

Abstract
5-aminosalicylic acid (5-ASA) is an antiinflammatory drug widely used in the treatment of inflammatory bowel diseases. It is known to inhibit the production of cytokines and inflammatory mediators, but the mechanism underlying the intestinal effects of 5-ASA remains unknown. Based on the common activities of peroxisome proliferator–activated receptor-γ (PPAR-γ) ligands and 5-ASA, we hypothesized that this nuclear receptor mediates 5-ASA therapeutic action. To test this possibility, colitis was induced in heterozygous PPAR-γ+/− mice and their wild-type littermates, which were then treated with 5-ASA. 5-ASA treatment had a beneficial effect on colitis only in wild-type and not in heterozygous mice. In epithelial cells, 5-ASA increased PPAR-γ expression, promoted its translocation from the cytoplasm to the nucleus, and induced a modification of its conformation permitting the recruitment of coactivators and the activation of a peroxisome-proliferator response element–driven gene. Validation of these results was obtained with organ cultures of human colonic biopsies. These data identify PPAR-γ as a target of 5-ASA underlying antiinflammatory effects in the colon.
5-aminosalicylic acid (5-ASA) is among the oldest antiinflammatory agents in use today for the treatment of Crohn's disease and ulcerative colitis, two major human forms of chronic inflammatory bowel diseases (IBDs). In 1942, 5-ASA was initially combined with an antibiotic sulfapyridine in a molecule named sulphasalazine (1). Thirty-five years later, Azad Khan et al. demonstrated that 5-ASA was the therapeutically active moiety of this drug with proven antiinflammatory effects in IBDs (2). After oral or rectal administration, 5-ASA acts locally in the colon and is absorbed by colonic epithelial cells (3–5). The effectiveness of the drug is related to its mucosal concentration (6), and systemic dosages remain low after oral sulphasalazine and rectal 5-ASA administration (7). The putative antiinflammatory actions of 5-ASA include modulation of inflammatory cytokine production (8), decreased transcriptional activity of NF-κB by modulating RelA/p65 phosphorylation (9), and inhibition of the biosynthesis of prostaglandins and leukotrienes (10). However, despite these numerous experimental studies, the basic mechanism of action of 5-ASA remains unknown.
Peroxisome proliferator–activated receptors (PPARs) are members of the nuclear receptor superfamily. They are activated by fatty acids and are involved in the transduction of metabolic and nutritional signals into transcriptional responses (11). Among these transcription factors, PPAR-γ plays an important role in the maintenance of mucosal integrity in the intestine (12). Expressed at high levels in the colonic epithelium, PPAR-γ and its high-affinity synthetic ligands such as thiazolidinediones are involved in the regulation of colon inflammation (12–15). Based on the common activities of 5-ASA and PPAR-γ ligands in colonic epithelial cells, we hypothesized that PPAR-γ may be the molecular target of 5-ASA. In the present study, we report that chemically induced colitis in mice heterozygous at the PPAR-γ locus (PPAR-γ+/−) was refractory to 5-ASA therapy. Using the HT-29 colon epithelial cell line, we found that 5-ASA induced PPAR-γ mRNA and protein expression. Furthermore, the ability of 5-ASA to bind and activate PPAR-γ in different experimental settings including receptor binding, transcriptional activation, and conformational changes indicated that 5-ASA exerts its effects in the colon through direct PPAR-γ activation.
RESULTS
Colon inflammation in PPAR-γ+/− mice is refractory to 5-ASA therapy
Intrarectal administration of 2,4,6-trinitrobenzene sulfonic acid (TNBS) to male 129/Sv mice provoked a severe colitis, which represents a well-validated model that has many macroscopic and histologic similarities to human IBDs (Fig. 1 A) (16–18). In nontreated animals killed 5 d after colitis induction, weight loss and severe macroscopic and histologic lesions that would have resulted in death were observed in 50% of mice (Fig. 1). These lesions, characterized by large areas of ulceration, necrosis, and a transparietal neutrophilic infiltration, were scored by macroscopic and histologic evaluation as 5.2 ± 0.8 and 2.6 ± 1.2, respectively, according to the multiparametric Wallace and Ameho criteria (19, 20). The most clinically effective dosage of a controlled-release 5-ASA preparation given orally was investigated in these conventional 129/Sv mice by a detailed dose–response study of dosages ranging from 50 mg/kg/d to 1,000 mg/kg/d (Fig. 1, B–D). Evolution of weight curves was similar in control mice without colitis and mice receiving TNBS and treated with 5-ASA at 100, 500, and 1,000 mg/kg, but not with 5-ASA at 50 mg/kg (Fig. 1 B). These data were used to eliminate the 5-ASA dosage of 50 mg/kg. Because 5-ASA at 100 mg/kg was the concentration of 5-ASA closest to that used to treat IBDs in humans (21), and because the therapeutic effects of 5-ASA on body-weight changes (Fig. 1 B), mortality rate (Fig. 1 C), and macroscopic Wallace scores (Fig. 1 D) were statistically similar for the dosages of 100, 500, and 1,000 mg/kg, we selected 100 mg/kg as the optimal concentration of 5-ASA for the following study in PPAR-γ+/− mice. At this concentration of 5-ASA, improvements in body-weight changes, mortality rate, and Wallace scores were similar those seen in the rosiglitazone-positive mice (Fig. 1, B–E). A significant decrease in colonic methyl peroxidase (MPO) and TNF-α mRNA concentrations used as biological markers of inflammation paralleled this improvement (Fig. 1 F). To determine whether the antiinflammatory effects of 5-ASA in the colon were mediated by PPAR-γ, colitis was induced in 129/Sv PPAR-γ+/− mice by intrarectal TNBS administration. Compared with their wild-type littermates, these heterozygous knockout mice had a mean 70 ± 8% decrease of PPAR-γ mRNA levels in the colon. As established previously (17), PPAR-γ+/− mice exhibited more pronounced macroscopic/histologic lesions and higher concentrations of biological markers of inflammation than wild-type animals (Fig. 1, E and F). In contrast with control mice, 5-ASA treatment did not affect the severity of TNBS colitis in PPAR-γ+/− mice, as assessed by macroscopic or histologic scores, colonic MPO, or TNF-α concentrations (Fig. 1, E and F), indicating PPAR-γ may have a major role in mediating the antiinflammatory effect of 5-ASA.
Figure 1.

Colon inflammation in PPAR-γ heterozygous mice is refractory to 5-ASA therapy. (A) Colitis was induced by intrarectal administration of TNBS in wild-type mice (black) and PPARγ+/− mice (gray). Animals were killed 5 d later to evaluate the intensity of colitis. (B–D) A detailed dose–response study was performed with oral 5-ASA given daily and compared with untreated animals with colitis (control) or receiving an optimal dosage (20 mg/kg) of rosiglitazone (rosi). 5-ASA effects were analyzed according to the evolution of (B) body weight, (C) mortality, and (D) macroscopic Wallace scores. (E and F) To evaluate whether the antiinflammatory role of 5-ASA in the colon was mediated by PPAR-γ, we compared the antiinflammatory effects of the most efficient dosage of 5-ASA in wild-type mice (black) and in PPARγ+/− mice (gray). In contrast with wild-type mice, 5-ASA was ineffective in PPARγ+/− mice and did not modify (E) macroscopic and histologic lesions evaluated by the Wallace and Ameho scores, respectively, or (F) the colonic molecular markers of inflammation such as myeloperoxidase (MPO) and the production of TNF-α. The number of mice and statistical significance are indicated. Results are expressed as the mean ± SEM. *, P < 0.05 compared with untreated mice with colitis; ○, P < 0.01 compared with untreated mice with colitis.
5-ASA induces differentiation of 3T3-L1 cells
Most ligands of PPAR-γ are potent and effective at stimulating adipogenesis in preadipocyte cell lines expressing PPAR-γ such as 3T3-L1 cells (22). To evaluate the role of 5-ASA as a PPAR-γ activator, we compared the effects of different concentrations of 5-ASA and of the thiazolidinedione rosiglitazone on confluent 3T3-L1 preadipocytes at day 8 after confluence. Optimal dosage of rosiglitazone (10−5 M) enhanced intracytosolic accumulation of lipid as monitored by Oil Red O staining in 94 ± 3% of cells (Fig. 2). Treatment with 5-ASA also induced a marked differentiation of preadipocytes in a dose-dependant manner leading to lipid accumulation in 98 ± 1% of cells (Fig. 2). Lactate dehydrogenase activity remained low and was not modified in the presence of rosiglitazone or 5-ASA (unpublished data), ensuring that 5-ASA did not exert a direct cytotoxic effect on 3T3-L1 cells. These data establish that 5-ASA has an adipogenic capacity similar to that of rosiglitazone.
