

Abstract
The mechanisms of tumor metastasis to the sentinel lymph nodes are poorly understood. Vascular endothelial growth factor (VEGF)-A plays a principle role in tumor progression and angiogenesis; however, its role in tumor-associated lymphangiogenesis and lymphatic metastasis has remained unclear. We created transgenic mice that overexpress VEGF-A and green fluorescent protein specifically in the skin, and subjected them to a standard chemically-induced skin carcinogenesis regimen. We found that VEGF-A not only strongly promotes multistep skin carcinogenesis, but also induces active proliferation of VEGF receptor-2–expressing tumor-associated lymphatic vessels as well as tumor metastasis to the sentinel and distant lymph nodes. The lymphangiogenic activity of VEGF-A–expressing tumor cells was maintained within metastasis-containing lymph nodes. The most surprising finding of our study was that even before metastasizing, VEGF-A–overexpressing primary tumors induced sentinel lymph node lymphangiogenesis. This suggests that primary tumors might begin preparing their future metastatic site by producing lymphangiogenic factors that mediate their efficient transport to sentinel lymph nodes. This newly identified mechanism of inducing lymph node lymphangiogenesis likely contributes to tumor metastasis, and therefore, represents a new therapeutic target for advanced cancer and/or for the prevention of metastasis.
Angiogenesis, the growth of new blood vessels from preexisting vessels, is essential for tumor growth (1). Vascular endothelial growth factor (VEGF)-A has been identified as the predominant tumor angiogenesis factor in the majority of human and experimental murine cancers (2), acting via VEGF receptor (VEGFR)-1 and VEGFR-2. Recent studies in mouse tumor models, however, have suggested that some types of malignant tumors also induce the formation of lymphatic vessels (lymphangiogenesis), and that this process promotes tumor spread to regional lymph nodes (3–5). Moreover, tumor lymphangiogenesis has been identified as a novel prognostic factor for determining the risk for human cutaneous melanomas to become metastatic (6).
In contrast to the detailed characterization of the molecular mechanisms that control (blood vascular) angiogenesis, the mechanisms underlying the pathologic growth and function of the lymphatic vascular system have remained poorly understood. Thus far, VEGF-C and VEGF-D are the only known lymphangiogenesis factors—acting predominantly through activation of VEGFR-3—which is expressed by lymphatic endothelium in vivo and in vitro (7). Several studies in animal tumor models have provided direct experimental evidence that increased levels of VEGF-C or VEGF-D promote tumor lymphangiogenesis and tumor spread via the lymphatic vessels to regional lymph nodes. Furthermore, these effects can be suppressed by blocking VEGFR-3 signaling (3–5, 8–10). Other factors, however, are likely to be involved in mediating tumor lymphangiogenesis and metastasis to the lymph nodes (11).
Generally, VEGF-A has been considered to only promote (blood vascular) angiogenesis; however, recent work from several laboratories revealed that VEGF-A promotes human lymphatic endothelial cell proliferation and migration in vitro (12–15), and that lymphatic endothelium expresses VEGFR-2 in situ and in vitro (12, 13, 16–18). Moreover, intradermal injection of mice with an adenoviral vector engineered to express mouse VEGF-A, but not with adenoviral human VEGF-A (18), resulted in the production of enlarged, proliferating lymphatic vessels (16). Our recent work has shown that chronic transgenic delivery of VEGF-A to mouse skin promoted lymphangiogenesis associated with chronic skin inflammation (19) and with cutaneous tissue repair (13).
To investigate whether VEGF-A also might be involved in mediating tumor lymphangiogenesis and metastasis to the lymph node, we generated a new transgenic mouse model to target expression of GFP to normal and malignant epidermal keratinocytes. GFP expression was controlled by a keratin 14 (K14) promoter-driven transgene cassette. This model enables the sensitive detection and quantification of lymph node metastasis during experimental, multi-step skin carcinogenesis. We then crossed these mice with our previously established K14/VEGF-A transgenic mice (20, 21), and subjected the offspring, which produce fluorescent keratinocytes in the skin that also overexpress VEGF-A, to a chemically induced, multi-step skin carcinogenesis regimen (22, 23).
We found that targeted overexpression of VEGF-A potently induced tumor lymphangiogenesis in cutaneous squamous cell carcinoma (SCC) and promoted tumor metastasis to sentinel lymph nodes. Importantly, VEGF-A–expressing tumor cells maintained their lymphangiogenic activity after metastasis to lymph nodes. Surprisingly, VEGF-A–overexpressing cutaneous SCC induced lymphangiogenesis in sentinel lymph nodes, even before the tumors had metastasized to these tissues. Together, these results identify VEGF-A as a novel tumor lymphangiogenesis factor. They also indicate that VEGF-A–induced lymph node lymphangiogenesis promotes metastasis, and therefore, is an important target for the treatment of advanced cancer and the prevention of metastasis.
RESULTS
Transgenic expression of GFP in the skin enables detection and quantification of SCC metastasis to lymph nodes
To facilitate detection and quantification of skin cancer metastases, we created a transgenic mouse model to selectively express GFP in epidermal keratinocytes, using a K14 promoter cassette (Fig. 1 A). Three transgenic founder lines expressed high levels of GFP in the skin; targeted expression of the K14/GFP transgene in the basal epidermal keratinocyte layer and in outer root sheath keratinocytes of hair follicles was confirmed by fluorescence microscopy (Fig. 1 B). Induction of epidermal hyperplasia by topical application of PMA to the ear skin of transgenic mice resulted in strong GFP expression throughout the hyperplastic epidermis at 1 and 4 d afterward (Fig. 1 C, D).
Figure 1.
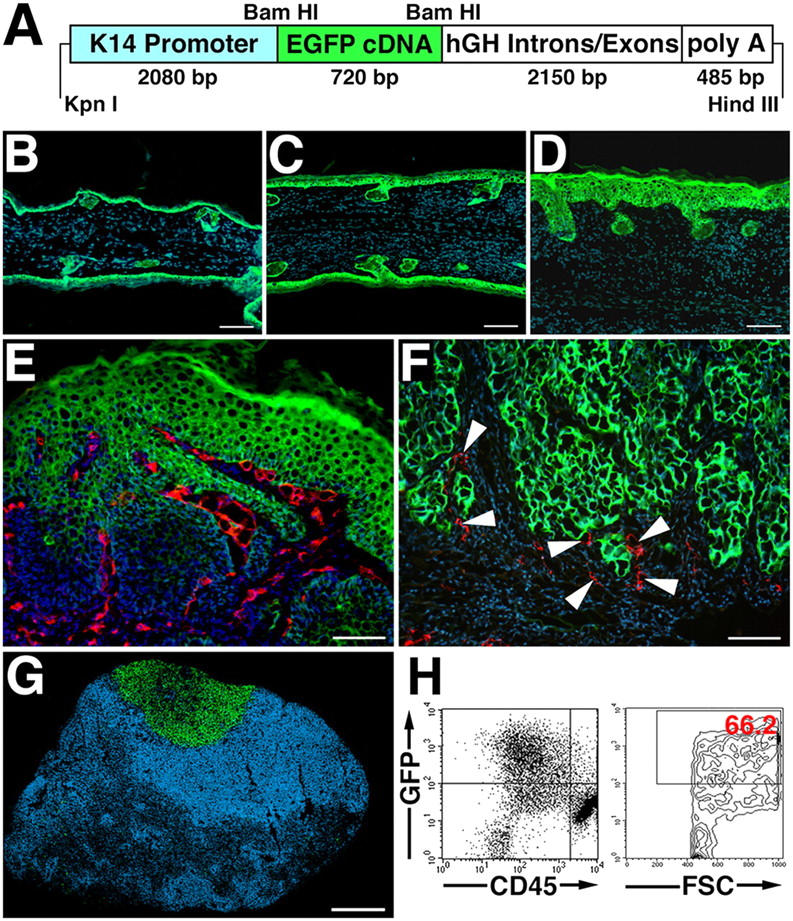
K14/GFP transgenic mice provide a reliable new model for the detection of skin cancer metastases. (A) Schematic representation of the K14-GFP transgenic construct. A 720-bp enhanced GFP (EGFP) cDNA fragment was ligated to the BamHI restriction site of the keratin 14 promoter cassette. (B) Targeted expression of the K14/GFP transgene in the basal epidermal keratinocyte layer and in outer root sheath keratinocytes of hair follicles was confirmed by fluorescence microscopy (green). (C and D) Induction of epidermal hyperplasia by topical application of PMA resulted in strong GFP expression throughout the epidermis. Nuclei are labeled blue (Hoechst stain). (E and F) High levels of GFP expression (green) were maintained in tumor cells of papillomas (E) and of SCC (F) induced by the chemical skin carcinogenesis regimen. Immunostains for the panvascular marker CD31 (E; red) revealed tumor-associated angiogenesis, whereas immunostains for the lymphatic-specific marker LYVE-1 (F; red; arrowheads) demonstrated lymphangiogenesis in close association with tumor cells at the advancing tumor edge. (G and H) Metastatic tumor cells in sentinel lymph nodes of K14/GFP transgenic mice maintained high levels of GFP expression, enabling simple detection of lymph node metastasis by fluorescence microscopy (G, green) and quantification of GFP-positive and CD45-negative metastatic tumor cells (66.2% of all cells in this representative case) by FACS analysis of single cell suspensions (H). Bars, 100 μm (B–F), 500 μm (G).
