

Abstract
Osteoporosis is a serious problem worldwide; it is characterized by bone fractures in response to relatively mild trauma. Osteoclasts originate from the fusion of macrophages and they play a central role in bone development and remodeling via the resorption of bone. Therefore, osteoclasts are important mediators of bone loss that leads, for example, to osteoporosis. Interleukin (IL)-1 receptor (IL-1R)–associated kinase M (IRAK-M) is only expressed in cells of the myeloid lineage and it inhibits signaling downstream of IL-1R and Toll-like receptors (TLRs). However, it lacks a functional catalytic site and, thus, cannot function as a kinase. IRAK-M associates with, and prevents the dissociation of, IRAK–IRAK-4–TNF receptor–associated factor 6 from the TLR signaling complex, with resultant disruption of downstream signaling. Thus, IRAK-M acts as a dominant negative IRAK. We show here that mice that lack IRAK-M develop severe osteoporosis, which is associated with the accelerated differentiation of osteoclasts, an increase in the half-life of osteoclasts, and their activation. Ligation of IL-1R or TLRs results in hyperactivation of NF-κB and mitogen-activated protein kinase signaling pathways, which are essential for osteoclast differentiation. Thus, IRAK-M is a key regulator of the bone loss that is due to osteoclastic resorption of bone.
Multinucleated osteoclasts originate from the fusion of mononuclear phagocytes and play a major role in the resorption of bone. Because osteoclasts are essential for the development and remodeling of bone, increases in their number and/or activity lead to diseases that are associated with generalized bone loss (e.g., osteoporosis) and others with localized bone loss (e.g., rheumatoid arthritis, periodontal disease).
The cytokines macrophage colony–stimulating factor (M-CSF) and receptor activator of NF-κB (RANKL) are necessary and sufficient for the induction of the differentiation of osteoclasts from mononuclear phagocytes (1–6), but agonists of IL-1R/Toll-like receptor (TLR) also play a critical role in the differentiation and activation of osteoclasts. IL-1 is a potent stimulator of the differentiation, activation and survival of osteoclasts that have been implicated in postmenopausal osteoporosis (7), whereas LPS, a ligand that binds to TLR4, is responsible for the inflammation-mediated loss of bone (8). Bone loss that is associated with estrogen deficiency in rodents can be prevented by administration of an antagonist of IL-1R (9, 10); however, bone loss that is associated with infection remains difficult to control, even in model systems.
IL-1R–associated kinase (IRAK) plays a central role in the signaling pathways that are initiated by members of the IL-1R/TLR family (11). Members of this family have a conserved “TLR- and IL-1R-related” intracytoplasmic domain. Thus, activation of the different members of this family induces similar signaling cascades that culminate in the activation of the IκB kinase complex and mitogen-activated protein kinases (MAPKs). This activation leads to the activation of NF-κB, c-Jun NH2-terminal kinase (JNK), p38, and extracellular signal-regulated kinase (ERK)1/2, as well as AP-1–dependent transcriptional responses (12, 13).
IRAKs are multidomain proteins with a similar central kinase domain and a death domain that interacts with myeloid differentiation factor 88. All IRAKs have a functional ATP-binding site, but IRAK-2 and IRAK-M lack a functional catalytic site because the critical aspartate residue is replaced by an asparagine and a serine residue, respectively. Hence, IRAK-2 and IRAK-M are inactive as kinases. In addition, IRAK-M is expressed predominantly in peripheral blood leukocytes and in macrophages (14). IRAK-M is expressed in monocytic cell lines and its expression is induced strongly upon the maturation of macrophages (14). We reported previously that IRAK-M acts as a negative regulator of IL-1R/TLR signaling by preventing the dissociation of IRAKs from myeloid differentiation factor 88 and maintaining the integrity of the complex between IRAK–TNF receptor–associated factor (TRAF)6 and TLR (15). By trapping both types of IRAK in the receptor complex, IRAK-M prevents the downstream activation of NF-κB and MAPK signaling pathways. Thus, IRAK-M seems to act as a dominant negative regulator downstream of IL-1R/TLR in macrophages (15). Therefore, we postulated that if the expression of IRAK-M is conserved in macrophages after the fusion that results in multinucleation, it should be a negative regulator of the differentiation and activation of osteoclasts, and thus, it should protect bone mass. To test our hypothesis, we examined mice with a homozygous deletion of the irak-M gene. We found that in normal mice, IRAK-M is expressed strongly in osteoclasts and that its absence is associated with accelerated osteoclastogenesis; an increase in the half-life of osteoclasts; and the hyperactivation of the NF-κB and MAKP signaling pathways, via ligation to IL-1R/TLR, with severe resultant osteoporosis. IRAK-M seems to be a key signaling molecule in the prevention of bone loss.
RESULTS
IL-1 receptor–associated kinase M–deficient mice develop osteoporosis
IRAK-M−/− mice are viable and fertile but seem to develop an accentuated spinal curvature or kyphosis at 4 mo of age (data not shown). At this age, IRAK-M−/− mice are smaller than wild-type mice (Fig. 1 A). To assess bone density in IRAK-M−/− mice, we subjected femurs from 4-mo-old IRAK-M−/− and IRAK-M+/+ mice to peripheral quantitative computed tomography analysis at the midshaft level. The cortical content was lower in IRAK-M−/− mice than in age-matched IRAK-M+/+ mice (Table I). This difference was associated with a decrease in the diameter of femurs, in males and females, and a decrease in cortical thickness in males. At the level of the epiphysis, the trabecular content and density were reduced in male and female IRAK-M−/− mice, as compared with sex- and age-matched IRAK-M+/+ mice. This decrease in trabecular bone density was associated with a 60% reduction in trabecular bone volume, as determined by microCT analysis (Fig. 1 B). Together, our data indicated that the deletion of the irak-M gene in mice leads to osteoporosis.