Figure 2.
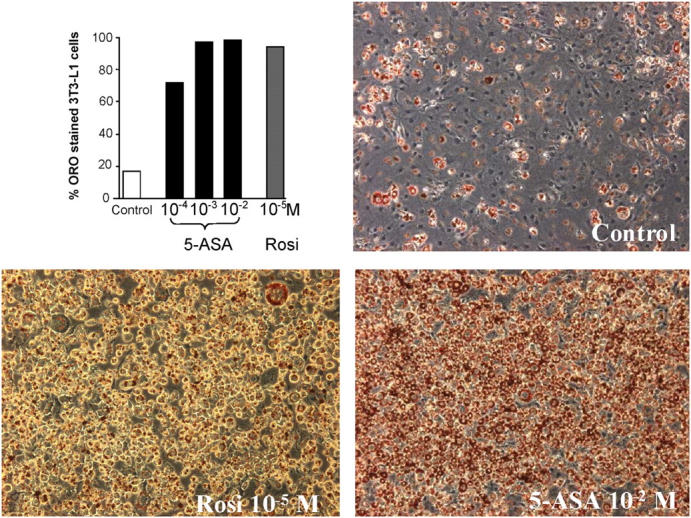
5-ASA induces differentiation of 3T3-L1 cells. The adipogenic effect of different concentrations of 5-ASA (from 10−4 M to 10−2 M) was evaluated in 3T3-L1 preadipocytes and compared with rosiglitazone (rosi, 10−5 M). After 4 d of culture, only 17 ± 4% of cells incubated with medium alone (control) showed enhanced intracytosolic accumulation of lipid droplets monitored by Oil Red O (ORO) staining. Treatment with the different concentrations of 5-ASA and rosiglitazone induced a marked differentiation of 3T3-L1 cells. Results are expressed as the mean ± SEM number of stained cells counted in four different experiments.
Induction and activation of PPAR-γ expression in 5-ASA–treated epithelial cells
Experiments in cultured preadipocyte suggest that activation of PPAR-γ by rosiglitazone modestly increases the expression of this receptor in a positive-feedback loop (23, 24). We then compared the capacity of 5-ASA and of rosiglitazone to induce PPAR-γ expression at the mRNA and protein levels in the HT-29 cell line. A threefold induction of PPAR-γ mRNA expression quantified by real-time PCR technique was found in colonic epithelial cells treated for 12 h with 5-ASA, 30 mM (Fig. 3 A). Western blot extended and confirmed these results, showing a threefold induction of PPAR-γ protein levels in cells treated by 5-ASA, 30 mM, and a twofold induction of PPAR-γ protein levels in cells treated by rosiglitazone, 10−5 M (Fig. 3 B).
Figure 3.
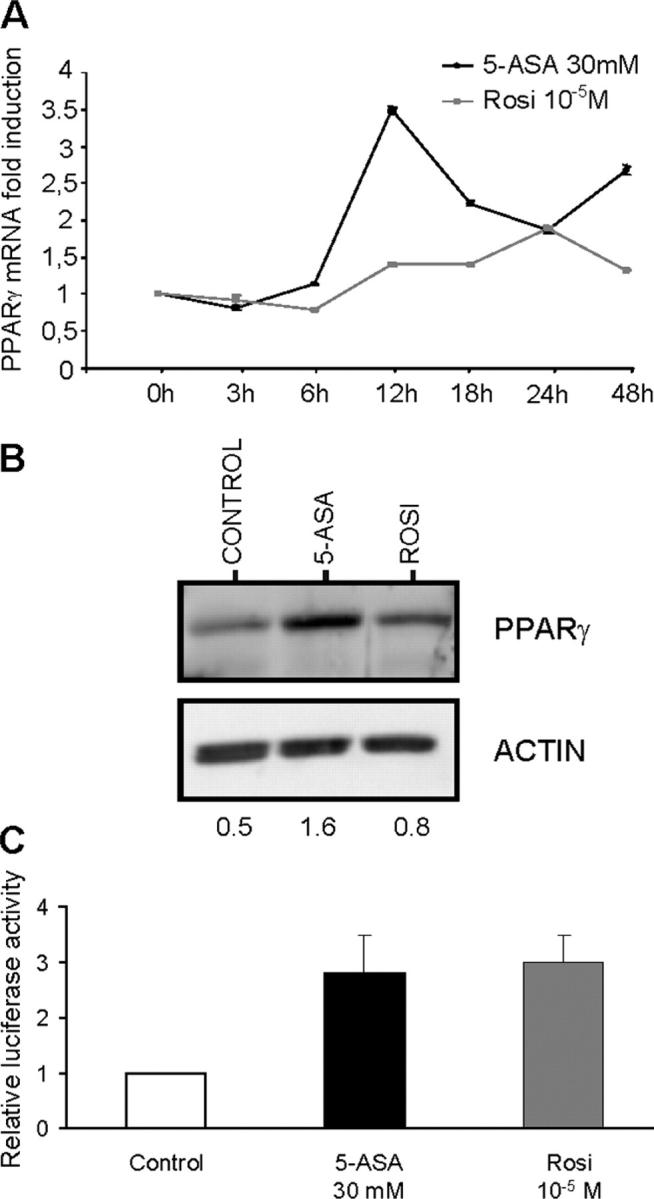
Induction and activation of PPAR-γ expression in 5-ASA–treated epithelial cells. (A) PPAR-γ mRNA expression was quantified by real-time PCR in HT-29 STD cells incubated for 3, 6, 12, 18, 24, and 48 h with 5-ASA (30 mM) or rosiglitazone (10−5 M). The main induction of PPAR-γ mRNA expression was observed at 12 h in cells incubated with 5-ASA. Results were expressed as the mean ± SEM of six different experiments. (B) The level of PPAR-γ protein expression was evaluated by Western blot assay in untreated HT-29 cells (control) and after 24 h of 5-ASA (30 mM) or rosiglitazone (rosi, 10−5 M) treatment. OD values of PPAR-γ were given for each condition in proportion to the quantity of the internal control β-actin in the same sample. (C) Activation of PPAR-γ by 5-ASA. HT-29 STD cells transfected with the response element for PPAR-γ (2XCYP) and treated by 5-ASA (30 mM) or rosiglitazone (10−5 M) showed a similar, approximately threefold induction of PPAR-γ reporter gene activity indicating the ability of 5-ASA to induce PPAR-γ activation. Results are expressed as fold activation (mean ± SEM) compared with untreated cells.
We also investigated PPAR-γ transcriptional activity by transient transfections (13). Analysis of PPAR-γ activity in transfected HT-29 cells showed that 5-ASA at 30 mM increased the reporter-gene activity by threefold, thereby displaying an activity similar to that of rosiglitazone (Fig. 3 C).
Binding of 5-ASA to PPAR-γ induces activation
Activation of PPAR-γ results in a cascade of reactions including translocation or redistribution of PPAR-γ in the cell nucleus (25, 26), conformational changes within PPAR-γ (27, 28), recruitment of coactivators, and binding to specific DNA sequence elements termed peroxisome-proliferator response elements (PPRE) (29–31). The effects of 5-ASA–bound PPAR-γ were examined at these different steps.
Intracellular localization of PPAR-γ in epithelial cells has not been characterized previously. In other cell types, distinct PPAR-γ localizations have been observed (25, 26, 32). Using fluorescence microscopy to visualize the cellular distribution of a GFP-tagged PPAR-γ, a predominantly cytoplasmic distribution of the fluorescent label was found in unstimulated HT-29 cells (Fig. 4 A). This cytoplasmic localization of PPAR-γ in epithelial cells has been noted previously ex vivo by immunohistochemistry in the colonic epithelial cells of patients (13). Exposure of epithelial cells to different concentrations of 5-ASA (1 mM, 5 mM, or 30 mM) for 24 h resulted in a similar redistribution of GFP-tagged PPAR-γ to the nucleus (Fig. 4, A and B). The translocation of GFP-tagged PPAR-γ from the cytoplasm to the nucleus was also observed with rosiglitazone.
