editor. Lippincott-Raven, Philadelphia. 1387–1425.
- 2.Ahmed, R., and D. Gray. 1996. Immunological memory and protective immunity: understanding their relation. Science. 272:54–60. [DOI] [PubMed] [Google Scholar]
- 3.Slifka, M.K., M. Matloubian, and R. Ahmed. 1995. Bone marrow is a major site of long-term antibody production after acute viral infection. J. Virol. 69:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachmann, M.F., T.M. Kundig, C.P. Kalberer, H. Hengartner, and R.M. Zinkernagel. 1994. How many specific B cells are needed to protect against a virus? J. Immunol. 152:4235–4241. [PubMed] [Google Scholar]
- 5.MacLennan, I.C. 1994. Germinal centers. Annu. Rev. Immunol. 12:117–139. [DOI] [PubMed] [Google Scholar]
- 6.Smith, K.G., A. Light, L.A. O'Reilly, S.M. Ang, A. Strasser, and D. Tarlinton. 2000. bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J. Exp. Med. 191:475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith, K.G., A. Light, G.J. Nossal, and D.M. Tarlinton. 1997. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 16:2996–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arpin, C., J. Dechanet, C. Van Kooten, P. Merville, G. Grouard, F. Briere, J. Banchereau, and Y.J. Liu. 1995. Generation of memory B cells and plasma cells in vitro. Science. 268:720–722. [DOI] [PubMed] [Google Scholar]
- 9.Zhang, X., L. Li, J. Jung, S. Xiang, C. Hollmann, and Y.S. Choi. 2001. The distinct roles of T cell-derived cytokines and a novel follicular dendritic cell-signaling molecule 8D6 in germinal center-B cell differentiation. J. Immunol. 167:49–56. [DOI] [PubMed] [Google Scholar]
- 10.Raman, V.S., R.S. Akondy, S. Rath, V. Bal, and A. George. 2003. Ligation of CD27 on B cells in vivo during primary immunization enhances commitment to memory B cell responses. J. Immunol. 171:5876–5881. [DOI] [PubMed] [Google Scholar]
- 11.Choe, J., and Y.S. Choi. 1998. IL-10 interrupts memory B cell expansion in the germinal center by inducing differentiation into plasma cells. Eur. J. Immunol. 28:508–515. [DOI] [PubMed] [Google Scholar]
- 12.Kawano, M.M., K. Mihara, N. Huang, T. Tsujimoto, and A. Kuramoto. 1995. Differentiation of early plasma cells on bone marrow stromal cells requires interleukin-6 for escaping from apoptosis. Blood. 85:487–494. [PubMed] [Google Scholar]
- 13.Shapiro-Shelef, M., K.I. Lin, L.J. McHeyzer-Williams, J. Liao, M.G. McHeyzer-Williams, and K. Calame. 2003. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 19:607–620. [DOI] [PubMed] [Google Scholar]
- 14.Reimold, A.M., N.N. Iwakoshi, J. Manis, P. Vallabhajosyula, E. Szomolanyi-Tsuda, E.M. Gravallese, D. Friend, M.J. Grusby, F. Alt, and L.H. Glimcher. 2001. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 412:300–307. [DOI] [PubMed] [Google Scholar]
- 15.Lin, K.I., C. Tunyaplin, and K. Calame. 2003. Transcriptional regulatory cascades controlling plasma cell differentiation. Immunol. Rev. 194:19–28. [DOI] [PubMed] [Google Scholar]
- 16.Fischer, M.B., S. Goerg, L. Shen, A.P. Prodeus, C.C. Goodnow, G. Kelsoe, and M.C. Carroll. 1998. Dependence of germinal center B cells on expression of CD21/CD35 for survival. Science. 280:582–585. [DOI] [PubMed] [Google Scholar]
- 17.Fearon, D.T., and R.H. Carter. 1995. The CD19/CR2/TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu. Rev. Immunol. 13:127–149. [DOI] [PubMed] [Google Scholar]
- 18.Fearon, D.T., and M.C. Carroll. 2000. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu. Rev. Immunol. 18:393–422. [DOI] [PubMed] [Google Scholar]
- 19.Chen, Z., S.B. Koralov, and G. Kelsoe. 2000. Regulation of humoral immune responses by CD21/CD35. Immunol. Rev. 176:194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahearn, J.M., M.B. Fischer, D. Croix, S. Goerg, M. Ma, J. Xia, X. Zhou, R.G. Howard, T.L. Rothstein, and M.C. Carroll. 1996. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 4:251–262. [DOI] [PubMed] [Google Scholar]