Figure 1.

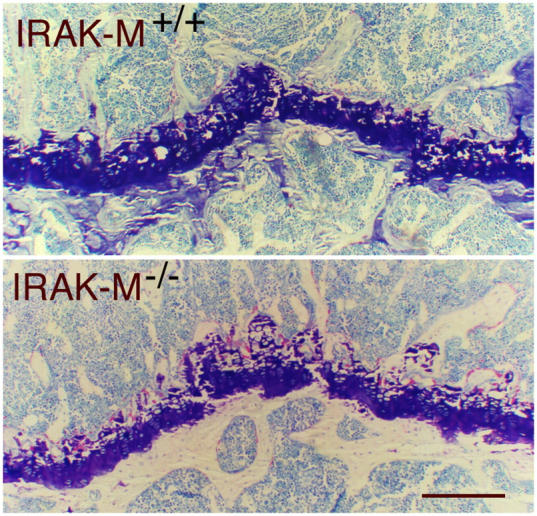
IRAK-M–deficient mice are smaller than wild-type mice and have reduced total bone mineral density. (A) Male and female IRAK-M−/− mice are smaller than IRAK-M+/+ mice; n = 10. (B) microCT analysis of distal femurs from 4-mo-old male IRAK-M−/− and IRAK-M+/+ mice. Note the paucity of the trabeculae inside the bone in IRAK-M−/− mice. The wider diameter of the bone sections measures ∼3 mm. Bar, 1 mm. (C) Toluidine blue staining of sections from epiphyseal bones from 4-mo-old male IRAK-M−/− and IRAK-M+/+ mice. Bar, 1 mm. (D) TRAP staining of sections from epiphyseal bones from 4-mo-old male IRAK-M−/− and IRAK-M+/+ mice. Bar, 200 μm.
Table I.
Peripheral quantitative computed tomography analysis of femurs from 4-mo-old IRAK-M−/− and IRAK-M+/+ male and female mice
| Femoral shaft
|
Distal femur
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Cortical content |
Cortical density |
Cortical thickness |
Periosteal circumference |
Endosteal circumference |
Trabecular content |
Trabecular density |
||
| mg/mm | mg/cm3 | mm | mm | mm | mg/mm | mg/cm3 | ||
| Male+/+ | mean | 1.985 | 1,235.6 | 0.375 | 5.462 | 3.107 | 0.72 | 291.65 |
| SD | 0.182 | 21.691 | 0.024 | 0.289 | 0.32 | 0.041 | 26.428 | |
| Male−/− | mean | 1.325 | 1,173.375 | 0.337 | 4.409 | 2.292 | 0.295 | 141.925 |
| SD | 0.061 | 13.579 | 0.015 | 0.075 | 0.124 | 0.093 | 46.187 | |
| −/−/+/+ | 0.7 | 0.9 | 0.9 | 0.8 | 0.7 | 0.4 | 0.5 | |
| p-value | <0.01 | <0.01 | <0.05 | <0.01 | <0.01 | <0.001 | <0.01 | |
| Female+/+ | mean | 1.51 | 1,179.08 | 0.347 | 4.801 | 2.625 | 0.338 | 177.04 |
| SD | 0.067 | 30.548 | 0.021 | 0.318 | 0.42 | 0.025 | 19.802 | |
| Female−/− | mean | 1.293 | 1,169.067 | 0.327 | 4.423 | 2.369 | 0.107 | 62.167 |
| SD | 0.035 | 18.003 | 0.01 | 0.077 | 0.135 | 0.078 | 47.218 | |
| −/−/+/+ | 0.9 | 1 | 0.9 | 0.9 | 0.9 | 0.3 | 0.4 | |
| p-value | <0.01 | NS | NS | <0.05 | NS | <0.01 | <0.05 | |
Increased numbers of osteoclasts in IL-1 receptor–associated kinase M–deficient mice
To examine the mechanism by which bone density was reduced in IRAK-M–deficient mice, we analyzed tibias from 4-mo-old IRAK-M−/− and IRAK-M+/+ mice by histomorphometry. Our analysis confirmed the reduction in trabecular bone volume in male and female IRAK-M−/− mice (Table II and Fig. 1 C). At the cellular level, we found an increase in the number of osteoclasts per unit area of bone surface (Fig. 1 D). This was associated, in males, with an increase in the number of osteoblasts, which indicated an elevated rate of bone turnover. Thus, the absence of IRAK-M led to a reduction in body size and bone mass that was associated with an increase in the number of osteoclasts.
Table II.