Figure 4.

Intracellular localization of PPAR-γ in epithelial cells. (A) HT-29 STD cells transfected with GFP-tagged PPAR-γ were incubated in the presence of medium alone (unstimulated cells), 5-ASA (30 mM), or rosiglitazone (rosi, 10−5 M) for 24 h. Nuclear staining in blue was performed with Hoescht 33342 solution. The intracellular distribution of the fluorescent tags was examined under a fluorescence microscope. (B) HT-29 STD cells transfected with GFP-tagged PPAR-γ incubated in the presence of 5-ASA (30 mM) for 24 h. Green color represents PPAR-γ (left); blue color represents the nucleus (middle); merged green and blue pictures illustrate the translocation of PPAR-γ into the cell nucleus (right).
Next, to evaluate the conformational changes induced by the binding of 5-ASA to PPAR-γ, we performed a protease protection assay of [35S]-PPAR-γ bound to either 5-ASA or rosiglitazone. Rosiglitazone binding did not lead to a radical change in the proteolytic pattern as compared with 5-ASA (Fig. 5). As previously described, rosiglitazone-positive control induced a conformational change in PPAR-γ as assessed by the generation of two protease-resistant bands following partial chymotrypsin digestion of [35S]-PPAR-γ (Fig. 5 and references 27, 28). 5-ASA also generated protease-resistant fragments different from those observed with rosiglitazone, indicating that 5-ASA induces structural transitions in PPAR-γ distinct from those induced by rosiglitazone.
Figure 5.

Protease protection assay of PPAR-γ in presence of 5-ASA or rosiglitazone. Difference in protease sensitivity of 5-ASA–bound and rosiglitazone-bound PPAR-γ compared with nonliganded PPAR-γ (control). Autoradiogram of an SDS-PAGE gel showing [35S]methionine-labeled full-length human PPAR-γ2 digested with chymotrypsin. Asterisks and arrow denote chymotrypsin-resistant and chymotrypsin-sensitive-protein fragments, respectively.
Recruitment of transcriptional coactivators plays a central role in mediating transcriptional activity of liganded PPAR-γ. Among these coactivators, vitamin D3 receptor–interacting protein (DRIP)-205 is an important coactivator interacting directly with PPAR-γ (31, 33). To study the effect of 5-ASA binding to PPAR-γ, we examined whether 5-ASA-bound PPAR-γ can bind to DRIP-205. Glutathione S-transferase (GST) pull-down assays with [35S]-PPAR-γ incubated with either 5-ASA (1 and 5 mM) or rosiglitazone and a GST-DRIP-205 fusion protein were performed. The results clearly showed that, like the control rosiglitazone, 5-ASA was able to bind and to activate PPAR-γ resulting in an increased interaction between [35S]-PPAR-γ and GST-DRIP (Fig. 6). The maximal efficacy of 5-ASA (5 mM) was ∼150% of that observed with rosiglitazone (10−5 M), suggesting that in these conditions the formation of PPAR-γ–DRIP complexes was more pronounced with 5-ASA than with rosiglitazone.
Figure 6.

GST pull-down assays with PPAR-γ incubated with 5-ASA or rosiglitazone. The GST-DRIP fusion protein bound to glutathione-sepharose beads was incubated with medium alone (control), 5-ASA (1 and 5 mM), or rosiglitazone (10−5 M) in the presence of [35S]methionine-labeled PPAR-γ. Autoradiograms of SDS-PAGE gel show higher amounts of the coactivator DRIP bound to PPAR-γ in the presence of 5-ASA and rosiglitazone than in the control. The amount of PPAR-γ bound to coactivator in the presence of the indicated ligands is expressed as fold induction relative to that measured in absence of ligand (control, defined as 1). Each sample was processed in duplicate.
Taken together, these observations point to the productive binding of 5-ASA to PPAR-γ, inducing a coordinated cascade of reactions leading to the translocation of PPAR-γ in the cell nucleus, a modification of its conformation permitting the recruitment of coactivators and activation of a PPRE-driven reporter gene.
5-ASA binding assay for PPAR-γ and docking studies
The direct binding of 5-ASA to PPAR-γ was explored by competitive ligand-binding assay using radioactive rosiglitazone. Because rosiglitazone is located in the ligand-binding pocket of the PPAR-γ ligand-binding domain (LBD) in the crystal structure of the complex (34), loss of rosiglitazone binding would demonstrate that 5-ASA displaces it. As previously described, in a competitive experiment the commercially available PPAR-γ ligand GW1929 displaced [3H]rosiglitazone used as positive control from the PPAR-γ LBD with a K i value of 60 nM (Fig. 7 A and reference 35). 5-ASA also displaced [3H]rosiglitazone from the PPAR-γ LBD with a K i value of 28.7 mM (Fig. 7 B). The concentration of 5-ASA that caused 50% inhibition (IC50) of rosiglitazone binding was 15.2 mM. This IC50 of 5-ASA corresponds to the efficient 5-ASA concentrations (1–50 mM) found in vivo in treated patients (36, 37).
Figure 7.
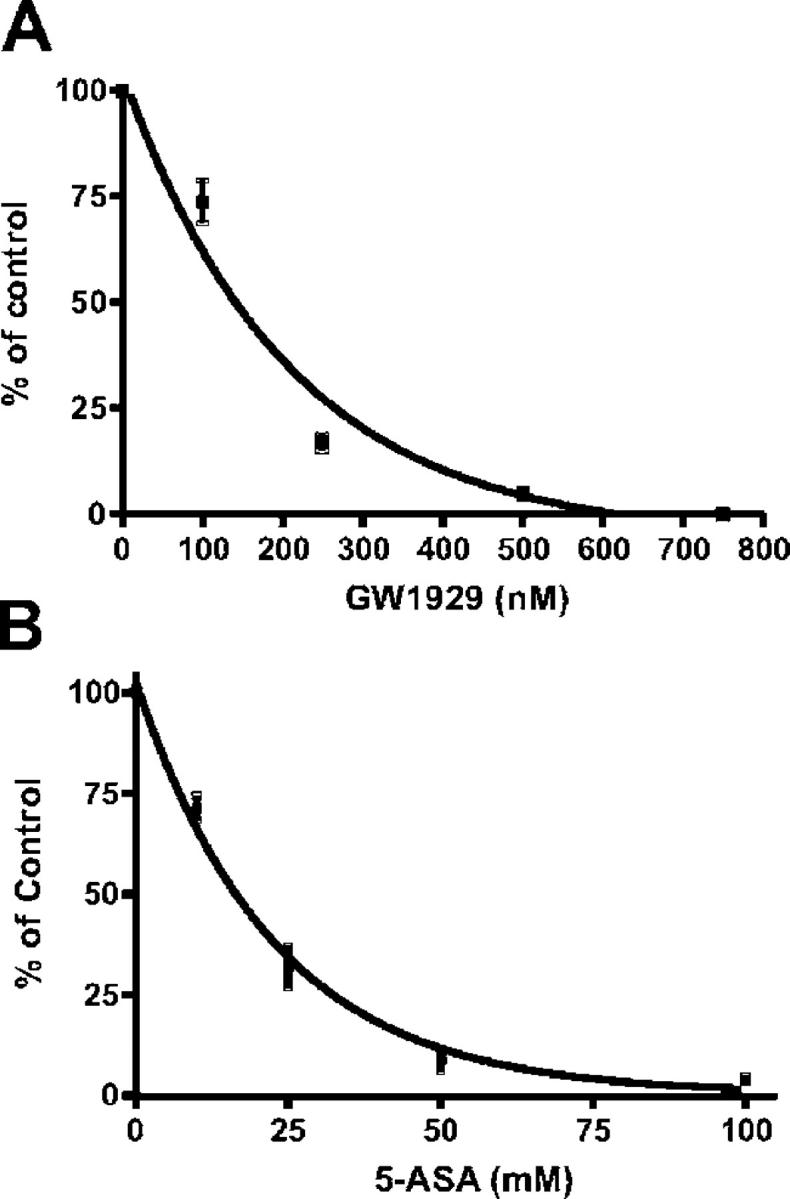
5-ASA binding assay for PPAR-γ. A competitive binding assay was performed with 40 nM [3H]-rosiglitazone in the presence or absence of (A) PPAR-γ agonist GW1929 (0–800 nM, Sigma-Aldrich) used as positive control or (B) increasing concentrations of nonradioactive 5-ASA (0–100 mM). 5-ASA competed with rosiglitazone for binding to PPAR-γ. The specific binding obtained with [3H]rosiglitazone alone was 100% of control. Results are expressed as the mean ± SEM in four different experiments.