To investigate whether GFP expression also was maintained during the successive stages of multi-step skin carcinogenesis and metastasis, the K14/GFP transgenic line that expressed the highest level of GFP was subjected to a standard 7,12-dimethylbenzanthracene (DMBA)/PMA chemical skin carcinogenesis regimen. This involved a single dose of DMBA to initiate tumor formation, followed by 20 weekly applications of the tumor promoter PMA. High levels of GFP expression were detected in all of the cells of early benign papillomas that already displayed marked tumor angiogenesis, as detected by stains for the vascular marker CD31, adjacent to the GFP-expressing tumor cells (Fig. 1 E). Similarly, SCC cells maintained strong GFP expression, and immunostains for the lymphatic-specific marker lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1) demonstrated lymphangiogenesis in close association with tumor cells at the advancing tumor edge (Fig. 1 F). Importantly, metastatic tumor cells in sentinel lymph nodes of K14/GFP transgenic mice also maintained strong GFP expression, enabling the simple detection of metastasis to the lymph node by fluorescence microscopy (Fig. 1 G).
We investigated whether the extent of metastasis to the lymph node might be quantified by flow cytometry analysis of homogenized lymph nodes. GFP-positive/CD45-negative tumor cells were easily detectable and represented up to ∼66.2% of all sorted lymph node cells (Fig. 1 H). Together, these results established the K14/GFP transgenic model as a suitable model for comparative studies of skin cancer metastasis.
Accelerated and increased skin carcinogenesis in VEGF-A transgenic mice
To investigate the effects of VEGF-A on tumor progression and metastasis, we crossed our line of K14/GFP mice that expressed the highest level of GFP with previously established and characterized heterozygous K14/VEGF-A transgenic mice (20). This led to the establishment of K14/GFP and K14/GFP/VEGF-A transgenic mice. These mice were then subjected to the skin carcinogenesis regimen described above. VEGF-A transgenic mice showed accelerated formation of skin papillomas, with an average latency period of 6 wk after the first PMA application, as compared with 12 wk for wild-type mice (Fig. 2 A). By 7 wk, 100% of VEGF-A transgenic mice had developed papillomas, whereas none of the wild-type mice had developed any visible tumor at this time point (Fig. 2 A). The number of papillomas also was increased dramatically in VEGF-A transgenic mice. After 20 wk of PMA treatment, VEGF-A overexpression resulted in the formation of more than 40 papillomas per mouse, as compared with less than 7 papillomas that formed, on the average, in wild-type mice (P < 0.001; Fig. 2 B).
Figure 2.
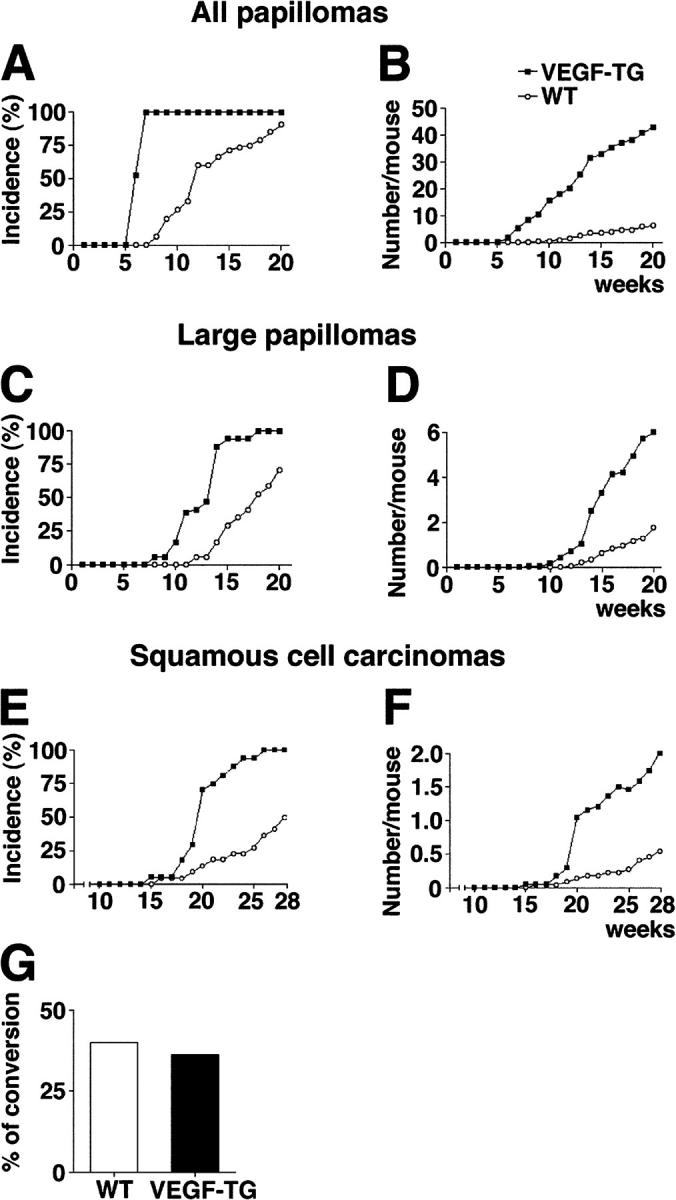
Accelerated and increased skin carcinogenesis in VEGF-A transgenic mice. (A) Accelerated development of skin papillomas in VEGF-A transgenic mice (n = 34; filled squares), as compared with wild-type mice (n = 31; open circles). Incidence is expressed as the percentage of mice with detectable papillomas (>1 mm) during the 20 wk of topical PMA application. (B) Significant increase in frequency of papilloma formation in VEGF-A transgenic mice, expressed as the average number of papillomas per mouse. P < 0.01 at wk 6, P < 0.001 from wk 7 to 20. (C) Accelerated development of large papillomas (>3 mm) in VEGF-A transgenic mice. (D) Increased frequency of large papillomas in VEGF-A transgenic mice. P < 0.01 from wk 11 to 13; P < 0.001 from wk 14 to 20. (E) Increased incidence of SCC in VEGF-A transgenic mice. (F) Increased number of SCC per mouse in VEGF-A transgenic mice. P < 0.01 from wk 19 to 22; P < 0.001 after 23 wk. (G) Comparable ratio of malignant conversion of large papillomas into SCC in VEGF-A transgenic and wild-type mice.
Similar, although less pronounced, effects were found when only larger papillomas (diameter >3 mm) were evaluated. In VEGF-A transgenic mice, large papillomas developed 4 wk earlier than in wild-type mice (Fig. 2 C), and the average number of large papillomas was increased by 3.4-fold in VEGF-A transgenic mice (Fig. 2 D; P < 0.001). The average latency period for the development of malignant SCC was 20 wk in VEGF-A transgenic mice, as compared with 28 wk in wild-type mice (Fig. 2 E). Furthermore, all of the VEGF-A transgenic mice had developed SCC by 28 wk after carcinogen treatment, whereas only 50% of the wild-type mice (Fig. 2 E) developed SCC by this time point. The average number of SCC was increased by 3.6-fold in VEGF-A transgenic mice (Fig. 2 F; P < 0.001). No significant differences in the ratio of malignant conversion of large papillomas to SCC were found between the two genotypes (Fig. 2 G), and no papillomas or carcinomas were observed in wild-type or VEGF-A transgenic mice treated with DMBA or with PMA alone (data not depicted).
Increased tumor lymphangiogenesis and angiogenesis in VEGF-A transgenic mice
We next investigated the effects of chronic VEGF-A overexpression on the tumor-associated formation of lymphatic and blood vessels, which can be detected with antibodies against LYVE-1 and CD31. We found that vascularization increased in papillomas and SCC in both genotypes, compared with their PMA-treated normal skin (Fig. 3 A, B); however, tumor angiogenesis was more prominent in VEGF-A transgenic mice (Fig. 3, D and F, and Fig. 5 B) than in wild-type mice (Fig. 3, C and E, and Fig. 5 A). Surprisingly, the benign papillomas that developed in VEGF-A transgenic mice also had increased numbers of greatly enlarged peritumoral lymphatic vessels, and peritumoral lymphangiogenesis was even more pronounced in the SCCs of VEGF-A transgenic mice, compared with tumors of wild-type mice (Fig. 3 C–F). Double-immunofluorescence analysis of SCC for the proliferation marker BrdU and for LYVE-1 revealed numerous proliferating lymphatic endothelial cells in VEGF-A transgenic mice (Fig. 3 H). Proliferating lymphatic endothelial cells were observed only occasionally in wild-type mice (Fig. 3 G). So VEGF-A seems to promote tumor lymphangiogenesis.