Histomorphometric analysis of proximal tibiae from 4-mo-old IRAK-M−/− and IRAK-M+/+ male and female mice
| BV/TV
|
BFR/BV
|
N.Oc/B.Pm
|
N.Ob./B.Pm
|
OV/BV
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| mean | SD | mean | SD | mean | SD | mean | SD | mean | SD | |
|
|
%
|
%/d
|
no./mm
|
no./mm
|
%
|
|||||
| Male+/+ | 12.35 | 2.59 | 2.01 | 0.40 | 2.65 | 0.49 | 28.87 | 6.86 | 5.14 | 1.53 |
| Male− /− | 6.72 | 0.16 | 4.38 | 1.46 | 10.50 | 1.33 | 40.34 | 5.60 | 6.40 | 0.71 |
| p-value | <0.01 | <0.05 | <0.001 | <0.05 | NS | |||||
| −/−/+/+ | 0.5 | 2.2 | 4.0 | 1.4 | 1.2 | |||||
| Female+/+ | 5.94 | 1.43 | 4.88 | 1.67 | 2.13 | 0.63 | 30.51 | 4.54 | 4.25 | 1.78 |
| Female− /− | 3.85 | 0.75 | 4.78 | 0.75 | 6.77 | 0.90 | 37.29 | 10.30 | 4.58 | 1.48 |
| p-value | <0.05 | NS | <0.001 | NS | NS | |||||
| −/−/+/+ | 0.6 | 0.9 | 3.2 | 1.2 | 1.1 | |||||
Accelerated osteoclastogenesis and an increase in the half-life of osteoclasts in IL-1 receptor–associated kinase M–deficient mice
To determine whether the increase in the number of osteoclasts in IRAK-M−/− mice might be the result of an increase in the rate of differentiation of their precursors, we treated bone marrow macrophages from male IRAK-M−/− and IRAK-M+/+ mice for increasing times with M-CSF to generate osteoclast precursors (16). To our surprise, IRAK-M−/− cells proliferated at a lower rate (Fig. 2 A) but fused more rapidly than IRAK-M+/+ cells (Fig. 2 B).
Figure 2.
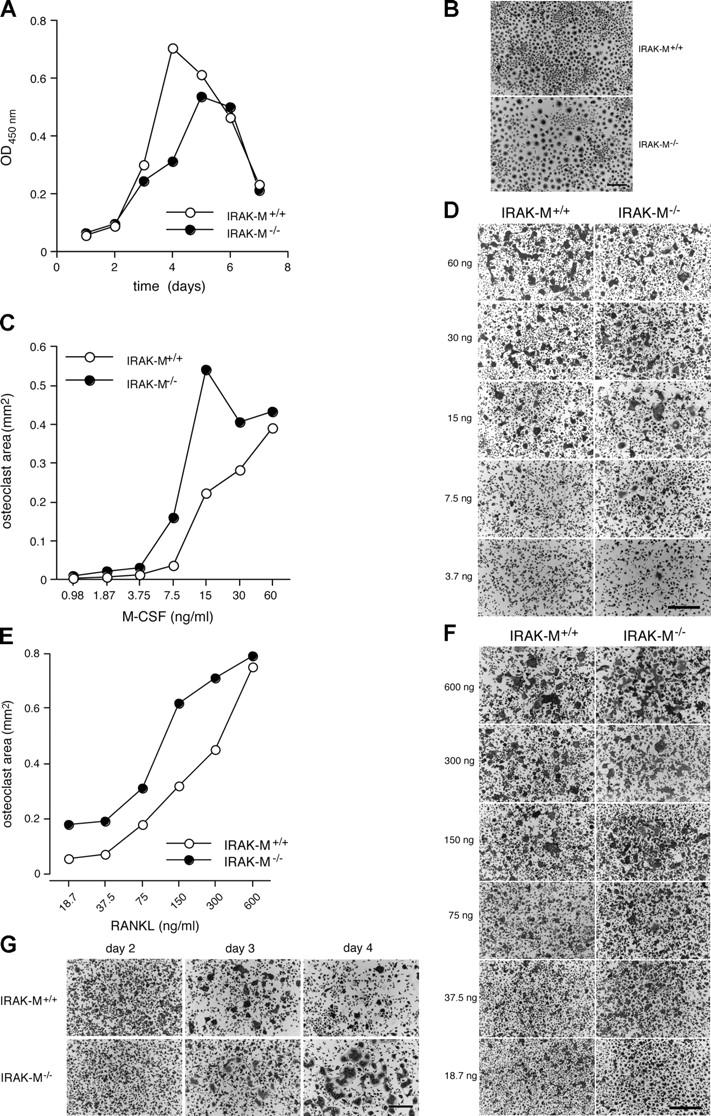
Absence of IRAK-M alters osteoclastogenesis in response to M-CSF and RANKL. (A) Bone marrow cells from IRAK-M−/− and IRAK-M+/+ male mice were cultured in the presence of M-CSF (5 ng/ml) for 12–18 h. Nonadherent cells were cultured further for the indicated times in 96-well dishes and their numbers determined in terms of OD450nm (n = 5; see Materials and methods for details). Standard deviations are too small to show. P < 0.001 for IRAK-M+/+ versus IRAK-M−/− macrophages treated for 3, 4, and 5 d; P < 0.01 for IRAK-M+/+ versus IRAK-M−/− macrophages treated for 6 d. (B) Cells from (A) were stained for TRAP. Bar, 100 μm. (C) Nonadherent cells, prepared as in (A), were treated further with 30 ng/ml M-CSF for 2 d, replated in 96-well dishes (2 × 105 cells/ml), and treated with 300 ng/ml RANKL plus with increasing concentrations of M-CSF for 3 d. The surface area occupied by TRAP+ multinucleated osteoclasts was recorded (n = 4). Standard deviations are too small to show. P < 0.001 for IRAK-M+/+ versus IRAK-M−/− macrophages treated with 7.5, 15, 30, and 60 ng/ml M-CSF. (D) Photographs of cells in (C). Bar, 100 μm. (E) Nonadherent cells, prepared as in (A), were treated further with 30 ng/ml M-CSF for 2 d, replated in 96-well dishes (2 × 105 cells/ml), and stimulated with 30 ng/ml M-CSF plus with increasing concentrations of RANKL for 3 d. The surface area occupied by TRAP+ multinucleated cells was recorded (n = 4). Standard deviations are too small to show. P < 0.001 for IRAK-M+/+ versus IRAK-M−/− macrophages at each RANKL concentration. (F) Cells from (E) were stained for TRAP. Bar,100 μm. (G) Osteoclasts were generated as in (E) but were treated with 300 ng/ml RANKL and 30 ng/ml M-CSF for the indicated times. Bar, 100 μm. Each experiment was repeated at least three times with similar results.
