

Abstract
High-mobility group box 1 (HMGB1) is a nuclear factor that is released extracellularly as a late mediator of lethality in sepsis as well as after necrotic, but not apoptotic, death. Here we demonstrate that in contrast to the delayed role of HMGB1 in the systemic inflammation of sepsis, HMGB1 acts as an early mediator of inflammation and organ damage in hepatic ischemia reperfusion (I/R) injury. HMGB1 levels were increased during liver I/R as early as 1 h after reperfusion and then increased in a time-dependent manner up to 24 h. Inhibition of HMGB1 activity with neutralizing antibody significantly decreased liver damage after I/R, whereas administration of recombinant HMGB1 worsened I/R injury. Treatment with neutralizing antibody was associated with less phosphorylation of c-Jun NH2-terminal kinase and higher nuclear factor–κB DNA binding in the liver after I/R. Toll-like receptor 4 (TLR4)-defective (C3H/Hej) mice exhibited less damage in the hepatic I/R model than did wild-type (C3H/HeOuj) mice. Anti-HMGB1 antibody failed to provide protection in C3H/Hej mice, but successfully reduced damage in C3H/Ouj mice. Together, these results demonstrate that HMGB1 is an early mediator of injury and inflammation in liver I/R and implicates TLR4 as one of the receptors that is involved in the process.
Ischemia reperfusion (I/R) injury is a pathophysiologic process whereby hypoxic organ damage is accentuated following return of blood flow and oxygen delivery. Transient episodes of ischemia are encountered during solid organ transplantation, trauma, hypovolemic shock, and elective liver resection, when inflow occlusion or total vascular exclusion is used to minimize blood loss. The pathophysiology of liver I/R injury includes direct cellular damage as the result of the ischemic insult as well as delayed dysfunction and damage that results from activation of inflammatory pathways. Histopathologic changes include cellular swelling, vacuolization, endothelial cell disruption, neutrophil infiltration, and hepatocellular necrosis (1, 2).
The distal cascade of inflammatory responses that result in organ damage after I/R injury has been studied extensively (3–8). Activation of Kupffer cells with production of reactive oxygen species, up-regulation of the inducible nitric oxide synthase, up-regulation of proinflammatory cytokines, and neutrophil accumulation have been identified as contributing events to the inflammation-associated damage. The extent to which the initial cellular injury contributes to propagation of the inflammatory response and further tissue damage is poorly understood.
We propose that a key link between the initial damage to cells and the activation of inflammatory signaling involves endogenous danger signals from ischemic cells. High-mobility group box 1 (HMGB1) recently was identified as an inflammatory cytokine that is involved as a late mediator of lethality in sepsis (9, 10). The observation that HMGB1 that is released from necrotic cells can serve as a mediator of inflammation in in vitro systems (11) points to this protein as a regulator for the inflammation that is seen following acute tissue damage. Recent in vitro studies suggests that some of the effects of HMGB1 result from its interaction with the individual members of the Toll-like receptor (TLR) family, TLR2 and TLR4 (12). Interaction of HMGB1 with TLR4, as we demonstrate here, could provide a critical link between tissue damage and activation of the innate immune response.
The aim of this study was to test the hypothesis that HMGB1 is an early mediator of inflammation and cell injury after hepatic I/R and that the actions of HMGB1 require TLR4. We show that HMGB1 is up-regulated in cultured hepatocytes by hypoxia and warm hepatic I/R in vivo. Neutralizing antibody to HMGB1 prevents hepatocellular damage and suppresses the activation of inflammatory cascades. In addition, we show that the TLR4 system plays a key role in the mechanism of hepatic I/R injury and implicate a HMGB1-TLR4 interaction in hepatic I/R.
RESULTS
Pretreatment with neutralizing antibody to HMGB1 protects against liver I/R injury
To determine if endogenous HMGB1 contributed to organ damage after liver I/R, neutralizing antibody to HMGB1 was administered to mice that were subjected to warm I/R. Animals were given anti-HMGB1 antibody (600 μg or 60 μg per mouse) or irrelevant IgG antibody 1 h before ischemia. Sixty minutes of warm hepatic ischemia followed by 6 h of reperfusion significantly increased serum alanine aminotransferase (sALT) levels in the IgG antibody control mice that were subjected to I/R. Treatment with 60 μg of anti-HMGB1 antibody did not confer any protection, whereas treatment with 600 μg of anti-HMGB1 antibody resulted in significant protection from hepatic injury (Fig. 1 a). This protection also was evident at 24 h after reperfusion in anti-HMGB1 antibody–treated mice (Fig. 1 b). Liver histology confirmed the sALT estimation of liver damage. Severe sinusoidal congestion and hepatocellular necrosis was present in liver tissue from mice that were treated with control IgG, whereas minimal damage was noted in samples from anti-HMGB1–treated mice (Fig. 1 c). Liver samples from control animals exhibited ∼26.9 ± 6.2% necrotic hepatocytes compared with 5.3 ± 1.1% necrotic cells in the anti-HMGB1–treated group (n = 6 per group; P < 0.05).
Figure 1.

Pretreatment with neutralizing antibody to HMGB1 protects against liver I/R injury. (a) Sham mice and mice that underwent ischemia and 6 h of reperfusion were treated with anti-HMGB1 antibody (60 μg or 600 μg) or control antibody i.p. 1 h before ischemia. Serum ALT levels were analyzed as a measure of hepatocellular injury. Data represent means ± SE, n = six mice per group. *P < 0.05 versus mice subjected to I/R given control antibody. (b) Sham mice and mice that underwent ischemia and 24 h of reperfusion were treated with anti-HMGB1 antibody (600 μg) or control antibody i.p. 1 h before ischemia. Data represent means ± SE, n = six mice per group. *P < 0.05 versus mice that were subjected to I/R and given control antibody. (c) Hematoxylin-eosin–stained liver sections from control antibody and anti-HMGB1 antibody–treated animals 24 h after reperfusion (original magnification ×200). Images are representative liver sections from six mice per group.
HMGB1 expression is up-regulated in hepatocytes after hypoxia in vitro and in the liver in vivo after I/R
To determine if the HMGB1-dependent injury was associated with changes in protein levels, Western blot analysis was performed on liver lysates from animals that were subjected to liver I/R (Fig. 2 a). Following 60 min of warm ischemia, HMGB1 protein expression was up-regulated as early as 1 h after reperfusion and then increased in a time-dependent manner up to 24 h. Previous studies have shown that HMGB1 localizes predominantly in the nucleus of macrophages (11). To determine the cellular localization of HMGB1 in the liver, immunofluorescence staining was performed in normal livers and livers that underwent I/R. Expression of HMGB1 was noted predominantly in the nucleus of hepatocytes in normal livers (Fig. 2 b). After ischemia and 6 h of reperfusion, HMGB1 expression was enhanced in the nucleus and cytoplasm of hepatocytes (Fig. 2 c). Although HMGB1 expression was increased in the liver, we were unable to detect increased HMGB1 levels in the serum of mice that underwent hepatic I/R injury (data not shown).