Figure 3.
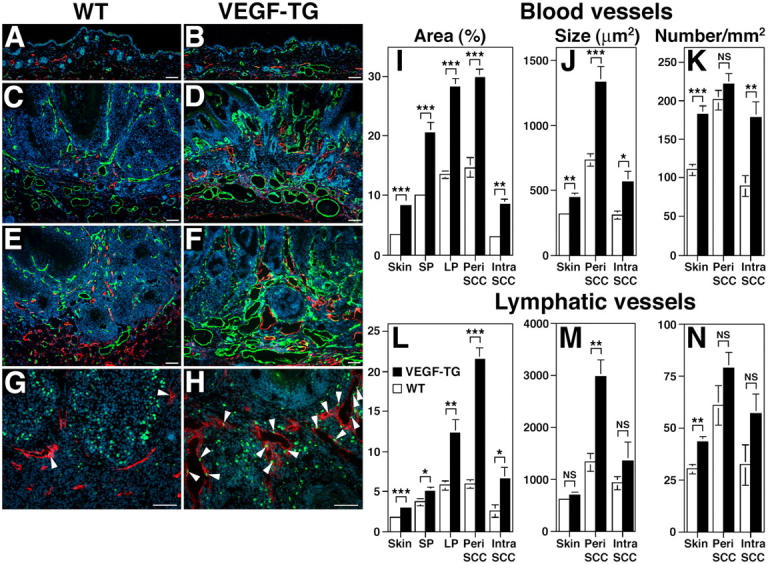
Enhanced tumor angiogenesis and lymphangiogenesis in VEGF-A transgenic mice. Immunofluorescence analysis with antibodies against CD31 (green) and LYVE-1 (red) of PMA-treated skin (A and B), early papillomas (C and D) and SCC (E and F) of wild-type (A,C,E) and VEGF-A transgenic mice (B,D,F) demonstrate highly increased vascularization of papillomas and SCC in both genotypes, as compared with PMA-treated skin. Tumor angiogenesis and lymphangiogenesis were more prominent in VEGF-A transgenic mice (D and F) than in wild-type mice (C and E). Note enlargement of blood vessels (green) and lymphatic vessels (red) in VEGF-A transgenic mice. (G and H) Double-immunofluorescence analysis of SCC for the proliferation marker BrdU (green; arrowheads) and the lymphatic marker LYVE-1 (red) revealed numerous proliferating lymphatic endothelial cells in VEGF-A transgenic mice (H), as compared with only occasional proliferating lymphatic endothelial cells observed in wild-type mice (G). This indicates that VEGF-A promotes tumor lymphangiogenesis. Nuclei are labeled blue (Hoechst stain). Scale bars = 100 μm. (I–N) Computer- assisted morphometric analysis of normal cutaneous and of tumor-associated lymphatic and blood vessels. (I) Significant increase of the relative area occupied by blood vessels in the peritumoral area (Peri SCC) as well as within SCC (Intra SCC), in VEGF-A transgenic mice (filled bars), as compared with wild-type mice (open bars). (J) The average blood vessel size was increased significantly in the intratumoral and the peritumoral areas of SCC in VEGF-A transgenic mice, as compared with wild-types, whereas the blood vessel density only was increased in the intratumoral areas (K). (L) Significant increase of the relative area occupied by lymphatic vessels in VEGF-A transgenic mice throughout all stages of skin carcinogenesis. (M) Significantly increased lymphatic vessel size in the peritumoral area of SCC, but not in the intratumoral area of VEGF-A transgenic mice. (N) No significant differences were found in tumor-associated lymphatic vessel density between the two genotypes. Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 5.
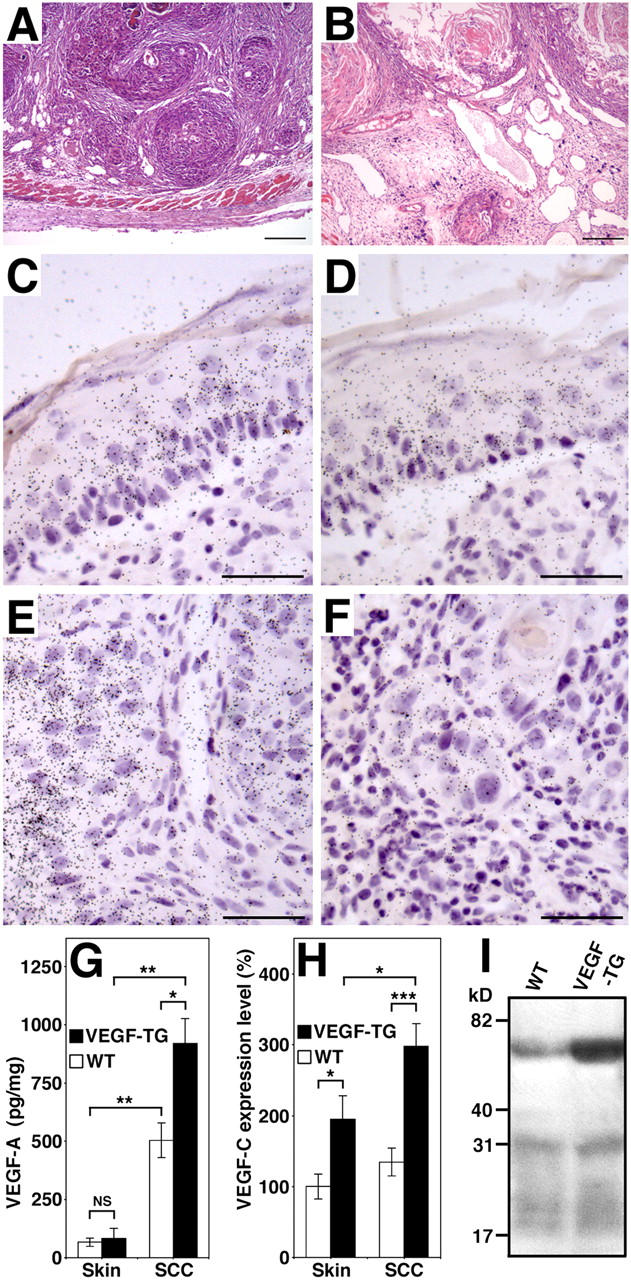
Expression of VEGF-A and VEGF-C by SCC of VEGF-A transgenic mice. (A) Representative histologic image (hematoxylin-eosin stain) of an SCC from a wild-type mouse shows nests of tumor cells surrounded by compact stroma that contain small vessels. (B) SCC observed in VEGF-A transgenic mice were characterized by tissue edema and vessel enlargement. Scale bars = 200 μm (A and B). (C–F) In situ hybridization for VEGF-A and VEGF-C mRNA expression in SCC of VEGF-A transgenic mice. Normal epidermal keratinocytes of the overlying epidermis (C) and SCC tumor cells (E) express high levels of VEGF-A mRNA, whereas stromal cells show little or no expression. Occasionally, low levels of focal VEGF-C expression were observed in the overlying epidermis (D) and in SCC cells (F). Scale bars = 50 μm (C–F). (G) ELISA analysis of VEGF-A expression in skin and tumor lysates (n = 5 per group) revealed significantly increased levels of VEGF-A protein in SCC of both genotypes, as compared with normal skin. VEGF-A levels were higher in VEGF-A transgenic mice than in wild-type mice. (H) Quantitative real-time TaqMan RT-PCR demonstrated increased levels of VEGF-C mRNA expression in the skin and in SCC of VEGF-A transgenic mice compared with wild-type mice. VEGF-C expression was increased significantly in SCC of VEGF-A transgenic mice, as compared with normal skin. *P < 0.05; **P < 0.01; ***P < 0.001. (I) Immunoprecipitation of SCC lysates reveals increased levels of the 58-kD VEGF-C propeptide in VEGF-A transgenic tumors, as compared with wild-type tumors. No major differences in the proteolytic processing of VEGF-C protein were found between the two genotypes. Results are representative for five independent experiments.
Computer-assisted morphometric analysis confirmed that the relative area occupied by CD31-positive vessels (blood vessels), as well as the average size of blood vessels, was increased significantly (P < 0.001) in the peritumoral area of VEGF-A transgenic mice—compared with wild-type mice—throughout the successive stages of skin carcinogenesis (Fig. 3 I, J). No significant difference was detected, however, in the number of blood vessels between the genotypes (Fig. 3 K). Intratumoral blood vessels also were increased in size and number in VEGF-A transgenic mice (P < 0.001; Fig. 3 I–K).