Next, to examine whether the differentiation of osteoclasts was affected by the absence of IRAK-M, we treated bone marrow macrophages with increasing concentrations of M-CSF in the presence of 300 ng/ml RANKL in order to generate tartrate-resistant acid phosphatase-positive (TRAP+) multinucleated macrophages with an osteoclast phenotype (16). IRAK-M−/− macrophages required as little as 15 ng/ml M-CSF to reach a maximally multinucleate state, whereas IRAK-M+/+ cells required 60 ng/ml to reach a maximal state that was less extensive (Fig. 2, C and D). However, higher concentrations of M-CSF tended to have a more limited effect on multinucleation in IRAK-M−/− macrophages, which suggested an increased sensitivity of IRAK-M−/− cells to M-CSF. When we treated macrophages with 30 ng/ml M-CSF in the presence of increasing concentrations of RANKL, IRAK-M−/− macrophages matured into osteoclasts at lower concentrations of RANKL than did IRAK-M+/+ macrophages (Fig. 2, E and F). However, this difference between IRAK-M−/− and IRAK-M+/+ macrophages remained relatively constant with increasing concentrations of RANKL, except at the highest dose when multinucleation of macrophages had reached its maximum. Thus, the absence of IRAK-M tended to favor the multinucleation of macrophages, a phenomenon that might explain, in part, the elevated numbers of osteoclasts in IRAK-M−/− mice.
To determine whether the half-life of osteoclasts also was altered in IRAK-M–deficient mice, we cultured osteoclast precursors in the presence of 30 ng/ml M-CSF plus 300 ng/ml RANKL for 2, 3, or 4 d, and then we stained the cells for TRAP. The IRAK-M−/− macrophages differentiated into multinucleated osteoclasts until day 4, by which time the IRAK-M+/+ osteoclasts had undergone apoptosis (Fig. 2 G). Together, these data indicate that the absence of IRAK-M is associated with an elevated rate of osteoclastogenesis in response to M-CSF alone and to M-CSF plus RANKL and to extension of the half-life of osteoclasts, which would explain the increased number of osteoclasts in IRAK-M−/− mice. Because M-CSF stimulates the production of cytokines by macrophages, which affects osteoclastogenesis (17), we postulated that the absence of IRAK-M might affect the response of macrophages to the M-CSF–induced production of IL-1.
To examine our hypothesis, we determined the relative amount of IL-1 that accumulated over the course of 12 and 24 h in the conditioned medium from IRAK-M+/+ and IRAK-M−/− bone marrow macrophages, with and without stimulation by M-CSF alone and by M-CSF plus RANKL. Our results confirmed that M-CSF stimulated the release of IL-1α and IL-1β by macrophages (Table III). RANKL did not alter that M-CSF–induced production of IL-1. Our analysis also revealed that the concentrations of IL-1α and IL-1β were higher in supernatants from macrophages that were deficient in IRAK-M than in those from wild-type cells; this supports the hypothesis that IRAK-M intercepts IL-1R signaling and prevents further production of IL-1. The lower abundance of IL-1 at 24 h might reflect the partial degradation of IL-1 or its binding to IL-1R.
Table III.
Deletion of the Irak-M gene in mice leads to increased production of IL-1α and IL-1β by bone marrow macrophages that are stimulated with M-CSF
| 12 h
|
24 h
|
||||||
|---|---|---|---|---|---|---|---|
| IRAK-M+/+ | IRAK-M−/− | −/− /+/+ | IRAK-M+/+ | IRAK-M−/− | −/− /+/+ | ||
| IL-1α | M-CSF | 1.09a | 1.26a | 1.15 | 0.53a | 1.09a | 2.06 |
| M-CSF + RANKL | 1.27a | 1.26a | 1.00 | 0.59a | 1.06a | 1.80 | |
| IL-1β | M-CSF | 1.59a | 2.08a | 1.31 | 0.52a | 0.91a | 1.75 |
| M-CSF + RANKL | 1.52a | 1.90a | 1.25 | 0.51a | 1.02a | 2.00 | |
Each experiment was repeated three times with similar results.
Values represent the ratios of 12-h or 24-h stimulation with M-CSF or M-CSF plus RANKL over controls (n = 2).
Next, we asked whether the expression of IRAK-M is preserved upon multinucleation of macrophages. We treated IRAK-M+/+ osteoclasts with 20 ng/ml IL-1α or with 100 μg/ml LPS for increasing times. To our surprise, we found that unlike macrophages, multinucleated osteoclasts expressed high levels of IRAK-M, independent of TLR/ IL-1R ligation (Fig. 3 A). Therefore, we asked whether RANKL itself could stimulate the expression of IRAK-M. RANKL induced the expression of IRAK-M in macrophages in a time-dependent manner (Fig. 3 B); this observation explained the high basal level of IRAK-M in osteoclasts.
Figure 3.