- 21.Molina, H., V.M. Holers, B. Li, Y. Fung, S. Mariathasan, J. Goellner, J. Strauss-Schoenberger, R.W. Karr, and D.D. Chaplin. 1996. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc. Natl. Acad. Sci. USA. 93:3357–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haas, K.M., M. Hasegawa, D.A. Steeber, J.C. Poe, M.D. Zabel, C.B. Bock, D.R. Karp, D.E. Briles, J.H. Weis, and T.F. Tedder. 2002. Complement receptors CD21/35 link innate and protective immunity during Streptococcus pneumoniae infection by regulating IgG3 antibody responses. Immunity. 17:713–723. [DOI] [PubMed] [Google Scholar]
- 23.Ochsenbein, A.F., D.D. Pinschewer, B. Odermatt, M.C. Carroll, H. Hengartner, and R.M. Zinkernagel. 1999. Protective T cell-independent antiviral antibody responses are dependent on complement. J. Exp. Med. 190:1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, Z., S.B. Koralov, M. Gendelman, M.C. Carroll, and G. Kelsoe. 2000. Humoral immune responses in Cr2−/− mice: enhanced affinity maturation but impaired antibody persistence. J. Immunol. 164:4522–4532. [DOI] [PubMed] [Google Scholar]
- 25.Wu, X., N. Jiang, Y.F. Fang, C. Xu, D. Mao, J. Singh, Y.X. Fu, and H. Molina. 2000. Impaired affinity maturation in Cr2−/− mice is rescued by adjuvants without improvement in germinal center development. J. Immunol. 165:3119–3127. [DOI] [PubMed] [Google Scholar]
- 26.Kozlovska, T.M., I. Cielens, D. Dreilinna, A. Dislers, V. Baumanis, V. Ose, and P. Pumpens. 1993. Recombinant RNA phage Q beta capsid particles synthesized and self-assembled in Escherichia coli. Gene. 137:133–137. [DOI] [PubMed] [Google Scholar]
- 27.Fehr, T., D. Skrastina, P. Pumpens, and R.M. Zinkernagel. 1998. T cell-independent type I antibody response against B cell epitopes expressed repetitively on recombinant virus particles. Proc. Natl. Acad. Sci. USA. 95:9477–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jegerlehner, A., T. Storni, G. Lipowsky, M. Schmid, P. Pumpens, and M.F. Bachmann. 2002. Regulation of IgG antibody responses by epitope density and CD21-mediated costimulation. Eur. J. Immunol. 32:3305–3314. [DOI] [PubMed] [Google Scholar]
- 29.Gatto, D., C. Ruedl, B. Odermatt, and M.F. Bachmann. 2004. Rapid response of marginal zone B cells to viral particles. J. Immunol. 173:4308–4316. [DOI] [PubMed] [Google Scholar]
- 30.Slifka, M.K., and R. Ahmed. 1998. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr. Opin. Immunol. 10:252–258. [DOI] [PubMed] [Google Scholar]
- 31.Zinkernagel, R.M., M.F. Bachmann, T.M. Kundig, S. Oehen, H. Pirchet, and H. Hengartner. 1996. On immunological memory. Annu. Rev. Immunol. 14:333–367. [DOI] [PubMed] [Google Scholar]
- 32.Iwakoshi, N.N., A.H. Lee, P. Vallabhajosyula, K.L. Otipoby, K. Rajewsky, and L.H. Glimcher. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4:321–329. [DOI] [PubMed] [Google Scholar]
- 33.Roberts, T., and E.C. Snow. 1999. Cutting edge: recruitment of the CD19/CD21 coreceptor to B cell antigen receptor is required for antigen-mediated expression of Bcl-2 by resting and cycling hen egg lysozyme transgenic B cells. J. Immunol. 162:4377–4380. [PubMed] [Google Scholar]
- 34.O'Connor, B.P., M. Cascalho, and R.J. Noelle. 2002. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J. Exp. Med. 195:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McHeyzer-Williams, L.J., M. Cool, and M.G. McHeyzer-Williams. 2000. Antigen-specific B cell memory: expression and replenishment of a novel b220− memory b cell compartment. J. Exp. Med. 191:1149–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Driver, D.J., L.J. McHeyzer-Williams, M. Cool, D.B. Stetson, and M.G. McHeyzer-Williams. 2001. Development and maintenance of a B220− memory B cell compartment. J. Immunol. 167:1393–1405. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto, M., S. Mariathasan, M.H. Nahm, F. Baranyay, J.J. Peschon, and D.D. Chaplin. 1996. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 271:1289–1291. [DOI] [PubMed] [Google Scholar]