Importantly, we observed that the size of peritumoral lymphatic vessels increased in the SCCs that formed in VEGF-A transgenic mice, as compared with wild-type mice (P < 0.01; Fig. 3 M). This resulted in a significant increase in the relative peritumoral area occupied by lymphatic vessels (P < 0.001; Fig. 3 L), whereas no significant difference was observed in the number of lymphatic vessels (Fig. 3 N). Similarly, the relative intratumoral area occupied by lymphatic vessels was increased in VEGF-A transgenic mice (Fig. 3 L).
Tumor-associated lymphatic vessels of VEGF-A transgenic mice express VEGFR-2
We further characterized tumor-associated lymphangiogenesis in VEGF-A transgenic mice by evaluating expression levels of other vessel-associated factors. In benign papillomas, CD31-positive blood vessels frequently were found to invade tumor nests, whereas LYVE-1–positive lymphatic vessels remained within the peritumoral stroma (Fig. 4 A). In contrast, lymphatic vessels frequently were found within malignant SCC (Fig. 4 B). Intratumoral LYVE-1–positive lymphatic vessels in SCCs of VEGF-A transgenic mice also expressed VEGFR-2 (Fig. 4 C–E). which indicated that they were responsive to VEGF-A stimulation. All LYVE-1–positive tumor-associated vessels also expressed the lymphatic-specific transcription factor Prox1 (Fig. 4 F), which confirmed their lymphatic identity. GFP-expressing SCC cells in VEGF-A transgenic mice sometimes were detected attached to LYVE-1–positive lymphatics (Fig. 4 G).
Figure 4.
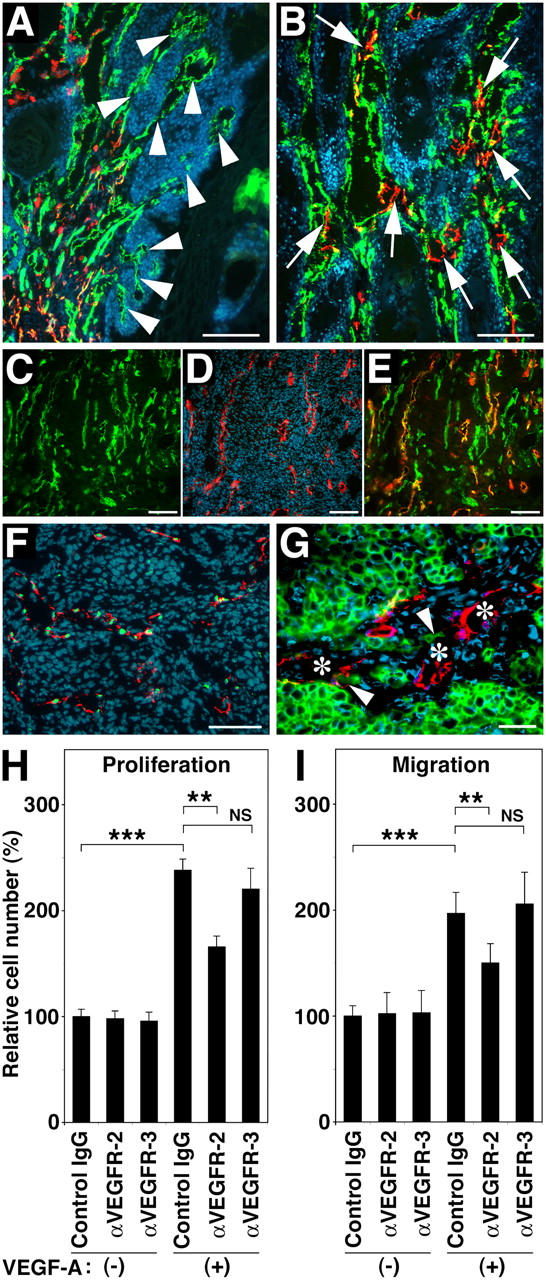
Formation of tumor-associated lymphatic vessels in VEGF-A transgenic mice. (A and B) Double immunofluorescence stains for CD31 (green) and LYVE-1 (red) reveal initial invasion of angiogenic blood vessels (green; arrowheads), but not of lymphatic vessels (red) into benign papillomas (A), whereas intratumoral blood vessels often are accompanied by newly formed lymphatic vessels (arrows) in primary cutaneous SCC (B) of VEGF-A transgenic mice. (C–E) Intratumoral LYVE-1–positive lymphatic vessels (D; red) in SCC express VEGFR-2 (C; green; E = merged image; orange/yellow). Green vessels in panel E are LYVE-1-negative blood vessels. (F) All tumor-associated LYVE-1–positive vessels (red) also express the lymphatic-specific transcription factor Prox1 (green). (G) GFP-expressing SCC cells (arrowheads) were found attached to LYVE-1–positive lymphatic vessels (red; asterisks). Nuclei are labeled blue (Hoechst stain). Scale bars = 200 μm (A and B); 100 μm (C–F); 50 μm (G). (H) VEGF-A induced LEC proliferation is inhibited significantly by antibody blockade of VEGFR-2, but not of VEGFR-3. (I) Blockade of VEGFR-2, but not of VEGFR-3, inhibits VEGF-A–induced LEC migration. Data are expressed as mean ± SD (n = 5 per group) and are representative for three independent experiments. **P < 0.01; ***P < 0.001.
We also investigated the mechanisms by which VEGF-A induces tumor lymphangiogenesis. To assess possible direct effects on lymphatic endothelial cell (LEC) proliferation and migration via activation of VEGFR-2 that is expressed by LECs, cultured human LECs were treated with VEGF-A165 in the presence or absence of blocking antibodies against VEGFR-2 or VEGFR-3. VEGF-A promoted LEC proliferation by 2.3-fold (Fig. 4 H). VEGF-A–induced proliferation was inhibited significantly by blockade of VEGFR-2 (P = 0.0011), but not by blockade of VEGFR-3 (Fig. 4 H). Similarly, VEGF-A–induced LEC migration was inhibited strongly by blockade of VEGFR-2 (P = 0.0047), but not of VEGFR-3 (Fig. 4 I). These results suggest that VEGF-A may be able to stimulate lymphangiogenesis directly via VEGFR-2.
It is possible that the tumor-associated lymphangiogenesis that was observed in VEGF-A transgenic mice also could be mediated, in part, by up-regulation of the known lymphangiogenesis factors VEGF-C or VEGF-D. SCC of VEGF-A transgenic mice, as well as the overlying epidermis, expressed high levels of VEGF-A mRNA (Fig. 5 C, E), as evaluated by in situ hybridization. In several tumors and in their overlying epidermis, weak to moderate expression of VEGF-C mRNA also was observed (Fig. 5 D, F). VEGF-C–expressing macrophages were observed only occasionally; this indicated that epidermal keratinocytes and tumor cells are the major source of VEGF-C production during skin carcinogenesis.
Quantitative RT-PCR of RNA isolated from skin and tumor lysates revealed that VEGF-C mRNA expression levels were significantly higher in SCCs of VEGF-A transgenic mice, compared with their normal skin (P = 0.015), and that VEGF-C expression levels were higher in SCCs of VEGF-A transgenic mice than of the SCCs that developed in wild-type mice (P = 0.0002; Fig. 5 H). These results were confirmed by immunoprecipitation of SCC lysates, which revealed increased levels of the 58-kD VEGF-C propeptide in SCCs of VEGF-A transgenic mice (Fig. 5 I). No major differences were observed in the proteolytic processing of VEGF-C protein between wild-type and VEGF-A transgenic tumors, however (Fig. 5 I). Little or no expression of VEGF-D mRNA was observed in all tissues by in situ hybridization, and no major differences of VEGF-D mRNA expression levels between the two genotypes were detected by real-time RT-PCR (data not depicted). ELISA analysis of tumor lysates confirmed that VEGF-A protein levels were higher in SCCs of VEGF-A transgenic mice (918.8 + 108.2 pg/mg) than in wild-type mice (503.9 + 74.1 pg/mg; P = 0.01; Fig. 5 G); this indicated that tumor-released VEGF-A might promote expression of VEGF-C, but not of VEGF-D.