Osteoclasts express IRAK-M, and demonstrate hyperactivation of signaling molecules downstream of IL-1R/TLR when IRAK-M is absent. (A) Bone marrow macrophages from wild-type mice were cultured in the presence of M-CSF (5 ng/ml) for 12–18 h. Nonadherent cells were cultured further for 2 d in 24-well dishes (macrophages) and treated for the indicated times with 20 ng/ml IL-1α or 1 mg/ml LPS (macrophages), or cultured for an additional 3 d in the presence of 30 ng/ml M-CSF and 300 ng/ml RANKL (osteoclasts, OC) before activation with IL-1α or LPS. Cells were lysed in Laemmli sample buffer supplemented with inhibitors of proteases and phosphatases and subjected to Western blotting analysis with anti–IRAK-M and anti-GADPH antibodies. (B) Bone marrow macrophages from wild-type mice were treated with 300 ng/ml RANKL for the indicated times, bone marrow macrophages from IRAK-M-deficient mice were treated similarly for 24 h and used as a negative control. Cells were analyzed as in (B). (C) IRAK-M+/+ and IRAK-M−/− osteoclasts were starved for 2 h, and then stimulated with 20 ng/ml IL-1α, IL-1β, or 1 mg/ml LPS for the indicated times. (D) IRAK-M+/+ and IRAK-M−/− osteoclasts were stimulated with 1 mg/ml LPS for the indicated times. The right lane shows a positive control for the phosphorylation of JNK. Each experiment was repeated three times with similar results. P-p38, phosphorylated p38.
Hyperactivation of NF-κB and mitogen-activated protein kinase signaling pathways in response to IL-1R/TLR ligation in IL-1 receptor–associated kinase M–deficient osteoclasts
We examined whether the absence of IRAK-M could lead to hyperactivation of the NF-κB and MAPK signaling pathways (15). Because IL-1R/TLR ligation affects the differentiation and activation of osteoclasts, we stimulated osteoclasts from IRAK-M−/− and IRAK-M+/+ mice for increasing times with IL-1α or IL-1β, both of which are potent agonists of osteoclastic bone resorption. We subjected cell lysates to Western blotting analysis with antibodies that are directed against IκB, and the MAPK signaling molecules, p38, ERK1/2, and JNK, and their phosphorylated, thus activated, form. Although p38 is not activated constitutively in macrophages (15), p38 was activated constitutively in IRAK-M−/− and IRAK-M+/+ osteoclasts (Fig. 3 C). Both IL-1α and IL-1β induced the hyperphosphorylation of ERK1/2 and JNK in a time-dependent manner in IRAK-M−/− osteoclasts. By contrast, only IL-1α induced the time-dependent hyperphosphorylation of I-κB in osteoclasts that lacked IRAK-M.
We next asked whether activation of osteoclasts by LPS, a ligand for TLR4, and a potent agonist of osteoclastic bone resorption in vivo, also might be affected by the absence of IRAK-M. We found that p38 was hyperphosphorylated constitutively in IRAK-M+/+ and IRAK-M−/− osteoclasts, independently of TLR4 ligation (Fig. 3 D). As did IL-1, LPS induced the hyperphosphorylation of ERK1/2 and IκB in IRAK-M−/− cells. However, in contrast to the results that were observed with IL-1, hyperphosphorylation of IκB in IRAK-M−/− osteoclasts occurred only 1 or 2 h after stimulation with LPS. LPS failed to activate JNK in IRAK-M−/− and wild-type osteoclasts.
Together, our results show that in macrophages and osteoclasts, the absence of IRAK-M leads to hyperactivation of the NF-κB and MAPK signaling pathways in response to IL-1R/TLR ligation.
DISCUSSION
IRAK-M is the first signaling molecule that lacks intrinsic enzymatic activity to be shown to regulate the differentiation, activation, and life span of osteoclasts. During signal transduction, phosphorylation of signaling molecules represents the sum, at any given time, of opposing effects of the kinases and phosphatases that determine the response. By virtue of its sequence homology to IRAKs, IRAK-M binds the IRAK/IRAK-4 complex, thereby acting as a dominant negative IRAK that intercepts signals downstream of IL-1R/TLR and whose effect is independent of phosphorylation. The induced expression of IRAK-M is sufficient to interfere with IL-1R/TLR signaling. In this report, we showed that the level of expression of IRAK-M in osteoclasts is high as compared with that in macrophages and that the absence of IRAK-M leads to severe osteoporosis which is associated with increased numbers of osteoclasts. These effects seem to be due, in part, to the M-CSF–induced production of IL-1, which accelerates the rate of differentiation, extends the half-life, and stimulates the activity of osteoclasts (7). Therefore, we propose the following model for IRAK-M signaling during osteoclastogenesis (Fig. 4).
Figure 4.

Hypothetical scheme for the role of IRAK-M in the differentiation and activation of osteoclasts. Ligation of c-Fms (1) stimulates the production of IL-1, which activates IL-1R. Ligation of RANK (2) leads to TRAF6-mediated activation of NF-κB (3), and induction of expression of IRAK-M, (4) IL-1 activates IL-1R (5), which stimulates osteoclastogenesis and the osteoclastic resorption of bone, and extends the half-life of osteoclasts. When IRAK-M is present, it prevents IL-1R/TLR-mediated downstream signaling and the activation of NF-κB. Thus, IRAK-M mediates a negative feedback mechanism that interrupts signaling downstream of IL-1R. In the absence of IRAK-M, the IL-1 cycle continues without interruption, leading to increased osteoclastogenesis, with the hyperactivation and prolonged half-life of osteoclasts.