Figure 2.

HMGB1 expression is up-regulated in hepatocytes by hypoxia and in the liver after I/R. (a) Mice underwent 60 min of ischemia and various lengths of reperfusion. Western blot analysis for cellular HMGB1 was performed for hepatic protein lysates of the ischemic lobes at the time points shown, with each lane representing a separate animal. Blot shown is representative of three experiments with similar results. (b) Immunofluorescent stain of HMGB1 from sections of normal liver and (c) liver subjected to 60 min of ischemia and 6 h of reperfusion (original magnification ×400). Images are representative liver sections from six mice per group. Red, HMGB1; blue, nuclei; green, F-actin. (d) Cultured rat hepatocytes were exposed to hypoxia (1% O2) from 0 to 24 h. Whole cell lystate and media were subjected to Western blot analysis of HMGB1. Blot shown is representative of three experiments with similar results.
Hypoxia is believed to be the initiating event in the I/R insult; therefore, we assessed the expression of HMGB1 in hepatocytes that were exposed to hypoxia in vitro. Primary rat hepatocytes that were exposed to normoxia (21% oxygen) had a basal level of HMGB1 expression which did not change significantly up to 24 h of incubation (data not shown). However, exposure of the hepatocytes to hypoxia (1% oxygen) resulted in a time-dependent increase in cellular HMGB1 expression (Fig. 2 d). HMGB1 also increased in the media of hepatocytes that were exposed to hypoxia. Cell viability was not different between hepatocytes that were exposed to hypoxia and normoxia at 24 h (data not shown).
Delayed administration of neutralizing antibody to HMGB1 protects against liver I/R injury
Although administration of neutralizing antibody to HMGB1 that was given as a pretreatment was protective in our model of liver I/R injury, strategies that use this antibody after the onset of injury would be clinically relevant. We next sought to determine the efficacy of the antibody given after the ischemic event. In I/R injury, the cascade of events that result in organ damage is initiated at the onset of ischemia; ischemia alone was shown to result in the activation of early signaling pathways in the liver (13). To determine whether antibody treatment could be delayed until after ischemia, anti-HMGB1 antibody (600 μg) was given to the animals immediately after reperfusion. As with pretreatment, the delayed treatment of neutralizing antibodies to HMGB1 also conferred significant protection against hepatic injury 6 h after reperfusion (Fig. 3).
Figure 3.

Delayed administration of neutralizing antibody to HMGB1 protects against liver I/R injury. Sham mice and mice that underwent ischemia and 6 hours of reperfusion were treated with anti-HMGB1 antibody (600 μg) or control antibody immediately after reperfusion. Serum ALT levels were analyzed as a measure of hepatocellular injury. Data represent means ± SE, n = six mice per group. *P < 0.05 versus mice that were subjected to I/R and given control antibody.
Neutralizing antibody to HMGB1 decreases production of inflammatory mediators
Inflammatory cytokines, such as TNF and IL-6, were shown to play key roles in the pathophysiology of hepatic I/R injury (14, 15). Using real-time RT-PCR, we measured steady-state mRNA levels for these cytokines in the liver after I/R. Compared with sham-treated animals, liver I/R in IgG-treated animals resulted in increased expression of TNF and IL-6 mRNA 1 h after reperfusion (Fig. 4, a and b). However, animals that were treated with anti-HMGB1 antibody exhibited minimal increases in hepatic TNF and IL-6 mRNA levels compared with control animals that were subjected to I/R. We showed previously that inducible NO synthase (iNOS) contributes to liver injury that is due to I/R (16, 17). Hepatic iNOS mRNA levels were increased 1 h after reperfusion in antibody control–treated mice compared with sham animals (Fig. 4 c). As with TNF and IL-6 expression, treatment with anti-HMGB1 prevented hepatic iNOS expression after I/R.
Figure 4.

Neutralizing antibody to HMGB1 decreases production of inflammatory mediators. Hepatic TNF-α (a), IL-6 (b), and iNOS (c) mRNA expression were measured after ischemia and 1 h of reperfusion in mice that were treated with anti-HMGB1 antibody or control antibody. Results were obtained using real time RT-PCR and expressed as relative increase of mRNA expression compared with sham animals. Data represents means ± SE, n = eight mice per group. *P < 0.05 versus mice that were subjected to I/R and given control antibody.
Neutralizing antibody to HMGB1 modulates inflammatory signaling pathways
Among the most proximal events in I/R is the activation of mitogen-activated protein (MAP) kinases. A role for c-Jun NH2-terminal kinase (JNK) activation in liver I/R injury has been demonstrated (18, 19). To determine if HMGB1 influenced MAP kinase activation, we assessed phosphorylation of JNK, p38, and extracellular signal-regulated kinase (ERK). After I/R, phosphorylation of JNK, p38, and ERK was increased in liver tissue from animals that were treated with the control IgG (Fig. 5 a). In contrast, when mice were treated with anti-HMGB1 antibody, phosphorylation of these MAP kinases was decreased. Treatment with anti-HMGB1 antibody did not affect total cellular levels of ERK, p38, or JNK. Thus, these results suggest that HMGB1 acts upstream of MAP kinase activation. NF-κB activation also was shown to be increased following hepatic I/R (20). When activated in hepatocytes, NF-κB prevents cell death (21). Using electrophorectic mobility shift assays, there was an increase in NF-κB DNA binding in the ischemic liver 1 h after reperfusion in control IgG-treated animals compared with sham-treated animals (Fig. 5 b). Animals that were treated with neutralizing anti-HMGB1 antibody had an even greater increase in NF-κB DNA binding. The NF-κB bands were specific as confirmed by cold competition in the presence of excess unlabeled NF-κB consensus motif. We previously performed supershift studies to determine that the NF-κB complex was a heterodimer composed of p65 and p50 subunits (22).
Figure 5.