Docking simulations were performed to predict the binding mode of 5-ASA into the active site of PPAR-γ formerly occupied by rosiglitazone (38). One of the most stable docking models showed a binding mode of 5-ASA similar to the crystal orientation of the thiazolidinedione head group of rosiglitazone (Fig. 8, A and B). 5-ASA fitted tightly with the PPAR-γ LBD interacting via hydrogen bonding with His-323, His-449, Tyr-473, and Ser-289, which are considered key determinants for molecular recognition and PPAR-γ activation (Fig. 8, C and D, and references 34, 39). The phenolic hydroxyl group of 5-ASA did not interact with the receptor, but its orientation favored an intramolecular hydrogen bond with the carboxyl group as shown in its crystal structure (40). Superimpositions of our 5-ASA model to other known PPAR-γ head group ligands such as the PPAR-γ–bound crystallographic conformations of tesaglitazar (AZ242) (39) and farglitazar (GI262570) (38) showed a similar PPAR-γ binding mode of 5-ASA and glitazars (Fig. 8 E).
Figure 8.

Docking simulation of the PPAR-γ binding mode of 5-ASA. Comparison of the interaction of (A) rosiglitazone and (B) 5-ASA colored by atom type in the ligand-binding domain (LBD) represented by a green surface in the X-ray crystal structure of PPAR-γ displayed as a blue ribbon. (C) 5-ASA displayed in space-filling representation colored by atom type fitted tightly within the PPAR-γ LBD represented as a white surface. (D) Key hydrogen binding interactions (dotted line) between 5-ASA and PPAR-γ. (E) Similar binding mode of 5-ASA model and crystallographic conformation of tesaglitazar, farglitazar, and rosiglitazone.
5-ASA induces PPAR-γ expression and activation in human colonic mucosa
To validate the previous results obtained in vitro and in vivo in mice, we evaluated the effects of 5-ASA in organ cultures of right colonic biopsies taken in healthy mucosa of 12 untreated patients (6 non-IBD patients, 3 patients with Crohn's disease, and 3 patients with ulcerative colitis). After 24 h in a 5% CO2 chamber, treatment of 5-ASA at 1, 30, and 50 mM increased expression of PPAR-γ mRNA and its target gene NGAL (Fig. 9 and reference 41). Release of lactic dehydrogenase from specimens in culture remained low and stable over the 24-h period. No change in lactic dehydrogenase release was induced by the presence of 5-ASA in the incubation medium.
Figure 9.

5-ASA induces PPAR-γ expression and activation in organ cultures of human colonic biopsies. Levels of PPAR-γ mRNA (red) and its target gene NGAL (blue) quantified by real-time PCR in human colon biopsies after 24 h of culture with medium alone and three concentrations of 5-ASA (1, 30, and 50 mM). Results are expressed as the fold induction (mean ± SEM) compared with control (medium). The number of patients and statistical significance are indicated. *, P < 0.05 compared with controls; ○, P < 0.05 compared with 5-ASA (1 mM);^, P < 0.05 compared with 5-ASA (30 mM).
DISCUSSION
Although 5-ASA therapy has been widely used in patients with IBD, the general mechanism underlying its antiinflammatory effects in the colon remain incompletely characterized. In the present study, we demonstrate that PPAR-γ is the key receptor for 5-ASA that mediates its main effects in the colon. This determination is based on multiple functional, pharmacological, and chemical lines of evidence and was validated in vivo in a murine model of IBD and with human clinical samples.
After oral or rectal administration into the colon, some 5-ASA is absorbed by colonic epithelial cells, but most remains within the lumen and is passed in the stool (3–5). In IBD patients receiving standard 5-ASA maintenance treatment, the median mucosal concentrations of 5-ASA are 16 ng/mg, ranging from 3 to 50 ng/mg of wet colonic tissues (2). The therapeutic effect of 5-ASA depends more on the direct contact of the molecule with the epithelium of the colon than on the tissue concentration of 5-ASA in the colon, indicating that a high perimucosal concentration of 5-ASA is a prerequisite for its action. It has been previously reported in patients conventionally treated with 5-ASA that stool concentrations of 5-ASA are in the median order of 30 mM, ranging from 10 to 100 mM; these concentrations correspond to luminal concentrations of 5-ASA 100 times greater than the concentrations in the colonic mucosa (2, 7, 34, 42, 43). In vitro, these millimolar concentrations of 5-ASA (1–50 mM) are able to inhibit signal transduction (44), cell proliferation (45), and expression of the inducible nitric oxide synthase (46). Taken together, these data indicate that the 5-ASA concentrations between 1 and 50 mM used in the present study are clinically and biologically relevant and may be even below the 5-ASA concentrations found intraluminally in IBD patients receiving 5-ASA maintenance treatment.
We have previously shown that PPAR-γ is expressed at the highest levels in adipose tissue and the colonic epithelium (12, 13, 47, 48). Best characterized as a regulator of cellular metabolism and adipocyte differentiation, PPAR-γ is also capable of inhibiting the inflammatory response in several experimental models of colitis induced either by the administration of dextran sodium sulfate (14, 15) or 2, 4, 6-TNBS (17) and by ischemia-reperfusion (49). Based on the common antiinflammatory functions of 5-ASA and PPAR-γ in epithelial cells and the high expression of PPAR-γ by the colon epithelial cells that 5-ASA targets, we hypothesized that 5-ASA may be a new functional ligand of PPAR-γ. Using clinically relevant 5-ASA concentrations (36, 37), we showed by transfection studies using GFP-tagged PPAR-γ, a PPAR-γ–sensitive reporter gene, protease protection assays, and GST pull-down assays that 5-ASA is able to bind PPAR-γ, to induce its translocation from the cytosol of epithelial cells to the nucleus, to promote a PPAR-γ conformational change, and to induce recruitment of a coactivator.
Evidence supporting 5-ASA as a direct functional agonist of PPAR-γ was obtained with a ligand-binding assay and by using 3T3-L1 cells. PPAR-γ is a master regulator of adipocyte differentiation, and this effect has been well demonstrated with the 3T3-L1 cells, which required PPAR-γ activation for their differentiation (22). We showed that, like the PPAR-γ ligand rosiglitazone, 5-ASA is an inducer of adipogenesis resulting in the conversion of 3T3-L1 preadipocytes into adipocytes. The relevance of these in vitro data was further demonstrated in an experimental model of colitis in mice treated with a controlled-release 5-ASA preparation developed to deliver the drug in the colon and to minimize its systemic absorption in the proximal intestine (3–5). Indeed, PPAR-γ+/− mice with colitis induced by intrarectal administration of TNBS were completely refractory to 5-ASA preventive treatment, whereas the same dosage of 5-ASA given to wild-type mice with the same genetic background significantly attenuated the inflammatory response in the colon. Besides this therapeutic effect of 5-ASA given preventively, we and others have also shown that, after induction of colitis by dextran sodium sulfate or TNBS, administration of PPAR-γ ligands is always effective in reducing lesion intensity and mortality compared with untreated mice, despite destruction of the epithelial cell layer (14, 17). The mechanisms involved in the therapeutic effects of PPAR-γ ligands in established intestinal lesions remain partially unknown. It is possible that expression of PPAR-γ in the colon by immune cells such as macrophages and lymphocytes residing in the lamina propria may play a role (50, 51). Because the spontaneous fluorescent properties of 5-ASA enable its localization in macrophages and lymphocytes of the lamina propria after oral or rectal administration (52), we can hypothesize that during 5-ASA administration, PPAR-γ expressed by macrophages and T cells may be activated in vivo and may induce antiinflammatory effects in the gut, as previously described (15, 53).