- 38.Karrer, U., C. Lopez-Macias, A. Oxenius, B. Odermatt, M.F. Bachmann, U. Kalinke, H. Bluethmann, H. Hengartner, and R.M. Zinkernagel. 2000. Antiviral B cell memory in the absence of mature follicular dendritic cell networks and classical germinal centers in TNFR1−/− mice. J. Immunol. 164:768–778. [DOI] [PubMed] [Google Scholar]
- 39.Hebell, T., J.M. Ahearn, and D.T. Fearon. 1991. Suppression of the immune response by a soluble complement receptor of B lymphocytes. Science. 254:102–105. [DOI] [PubMed] [Google Scholar]
- 40.van Noesel, C.J., A.C. Lankester, and R.A. van Lier. 1993. Dual antigen recognition by B cells. Immunol. Today. 14:8–11. [DOI] [PubMed] [Google Scholar]
- 41.Bachmann, M.F., U.H. Rohrer, T.M. Kundig, K. Burki, H. Hengartner, and R.M. Zinkernagel. 1993. The influence of antigen organization on B cell responsiveness. Science. 262:1448–1451. [DOI] [PubMed] [Google Scholar]
- 42.Bachmann, M.F., R.M. Zinkernagel, and A. Oxenius. 1998. Immune responses in the absence of costimulation: viruses know the trick. J. Immunol. 161:5791–5794. [PubMed] [Google Scholar]
- 43.Bachmann, M.F., and R.M. Zinkernagel. 1997. Neutralizing antiviral B cell responses. Annu. Rev. Immunol. 15:235–270. [DOI] [PubMed] [Google Scholar]
- 44.Kopf, M., B. Abel, A. Gallimore, M. Carroll, and M.F. Bachmann. 2002. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat. Med. 8:373–378. [DOI] [PubMed] [Google Scholar]
- 45.O'Connor, B.P., M.W. Gleeson, R.J. Noelle, and L.D. Erickson. 2003. The rise and fall of long-lived humoral immunity: terminal differentiation of plasma cells in health and disease. Immunol. Rev. 194:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angelin-Duclos, C., G. Cattoretti, K.I. Lin, and K. Calame. 2000. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J. Immunol. 165:5462–5471. [DOI] [PubMed] [Google Scholar]
- 47.Falini, B., M. Fizzotti, A. Pucciarini, B. Bigerna, T. Marafioti, M. Gambacorta, R. Pacini, C. Alunni, L. Natali-Tanci, B. Ugolini, et al. 2000. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 95:2084–2092. [PubMed] [Google Scholar]
- 48.Barrington, R.A., O. Pozdnyakova, M.R. Zafari, C.D. Benjamin, and M.C. Carroll. 2002. B lymphocyte memory: role of stromal cell complement and FcγRIIB receptors. J. Exp. Med. 196:1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klaus, G.G., J.H. Humphrey, A. Kunkl, and D.W. Dongworth. 1980. The follicular dendritic cell: its role in antigen presentation in the generation of immunological memory. Immunol. Rev. 53:3–28. [DOI] [PubMed] [Google Scholar]
- 50.Croix, D.A., J.M. Ahearn, A.M. Rosengard, S. Han, G. Kelsoe, M. Ma, and M.C. Carroll. 1996. Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J. Exp. Med. 183:1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fang, Y., C. Xu, Y.X. Fu, V.M. Holers, and H. Molina. 1998. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J. Immunol. 160:5273–5279. [PubMed] [Google Scholar]
- 52.Pfeffer, K., T. Matsuyama, T.M. Kundig, A. Wakeham, K. Kishihara, A. Shahinian, K. Wiegmann, P.S. Ohashi, M. Kronke, and T.W. Mak. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 73:457–467. [DOI] [PubMed] [Google Scholar]
- 53.Cielens, I., V. Ose, I. Petrovskis, A. Strelnikova, R. Renhofa, T. Kozlovska, and P. Pumpens. 2000. Mutilation of RNA phage Qbeta virus-like particles: from icosahedrons to rods. FEBS Lett. 482:261–264. [DOI] [PubMed] [Google Scholar]
- 54.Klovins, J., G.P. Overbeek, S.H. van den Worm, H.W. Ackermann, and J. van Duin. 2002. Nucleotide sequence of a ssRNA phage from Acinetobacter: kinship to coliphages. J. Gen. Virol. 83:1523–1533. [DOI] [PubMed] [Google Scholar]
- 55.Sedgwick, J.D., and P.G. Holt. 1983. A solid-phase immunoenzymatic technique for the enumeration of specific antibody-secreting cells. J. Immunol. Methods. 57:301–309. [DOI] [PubMed] [Google Scholar]