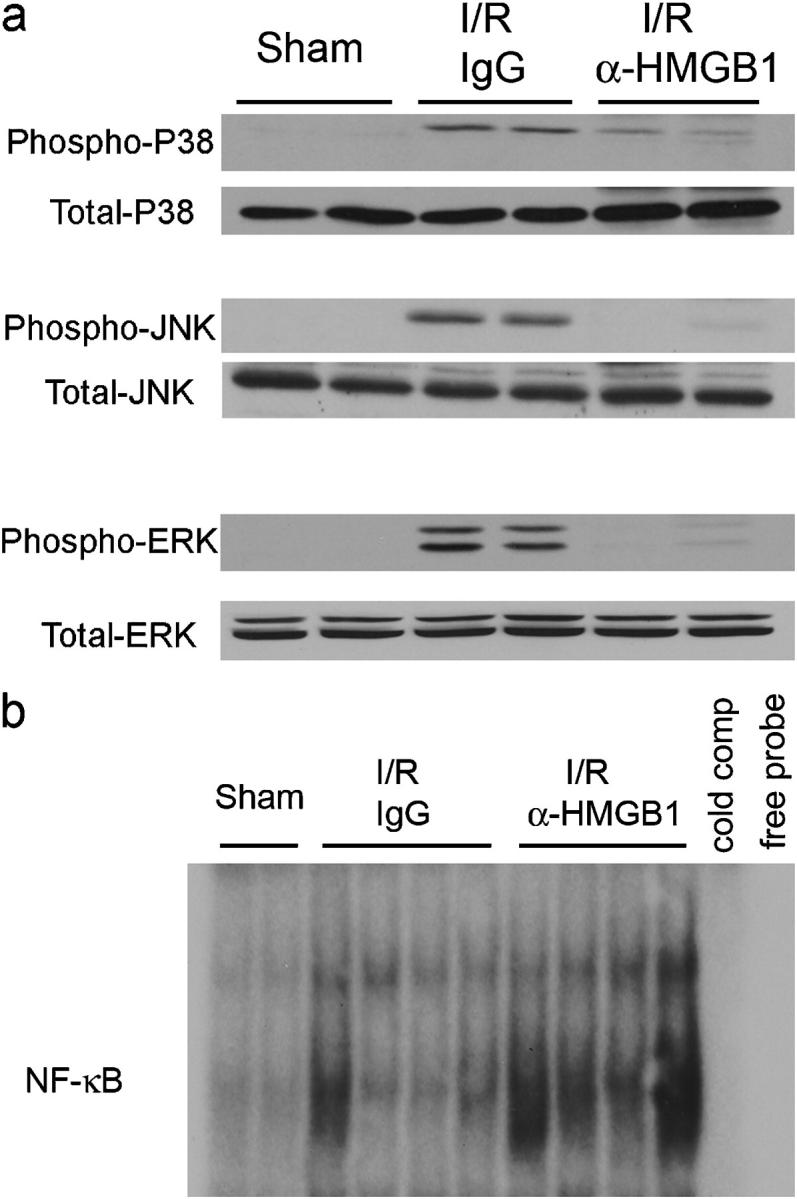
Neutralizing antibody to HMGB1 modulates inflammatory signaling pathways. (a) Mitogen-activated protein kinase activation was determined in sham mice and mice that underwent ischemia and 1 h of reperfusion. Animals were treated with anti-HMGB1 antibody or control antibody. Western blot analysis for phosphorylated and total ERK, p38, and JNK was performed. Hepatic protein lysates from ischemic lobes were obtained; each lane represents a separate animal. Blot shown is representative of three experiments with similar results. (b) NF-κB activation during hepatic I/R injury was assessed. Mice that underwent ischemia and 1 h of reperfusion were treated with anti-HMGB1 antibody or control antibody. Nuclear extracts were prepared from the ischemic livers and subjected to electrophoretic mobility shift assay. Assay shown is representative of three experiments with similar results.
Administration of recombinant HMGB1 worsens liver I/R injury
To determine if exogenous HMGB1 would alter hepatic injury in I/R, a nonlethal dose (20 μg) of recombinant HMGB1 (rHMGB1)was given to mice immediately after reperfusion. There was no difference in liver injury as assessed by sALT levels between sham animals that were given either vehicle (PBS) or this low dose of rHMGB1 (Fig. 6). As expected, I/R increased sALT levels in PBS-treated animals; however, animals that were given rHMGB1 had a significantly greater increase in liver enzyme levels than controls.
Figure 6.

Administration of recombinant HMGB1 worsens liver I/R injury. Serum ALT levels were measured in sham mice and mice that underwent ischemia and 6 h of reperfusion. Animals were treated with a nonlethal dose of recombinant HMGB1 (20 μg) or vehicle PBS immediately after reperfusion. Data represent means ± SE, n = four mice per group. *P < 0.05 versus mice that were subjected to I/R and given vehicle PBS.
HMGB1-mediated I/R injury involves TLR4 system
Recent in vitro evidence suggest that TLR4 acts as a receptor to HMGB1 (12). To determine if functional TLR4 was required for I/R-induced injury in the liver, TLR4-defective (C3H/Hej) mice and wild-type control (C3H/HeOuj) mice were subjected to liver I/R. TLR4-defective mice were protected from hepatic I/R injury compared with TLR4-intact mice (Fig. 7 a). To shed some light on whether the reduction in hepatic injury was due to LPS-TLR4 interaction, CD14−/− mice were also subjected to warm I/R. No difference in liver injury was seen between CD14−/− and CD14+/+ mice (Fig. 7 b). In addition, treatment of anti-HMGB1 antibody to CD14−/− and CD14+/+ mice protected the animals from I/R injury. To determine if a HMGB1-TLR4 interaction accounted for the HMGB1-dependent effect in liver injury from I/R, TLR4-defective and TLR4-intact animals were pretreated with anti-HMGB1 antibody. Similar to that seen with C57BL/6 mice, blockade of HMGB1 in TLR4 wild-type animals led to protection from hepatic I/R damage (Fig. 7 a). However, this protection was not seen in TLR4-defective mice who underwent hepatic I/R. We also examined the expression of TNF and IL-6 mRNA in the liver after I/R for these animals. Whereas TLR4 wild-type mice that were treated with anti-HMGB1 antibody exhibited decreased hepatic TNF and IL-6 mRNA levels compared with control I/R animals, there was no difference in expression of these cytokines after I/R in TLR4-defective mice that were treated with control antibody or neutralizing antibodies to HMGB1 (Fig. 8 a). To confirm the role of TLR4 in HMGB1-mediated liver injury, we administered rHMGB1 to TLR4-defective and wild-type animals. There was no difference in serum ALT levels between sham animals that were treated with PBS or rHMGB1 (Fig. 8 b). rHMGB1 worsened liver injury after I/R in TLR4 wild-type mice but had no effect in TLR4-defective mice.
Figure 7.