Regulation of PPAR-γ expression in epithelial cells remains completely unknown. There is evidence that activation of PPAR-γ by thiazolidinedione in preadipocytes can increase modestly the expression of this receptor in a positive-feedback loop (23, 24). In HT-29 colonic epithelial cells treated with 5-ASA, we observed a threefold induction of PPAR-γ expression at the mRNA and protein levels. These results obtained by quantitative real-time PCR paralleled those found in organ cultures of human colonic biopsies and by Western blot analysis at the protein level in epithelial cell line. These data suggest that 5-ASA is able to bind and activate PPAR-γ and also may enhance its expression. This point is particularly important in patients with ulcerative colitis characterized by a chronic inflammation of the colon in which PPAR-γ expression is decreased in colonic epithelial cells (13).
Taken together, these data make a compelling case that 5-ASA is a novel PPAR-γ agonist and show that PPAR-γ is the major functional receptor mediating the common 5-ASA activities in IBD. Several X-ray studies of PPAR-γ crystal structures revealed the binding mode of synthetic PPAR-γ agonists such as glitazones and glitazars in the LBD of the receptor (38, 39). In our study, we identified 5-ASA as a new synthetic PPAR-γ ligand interacting with the LBD. Because 5-ASA was originally developed without any knowledge of its molecular target, there is hope that the research described here will lead to the rational optimization or development of better PPAR-γ ligands. Improvements in efficacy may reside in a new compound with higher affinity or an association of agents with additive or synergic effects on the PPAR-γ/retinoid x receptor (RXR) heterodimer. Studies are also in progress to evaluate if quantification of PPAR-γ expression in the intestinal mucosa of patients may be a marker for monitoring therapeutic effectiveness of 5-ASA. By analogy with glitazones or glitazars, other studies should also be done to assess whether 5-ASA could have therapeutic effects in patients with metabolic disorders.
MATERIALS AND METHODS
5-ASA, GW1929 (N-aryl tyrosine activator), and 2, 4, 6-TNBS were purchased at Sigma-Aldrich. Rosiglitazone was acquired at Spi Bio. [3H]rosiglitazone was synthesized at Isobio.
Induction of TNBS colitis and study design.
Animal experiments were performed in accredited establishments at the Institut de Génétique et de Biologie Moléculaire et Cellulaire in Strasbourg (B59-108 and B 67–218-5) and at the Institut Pasteur from Lille (86/609/CEE) according to governmental guidelines. Animals were housed five per cage and had free access to standard mouse chow and tap water. For colitis induction, mice were anesthetized for 90–120 min and received an intrarectal administration of TNBS (40 μl, 150 mg/kg) dissolved in a 1:1 mixture of 0.9% NaCl with 100% ethanol (13, 18). Control mice received a 1:1 mixture of 0.9% NaCl with 100% ethanol or a saline solution using the same technique. Animals were killed 5 d after TNBS administration. The antiinflammatory effects of 5-ASA were evaluated by administration of different dosages of a controlled-release 5-ASA preparation (50 mg/kg/d–1,000 mg/kg/d) and compared with the PPAR-γ ligand rosiglitazone used at the optimal dosage of 20 mg/kg in 129/Sv wild-type and PPAR-γ+/− mice (Fig. 1 and reference 17). 5-ASA was administered orally once daily, starting 2 d before colitis induction. Body-weight changes and macroscopic and histological indications of colitis were evaluated blindly by two investigators. The colon of each mouse was examined under a dissecting microscope (at a magnification of 5) to evaluate the macroscopic lesions according to the Wallace criteria. The Wallace score rates macroscopic lesions on a scale from 0 to 10 based on features reflecting inflammation, such as hyperemia, thickening of the bowel, and extent of ulceration (19). A colon specimen located precisely 2 cm above the anal canal was used for histological evaluation according to the Ameho criteria (20). This grading on a scale from 0 to 6 takes into account the degree of inflammation infiltrate, the presence of erosion, ulceration, or necrosis, and the depth and surface extension of lesions. The other parts of the colon were frozen and used to quantify PPAR-γ, MPO, and TNF-α mRNA levels (16, 17).
Cell lines.
3T3-L1 preadipocytes (ATCC CL-173) were grown to confluence in DME (Invitrogen) with 10% FCS and antibiotics. The medium was supplemented with 1 μM dexamethasone, 0.2 mM isobutyl methyl xanthine, 10 μg/ml insulin, and 10% FCS for 2 d (27). The effects of 5-ASA (10−4 M–10−2 M) on the differentiation of 3T3-L1 preadipocytes into adipocytes were compared with the effects of rosiglitazone (10−5 M) on confluent cells over 8 d. Medium, 5-ASA, and rosiglitazone were changed every 2 d. Cell viability was assessed by testing lactate dehydrogenase activity in the culture medium taken during the course of the differentiation process or on mature 3T3-L1 cells, treated or not with rosiglitazone and 5-ASA. Adipogenesis was scored by staining lipids with Oil Red O. Counts of at least 500 cells/sample were systematically performed blindly in four different experiments. Results were expressed as the mean ± SEM number of stained cells per experiment.
The colon carcinoma cell line HT-29 STD (American Type Culture Collection; HTB-38) was routinely grown in DME supplemented with 10% heat-FCS and antibiotics. Cells were grown in monolayers, incubated in 5% CO2 at 37°C and 95% relative humidity.
Real-time mRNA quantification.
Total RNA was isolated from cells and colonic tissues using Rneasy kit (Macherey Nagel) according to the manufacturer's instructions. RNA quantification was performed using spectrophotometry. After treatment at 37°C for 30 min with 20–50 units of RNase-free DNase I (Roche Diagnostics Corp.), oligo-dT primers (Roche Diagnostics Corp.) were used to synthesize single-stranded cDNA. mRNAs were quantified using SYBR green Master Mix (Applera Corp.) with specific mouse oligonucleotides: TNF-α, forward, 5′-TGGGAGTAGACAAGGTACAACCC-3′, reverse, 5′-CATCTTCTCAAAATTCGAGTGACAA-3′; and human oligonucleotides: PPAR-γ, forward, 5′-CCTGATAGGCCCCACTGTGT-3′, reverse, 5′-CAGGTGGGAGTGGAA-CAAT-3′ and NGAL, forward, 5′-TTCAGGGAGGCCCAAAGA-3′, reverse, 5′-CCTCTACGGGAGAACCAAGGA-3′ in a GeneAmp Abiprism 7000 (Applera Corp.). In each assay, calibrated and no-template controls were included. Each sample was run in triplicate. SYBR green dye intensity was analyzed using the Abiprism 7000 SDS software (Applera Corp.). All results were normalized to the unaffected housekeeping gene β-actin: mouse β-actin, forward, 5′-GGGTCAGAAGGATTCCTATG-3′, reverse, 5′-GGTCTCAAACATGATCTGGG-3′ and human β-actin, forward, 5′-TCACCCACACTGTGCCCATCTACG-3′, reverse, 5′-CAGCGGAACCGCTCATTGCCAATG-3′ (16).
Evaluation of PPARγ, β-actin, and MPO by Western blot analysis.
Total proteins were obtained by cell or colon homogenization in an extraction buffer consisting of PBS with 2% triton, 100 mM PMSF, and a classical protease-inhibitor cocktail. Total proteins were then separated by PAGE and electroblotted. Polyvinylidendifluoride membranes were incubated overnight with rabbit polyclonal primary antibody directed against PPAR-γ (dilution 1/500, Tebu Bio) or MPO (dilution 1/500; DakoCytomation) (13, 17). β-actin was detected using a rabbit monoclonal primary antibody diluted at 1/10,000 (Sigma-Aldrich). Immunodetection with a swine secondary peroxidase-conjugated antibody (1/1,000; DakoCytomation) and chemiluminescence were performed according to manufacturer's protocol (ECL, Amersham Biosciences). MPO results were expressed as units of OD per quantity of total protein. OD values of PPAR-γ were given for each condition in proportion to the quantity of the internal control β-actin in the sample.
GFP-tagged PPAR-γ.
1 μg of GFP-tagged PPAR-γ vector was transfected in HT-29 STD cells using the Effectene transfection reagent (QIAGEN). After 24 h incubation, cells were treated with 5-ASA (1 mM, 5 mM, or 30 mM) or with rosiglitazone (10−5 M) for 1 d. After two PBS washes, cells were fixed by 4% paraformaldehyde. Nuclei were stained with Hoescht 33342 solution, 0.125 mg/ml (Sigma-Aldrich). Slides were mounted and visualized under a fluorescence microscope (Leica).