As shown schematically in Fig. 4, M-CSF that is expressed by bone marrow stromal cells and osteoblasts induces the clonal expansion of macrophages and the release by them of IL-1, with resultant activation of IL-1R/TLR. Activation of IL-1R leads to an elevated rate of differentiation, an extended half-life, and the activation of osteoclasts. RANKL, expressed by bone marrow cells and osteoblasts, binds receptor activator of NF-κB (RANK) and activates NF-κB via TRAF6, which promotes the expression of IRAK-M. Upon religation of IL-1R, IRAK-M prevents the dissociation of the IRAK/IRAK-4–TRAF6 complex and, hence, the downstream activation of NF-κB. Overall, IRAK-M ensures the ability of osteoclasts to tolerate the effects of IL-1. M-CSF, RANK, TRAF6, and NF-κB are required for osteoclastogenesis (1, 5, 18–20). Hence, IRAK-M–mediated deactivation of IL-1R allows the transient activation and survival of osteoclasts. The absence of IRAK-M leads to constitutive activation of IL-1R/TLR, constitutive production of IL-1, and the prolonged survival and activation of osteoclasts. Moreover, IRAK-M also mediates tolerance to LPS. Thus, IRAK-M, whose expression is restricted to myeloid cells and is induced strongly by RANKL, seems to be a novel and critical down-regulator of the differentiation and activation of osteoclasts. Because the expression of IRAK-M is induced by RANKL, which is required for the maturation, fusion, and differentiation of macrophages into osteoclasts, expression of IRAK-M provides a powerful negative feedback mechanism.
Fusion of mononuclear phagocytes is the first step in osteoclastogenesis. This explains why mononucleate macrophages cannot resorb bone efficiently and leads to osteopetrosis. Hence, inhibition of the production of IL-1 is an essential step in the prevention of osteoclastic bone resorption.
Together, increased osteoclastogenesis and the extended survival of osteoclasts explain the elevated number of osteoclasts in IRAK-M−/− mice, and, hence, their low bone mass. It is unclear whether the osteoclasts are individually more active and resorb bone more efficiently. Although we have recorded the numbers and the sizes of the “pits” that are formed in dentin slices by osteoclasts as a function of time, we have not been able to detect differences between pits of IRAK-M+/+ and IRAK-M−/− osteoclasts (unpublished data). However, because numbers and half-life of osteoclasts are increased in IRAK-M−/− mice, further increases in their activity would aggravate osteoporosis even further.
It also is unclear whether the osteoporotic phenotype of mice that lack IRAK-M is mediated solely by osteoclasts. Macrophages are present in all tissues and might well contribute, via the cytokines and growth factors that they produce (e.g., IL-1), to the differentiation and activation of osteoclasts that leads to osteoporosis. Osteoclasts and macrophages share a common precursor and numerous functions. Thus, there might be cross talk between them.
In terms of phenotype, male mice were affected more severely than female mice by the absence of IRAK-M. Possible associations between sex hormones and the expression of IRAK-M remain to be investigated. To avoid confounding effects that were due to gender, we performed our studies using cells that originated exclusively from same-sex mice, namely male mice.
Osteoclasts, which originate from the fusion of mononuclear phagocytes, as do the giant cells that are found at sites of chronic inflammatory reactions and granulomas, not only express IRAK-M, but express it at higher levels than mononuclear cells. This supports the possibility that multinucleation might represent a primitive defense mechanism that evolved to protect organisms against invasive pathogens. That same developmental process has endowed macrophages with the ability to resorb bone by differentiating into osteoclasts. Moreover, macrophage fusion seems to involve the macrophage fusion receptor/signal regulatory protein α-CD47 (21, 22) axis, and macrophage fusion receptor shares a common origin with antigen receptors. Thus, macrophage fusion might have evolved before jawed vertebrates (23).
IRAK-M is a noncatalytic signaling molecule whose expression is induced by RANKL. Its potency seems to depend upon its level of expression. Thus, if we could develop methods for regulating the expression of IRAK-M, we might be able to control bone mass and move toward prevention of osteoporosis.
MATERIALS AND METHODS
Animals.
IRAK-M−/− mice were produced by homologous recombination as described previously (17). Mice were screened by PCR using the IRAK-M+/+ forward primer (exon 4–5) 5′-gccagaagaatacatcagacaggg-3′ and reverse primer 5′-tgtttcgggtcatccagcac-3′; and the IRAK-M−/− forward primer 5′-cgttccataacacacctctctgc-3′ and reverse primer 5′-ttctatcgccttcttgacgagttc-3′. Animals were housed and bred at the Yale Animal Care facility, under sterile conditions reserved for immuno-deficient mice, which include autoclaved caging and food, as well as changing of cages in a clean-air cabinet/change station using sterile techniques. Mice whose bones were subjected to histomorphometric analysis received two i.p. injections of calcein (3 μg/g body weight; Merck) on days 1 and 6 before being killed. All experiments were approved by the Yale Animal Care and Use Committee.
Computed tomography on a microscale.
The proximal tibiae from 4-mo-old male IRAK-M−/− and IRAK-M+/+ mice were scanned with a MicroCT 40 scanner (Scanco) with a 2,048 × 2,048 matrix and isotropic resolution of 5 μm3; three-dimensional trabecular measurements in the secondary spongiosa were made directly.
Bone densitometry.
Bone density was determined as described previously (24) by peripheral quantitative computed tomography with a Stratec scanner model XCT 960M (Norland Medical Systems). Routine calibration was performed daily with a defined standard that contained hydroxyapatite crystals embedded in lucite, provided by Norland Medical Systems. We scanned 1-mm–thick slices located at a distance of 3 mm, proximally, from the distal end of distal femoral metaphyses. The voxel size was set at 0.15 mm. Scans were analyzed with a software program that was supplied by the manufacturer (XMICE, version 5.1). Bone density and geometric parameters were estimated by Loop analysis. The low- and high-density threshold settings were 1,300 and 2,000, respectively. Separation of soft tissue from the outer edge of bone was achieved using contour mode 1. Cortical (high bone density) and trabecular (low bone density) bone were separated to obtain trabecular data using peel mode 3. Cortical and trabecular bone were separated to obtain cortical data using cortical mode 1.