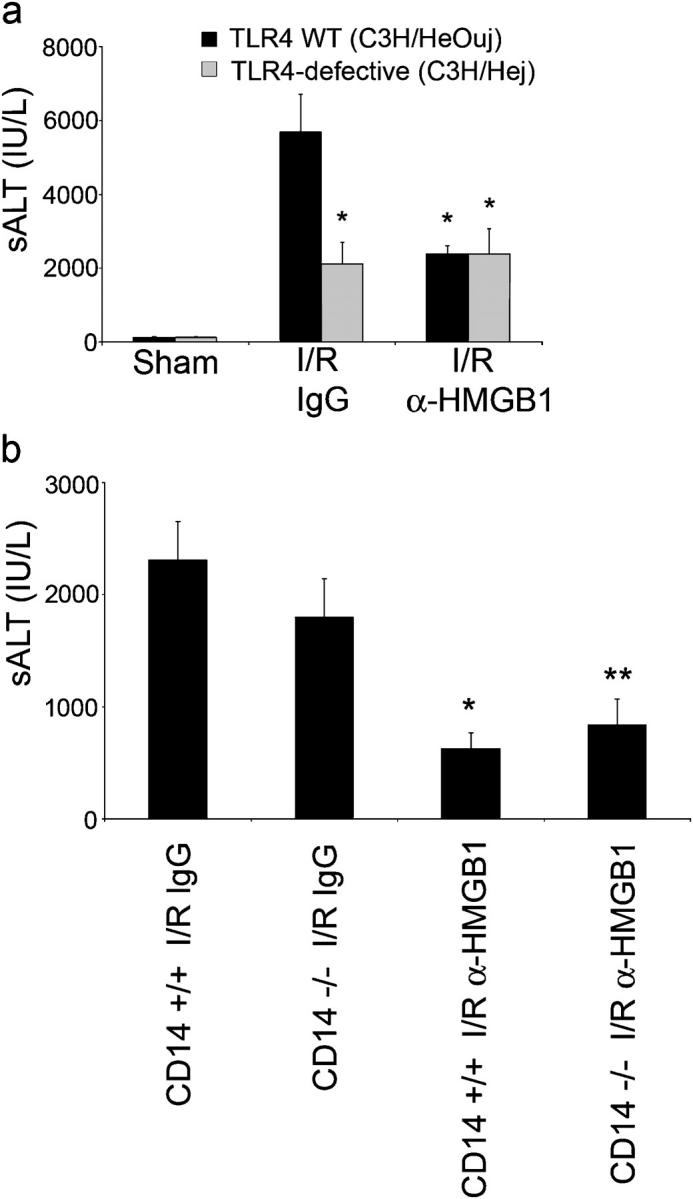
Hepatic I/R injury involves TLR4 system and is independent of LPS-TLR4 interaction. (a) TLR4-defective mice (C3H/Hej) and their wild-type (WT) mice counterparts (C3H/HeOuj) were subjected to liver ischemia and 6 h of reperfusion. Animals were treated with anti-HMGB1 antibody or control antibody 1 h before ischemia and sALT levels were measured. Data represent means ± SE, n = four to six mice per group. *P < 0.05 versus TLR4 wild-type mice that were subjected to I/R and given control antibody. (b) CD14+/+ and CD14−/− mice were subjected to liver ischemia and 6 h of reperfusion. Animals were treated with anti-HMGB1 antibody or control antibody at reperfusion. Data represent means ± SE, n = 5 mice per group. *, P < 0.05 versus CD14+/+ mice subjected to I/R given control antibody. **, P < 0.05 versus CD14−/− mice subjected to I/R given control antibody.
Figure 8.

HMGB1-mediated I/R injury involves TLR4 system. (a) Hepatic TNF-α and IL-6 mRNA expression were obtained in TLR4-defective mice and wild-type mice after ischemia and 6 h of reperfusion. Mice were treated with anti-HMGB1 antibody or control antibody. Results are expressed as relative increase of mRNA expression compared with sham animals. Data represent means ± SE, n = four to six mice per group. *P < 0.05 versus TLR4 wild-type mice that were subjected to I/R and given control antibody. (b) Serum ALT levels were determined in TLR4-defective mice and wild-type mice after ischemia and 6 h of reperfusion. Animals were treated with recombinant HMGB1 or vehicle PBS immediately after reperfusion. Data represent means ± SE, n = four to six mice per group. *P < 0.05 versus TLR4 wild-type mice that were subjected to I/R and given vehicle PBS.
DISCUSSION
HMGB1 is an ancient protein that predates speciation with extraordinary conservation among species, and acts as a nuclear protein that promotes transcriptional activation and access. Prototypic of other damage-associated molecular pattern molecules, it acts late as a downstream mediator of systemic inflammation in sepsis (9). Macrophages that are stimulated with endotoxin secrete HMGB1 after a significant delay compared with the release of other cytokines, such as TNF and IL-1, which are much more immediate (23). Release of HMGB1 also is released readily from necrotic or damaged cells, and serves as a signal for inflammation (11, 24). Although blockade of HMGB1 in a model of massive hepatocyte necrosis from acetaminophen in vivo did not protect against liver damage, inflammation was reduced as seen by reduced myeloperoxidase activity in total liver extracts (11). The purpose of this study was to test the notion that HMGB1 acts as an early mediator of inflammation and injury in hepatic I/R injury. The major and novel findings of this investigation are: (a) HMGB1 levels are increased by hypoxia in cultured hepatocytes and by I/R in the liver in vivo; (b) blockade of HMGB1 protects against warm hepatic I/R injury; (c) the protection is associated with a decrease in MAP kinase activation, augmented NF-κB activation, and a decrease in local hepatic proinflammatory cytokine expression; and (d) evidence for HMGB1–TLR4 interaction in the mechanism of HMGB1-mediated damage in hepatic I/R.
Whereas HMGB1 is a late mediator of systemic inflammation in sepsis, our findings implicate HMGB1 as an early mediator following acute, local organ injury. This role for HMGB1 may include a rapid increase in HMGB1 levels in the liver. We showed that hepatic HMGB1 expression increases as early as 1 h after reperfusion of ischemic liver. That hypoxia alone is adequate to increase HMGB1 levels is shown by our studies with cultured hepatocytes. It is unknown whether the increases in HMGB1 levels are essential to the proinflammatory effects. Our observation that even early, proximal signaling events (i.e., MAP kinase activation) are partially HMGB1-dependent suggests that increases in HMGB1 levels are not essential. It is not known from our studies whether HMGB1 is secreted or passively released from damaged cells after I/R injury. It could be that severe ischemic stress leads to a regulated release or surface expression of HMGB1 as a danger signaling event. As the injury progresses and cells begin to die, the release may then be passive; however, this is yet to be proven. It is notable that circulating levels of HMGB1 did not increase; this is consistent with a local or paracrine-like action for HMGB1 in this organ-specific acute injury model.