Protease digestion assay.
[35S]Methionine-labeled full-length human PPAR-γ2 was generated using transcription/translation system (Promega) according to the manufacturer's instructions. 1 μl of labeled PPAR-γ2, diluted in binding buffer (10 mM Hepes, pH 7.4; 100 mM KCl; 2 mM dithiothreitol; 10% glycerol), was incubated with 5-ASA (5 mM) or rosiglitazone (10−5 M) for 120 min at 4°C and submitted to chymotrypsin digestion as previously described (54). This high concentration of rosiglitazone was used to ensure complete receptor saturation. Different concentrations of chymotrypsin (0, 1, 3, 6, 9, and 12 U; Sigma-Aldrich) were added to the mix for 30 min at 25°C. The reaction was terminated by the addition of 15 μl of 3× denaturing gel loading buffer and by boiling for 1 min. Proteolysis products were separated by electrophoresis through a 12% SDS-PAGE gel. After electrophoresis, gels were dried, and radioactivity was detected with a Storm 860 phosphoimager (Molecular Dynamics).
GST pull-down experiments.
The protocol used has been published elsewhere (55). In brief, 5–10 pmol (5 μl) of [35S]-labeled human PPAR-γ was incubated with the indicated concentration of ligand in a 200-μl final volume. The binding buffer consisted of 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 0.1% Triton X-100. After 1 h incubation at 20°C, 40 μl of GST-DRIP slurry was added to the mix and agitated slowly on a rotating wheel for 90 min at 20°C. Unbound material was removed by three successive washes of sepharose beads by 10 volumes of 1× phosphate-buffered saline 0.1% Triton X-100. Resin-bound receptors were then resolved by SDS-PAGE on a 10% gel and detected by autoradiograph with a Storm 860 phosphoimager (Molecular Dynamics). The amount of PPAR-γ bound to coactivator in the presence of the indicated ligands was expressed as fold-induction relative to that measured in the absence of ligand (defined as 1). Each sample was processed in duplicate.
Transient transfection with PPAR-γ and stimulation of cells.
HT-29 STD cells were transiently transfected using the Effectene transfection reagent (QIAGEN) according to instructions from the manufacturer. To test PPAR-γ activation, we performed transfection with 500 ng of a minimal promoter construct containing two copies of PPRE obtained from the cytochrome p450 4A (2XCYP) (13). The renilla luciferase plasmid (0.1 μg/well) was also transfected as an internal control for monitoring transfection efficiency and for normalizing the firefly luciferase activity. Transfected cells were incubated for 48 h at 37°C. Stimulations were performed for 6 h with 5-ASA 30 mM or with the PPAR-γ synthetic ligand rosiglitazone 10−5 M used as positive control. Total cell extracts were prepared using the Passive Lysis Buffer (Promega). Luciferase activity was assayed in 20 μl of the extract using Promega's Dual Luciferase assay system according to the manufacturer's protocol. Transfections were assayed in triplicate in at least three separate experiments. The luciferase activity was expressed as fold of the activity obtained in cells treated with 5-ASA dividing by luciferase activity from nonstimulated cells.
Competition binding assay for PPAR-γ.
Ligand-binding assays were processed as described previously (56). In brief, purified PPAR-γ–LBD (Invitrogen) was incubated at 4°C for 2–3 h in 100 μl of buffer A (50 mM Tris, pH 8.0, 100 mM KCl, 100 μg/ml OVA, 0.3% ([3-cholamidopropyl-dimethylammonio]-1-propanesulfonate), and 10 mM dithiothreitol) with 40 nM [3H]rosiglitazone in the presence of increasing concentrations of unlabeled 5-ASA (0–100 mM) or the known PPAR-γ agonist GW1929 (0–800 nM, Sigma-Aldrich) used as positive control. 40 nM is the concentration of [3H]rosiglitazone usually required for competition experiments, yielding a reversible 80% saturation of the receptor (27). Bound radioactive ligand was separated from free radioactive ligand by incubating samples for 10 min at 4°C with 50 μl of a charcoal-dextran suspension (3% charcoal, 0.3% dextran in buffer A) and was quantified by liquid scintillation counting. Each assay was performed in triplicate. Ki values were calculated according to the equation of Cheng and Prusoff corresponding to Ki = IC50/(1+ [L]/Kd) where L is the concentration of [3H]rosiglitazone (40 nM), and the Kd for rosiglitzone is 45 nM (57).
Molecular modeling.
Molecular modeling studies were performed using SYBYL software version 6.9 (Tripos Associates Inc.) running on Silicon Graphics workstations. A three-dimensional model of the zwitterion form of 5-ASA was built from a standard fragments library, and its geometry was subsequently optimized using the Tripos force field (58) including the electrostatic term calculated from Gasteiger and Hückel atomic charges. Powell's method, available in Maximin 2 (SYBYL 6.9; Tripos Associates) procedure, was used for energy minimization until the gradient value was smaller than 0.001 kcal/mol Å. The structure of the human PPAR-γ ligand-binding domain was obtained from its complexed X-ray crystal structure with the rosiglitazone available in the Research Collaboratory for Structural Bioinformatics Protein Data Bank (1FM6) (38). Flexible docking of 5-ASA into the receptor active site was performed using GOLD software (Cambridge Crystallographic Data Centre; reference 59). The most stable docking models were selected according to the best-scored conformation predicted by the GoldScore (59) and X-Score scoring functions (60). The complexes were energy-minimized using the Powell method available in Maximin2 procedure with the Tripos force field and a dielectric constant of 4.0 until the gradient value reached 0.01 kcal/mol Å. The anneal function was used to define a 10-Å hot region and a 15-Å region of interest around the ligand.
Patients and organ culture.
Six endoscopically normal right colonic biopsies were taken with an endoscopic forceps on surgical specimens from 12 untreated patients operated on for Crohn's disease (n = 3), ulcerative colitis (n = 3), or colon cancer (n = 6). The study was approved by the local ethics committee. Biopsies were immediately placed in HBSS–calcium or magnesium free supplemented with 100 IU/ml penicillin and 100 mg/ml streptomycin. After washing, biopsies were incubated in a humidified atmosphere for 1 d at 37°C in six-well tissue-culture plates containing 2 ml of RPMI 1640 supplemented with 1, 30, or 50 mM 5-ASA (61). Supernatants were removed, filtered, and stored at −80°C for determination of lactate dehydrogenase release, and biopsies were processed to quantify PPAR-γ and NGAL mRNA.
Acknowledgments
This work was supported by grants from Giuliani S.p.A, which registered a patent concerning this work in Italy in January 2004, and from the Association François Aupetit.
The authors have no conflicting financial interests.
Abbreviations used: 5-ASA, 5-aminosalicylic acid; DRIP, vitamin D3 receptor–interacting protein; GST, glutathione S-transferase; IBD, inflammatory bowel disease; IC50, concentration resulting in 50% inhibition; LBD, ligand-binding domain; MPO, methyl peroxidase; PPAR, peroxisome proliferator–activated receptor; PPRE, peroxisome-proliferator response element; RXR, retinoid x receptor; TNBS, trinitrobenzene sulfonic acid.