Increased metastasis to the lymph nodes of VEGF-A transgenic mice
We next investigated whether the increased levels of tumor lymphangiogenesis that were observed in the SCCs of VEGF-A transgenic mice were associated with increases in tumor metastasis to regional (sentinel) lymph nodes. In situ hybridization studies confirmed that all metastatic tumors that were found in VEGF-A transgenic mice maintained high levels of VEGF-A mRNA expression (data not depicted). As assessed by fluorescence microscopy, GFP-expressing cells were found in the sentinel lymph nodes of 100% of SCC-bearing VEGF-A transgenic mice, but only in the sentinel lymph nodes of 53.8% of SCC-bearing wild-type mice. Flow cytometry analysis of primary SCC and of sentinel lymph nodes at 8 wk after the first diagnosis of SCC revealed that 5.9 + 2.1% of all sorted lymph node cells that were isolated from SCC-bearing wild-type mice were GFP-positive/CD45-negative metastatic tumor cells (Fig. 6 C, D), compared with 51.54 + 7.45% for the primary SCC (Fig. 6 A, B). In contrast, VEGF-A transgenic mice demonstrated a significant increase in the percentage of GFP-expressing tumor cells within sentinel lymph nodes (44.61 + 4.32%; P < 0.0001; Fig. 6 C, D), whereas the percentage of GFP-positive cells within primary SCC (55.93 + 2.44%; Fig. 6 A, B) was comparable to the findings in wild-type mice. Nontransgenic mice did not show detectable GFP expression in primary lesions or in lymph nodes (Fig. 6 A, C).
Figure 6.

Increased metastasis to the sentinel lymph nodes in VEGF-A transgenic mice. (A and C) Representative flow cytometry analysis of GFP-expressing, CD45-negative cells in primary SCC and in sentinel lymph node (LN). Whereas no GFP-positive cells were found in nontransgenic control mice at 8 wk after the first detection of cutaneous SCC, 53.5% of all SCC-derived cells in K14/GFP transgenic mice and 67.9% in K14/GFP/VEGF-A transgenic mice were GFP-positive (A). In contrast, the percentage of GFP-expressing tumor cells was significantly higher in metastases to the sentinel lymph nodes of K14/GFP/VEGF-A transgenic mice (50.2%) than of K14/GFP transgenic mice (4.9%; C). (B and D) Statistical analysis revealed a significant increase of GFP-positive tumor cells in the sentinel lymph node metastases of VEGF-A/GFP double transgenic mice, compared with GFP transgenic mice (D), whereas no significant differences were found in the primary lesions (B). Data are expressed as mean ± SEM (n = 5 per group). ***P < 0.001.
Importantly, a greater number of distant lymph nodes contained SCC metastases in VEGF-A transgenic mice (57.1% of all SCC-bearing VEGF-A transgenic mice; P = 0.028), compared with wild-type mice with SCC (15.4%). The percentage of mice with sentinel lymph node metastasis that also developed distant lymph node metastasis was twofold greater in VEGF-A transgenic mice (57.1%) than in wild-type mice (28.5%). Moreover, the incidence of lung metastases was significantly higher in VEGF-A transgenic mice (64.3%; P = 0.035) than in wild-type mice (23.1%). No lung metastases were found in mice without lymph node metastasis. Together, these results reveal that VEGF-A not only induces tumor lymphangiogenesis, but also promotes tumor metastasis to regional and distal lymph nodes and organs.
VEGF-A promotes lymphangiogenesis within sentinel lymph nodes
Immunofluorescence analysis of the lymph nodes of nontumor-bearing mice revealed comparable numbers of CD31-positive postcapillary high endothelial venules and LYVE-1–positive sinusoids in wild-type and transgenic genotypes (Fig. 7 A, B). Importantly, however, increased numbers of blood vessels and of LYVE-1–positive lymphatic vessels were found in the metastasis-containing lymph nodes of VEGF-A transgenic mice (Fig. 7 F, H), as compared with wild-type mice (Fig. 7 E, G). The cells of the tumor-associated LYVE-1–positive vessels in VEGF-A transgenic mice were undergoing high levels of proliferation, as revealed by 5-bromo-2′-deoxyuridine (BrdU) staining (Fig. 7 I). All LYVE-1–positive vessels in VEGF-A transgenic mice also expressed Prox1, which confirmed their lymphatic identity (Fig. 7 J). These results demonstrate that overexpression of VEGF-A in the skin promotes tumor-associated active lymphangiogenesis in the metastatic lymph nodes, and that the tumor-associated lymphatic vessels maintain their lymphatic phenotype.
Figure 7.
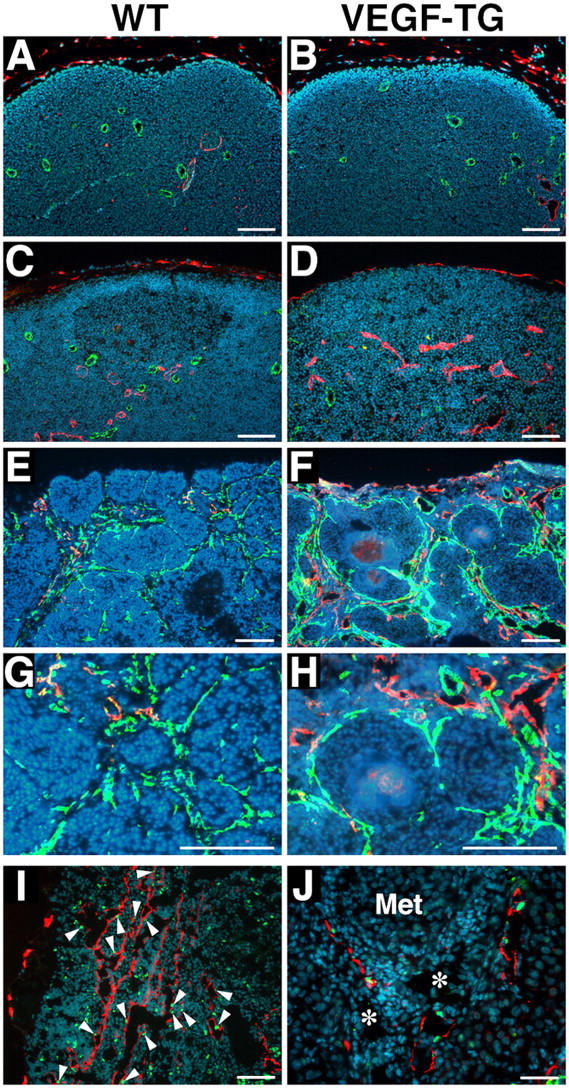
Increased lymph node lymphangiogenesis in VEGF-A transgenic mice. Double immunofluorescence stains of lymph nodes of nontumor-bearing mice demonstrate a comparable pattern of CD31-positive postcapillary high endothelial venules (green) and LYVE-1–positive sinusoids (red) in wild-type (WT) mice (A) and in VEGF-A transgenic (VEGF-TG) mice (B). Nonmetastatic sentinel lymph nodes of SCC-bearing VEGF-A transgenic mice have increased numbers of enlarged LYVE-1–positive sinusoids (red; D), as compared with wild-type mice (C). Increased numbers of enlarged LYVE-1-positive lymphatic vessels (red) and of CD31-positive blood vessels (green) were found in the metastatic sentinel lymph nodes of VEGF-A transgenic mice (F), as compared with wild-type mice (E). (G and H) Higher magnification of panels E and F, respectively. The tumor-associated LYVE-1–positive vessels (red) in VEGF-A transgenic mice had high levels of BrdU staining in lymphatic endothelial cells (I; green), indicating active lymphatic proliferation (arrowheads) within the sentinel lymph node. These cells also expressed Prox1 (J; green). Blood vessels (J; asterisks) do not express LYVE-1 or Prox1. ‘Met’ indicates a metastasizing cutaneous SCC. Nuclei are labeled blue (Hoechst stain). Scale bars = 100 μm (A–D and I); 200 μm (E–H); 50 μm (J).
We next analyzed the nonmetastasis–containing lymph nodes of tumor-bearing mice of both genotypes. Surprisingly, we found increased numbers of enlarged LYVE-1–positive sinusoidal vessels in nonmetastasis–containing draining lymph nodes of tumor-bearing VEGF-A transgenic mice (Fig. 7 D), as compared with tumor-bearing wild-type mice (Fig. 7 C). In contrast, no major difference was found in the number of CD31-positive vessels (Fig. 7 C, D). These intriguing findings reveal that VEGF-A, which is secreted by primary skin tumors, induces lymphangiogenesis within the draining lymph nodes, even before the tumor has metastasized to these tissues; this may facilitate future metastatic tumor spread within the lymphatic system.
DISCUSSION
Our study identifies VEGF-A as a novel tumor lymphangiogenesis factor. Consistent transgenic overexpression of mouse VEGF-A164, specifically in the skin, resulted in enhanced numbers of enlarged, actively proliferating, tumor-associated lymphatic vessels during multi-step skin carcinogenesis. Previously, VEGF-A was considered to be only a (blood vascular) tumor angiogenesis factor, signaling through VEGFR-2 and possibly VEGFR-1, which are expressed on tumor-associated blood vessels (2, 28, 29). Indeed, a large number of studies showed that overexpression of VEGF-A in tumor xenotransplants promotes tumor angiogenesis (30), and that systemic inhibition of VEGF-A, VEGFR-1, or VEGFR-2 inhibits tumor growth and angiogenesis in experimental mouse tumor models and in human cancers (30–32).