Histomorphometry.
Tibiae from IRAK-M+/+ and IRAK-M−/− mice were dehydrated in a graded ethanol series and embedded without decalcification in methylmethacrylate, as we described previously (25). Longitudinal sections were cut with an Autocut microtome with a tungsten carbide blade (Jung). 4-μm-thick sections were stained with toluidine blue (pH 3.7) and subjected to static histomorphometric analysis, whereas 8-μm–thick sections were mounted (unstained) for dynamic histomorphometric analysis, which was performed at a constant distance from the growth plate (including trabecular bone), with an image analysis system (Osteomeasure; Osteometrics). The measured parameters included the bone volume relative to the total volume (BV/TV); the rate of bone formation (BFR/BV), which takes into account the mineral apposition rate; the number of osteoclasts per active resorption perimeter (N.Oc/B.Pm); the number of osteoblasts per active formation perimeter (N.Ob/B.Pm); and the osteoid volume relative to bone volume (OV/BV).
Reagents.
The soluble form of recombinant TRANCE/RANKL that we used was described previously (16). Recombinant mouse M-CSF, IL-1α, and IL-1β were obtained from R & D Systems. A cell proliferation (WST-1) kit was purchased from Roche Molecular Biochemicals. Unless otherwise stated, all chemicals were from Sigma-Aldrich. A polyclonal antibody raised in rabbits and directed against IRAK-M was purchased from Chemicon International; rabbit polyclonal antibodies directed against p38, phosphorylated-p38, ERK1/2, P-ERK1/2, JNK, and mouse monoclonal antibodies directed against P-IκB and P-JNK were obtained from Cell Signaling. A mouse monoclonal antibody directed against GAPDH was purchased from Novus Biologicals, Inc. Horseradish peroxidase-conjugated F(ab′)2 directed against rabbit and mouse IgG was purchased from Jackson ImmunoResearch Laboratories. All supplies and reagents for tissue culture were endotoxin-free. Some bone marrow cells were treated with polymyxin B sulfate for 24 h to avoid the effects of the endotoxin before treatment.
Bone marrow macrophages and osteoclasts.
Bone marrow cells from 6- to 12-wk-old IRAK-M−/− and IRAK-M+/+ male mice were plated in 10 cm dishes and cultured in α-MEM (Life Technologies) supplemented with 10% FBS in the presence of M-CSF (5 ng/m; 107 cells/10-cm dish) for 12–18 h. Nonadherent cells were harvested and cultured with M-CSF (30 ng/ml) in 10 cm dishes, at the same density as before, for an additional 48 h. Floating cells were removed and attached cells, which are TRAP+ macrophages were used as osteoclast precursors (16).
To generate osteoclasts, we cultured bone marrow macrophages in the presence of RANKL (300 ng/ml) and M-CSF (30 ng/ml) or a 30% (vol/vol) dilution of the supernatant from a culture of L929 cells, in 96-well, 24-well, or 60 mm dishes at a density of 0.5 × 106 cells/ml.
Cytokine arrays.
Bone marrow macrophages from IRAK-M+/+ and IRAK-M−/− mice were plated, in duplicate wells, in 24-well dishes (0.5 × 106 cells/well) and treated with 20 ng/ml M-CSF for 10–12 h. Cells were starved for 4 h and then cultured unstimulated or stimulated for 24 h with 20 ng/ml M-CSF alone or in combination with 300 ng/ml RANKL in 0.5 ml of serum-free medium. Supernatants were collected and analyzed for cytokines using mouse cytokine antibody arrays (Raybiotech, Inc.). Signals on films were quantitated with National Institutes of Health Image software.
Western blotting analysis.
Cultured cells were lysed directly in Laemmli's sample buffer supplemented with a cocktail of protease inhibitors (Complete Tablets, Roche Molecular Biochemicals) and phosphatase inhibitors (sodium fluoride and sodium vanedate). The lysates were sonicated before analysis by electrophoresis and Western blotting with ECL (Pierce Chemical Co.).
Statistical analysis.
Data in Tables I and II are means ± 1 SD. Statistically significant differences among experimental groups were evaluated by the analysis of variances (26). The significance of mean changes was determined by an unpaired Student's two-tailed t test, and significance was recognized when P < 0.05.
Acknowledgments
The authors are deeply grateful to Dr. A. Körner for her careful editing of this text.
This work was supported by grants from the National Institutes of Health (DE12110) to A. Vignery.
The authors have no conflicting financial interests.
Abbreviations used: ERK, extracellular signal-regulated kinase; IRAK, IL-1R–associated kinase; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; M-CSF, macrophage colony–stimulating factor; RANK, receptor activator of NF-κB; RANKL, RANK ligand; TLR, Toll-like receptor; TRAF, TNF receptor–associated factor.