C. Rousseaux and B. Lefebvre contributed equally to this work.
References
- 1.Svartz, N. 1941. Ett nytt sulfonamidpreparat? Forelopande meddelande [Swedish]. Nord. Med. 9:544. [Google Scholar]
- 2.Azad Khan, A.K., J. Piris, and S.C. Truelove. 1977. An experiment to determine the active therapeutic moiety of sulphasalazine. Lancet. 2:892–895. [DOI] [PubMed] [Google Scholar]
- 3.Brogden, R.N., and E.M. Sorkin. 1989. Mesalazine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in chronic inflammatory bowel disease. Drugs. 38:500–523. [DOI] [PubMed] [Google Scholar]
- 4.Greenfield, S.M., N.A. Punchard, J.P. Teare, and R.P. Thompson. 1993. The mode of action of the aminosalicylates in inflammatory bowel disease. Aliment. Pharmacol. Ther. 7:369–383. [DOI] [PubMed] [Google Scholar]
- 5.Zhou, S.Y., D. Fleisher, L.H. Pao, C. Li, B. Winward, and E.M. Zimmermann. 1999. Intestinal metabolism and transport of 5-aminosalicylate. Drug Metab. Dispos. 27:479–485. [PubMed] [Google Scholar]
- 6.Frieri, G., R. Giacomelli, M. Pimpo, G. Palumbo, A. Passacantando, G. Pantaleoni, and R. Caprilli. 2000. Mucosal 5-aminosalicylic acid concentration inversely correlates with severity of colonic inflammation in patients with ulcerative colitis. Gut. 47:410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christensen, L.A., J. Fallingborg, K. Abildgaard, B.A. Jacobsen, G. Sanchez, S.H. Hansen, S. Bondesen, E.F. Hvidberg, and S.N. Rasmussen. 1990. Topical and systemic availability of 5-aminosalicylate: comparisons of three controlled release preparations in man. Aliment. Pharmacol. Ther. 4:523–533. [DOI] [PubMed] [Google Scholar]
- 8.Kaiser, G.C., F. Yan, and D.B. Polk. 1999. Mesalamine blocks tumor necrosis factor growth inhibition and nuclear factor kappaB activation in mouse colonocytes. Gastroenterology. 116:602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egan, L.J., D.C. Mays, C.J. Huntoon, M.P. Bell, M.G. Pike, W.J. Sandborn, J.J. Lipsky, and D.J. McKean. 1999. Inhibition of interleukin-1-stimulated NF-kappaB RelA/p65 phosphorylation by mesalamine is accompanied by decreased transcriptional activity. J. Biol. Chem. 274:26448–26453. [DOI] [PubMed] [Google Scholar]
- 10.Sharon, P., M. Ligumsky, D. Rachmilewitz, and U. Zor. 1978. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology. 75:638–640. [PubMed] [Google Scholar]
- 11.Walczak, R., and P. Tontonoz. 2002. PPARadigms and PPARadoxes: expanding roles for PPARgamma in the control of lipid metabolism. J. Lipid Res. 43:177–186. [PubMed] [Google Scholar]
- 12.Dubuquoy, L., S. Dharancy, S. Nutten, S. Pettersson, J. Auwerx, and P. Desreumaux. 2002. Role of peroxisome proliferator-activated receptor gamma and retinoid X receptor heterodimer in hepatogastroenterological diseases. Lancet. 360:1410–1418. [DOI] [PubMed] [Google Scholar]
- 13.Dubuquoy, L., E.A. Jansson, S. Deeb, S. Rakotobe, M. Karoui, J.F. Colombel, J. Auwerx, S. Pettersson, and P. Desreumaux. 2003. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology. 124:1265–1276. [DOI] [PubMed] [Google Scholar]
- 14.Su, C.G., X. Wen, S.T. Bailey, W. Jiang, S.M. Rangwala, S.A. Keilbaugh, A. Flanigan, S. Murthy, M.A. Lazar, and G.D. Wu. 1999. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J. Clin. Invest. 104:383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katayama, K., K. Wada, A. Nakajima, H. Mizuguchi, T. Hayakawa, S. Nakagawa, T. Kadowaki, R. Nagai, Y. Kamisaki, R.S. Blumberg, et al. 2003. A novel PPAR gamma gene therapy to control inflammation associated with inflammatory bowel disease in a murine model. Gastroenterology. 124:1315–1324. [DOI] [PubMed] [Google Scholar]
- 16.Neurath, M.F., S. Pettersson, K.H. Meyer zum Buschenfelde, and W. Strober. 1996. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat. Med. 2:998–1004. [DOI] [PubMed] [Google Scholar]
- 17.Desreumaux, P., L. Dubuquoy, S. Nutten, M. Peuchmaur, W. Englaro, K. Schoonjans, B. Derijard, B. Desvergne, W. Wahli, P. Chambon, et al. 2001. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J. Exp. Med. 193:827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Philippe, D., L. Dubuquoy, H. Groux, V. Brun, M.T. Chuoi-Mariot, C. Gaveriaux-Ruff, J.F. Colombel, B.L. Kieffer, and P. Desreumaux. 2003. Anti-inflammatory properties of the mu opioid receptor support its use in the treatment of colon inflammation. J. Clin. Invest. 111:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace, J.L., W.K. MacNaughton, G.P. Morris, and P.L. Beck. 1989. Inhibition of leukotriene synthesis markedly accelerates healing in a rat model of inflammatory bowel disease. Gastroenterology. 96:29–36. [DOI] [PubMed] [Google Scholar]
- 20.Ameho, C.K., A.A. Adjei, E.K. Harrison, K. Takeshita, T. Morioka, Y. Arakaki, E. Ito, I. Suzuki, A.D. Kulkarni, A. Kawajiri, and S. Yamamoto. 1997. Prophylactic effect of dietary glutamine supplementation on interleukin 8 and tumour necrosis factor alpha production in trinitrobenzene sulphonic acid induced colitis. Gut. 41:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutherland, L., and J.K. MacDonald. 2003. Oral 5-aminosalicylic acid for induction of remission in ulcerative colitis. Cochrane Database Syst. Rev. CD000543. [DOI] [PubMed]
- 22.Spiegelman, B.M. 1998. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 47:507–514. [DOI] [PubMed] [Google Scholar]
- 23.Tontonoz, P., E. Hu, and B.M. Spiegelman. 1994. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 79:1147–1156. [DOI] [PubMed] [Google Scholar]
- 24.Li, Y., and M.A. Lazar. 2002. Differential gene regulation by PPARgamma agonist and constitutively active PPARgamma2. Mol. Endocrinol. 16:1040–1048. [DOI] [PubMed] [Google Scholar]
- 25.Shibuya, A., K. Wada, A. Nakajima, M. Saeki, K. Katayama, T. Mayumi, T. Kadowaki, H. Niwa, and Y. Kamisaki. 2002. Nitration of PPARgamma inhibits ligand-dependent translocation into the nucleus in a macrophage-like cell line, RAW 264. FEBS Lett. 525:43–47. [DOI] [PubMed] [Google Scholar]
- 26.Han, J., D.P. Hajjar, X. Zhou, A.M. Gotto Jr., and A.C. Nicholson. 2002. Regulation of peroxisome proliferator-activated receptor-gamma-mediated gene expression. A new mechanism of action for high density lipoprotein. J. Biol. Chem. 277:23582-23586. [DOI] [PubMed] [Google Scholar]
- 27.Rocchi, S., F. Picard, J. Vamecq, L. Gelman, N. Potier, D. Zeyer, L. Dubuquoy, P. Bac, M.F. Champy, K.D. Plunket, et al. 2001. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol. Cell. 8:737–747. [DOI] [PubMed] [Google Scholar]
- 28.Berger, J., P. Bailey, C. Biswas, C.A. Cullinan, T.W. Doebber, N.S. Hayes, R. Saperstein, R.G. Smith, and M.D. Leibowitz. 1996. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology. 137:4189–4195. [DOI] [PubMed] [Google Scholar]
- 29.Westin, S., R. Kurokawa, R.T. Nolte, G.B. Wisely, E.M. McInerney, D.W. Rose, M.V. Milburn, M.G. Rosenfeld, and C.K. Glass. 1998. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature. 395:199–202. [DOI] [PubMed] [Google Scholar]