To elucidate the molecular basis of protection in blocking HMGB1, we investigated its effect on the MAP kinase and NF-κB signaling pathways. HMGB1 activates one or more of these signaling pathways in endothelial cells, smooth muscle cells, enterocytes, and neutrophils (24–27). The MAP kinase family represents a group of proteins that is involved in signal transduction of a variety of cellular stimuli. The JNK subgroup of MAP kinases, also known as stress-activated protein kinases, is activated in response to environmental stresses (28). JNK phosphorylation occurs in cultured rat hepatocytes following hypoxia alone, with further increases upon reoxygenation (13, 29). JNK activation also has been found in the liver after I/R (17–19, 30) and hemorrhagic shock (13). The reduction in JNK activation, as well as p38 and ERK, suggests that HMGB1 accounts, in part, for MAP kinase activation following I/R. Such is not the case for NF-κB, where neutralization increased NF-κB DNA binding activity. Thus, the actions of HMGB1 are selective in liver I/R. It is possible that the enhanced NF-κB activation contributed to the protection that was afforded by HMGB1 neutralization. Although NF-κB activation leads to proinflammatory gene expression in leukocytes and endothelial cells, it is known to protect hepatocytes from cell death (21). In vivo, inhibition of NF-κB after partial hepatectomy results in massive hepatocyte apoptosis associated with impaired liver function and decreased survival (31). Following I/R that is associated with liver transplantation, two peaks of NF-kB activation were observed, and blockade of NF-kB activation worsened the liver injury (22). Further work is required to elucidate the mechanisms that are involved in the differential effects of HMGB1 on MAP kinase and NF-κB. Enhanced NF-κB activation is seen in mice that are protected from hepatic I/R following blockade of the receptor for advanced glycation end product (30).
To study further the mechanism of HMGB1-mediated injury in hepatic I/R, we explored the TLR4 system as a possible signal transduction pathway for extracellular HMGB1. HMGB1, in vitro, interacts with TLR2, TLR4, and the receptor for advanced glycation end products for HMGB1-induced activation of inflammatory signaling (12, 32, 33). The TLRs are one of the components by which the innate immune system senses the invasion of pathogenic microorganisms by recognizing specific molecular patterns that are present in microbial products (34). TLR4 plays a critical role in endotoxin signaling and was shown to participate in the recognition of several endogenous ligands, such as hyaluronic acid, heparin sulfate, fibrinogen, and perhaps, heat shock proteins (35–38). The release of these ligands stimulates inflammatory activity through TLR4 signaling. Our data suggest that the TLR4 system also may play a key role in HMGB1-mediated hepatic I/R injury; TLR4-defective mice who undergo hepatic I/R are not affected by the administration of rHMGB1 or neutralizing antibodies to HMGB1, compared with TLR4-intact mice. We hypothesize that the release of HMGB1 from damaged or necrotic cells may produce an early inflammatory response by activating the TLR4 system. Further, the protection from hepatic I/R in TLR4-defective mice may be due to their diminished ability to respond to HMGB1.
In summary, this study documents the contribution of HMGB1 to hepatic I/R injury. In this model of acute local tissue injury, HMGB1 seems to act as an early mediator of inflammation. This is in clear contrast to sepsis, in which its action is delayed. Our results also suggest that the protective effects of blocking HMGB1 and the detrimental effects of rHMGB1 in hepatic I/R are dependent on the activation of TLR4 signaling. Thus, interventions that inhibit HMGB1 activity may be effective in settings of ischemic liver injury to minimize organ damage and may be useful in other clinical settings that are associated with inflammation and cellular necrosis (39–41).
MATERIALS AND METHODS
Materials.
Polyclonal antibodies against HMGB1 and recombinant HMGB1 were prepared as described previously (10). Polyclonal antibodies against HMGB1 B box were raised in rabbits and titers were determined by immunoblotting. Anti-HMGB1 B box antibodies were affinity-purified by using cyanogens bromide-activated Sepharose beads following standard procedures. Neutralizing activity of anti-HMGB1 was confirmed in HMGB1-stimulated macrophage cultures by assay of TNF release. In the presence of anti-HMGB1 antibody, neutralizing antibody was defined as inhibiting >80% of HMGB1-induced TNF release. Nonimmune rabbit IgG (item I5006) was purchased from Sigma-Aldrich.
Animals.
Male wild-type (C57BL/6; C3H/HeOuj) and TLR4-defective (C3H/Hej) mice (8–12-wk-old) were purchased from Jackson ImmunoResearch Laboratories. Male Sprague-Dawley rats, also pathogen-free and weighing 350–400 g, were obtained from Charles River Laboratories. CD14−/− mice were provided by Mason Freeman (Harvard University, Boston, MA). All animals were maintained in a laminar-flow, specific pathogen-free atmosphere at the University of Pittsburgh. Animal protocols were approved by the Animal Care and Use Committee of the University of Pittsburgh and the experiments were performed in adherence to the National Institutes of Health Guidelines for the Use of Laboratory Animals.
Liver ischemia.
A nonlethal model of segmental (70%) hepatic warm ischemia was used. The I/R protocol was initiated with the abdominal wall being clipped of hair and cleansed with betadine. Under sodium pentobarbital (40 mg/kg, i.p.) and methoxyflurane (inhalation) anesthesia, a midline laparotomy was performed. With the use of an operating microscope, the liver hilum was dissected free of surrounding tissue. All structures in the portal triad (hepatic artery, portal vein, bile duct) to the left and median liver lobes were occluded with a microvascular clamp (Fine Science Tools) for 60 min; reperfusion was initiated by removal of the clamp. This method of segmental hepatic ischemia prevents mesenteric venous congestion by permitting portal decompression through the right and caudate lobes. The abdomen was covered with a sterile plastic wrap to minimize evaporative loss. Throughout the ischemic interval, evidence of ischemia was confirmed by visualizing the pale blanching of the ischemic lobes. The clamp was removed and gross evidence of reperfusion that was based on immediate color change was assured before closing the abdomen with continuous 4–0 polypropelene suture. The absence of ischemic color changes or the lack of response to reperfusion was a criterion for immediate sacrifice and exclusion from further analysis. Temperature was monitored by rectal temperature probe and was maintained at 37°C by means of a warming pad and heat lamp. At the end of the observation period following reperfusion, the mice were anesthetized with inhaled methoxyflurane and were killed by exsanguination.
Experimental design.
Mice received anti-HMGB1 antibody (600 or 60 μg per mouse) or nonimmune control IgG (Sigma-Aldrich) by i.p. injection 1 h before ischemia, or recombinant HMGB1 (10 μg per mouse) or vehicle PBS i.p. immediately after reperfusion. Previous studies showed that anti-HMGB1 antibody at 600 μg/mouse protects against sepsis-induced lethality (10). The HMGB1 that was used for these studies contained 219 pg/μg protein of LPS as measured by the chromogenic Limulus amoebocyte lysate assay (Associates of Cape Cod or BioWhittaker). Sham animals underwent anesthesia, laparotomy, and exposure of the portal triad without hepatic ischemia. Animals were killed at predetermined time points (1–24 h) after reperfusion for serum and liver samples.