Only one of these studies (4), however, investigated the effects of VEGF-A on tumor-associated lymphatic vessel formation—largely because of the absence of specific markers for lymphatic vessels. We have used the lymphatic endothelium-specific hyaluronan receptor, LYVE-1 (25, 33), as a marker to detect and quantify tumor-associated lymphatic vessels. LYVE-1 also is expressed by some nonlymphatic endothelial cells, such as those of liver sinusoids (34) and of embryonic veins (35); therefore, to increase specificity we also monitored expression of the transcription factor Prox1, which is expressed specifically by lymphatic, but not by blood, vessels (35). Using these two markers, we were able to quantify carefully lymphatic vessels that are associated with tumors and metastases.
The role of active lymphangiogenesis in tumor growth has been questioned, as it has been proposed that tumor-associated lymphatic vessels might represent preexisting lymphatics within or surrounding rapidly-invading tumor xenotransplants (36). Through analysis of BrdU and LYVE-1 levels, however, our study reveals that active proliferation of lymphatic endothelial cells occurs within and surrounding the slowly growing, orthotopic cutaneous SCC, and that overexpression of VEGF-A increases proliferation of cells in the lymphatic vessels. Together, these results confirm that tumors can induce the growth of tumor-associated lymphatic vessels actively, and that VEGF-A promotes tumor lymphangiogenesis. In contrast, a previous study of VEGF-A–transfected human 293 cell xenotransplants did not detect promotion of tumor lymphangiogenesis (4). These findings might be due to different levels of VEGF-A expression, the significantly shorter observation period, and a possible different genetic lymphangiogenic disposition between the different mouse strains that were used. The multi-step skin carcinogenesis model that was used in our study—with autochtonous tumors developing slowly over a period of several months—likely resembles the pathogenesis of human tumors more closely than the widely used xenotransplant models of human cancer cell lines in immunodeficient mice.
There also is controversy over whether VEGF-A can induce lymphangiogenesis, and, if so, whether VEGF-A induces lymphatic vessel growth directly or via up-regulation of the thus far only known direct lymphangiogenesis factors, VEGF-C and VEGF-D. VEGF-C and -D also were shown to induce tumor lymphangiogenesis potently and to be up-regulated in a large number of metastatic human cancers (3–6, 8, 9, 37). Whereas injection of adenoviral human VEGF-A165 into murine skin did not result in increased sprouting of lymphatic vessels, although lymphatic enlargement was observed (18), injection of the ears of mice with an adenoviral vector that expressed mouse VEGF-A164 actively promoted new lymphatic vessel formation and proliferation of lymphatic vessel cells. This occurred without any detectable up-regulation of VEGF-C or VEGF-D (16).
We also showed previously that overexpression of murine VEGF-A164 in the skin promoted lymphatic vessel formation during cutaneous tissue repair (13) and during chronic skin inflammation (19), without detectable up-regulation of VEGF-C or VEGF-D. Moreover, cultured dermal LECs express VEGFR-2, and we found that treatment of LECs with human VEGF-A165 potently promoted LEC proliferation and migration that was inhibited by blockade of VEGFR-2, but not VEGFR-3. Together with our findings that tumor-associated lymphatic vessels in VEGF-A transgenic mice expressed VEGFR-2 during skin carcinogenesis, it is conceivable that VEGF-A directly induces the formation of lymphatic vessels.
In this study, we observed that levels of VEGF-C (but not of VEGF-D) mRNA and protein expression were increased in VEGF-A–overexpressing tumors; this is in agreement with the finding that VEGF-A treatment up-regulates VEGF-C expression in cultured endothelial cells (38); however, the proteolytic processing of VEGF-C protein was not altered in VEGF-A–overexpressing tumors. So the relative importance of different mechanisms by which VEGF-A promotes tumor lymphangiogenesis—whether via direct or indirect up-regulation of VEGF-C, or other factors, such as fibroblast growth factors or angiopoietin—remains to be elucidated. Once they become available, studies with reagents that block VEGFR-3 signaling in mice or that specifically neutralize VEGF-C might answer these questions.
Our findings that chronic transgenic overexpression of VEGF-A in the skin promoted skin carcinogenesis and tumor angiogenesis are in agreement with a large number of studies in tumor xenotransplants (30) and with a previous study in transgenic mice with skin-specific overexpression of the mouse VEGF120 isoform (39). It is remarkable that despite the fact that VEGF-A transgenic mice developed more benign papillomas and more malignant SCCs, the ratio of malignant conversion of large papillomas into SCC was comparable in VEGF-A transgenic mice and in wild-type mice. These findings are in agreement with previous findings in thrombospondin-2–deficient mice (23), and they indicate that increased levels of VEGF-A did not result in major additional genetic events. Instead, the increased number of SCCs in VEGF-A transgenic mice most likely reflects the acceleration in the early stages of skin carcinogenesis, because comparable latency periods between papilloma and SCC formation were observed in both genotypes.
Importantly, we found that tumor metastasis to regional (sentinel) lymph nodes occurred more frequently in VEGF-A transgenic mice than in wild-type mice; this indicated that VEGF-A–induced tumor lymphangiogenesis of primary cutaneous SCC promoted metastasis to these tissues. In studies in human cutaneous melanomas, the degree of tumor-associated lymphangiogenesis was correlated with future lymph node metastasis and with reduced overall survival (6), and indicated the clinical relevance of tumor lymphangiogenesis in human cancer. The establishment of the K14/GFP transgenic model has allowed us to quantify the incidence of SCC metastasis to sentinel and distant lymph nodes in response to increased levels of lymphangiogenesis. This model also might be used to study other factors that contribute to multi-step skin carcinogenesis and metastasis.
One of the most important and novel findings of our study was that VEGF-A–overexpressing SCC cells maintained their lymphangiogenic activity after metastasizing to sentinel lymph nodes; this resulted in increased numbers of proliferating LYVE-1– and Prox1-positive lymphatic vessels within lymph nodes. These results indicate that VEGF-A not only promotes primary tumor lymphangiogenesis, but also induces lymph node lymphangiogenesis. It is tempting to speculate that this newly identified phenomenon of “lymph node lymphangiogenesis” might facilitate further metastatic tumor spread throughout the lymphatic system, because the percentage of mice with sentinel lymph node metastasis that also developed distant lymph node metastasis was twofold greater in VEGF-A transgenic mice than in wild-type mice.
Therefore, VEGF-A might represent a novel target for treating patients who have advanced forms of SCC or other cancers, as well as to prevent metastasis to sentinel lymph nodes. Systemic inhibition of VEGF-A by a blocking antibody (bevacizumab) recently was shown to retard tumor progression in patients who had metastatic colorectal or renal cancer. Our present study suggests that systemic anti–VEGF-A treatment also might prevent the systemic spread of an early-stage malignancy by inhibiting tumor lymphangiogenesis and/or lymph node lymphangiogenesis. Future studies that use long-term neutralization of VEGF-A during multi-step carcinogenesis in mice or in novel models of lymph node metastasis are needed to determine whether VEGF-A blockade might inhibit the further metastatic spread of already existing, potentially dormant micrometastases within sentinel (or distant) lymph nodes.
Perhaps the most surprising finding of our study was that even before metastasizing to the lymph nodes, VEGF-A–overexpressing primary tumors induced sentinel lymph node lymphangiogenesis. This suggests that primary tumors might begin preparing their future metastatic site by producing lymphangiogenic factors that mediate their efficient transport to sentinel lymph nodes. This concept is an important new twist to the previously proposed “seed-and-soil” hypothesis for the preferential organ metastasis pattern of distinct human tumors.
The induction of lymphangiogenesis within the premetastatic lymph nodes was much more dramatic than the induction of angiogenesis. This suggests that the fluid which is drained from the primary tumors into the sentinel lymph node promotes their further metastatic spread from sentinel lymph nodes. Initiation of antilymphangiogenic (or anti–VEGF-A) therapy during the presurgery period, immediately after the diagnosis of cancer, might reduce the future risk for metastasis of SCC or other tumor types to lymph nodes in patients.
Materials and methods
Generation of K14/GFP and of K14/GFP/VEGF-A transgenic mice.
A 787-bp fragment that encodes enhanced GFP cDNA was obtained by PCR amplification from a pEGFP vector template (BD Biosciences) and was cloned into the BamHI site of a K14 promoter/pGEM-3Z transgene expression cassette ([24]; provided by E. Fuchs, Rockefeller University, New York, NY). A 5.4-kb KpnI–HindIII fragment (Fig. 1 A) was used to generate transgenic mice on the FVB genetic background as described (20). Transgenic founders were detected by Southern blot analysis. For rapid identification, GFP expression was analyzed in the tails of 12-day-old mice, using a Nikon TE-300 fluorescence microscope. Three independent homozygous transgenic lines were established. After confirmation of specific GFP expression in sites that previously were shown to express K14 (24), the transgenic line with the highest level of GFP expression was crossed with previously established and characterized heterozygous K14/VEGF-A transgenic mice (20), to establish K14/GFP and K14/GFP/VEGF-A transgenic mice. All animal studies were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Chemical skin carcinogenesis regimen.