- 30.Mangelsdorf, D.J., C. Thummel, M. Beato, P. Herrlich, G. Schutz, K. Umesono, B. Blumberg, P. Kastner, M. Mark, P. Chambon, et al. 1995. The nuclear receptor superfamily: the second decade. Cell. 83:835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Misra, P., E.D. Owuor, W. Li, S. Yu, C. Qi, K. Meyer, Y.J. Zhu, M.S. Rao, A.N. Kong, and J.K. Reddy. 2002. Phosphorylation of transcriptional coactivator peroxisome proliferator-activated receptor (PPAR)-binding protein (PBP). Stimulation of transcriptional regulation by mitogen-activated protein kinase. J. Biol. Chem. 277:48745-48754. [DOI] [PubMed] [Google Scholar]
- 32.Akiyama, T.E., C.T. Baumann, S. Sakai, G.L. Hager, and F.J. Gonzalez. 2002. Selective intranuclear redistribution of PPAR isoforms by RXR alpha. Mol. Endocrinol. 16:707–721. [DOI] [PubMed] [Google Scholar]
- 33.Zhu, Y., C. Qi, S. Jain, M.S. Rao, and J.K. Reddy. 1997. Isolation and characterization of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J. Biol. Chem. 272:25500–25506. [DOI] [PubMed] [Google Scholar]
- 34.Nolte, R.T., G.B. Wisely, S. Westin, J.E. Cobb, M.H. Lambert, R. Kurokawa, M.G. Rosenfeld, T.M. Willson, C.K. Glass, and M.V. Milburn. 1998. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 395:137–143. [DOI] [PubMed] [Google Scholar]
- 35.Brown, K.K., B.R. Henke, S.G. Blanchard, J.E. Cobb, R. Mook, I. Kaldor, S.A. Kliewer, J.M. Lehmann, J.M. Lenhard, W.W. Harrington, et al. 1999. A novel N-aryl tyrosine activator of peroxisome proliferator-activated receptor-gamma reverses the diabetic phenotype of the Zucker diabetic fatty rat. Diabetes. 48:1415–1424. [DOI] [PubMed] [Google Scholar]
- 36.Staerk Laursen, L., M. Stokholm, K. Bukhave, J. Rask-Madsen, and K. Lauritsen. 1990. Disposition of 5-aminosalicylic acid by olsalazine and three mesalazine preparations in patients with ulcerative colitis: comparison of intraluminal colonic concentrations, serum values, and urinary excretion. Gut. 31:1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frieri, G., M.T. Pimpo, G.C. Palumbo, L. Onori, A. Viscido, G. Latella, B. Galletti, G.C. Pantaleoni, and R. Caprilli. 1999. Rectal and colonic mesalazine concentration in ulcerative colitis: oral vs. oral plus topical treatment. Aliment. Pharmacol. Ther. 13:1413–1417. [DOI] [PubMed] [Google Scholar]
- 38.Gampe, R.T. Jr., V.G. Montana, M.H. Lambert, A.B. Miller, R.K. Bledsoe, M.V. Milburn, S.A. Kliewer, T.M. Willson, and H.E. Xu. 2000. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol. Cell. 5:545–555. [DOI] [PubMed] [Google Scholar]
- 39.Xu, H.E., M.H. Lambert, V.G. Montana, K.D. Plunket, L.B. Moore, J.L. Collins, J.A. Oplinger, S.A. Kliewer, R.T. Gampe Jr., D.D. McKee, et al. 2001. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 98:13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banié-Tomis̆ic, Z., B. Kojic-Prodic, and I. S̆irola. 1997. Hydrogen bonds in the crystal packings of mesalazine and mesalazine hydrochloride. J. Mol. Struct. 416:209–220. [Google Scholar]
- 41.Gupta, R.A., J.A. Brockman, P. Sarraf, T.M. Willson, and R.N. DuBois. 2001. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J. Biol. Chem. 276:29681–29687. [DOI] [PubMed] [Google Scholar]
- 42.Jacobsen, B.A., K. Abildgaard, H.H. Rasmussen, L.A. Christensen, J. Fallingborg, S.H. Hansen, and S.N. Rasmussen. 1991. Availability of mesalazine (5-aminosalicylic acid) from enemas and suppositories during steady-state conditions. Scand. J. Gastroenterol. 26:374–378. [DOI] [PubMed] [Google Scholar]
- 43.Riley, S.A. 1998. What dose of 5-aminosalicylic acid (mesalazine) in ulcerative colitis? Gut. 42:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan, F., and D.B. Polk. 1999. Aminosalicylic acid inhibits IkappaB kinase alpha phosphorylation of IkappaBalpha in mouse intestinal epithelial cells. J. Biol. Chem. 274:36631–36636. [DOI] [PubMed] [Google Scholar]
- 45.Reinacher-Schick, A., A. Schoeneck, U. Graeven, I. Schwarte-Waldhoff, and W. Schmiegel. 2003. Mesalazine causes a mitotic arrest and induces caspase-dependent apoptosis in colon carcinoma cells. Carcinogenesis. 24:443–451. [DOI] [PubMed] [Google Scholar]
- 46.Kennedy, M., L. Wilson, C. Szabo, and A.L. Salzman. 1999. 5-aminosalicylic acid inhibits iNOS transcription in human intestinal epithelial cells. Int. J. Mol. Med. 4:437–443. [DOI] [PubMed] [Google Scholar]
- 47.Fajas, L., D. Auboeuf, E. Raspe, K. Schoonjans, A.M. Lefebvre, R. Saladin, J. Najib, M. Laville, J.C. Fruchart, S. Deeb, et al. 1997. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 272:18779–18789. [DOI] [PubMed] [Google Scholar]
- 48.Lefebvre, M., B. Paulweber, L. Fajas, J. Woods, C. McCrary, J.F. Colombel, J. Najib, J.C. Fruchart, C. Datz, H. Vidal, et al. 1999. Peroxisome proliferator-activated receptor gamma is induced during differentiation of colon epithelium cells. J. Endocrinol. 162:331–340. [DOI] [PubMed] [Google Scholar]
- 49.Nakajima, A., K. Wada, H. Miki, N. Kubota, N. Nakajima, Y. Terauchi, S. Ohnishi, L.J. Saubermann, T. Kadowaki, R.S. Blumberg, et al. 2001. Endogenous PPAR gamma mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology. 120:460–469. [DOI] [PubMed] [Google Scholar]
- 50.Clark, R.B., D. Bishop-Bailey, T. Estrada-Hernandez, T. Hla, L. Puddington, and S.J. Padula. 2000. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J. Immunol. 164:1364–1371. [DOI] [PubMed] [Google Scholar]
- 51.Ricote, M., A.C. Li, T.M. Willson, C.J. Kelly, and C.K. Glass. 1998. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 391:79–82. [DOI] [PubMed] [Google Scholar]
- 52.de Vos, M. 1992. Concentrations of 5-ASA and its major metabolite Ac 5-ASA in human intestinal biopsy homogenates. Ph.D. thesis. University of Ghent, Ghent, Belgium. 205 pp.
- 53.Bassaganya-Riera, J., K. Reynolds, S. Martino-Catt, Y. Cui, L. Hennighausen, F. Gonzalez, J. Rohrer, A.U. Benninghoff, and R. Hontecillas. 2004. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 127:777–791. [DOI] [PubMed] [Google Scholar]
- 54.Benkoussa, M., B. Nomine, A. Mouchon, B. Lefebvre, J.M. Bernardon, P. Formstecher, and P. Lefebvre. 1997. Limited proteolysis for assaying ligand binding affinities of nuclear receptors. Recept. Signal Transduct. 7:257–267. [PubMed] [Google Scholar]
- 55.Lefebvre, B., A. Mouchon, P. Formstecher, and P. Lefebvre. 1998. Distinct modes of interaction of the retinoic acid receptor alpha with natural and synthetic retinoids. Mol. Cell. Endocrinol. 139:161–169. [DOI] [PubMed] [Google Scholar]
- 56.Lefebvre, B., C. Rachez, P. Formstecher, and P. Lefebvre. 1995. Structural determinants of the ligand-binding site of the human retinoic acid receptor alpha. Biochemistry. 34:5477–5485. [DOI] [PubMed] [Google Scholar]
- 57.Cheng, Y., and W.H. Prusoff. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- 58.Clark, M., R. Cramer III, and N. Van Opdenbosch. 1989. Validation of the general purpose Tripos 5.2 field. J. Comput Chem. 10:982-1012. [Google Scholar]
- 59.Jones, G., P. Willett, R.C. Glen, A.R. Leach, and R. Taylor. 1997. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267:727–748. [DOI] [PubMed] [Google Scholar]
- 60.Wang, R., L. Lai, and S. Wang. 2002. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 16:11–26. [DOI] [PubMed] [Google Scholar]
- 61.Borruel, N., M. Carol, F. Casellas, M. Antolin, F. de Lara, E. Espin, J. Naval, F. Guarner, and J.R. Malagelada. 2002. Increased mucosal tumour necrosis factor alpha production in Crohn's disease can be downregulated ex vivo by probiotic bacteria. Gut. 51:659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]