Hepatocyte isolation.
Hepatocytes were isolated from normal rats by an in situ collagenase (type IV, Sigma-Aldrich) perfusion technique, modified as described previously (42). Hepatocytes were separated from the nonparenchymal cells by two cycles of differential centrifugation (50 g for 2 min) and further purified over a 30% Percoll gradient. Hepatocyte purity exceeded 98% as assessed by light microscopy, and viability typically was >95% as determined by trypan blue exclusion assay.
Cell culture and treatment.
Hepatocytes (3 × 106) were plated onto 6-cm gelatin-coated plastic tissue culture dishes. The initial culture medium was Williams medium E containing 10% calf serum, 15 mM Hepes, 2 mM L-glutamine, and 100 U/ml of penicillin and streptomycin. Hepatocytes were allowed to attach to plates overnight. The medium was replaced with hypoxic medium (equilibrated with 1% O2, 5% CO2, and 94% N2), and placed into a modular incubator chamber (Billups-Rothenburg), which was flushed with the same hypoxic gas mixture. All cells were washed with ice-cold PBS and lysed with 1× lysis buffer that contained freshly added 0.5 mM PMSF, and protein was extracted as described below.
Isolation of nuclear and cytoplasmic proteins.
Frozen liver tissues were suspended in a buffer that contained 10 mM Tris, pH 7.5, 1.5 mM MgCl2, 10 mM KCl, and 0.1% Triton X-100 and lysed by homogenization. Nuclei were recovered by microcentrifugation at 7,500 rpm for 5 min. The supernatant that contained cytoplasmic and membrane protein was collected and stored at −80°C for Western blot analysis. Nuclear proteins were extracted at 4°C by gently resuspending the nuclei pellet in buffer that contained 20 mM Tris, pH 7.5, 20% glycerol, 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, and 0.1% Triton X-100, followed by 1 h incubation at 4°C with occasional vortexing. After microcentrifugation at 13,000 revolutions/min for 15 min at 4°C, the supernatant that contained nuclear protein was collected. Protein concentration was quantitated with bicinchoninic acid protein assay reagent (Pierce Chemical Co.).
Liver damage assessment.
To assess hepatic function and cellular injury following liver ischemia, sALT levels were measured using the Opera Clinical Chemistry System (Bayer Co.).
Histopathology.
Formalin-fixed liver samples were embedded in paraffin and cut to 6-μm-thick sections. Tissues were stained with hematoxylin-eosin, and slides were assessed for inflammation and tissue damage. The percent necrotic area was estimated by random evaluation of 10 low-power fields (×100) of each hematoxylin-eosin–stained section; the observer was blinded to treatment group.
Immunohistochemical staining.
A segment of the left lobe of the liver was cryoprotected in 2% paraformaldehyde in PBS overnight and then 30% sucrose in PBS for another 24 h. It was then embedded and frozen in optimal cutting temperature compound using liquid nitrogen–cooled isopentane. These samples were stored at −80°C until ready to be cut. To prepare the slides, the samples were cut into 6-μ sections using a cryostat machine and placed onto slides. They were left to dry at room temperature overnight and then stored at −20°C. Immunofluorescent staining for HMGB was begun by first rehydrating the slides with PBS. Then, nonspecific proteins were blocked using 2% BSA for 45 min followed by rinses with 0.5% BSA. This was followed by placing the primary antibody (anti-HMGB1, BD Biosciences, 1:1,000 in 0.5% BSA) for 60 min. The slides were washed with 0.5% BSA and then the secondary antibody (goat anti–rabbit cy3, Jackson ImmunoResearch Laboratories, 1:1,000 in 0.5% BSA) was applied for another 60 min. The slides were rinsed again in BSA and stained for F-actin (phalloidin cy5, Jackson ImmunoResearch Laboratories, 1:250 in PBS) for 30 min. A nuclear stain, hoechst dye (bisbenzimide, 1 mg/100 ml), was applied for 30 s. The slides were rinsed with PBS and a coverslip with gelvatol, a water-soluble mounting media (21 g polyvinylalcohol, 52 ml water, sodium azide, 106 ml 0.2M Tris buffer), was placed. Slides were visualized using an Olympus BX51 epi-fluorescence microscope and digitized with an Olympus color videocamera.
Sodium dodecyl sulfate/polyacrylamide gel electrophoresis and Western blotting.
Western blot analysis for HMGB1, phosphorylated and total kinase forms of ERK, p38, and JNK were performed as described (43). Primary polyclonal rabbit antibody to HMGB1 (1:5,000; BD Biosciences), primary polyclonal antibody to phosphorylated ERK, p38, and JNK (1:1,000; Cell Signaling Technology), and primary polyclonal rat antibody to total ERK, p38, and JNK (1:1,000; Santa Cruz Biotechnology, Inc.) were used for Western blot. Membranes were developed with the Super Signal West Pico chemiluminescent kit (Pierce Chemical Co.) and exposed to film.
SYBR green real-time RT-PCR.
Total RNA was extracted from the liver using the TRIzol reagent (Life Technologies) according to the manufacturer's instruction. mRNA for TNF-α, IL-6, and iNOS, and GAPDH was quantified in duplicate by SYBR Green two-step, real-time RT-PCR. After removal of potentially contaminating DNA with DNase I (Life Technologies), 1 μg of total RNA from each sample was used for RT with an oligo dT (Life Technologies) and a Superscript II (Life Technologies) to generate first-strand cDNA. PCR reaction mixture was prepared using SYBR Green PCR Master Mix (PE Applied Biosystems) using the primers as described previously (44, 45). Thermal cycling conditions were 10 min at 95°C followed by 40 cycles of 95°C for 15 s and 60°C for 1 min on an ABI PRISM 7000 Sequence Detection System (PE Applied Biosystems). Each gene expression was normalized with GAPDH mRNA content.
Electrophoretic mobility shift assays.
NF-κB DNA binding activity was measured by electrophoretic mobility shift assays using nuclear extracts from liver tissues. The NF-κB oligonucleotide (Promega) was based on the NF-κB sequence in the immunoglobulin light-chain enhancer. DNA probes were prepared by end labeling with γ-[32P]dATP (Dupont, Merck Pharmaceuticals) and T4 polynucleotide kinase (Boehringer) and was purified in Tris-EDTA buffer that contained NaCl (100 mM) using G-50 resin columns (Whatman). Typically, 5 μl (5–10 μg) of hepatic nuclear extract were incubated with 100,000 counts/min of 32P-labeled oligonucleotides (0.5 ng) for 1–2 h at room temperature in a buffer that contained 10 mM Tris, pH 7.6, 10% glycerol, 1 mM EDTA, 1 mg/ml BSA, and 0.2% Nonidet P-40. Additionally, 2–4 μg of poly(dI-dC) (Boehringer) were added to the mixture as nonspecific competitor DNA. Protein-DNA complexes were resolved on 4% nondenaturing polyacrylamide gels in 0.4× running buffer that contained 450 mM Tris borate and 1 μM EDTA, pH 8.0. Gels were dried after electrophoresis and subjected to autoradiography.