For tumor initiation, 50 μg of DMBA (Sigma-Aldrich) was applied topically to the shaved back skin of 8-wk-old female K14/GFP/VEGF-A transgenic mice (n = 34), K14/GFP transgenic mice (n = 31), and FVB wild-type mice (n = 29), followed by weekly topical application of 5 μg of the tumor promoter PMA (Sigma-Aldrich) over 20 wk as described (22, 23). Raised lesions of a minimum diameter of 1 mm that had been present for at least 1 wk were recorded as tumors. Mice were killed after 35 wk, or at 8 wk after the first diagnosis of SCC. The ratio of malignant conversion was calculated for each group of mice as the total number of SCC divided by the number of large papillomas, expressed as a percentage. The two-sided unpaired Student's t test was used to analyze differences in the number of tumors per mouse between VEGF-A transgenic and wild-type mice. Regional and distal lymph node involvement was evaluated clinically and by fluorescence microscopy of 100-μm step sections. Lung metastases were evaluated on 100-μm step sections. Metastasis data were analyzed by the Mann-Whitney test.
Immunofluorescence, in situ hybridization, ELISA, and immunoprecipitation.
Tumors or skin samples were fixed for 2 h in 4% paraformaldehyde and were embedded in paraffin or in optimal cutting temperature compound (Sakura Finetek) and snap-frozen, or were freshly frozen. At autopsy, all axillary and inguinal lymph nodes of SCC-bearing mice were examined for the presence of metastases. The presence of metastases was confirmed further by fluorescence microscopic analysis of GFP expression in five 100-μm-thick serial sections for each lymph node. Immunostains were performed on 6-μm paraffin sections or cryostat sections as described previously (23), using a rat mAb against CD31 (BD Biosciences), a goat polyclonal antibody to mouse VEGFR-2/Flk-1 (R&D Systems), a rat monoclonal and a rabbit polyclonal antibody to mouse LYVE-1 (a gift from D. Jackson, University of Oxford, Oxford, England; reference 25), a rabbit anti-Prox1 antibody (Covance), and corresponding secondary antibodies labeled with Alexa Fluor 488 or 594 (Molecular Probes). Before most immunostains, frozen sections were photobleached by exposure to ultraviolet B irradiation. Nuclei were counterstained with Hoechst bisbenzimide (Molecular Probes). Mice were injected with BrdU, 50 mg/kg (Sigma-Aldrich) 2 h before sacrifice, and a FITC-conjugated mouse mAb to BrdU (BD Biosciences) was used for detection of proliferating cells. In situ hybridization was performed as described (20). Antisense and sense single-stranded 35S-labeled RNA probes for VEGF-A, VEGF-C, and VEGF-D were as described previously (16). Murine VEGF-A protein levels were quantified by ELISA (Quantikine M; R&D Systems) of skin or tumor lysates as described (19). Statistical analysis was performed using the unpaired Student's t test. SCC lysates (n = 5 per genotype) also were immunoprecipitated using a rabbit anti-VEGF-C antibody (Abcam Inc.), followed by SDS-PAGE under reducing conditions and Western blotting using a goat anti-VEGF-C antibody (R&D Systems).
Computer-assisted morphometric vessel analysis.
Representative LYVE-1 and CD31 double-stained sections, obtained from biopsies of PMA-treated skin (n = 5), small papillomas (1–3 mm in diameter; n = 5), large papillomas (>3 mm in diameter; n = 6), and SCC (n = 6) were analyzed for each genotype, using a Nikon E-600 microscope. Images were captured with a Spot digital camera (Diagnostic Instruments), and computer-assisted morphometric analyses of blood vessels and lymphatic vessels were performed as described (22), using the IP-LAB software (Scanalytics). Statistical analyses were performed using the two-sided, unpaired Student's t test.
Flow cytometry analysis.
Tissue samples of SCC and of sentinel lymph nodes were minced gently in PBS, followed by incubation with 0.25% trypsin (Invitrogen) for 10 min at 37°C. Single-cell suspensions were prepared as described (26), and were passed through 40-μm pore size cell strainers (BD Biosciences), washed in PBS, and stained with a PE-labeled mouse anti-CD45 antibody (BD Biosciences) and with propidium iodide. The stained cells (>10,000 cells per sample) were subjected to flow cytometry with a FACScan and a FACSCalibur instrument (BD Biosciences). GFP expression was detected under FITC settings (26).
Quantitative real-time RT-PCR.
Total RNA was isolated from mouse skin or SCC by using Trizol reagent (Sigma-Aldrich). Taqman-based real-time RT-PCR reactions for murine VEGF-C, VEGF-D, and GAPDH were performed as described (19). GAPDH transcripts were measured simultaneously in all reactions as internal controls. Total RNA was treated with RNase-free RNA qualified–DNase (Promega) before analysis, and 50 ng of total RNA was used for each reaction. Data were normalized based on the expression levels of GAPDH.
Cell proliferation and migration assays.
Human dermal LECs were isolated from neonatal foreskins as described (12). For proliferation assays, cells (2 × 103) were seeded onto fibronectin-coated 96-well plates and were pretreated (n = 5 per treatment group) with 5 μg/ml of anti–human VEGFR-2 mAb (89106, R&D Systems), anti–human VEGFR-3 mAb (hF4-3C5, Imclone), or control IgG for 16 h, followed by incubation with or without recombinant human VEGF-A165 (10 ng/ml; R&D Systems) in endothelial cell basic medium containing 2% FBS. After 48 h, cells were incubated with 4-methylumbelliferylheptanoate (Sigma-Aldrich) and resulting fluorescence—which is correlated to the number of living cells (27)—was quantified using a Victor2 Fluorometer (PerkinElmer). Haptotactic LEC migration was studied as described (13). Cells were seeded in serum-free endothelial cell basic medium containing 0.2% delipidized BSA into the upper chambers of FluoroBlok inserts (Falcon) in the presence of anti–human VEGFR-2 mAb (10 μg/ml), anti–human VEGFR-3 mAb (5 μg/ml), or corresponding control IgG, and were incubated for 3 h at 37°C in the presence or absence of VEGF-A165 (10 ng/ml). Cells on the underside of inserts were stained with Hoechst 33342 (Molecular Probes), and the number of migrated cells was determined by computer-assisted image analysis of three random 20× fields per well. Three independent experiments were performed for each assay. Statistical analyses were performed by using the unpaired Student's t test.
Acknowledgments
We thank M. Constant, L. Wu, L. Janes, A. Skrzypek, and M. Min for expert technical assistance.
This work was supported by grants from the National Institutes of Health (CA69184, CA86410, CA92644), American Cancer Society Research Project (99-23901), and the Cutaneous Biology Research Center through the Massachusetts General Hospital/Shiseido Co. Ltd. Agreement to M. Detmar; and the Max Kade Foundation to R. Kunstfeld.
The authors have no conflicting financial interests.