H. Li and E. Cuartas contributed equally to this work.
References
- 1.Yoshida, H., S.-I. Hayashi, T. Kunisada, M. Ogawa, S. Nishikawa, H. Okamura, T. Sudo, L.D. Shultz, and S.-I. Nishikawa. 1990. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 345:442–444. [DOI] [PubMed] [Google Scholar]
- 2.Yasuda, H., N. Shima, N. Nakagawa, K. Yamaguchi, M. Kinosaki, S. Mochizuki, A. Tomoyasu, K. Yano, M. Goto, A. Murakami, et al. 1998. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 31:3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lacey, D.L., E. Timms, H.L. Tan, M.J. Kelley, C.R. Dunstan, T. Burgess, R. Elliott, A. Colombero, G. Elliott, S. Scully, et al. 1998. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 93:165–176. [DOI] [PubMed] [Google Scholar]
- 4.Simonet, W.S., D.L. Lacey, C.R. Dunstan, M. Kelley, M.-S. Chang, R. Lüthy, H.Q. Nguyen, S. Wooden, L. Bennett, T. Boone, et al. 1997. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 18:309–319. [DOI] [PubMed] [Google Scholar]
- 5.Dougall, W.C., M. Glaccum, K. Charrier, K. Rohrbach, K. Brasel, T. De Smedt, E. Daro, J. Smith, M.E. Tometsko, C.R. Maliszewski, et al. 1999. RANK is essential for osteoclast and lymph node development. Genes Dev. 15:2412–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li, J., I. Sarosi, X.Q. Yan, S. Morony, C. Capparelli, H.L. Tan, S. McCabe, R. Elliott, S. Scully, G. Van, et al. 2000. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA. 15:1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jimi E., I. Nakamura, L.T. Duong, T. Ikebe, N. Takahashi, G.A. Rodan, and T. Suda. 1999. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp. Cell Res. 247: 84-93. [DOI] [PubMed] [Google Scholar]
- 8.Nair, S.P., S. Meghji, M. Wilson, K. Reddi, P. White, and B. Henderson. 1996. Bacterially induced bone destruction: mechanism and misconceptions. Infect. Immun. 64:2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitazawa, R., R.B. Kimble, J.L. Vannice, V.T. Kung, and R. Pacifici. 1994. Interleukin-1 receptor antagonist and tumor necrosis factor binding protein decrease osteoclast formation and bone resorption in ovariectomized mice. J. Clin. Invest. 94:2397–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimble, R.B., A.B. Matayoshi, J.L. Vannice, V.T. Kung, C. Williams, and R. Pacifici. 1995. Simultaneous block of interleukin-1 and tumor necrosis factor is required to completely prevent bone loss in the early postovariectomy period. Endocrinology. 136:3054–3061. [DOI] [PubMed] [Google Scholar]
- 11.Janssens, S., and R. Beyaert. 2003. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol. Cell. 11:293–302. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-κB puzzle. Cell Suppl. 109:S81–S96. [DOI] [PubMed] [Google Scholar]
- 13.Kyriakis, J.M., and J. Avruch. 2001. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81:807–809. [DOI] [PubMed] [Google Scholar]
- 14.Wesche, H., X. Gao, X. Li, C.J. Kirschning, G.R. Stark, and Z. Cao. 1999. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 274:19403–19410. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi, K., L.D. Hernandez, J.E. Galan, C.A. Janeway Jr, R. Medzhitov, and R.A. Flavell. 2002. IRAK-M is a negative regulation of Toll-like receptor signaling. Cell. 110:191–202. [DOI] [PubMed] [Google Scholar]
- 16.Takami, M., N. Kim, J. Rho, and Y. Choi. 2002. Stimulation by toll-like receptors inhibits osteoclast differentiation. J. Immunol. 169:1516–1523. [DOI] [PubMed] [Google Scholar]
- 17.Cappellen, D., N.H. Luong-Nguyen, S. Bongiovanni, O. Grenet, C. Wanke, and M. Susa. 2002. Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NF-κB. J. Biol. Chem. 277:21971–21982. [DOI] [PubMed] [Google Scholar]
- 18.Naito, A., S. Azuma, S. Tanaka, T. Miyazaki, S. Takaki, K. Takatsu, K. Nakao, K. Nakamura, M. Katsuki, T. Yamamoto, and J. Inoue. 1999. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 4:353–362. [DOI] [PubMed] [Google Scholar]
- 19.Lomaga, M.A., W.C. Yeh, I. Sarosi, G.S. Duncan, C. Furlonger, A. Ho, S. Morony, C. Capparelli, G. Van, S. Kaufman, et al. 1999. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13:1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lotsova, V., J. Caamano, J. Loy, Y. Yang, A. Lewin, and R. Bravo. 1997. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat. Med. 3:1285–1289. [DOI] [PubMed] [Google Scholar]
- 21.Saginario, C., H. Sterling, C. Beckers, R. Kobayashi, M. Solimena, E. Ullu, and A. Vignery. 1998. MFR, a putative receptor mediating the fusion of macrophages. Mol. Cell. Biol. 18:6213–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han, X., H. Sterling, Y. Chen, C. Saginario, E.J. Brown, W.A. Frazier, F. Lindberg, A. Vignery, J.A. Yoder, and G.W. Litman. 2004. On the origins of adaptive immunity: innate immune receptors join the tale. Trends Immunol. 25:11–16. [DOI] [PubMed] [Google Scholar]
- 23.Ballica, R., K. Valentijn, A. Khachatryan, S. Guerder, S. Kapadia, C. Gundberg, J. Gilligan, R.A. Flavell, and A. Vignery. 1999. Targeted expression of calcitonin gene related peptide to osteoblasts increases bone density in mice. J. Bone Miner. Res. 14:1067–1074. [DOI] [PubMed] [Google Scholar]
- 24.Baron, R., A. Vignery, L. Neff, A. Silvergate, and A. Santa Maria. 1982. Processing of undecalcified bone specimens for bone histomorphometry. Bone Histomorphometry: Techniques and Interpretation. R.R. Recker, H.M. Frost, editors. CRC Press, Inc., Boca Raton, FL. 13-35.
- 25.Zar, J.H. 1974. Biostatistical Analysis. Prentice-Hall, Englewood Cliffs, NJ. 620 pp.