Statistical analysis.
Results are expressed as the mean ± SEM. Group comparisons were performed using Student's t test or analysis of variance. Differences were considered significant at P < 0.05.
Acknowledgments
This work was supported by American College of Surgeons Resident Scholarship (to A. Tsung), National Institutes of Health grant nos. R01-GM37631 and R01-GM53789 (to M.P. Fink), and National Institutes of Health grant nos. R01-GM52021 (to D.A Geller) and R01-GM50441 and P50-GM53789 (to T.R. Billiar).
K.J. Tracey and M.P. Fink are cofounders of Critical Therapeutics, a biotechnology company that is developing antibodies against HMGB1 as therapeutic agents. The authors have no other potential conflicting financial interests.
Abbreviations used: ERK, extracellular signal-regulated kinase; HMGB1, high-mobility group box 1; iNOS, inducible NO synthase; I/R, ischemia/reperfusion; JNK, c-Jun NH2-terminal kinase; MAP, mitogen-activated protein; rHMGB1, recombinant HMGB1; sALT, serum alanine aminotransferase; TLR, Toll-like receptor.
References
- 1.Selzner, N., H. Rudiger, R. Graf, and P.A. Clavien. 2003. Protective strategies against ischemic injury of the liver. Gastroenterology. 125:917–936. [DOI] [PubMed] [Google Scholar]
- 2.Fondevila, C., R.W. Busuttil, and J.W. Kupiec-Weglinski. 2003. Hepatic ischemia/reperfusion injury—a fresh look. Exp. Mol. Pathol. 74:86–93. [DOI] [PubMed] [Google Scholar]
- 3.Clavien, P.A., H.A. Rudiger, and M. Selzner. 2001. Mechanism of hepatocyte death after ischemia: apoptosis versus necrosis. Hepatology. 33:1555–1557. [DOI] [PubMed] [Google Scholar]
- 4.Kohli, V., M. Selzner, J.F. Madden, C. Bentley, and P.A. Clavien. 1999. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia-reperfusion injury in the rat liver. Transplantation. 67:1099–1105. [DOI] [PubMed] [Google Scholar]
- 5.Fan, C., R.M. Zwacka, and J.F. Engelhardt. 1999. Therapeutic approaches for ischemia/reperfusion injury in the liver. J. Mol. Med. 77:577–592. [DOI] [PubMed] [Google Scholar]
- 6.Jaeschke, H., and J.J. Lemasters. 2003. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 125:1246–1257. [DOI] [PubMed] [Google Scholar]
- 7.Jaeschke, H. 2003. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am. J. Physiol. Gastrointest. Liver Physiol. 284:G15–G26. [DOI] [PubMed] [Google Scholar]
- 8.Mavier, P., A.M. Preaux, B. Guigui, M.C. Lescs, E.S. Zafrani, and D. Dhumeaux. 1988. In vitro toxicity of polymorphonuclear neutrophils to rat hepatocytes: evidence for a proteinase-mediated mechanism. Hepatology. 8:254–258. [DOI] [PubMed] [Google Scholar]
- 9.Wang, H., O. Bloom, M. Zhang, J.M. Vishnubhakat, M. Ombrellino, J. Che, A. Frazier, H. Yang, S. Ivanova, L. Borovikova, et al. 1999. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–251. [DOI] [PubMed] [Google Scholar]
- 10.Yang, H., M. Ochani, J. Li, X. Qiang, M. Tanovic, H.E. Harris, S.M. Susarla, L. Ulloa, H. Wang, R. DiRaimo, et al. 2004. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. USA. 101:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaffidi, P., T. Misteli, and M.E. Bianchi. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418:191–195. [DOI] [PubMed] [Google Scholar]
- 12.Park, J.S., D. Svetkauskaite, Q. He, J. Kim, D. Strassheim, A. Ishizaka, and E. Abraham. 2004. Involvement of Toll-like receptors 2 and 4 in cellular activation by high-mobility group box 1 protein. J. Biol. Chem. 279:7370–7377. [DOI] [PubMed] [Google Scholar]
- 13.McCloskey, C.A., M.V. Kameneva, A. Uryash, D.J. Gallo, and T.R. Billiar. 2004. Tissue hypoxia activates JNK in the liver during hemorrhagic shock. Shock. 22:380–386. [DOI] [PubMed] [Google Scholar]
- 14.Colletti, L.M., S.L. Kunkel, A. Walz, M.D. Burdick, R.G. Kunkel, C.A. Wilke, and R.M. Strieter. 1996. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 23:506–514. [DOI] [PubMed] [Google Scholar]
- 15.Wanner, G.A., P.E. Muller, W. Ertel, M. Bauer, M.D. Menger, and K. Messmer. 1999. Differential effect of anti-TNF-alpha antibody on proinflammatory cytokine release by Kupffer cells following liver ischemia and reperfusion. Shock. 11:391–395. [PubMed] [Google Scholar]
- 16.Lee, V.G., M.L. Johnson, J. Baust, V.E. Laubach, S.C. Watkins, and T.R. Billiar. 2001. The roles of iNOS in liver ischemia-reperfusion injury. Shock. 16:355–360. [DOI] [PubMed] [Google Scholar]
- 17.Tsung, A., T. Kaisu, A. Nakao, L. Shao, B. Bucher, M.P. Fink, N. Murase, and D.A. Geller. 2005. Ethyl pyruvate ameliorates liver ischemia-reperfusion injury by decreasing hepatic necrosis and apoptosis. Transplantation. 79:196–204. [DOI] [PubMed] [Google Scholar]
- 18.Bendinelli, P., R. Piccoletti, P. Maroni, and A. Bernelli-Zazzera. 1996. The MAP kinase cascades are activated during post-ischemic liver perfusion. FEBS Lett. 398:193–197. [DOI] [PubMed] [Google Scholar]
- 19.Bradham, C.A., R.F. Stachlewitz, W. Gao, T. Qian, S. Jayadev, G. Jenkins, Y. Hannun, J.J. Lemasters, R.G. Thurman, and D. Brenner. 1997. Reperfusion after liver transplantation in rats differentially activates the mitogen-activated protein kinases. Hepatology. 25:1128–1135. [DOI] [PubMed] [Google Scholar]
- 20.Yoshidome, H., A. Kato, M.J. Edwards, and A.B. Lentsch. 1999. Interleukin-10 suppresses hepatic ischemia/reperfusion injury in mice: implications of a central role for nuclear factor kappaB. Hepatology. 30:203–208. [DOI] [PubMed] [Google Scholar]