Abbreviations used: K14, keratin 14; BrdU, 5-bromo-2′deoxyuridine; DMBA, 7,12-dimethylbenzanthracene; LEC, lymphatic endothelial cell; LYVE-1, lymphatic vessel endothelial hyaluronan receptor 1; SCC, squamous cell carcinoma; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
References
- 1.Folkman, J. 1992. The role of angiogenesis in tumor growth. Semin. Cancer Biol. 3:65–71. [PubMed] [Google Scholar]
- 2.Ferrara, N., H.P. Gerber, and J. LeCouter. 2003. The biology of VEGF and its receptors. Nat. Med. 9:669–676. [DOI] [PubMed] [Google Scholar]
- 3.Skobe, M., T. Hawighorst, D.G. Jackson, R. Prevo, L. Janes, P. Velasco, L. Riccardi, K. Alitalo, K. Claffey, and M. Detmar. 2001. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 7:192–198. [DOI] [PubMed] [Google Scholar]
- 4.Stacker, S.A., C. Caesar, M.E. Baldwin, G.E. Thornton, R.A. Williams, R. Prevo, D.G. Jackson, S. Nishikawa, H. Kubo, and M.G. Achen. 2001. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 7:186–191. [DOI] [PubMed] [Google Scholar]
- 5.Mandriota, S.J., L. Jussila, M. Jeltsch, A. Compagni, D. Baetens, R. Prevo, S. Banerji, J. Huarte, R. Montesano, D.G. Jackson, et al. 2001. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 20:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dadras, S.S., T. Paul, J. Bertoncini, L.F. Brown, A. Muzikansky, D.G. Jackson, U. Ellwanger, C. Garbe, M.C. Mihm, and M. Detmar. 2003. Tumor lymphangiogenesis: a novel prognostic indicator for cutaneous melanoma metastasis and survival. Am. J. Pathol. 162:1951–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alitalo, K., and P. Carmeliet. 2002. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell. 1:219–227. [DOI] [PubMed] [Google Scholar]
- 8.Karpanen, T., M. Egeblad, M.J. Karkkainen, H. Kubo, S. Yla-Herttuala, M. Jaattela, and K. Alitalo. 2001. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 61:1786–1790. [PubMed] [Google Scholar]
- 9.Skobe, M., L.M. Hamberg, T. Hawighorst, M. Schirner, G.L. Wolf, K. Alitalo, and M. Detmar. 2001. Concurrent induction of lymphangiogenesis, angiogenesis, and macrophage recruitment by vascular endothelial growth factor-C in melanoma. Am. J. Pathol. 159:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattila, M.M., J.K. Ruohola, T. Karpanen, D.G. Jackson, K. Alitalo, and P.L. Harkonen. 2002. VEGF-C induced lymphangiogenesis is associated with lymph node metastasis in orthotopic MCF-7 tumors. Int. J. Cancer. 98:946–951. [DOI] [PubMed] [Google Scholar]
- 11.Detmar, M., and S. Hirakawa. 2002. The formation of lymphatic vessels and its importance in the setting of malignancy. J. Exp. Med. 6:713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirakawa, S., Y.K. Hong, N. Harvey, V. Schacht, K. Matsuda, T. Libermann, and M. Detmar. 2003. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am. J. Pathol. 162:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong, Y.K., B. Lange-Asschenfeldt, P. Velasco, S. Hirakawa, R. Kunstfeld, L.F. Brown, P. Bohlen, D.R. Senger, and M. Detmar. 2004. VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J. 18:1111–1113. [DOI] [PubMed] [Google Scholar]
- 14.Makinen, T., T. Veikkola, S. Mustjoki, T. Karpanen, B. Catimel, E.C. Nice, L. Wise, A. Mercer, H. Kowalski, D. Kerjaschki, et al. 2001. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 20:4762–4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veikkola, T., M. Lohela, K. Ikenberg, T. Makinen, T. Korff, A. Saaristo, T. Petrova, M. Jeltsch, H.G. Augustin, and K. Alitalo. 2003. Intrinsic versus microenvironmental regulation of lymphatic endothelial cell phenotype and function. FASEB J. 17:2006–2013. [DOI] [PubMed] [Google Scholar]
- 16.Nagy, J.A., E. Vasile, D. Feng, C. Sundberg, L.F. Brown, M.J. Detmar, J.A. Lawitts, L. Benjamin, X. Tan, E.J. Manseau, et al. 2002. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J. Exp. Med. 196:1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kriehuber, E., G.S. Breiteneder, M. Groeger, A. Soleiman, S.F. Schoppmann, G. Stingl, D. Kerjaschki, and D. Maurer. 2001. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J. Exp. Med. 194:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saaristo, A., T. Veikkola, B. Enholm, M. Hytonen, J. Arola, K. Pajusola, P. Turunen, M. Jeltsch, M.J. Karkkainen, D. Kerjaschki, et al. 2002. Adenoviral VEGF-C overexpression induces blood vessel enlargement, tortuosity, and leakiness but no sprouting angiogenesis in the skin or mucous membranes. FASEB J. 16:1041–1049. [DOI] [PubMed] [Google Scholar]
- 19.Kunstfeld, R., S. Hirakawa, Y.K. Hong, V. Schacht, B. Lange-Asschenfeldt, P. Velasco, C. Lin, E. Fiebiger, X. Wei, Y. Wu, et al. 2004. VEGF-A plays a key role in the induction of chronic inflammation and the associated lymphangiogenic response. Blood. 104:1048–1057. [DOI] [PubMed] [Google Scholar]
- 20.Detmar, M., L.F. Brown, M.P. Schön, B.M. Elicker, P. Velasco, L. Richard, D. Fukumura, W. Monsky, K.P. Claffey, and R.K. Jain. 1998. Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J. Invest. Dermatol. 111:1–6. [DOI] [PubMed] [Google Scholar]
- 21.Yano, K., L.F. Brown, and M. Detmar. 2001. Control of hair growth and follicle size by VEGF-mediated angiogenesis. J. Clin. Invest. 107:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawighorst, T., H. Oura, M. Streit, L. Janes, L. Nguyen, L.F. Brown, G. Oliver, D.G. Jackson, and M. Detmar. 2002. Thrombospondin-1 selectively inhibits early-stage carcinogenesis and angiogenesis but not tumor lymphangiogenesis and lymphatic metastasis in transgenic mice. Oncogene. 21:7945–7956. [DOI] [PubMed] [Google Scholar]
- 23.Hawighorst, T., P. Velasco, M. Streit, T.R. Kyriakides, L.F. Brown, P. Bornstein, and M. Detmar. 2001. Thrombospondin-2 plays a protective role in multistep carcinogenesis: a novel host anti-tumor defense mechanism. EMBO J. 20:2631–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vassar, R., M. Rosenberg, S. Ross, A. Tyner, and E. Fuchs. 1989. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proc. Natl. Acad. Sci. USA. 86:1563–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prevo, R., S. Banerji, D.J. Ferguson, S. Clasper, and D.G. Jackson. 2001. Mouse LYVE-1 is an endocytic receptor for hyaluronan in lymphatic endothelium. J. Biol. Chem. 276:19420–19430. [DOI] [PubMed] [Google Scholar]
- 26.Kodama, S., W. Kuhtreiber, S. Fujimura, E.A. Dale, and D.L. Faustman. 2003. Islet regeneration during the reversal of autoimmune diabetes in NOD mice. Science. 302:1223–1227. [DOI] [PubMed] [Google Scholar]
- 27.Detmar, M., S. Tenorio, U. Hettmannsperger, Z. Ruszczak, and C.E. Orfanos. 1992. Cytokine regulation of proliferation and ICAM-1 expression of human dermal microvascular endothelial cells in vitro. J. Invest. Dermatol. 98:147–153. [DOI] [PubMed] [Google Scholar]
- 28.Carmeliet, P. 2003. Angiogenesis in health and disease. Nat. Med. 9:653–660. [DOI] [PubMed] [Google Scholar]
- 29.Dvorak, H.F., L.F. Brown, M. Detmar, and A.M. Dvorak. 1995. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrara, N. 2002. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer. 2:795–803. [DOI] [PubMed] [Google Scholar]
- 31.Malik, A.K., and H.P. Gerber. 2003. Targeting VEGF ligands and receptors in cancer. Targets. 2:48–57. [Google Scholar]
- 32.Ferrara, N., K.J. Hillan, H.P. Gerber, and W. Novotny. 2004. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 3:391–400. [DOI] [PubMed] [Google Scholar]
- 33.Banerji, S., J. Ni, S.X. Wang, S. Clasper, J. Su, R. Tammi, M. Jones, and D.G. Jackson. 1999. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J. Cell Biol. 144:789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mouta Carreira, C., S.M. Nasser, E. di Tomaso, T.P. Padera, Y. Boucher, S.I. Tomarev, and R.K. Jain. 2001. LYVE-1 is not restricted to the lymph vessels: expression in normal liver blood sinusoids and down-regulation in human liver cancer and cirrhosis. Cancer Res. 61:8079–8084. [PubMed] [Google Scholar]
- 35.Oliver, G., and M. Detmar. 2002. The rediscovery of the lymphatic system. Old and new insights into the development and biological function of lymphatic vascular system. Genes Dev. 16:773–783. [DOI] [PubMed] [Google Scholar]
- 36.Leu, A.J., D.A. Berk, A. Lymboussaki, K. Alitalo, and R.K. Jain. 2000. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 60:4324–4327. [PubMed] [Google Scholar]
- 37.Stacker, S.A., M.G. Achen, L. Jussila, M.E. Baldwin, and K. Alitalo. 2002. Lymphangiogenesis and cancer metastasis. Nat. Rev. Cancer. 2:573–583. [DOI] [PubMed] [Google Scholar]
- 38.Skobe, M., and M. Detmar. 2000. Structure, function and molecular control of the skin lymphatic system. J. Invest. Dermatol. Symp. Proc. 5:14–19. [DOI] [PubMed] [Google Scholar]
- 39.Larcher, F., R. Murillas, M. Bolontrade, C.J. Conti, and J.L. Jorcano. 1998. VEGF/VPF overexpression in skin of transgenic mice induces angiogenesis, vascular hyperpermeability and accelerated tumor development. Oncogene. 17:303–311. [DOI] [PubMed] [Google Scholar]