- 21.Hitano, E., B.L. Bennett, A.M. Manning, T. Qian, J.J. Lemasters, and D.A. Brenner. 2001. NF-κB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-α- and Fas-mediated apoptosis. Gastroenterology. 120:1251–1262. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi Y., R.W. Ganster, A. Gambotto, L. Shao, T. Kaizu, T. Wu, G.P. Yagnik, A. Nakao, G. Tsoulfas, T. Ishikawa, et al. 2002. Role of NF-kappa on liver cold ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G1175-G1184. [DOI] [PubMed] [Google Scholar]
- 23.Andersson, U., H. Wang, K. Palmblad, A. Aveberger, O. Bloom, H. Erlandsson-Harris, A. Janson, R. Kokkola, M. Zhang, H. Yang, and K.J. Tracey. 2000. High Mobility Group 1 Protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 192:565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degryse, B., T. Bonaldi, P. Scaffidi, S. Muller, M. Resnati, F. Sanvito, G. Arrigoni, and M.E. Bianchi. 2001. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell Biol. 152:1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiuza, C., M. Bustin, S. Talwar, M. Tropea, E. Gerstenberger, J.H. Shelhamer, and A.F. Suffredini. 2003. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 101:2652–2660. [DOI] [PubMed] [Google Scholar]
- 26.Sappington, P.L., R. Yang, H. Yang, K.J. Tracey, R.L. Delude, and M.P. Fink. 2002. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 123:790–802. [DOI] [PubMed] [Google Scholar]
- 27.Park, J.S., J. Arcaroli, H.K. Yum, H. Yang, H. Wang, K.Y. Yang, K.H. Choe, D. Strassheim, T.M. Pitts, K.J. Tracey, and E. Abraham. 2003. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am. J. Physiol. Cell Physiol. 284:C870–C879. [DOI] [PubMed] [Google Scholar]
- 28.Whitmarsch, A.J., and R.J. Davis. 1996. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 74:589–607. [DOI] [PubMed] [Google Scholar]
- 29.Crenesse, D., A. Schmid-Alliana, M. Laurens, R. Cursio, and J. Gugenheim. 2001. JNK(1)/SAPK(1) involvement in hypoxia-reoxygenation-induced apoptosis in rat hepatocytes. Transplant. Proc. 33:260–261. [DOI] [PubMed] [Google Scholar]
- 30.Zeng, S., N. Feirt, M. Goldstein, J. Guarrera, N. Ippagunta, U. Ekong, H. Dun, Y. Lu, W. Qu, A. Schmidt, and J.S. Emond. 2004. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology. 39:422–432. [DOI] [PubMed] [Google Scholar]
- 31.Iimuro, Y., T. Nishiura, C. Hellerbrand, K.E. Behrns, R. Schoonhoven, J.W. Grisham, and D.A. Brenner. 1998. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. J. Clin. Invest. 101:802–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huttunen, H.J., J. Kuja-Panula, and H. Rauvala. 2002. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J. Biol. Chem. 277:38635–38646. [DOI] [PubMed] [Google Scholar]
- 33.Huttunen, H.J., J. Kuja-Panula, G. Sorci, A.L. Agneletti, R. Donato, and H. Rauvala. 2000. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 275:40096–40105. [DOI] [PubMed] [Google Scholar]
- 34.Akira, S., and K. Takeda. 2004. Toll-like receptor signaling. Nature. 4:499–511. [DOI] [PubMed] [Google Scholar]
- 35.Termeer, C., F. Benedix, J. Sleeman, C. Fieber, U. Voith, T. Ahrens, K. Miyake, M. Freudenberg, C. Galanos, and J.C. Simon. 2002. Oligosaccharides of hyaluronan activate dendritic cells via Toll-like receptor 4. J. Exp. Med. 195:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson, G.B., G.J. Brunn, Y. Kodaira, and J.L. Platt. 2002. Receptor-mediated monitoring of tissue well-being via detection of soluble heparin sulfate by Toll-like receptor 4. J. Immunol. 168:5233–5239. [DOI] [PubMed] [Google Scholar]
- 37.Smiley, S.T., J.A. King, and W.W. Hancock. 2001. Fibrinogen stimulates macrophage chemokines secretion through Toll-like receptor 4. J. Immunol. 167:2887–2894. [DOI] [PubMed] [Google Scholar]
- 38.Vabulas, R.M., P. Ahmad-Nejad, S. Ghose, C.J. Kirschning, R.D. Issels, and H. Wagner. 2002. HSP70 as endogenous stimulus of the Toll/Interleukin-1 receptor signal pathway. J. Biol. Chem. 277:15107–15112. [DOI] [PubMed] [Google Scholar]
- 39.DeMarco, R.A., M.P. Fink, and M.T. Lotze. 2004. Monocytes promote natural killer cell interferon gamma production in response to the endogenous danger signal HMGB1. Mol. Immunol. 42:433-444. [DOI] [PubMed] [Google Scholar]
- 40.Lotze, M.T., and R.A. DeMarco. 2003. Dealing with death: HMGB1 as a novel target for cancer therapy. Curr. Opin. Investig. Drugs. 4:1405–1409. [PubMed] [Google Scholar]
- 41.Lotze, M.T., and K.J. Tracey. 2005. HMGB1: nuclear weapon in the immune arsenal. Nat. Rev. Immunol. In press. [DOI] [PubMed] [Google Scholar]
- 42.West, M.A., T.R. Billiar, R.D. Curran, B.J. Hyland, and R.L. Simmons. 1989. Evidence that rat Kupffer cells stimulate and inhibit hepatocyte protein synthesis in vitro by different mechanism. Gastroenterology. 96:1572–1582. [DOI] [PubMed] [Google Scholar]
- 43.Morris, S.M., Jr., D. Kepka-Lenhart, and L.C. Chen. 1998. Differential regulation of arginases and inducible nitric oxide synthase in murine macrophage cells. Am. J. Physiol. 275:E740–E747. [DOI] [PubMed] [Google Scholar]
- 44.Overbergh, L., D. Valckx, M. Waer, and C. Mathieu. 1999. Quantification of murine cytokine mRNAs using real time quantitative reverse transcriptase PCR. Cytokine. 11:305–312. [DOI] [PubMed] [Google Scholar]
- 45.Moore, B.A., L.E. Otterbein, A. Turler, A.M. Choi, and A.J. Bauer. 2003. Inhaled carbon monoxide suppresses the development of postoperative ileus in the murine small intestine. Gastroenterology. 124:377–391. [DOI] [PubMed] [Google Scholar]