

Abstract
The effects of a chimeric monoclonal antibody (chA6 mAb) that recognizes both the RO and RB isoforms of the transmembrane protein tyrosine phosphatase CD45 on human T cells were investigated. Chimeric A6 (chA6) mAb potently inhibited antigen-specific and polyclonal T cell responses. ChA6 mAb induced activation-independent apoptosis in CD4+CD45RO/RBhigh T cells but not in CD8+ T cells. In addition, CD4+ T cell lines specific for tetanus toxoid (TT) generated in the presence of chA6 mAb were anergic and suppressed the proliferation and interferon (IFN)-γ production by TT-specific effector T cells by an interleukin-10–dependent mechanism, indicating that these cells were equivalent to type 1 regulatory T cells. Similarly, CD8+ T cell lines specific for the influenza A matrix protein-derived peptide (MP.58-66) generated in the presence of chA6 mAb were anergic and suppressed IFN-γ production by MP.58-66–specific effector CD8+ T cells. Furthermore, chA6 mAb significantly prolonged human pancreatic islet allograft survival in nonobese diabetic/severe combined immunodeficiency mice injected with human peripheral blood lymphocytes (hu-PBL-NOD/SCID). Together, these results demonstrate that the chA6 mAb is a new immunomodulatory agent with multiple modes of action, including deletion of preexisting memory and recently activated T cells and induction of anergic CD4+ and CD8+ regulatory T cells.
The transmembrane protein tyrosine phosphatase (PTPase) CD45 plays a critical role in lymphocyte activation. Alternative splicing of exons 4–6 generates eight different CD45 isoforms in humans which differ in the size of their extracellular domains while sharing identical cytoplasmic PTPase domains (1). Although the function of the extracellular domain of each CD45 isoform remains to be defined, it is well established that the cytoplasmic PTPase domain acts as a positive regulator of T cell receptor–mediated signaling, which is essential for activation and development of lymphocytes (2).
Individual lymphocytes simultaneously express multiple isoforms of CD45 (1, 3). However, the highest, intermediate, and lowest molecular weight isoforms recognized by CD45RABC-, CD45RB-, and CD45RO-specific mAbs, respectively, are differentially expressed on T and B cells as well as on functionally different subsets of CD4+ T cells (4–6). In mice, mAbs recognizing CD45RB isoforms are used to differentiate two populations of CD4+ T cells, CD4+CD45RBhigh and CD4+CD45RBlow (5, 6), that secrete different cytokines and have distinct functional properties. The CD4+CD45RBhigh population contains effector T cells, which have been shown to induce autoimmunity (7) or inflammatory bowel disease (8), whereas the CD4+CD45RBlow population contains regulatory T (T reg) cells, which prevent the induction of T cell–mediated diseases (8) including acute allograft rejection (9).
Several studies demonstrated that a mAb specific for the CD45RB isoform is a potent immunomodulator that prolongs allograft survival in several murine transplantation models (10–14) and induces long-term engraftment and donor-specific tolerance in murine renal and islet allografts (11, 13). The exact mechanism underlying tolerance mediated by anti-CD45RB mAb is still unclear. It has been suggested that anti-CD45RB mAb interferes with T cell activation and causes a shift toward the expression of the low isoform (CD45RBlow) on CD4+ T cells (10–12, 15). This inversion of the CD45RBhigh/CD45RBlow T cell subset ratio is caused by selective depletion of CD45RBhigh effector cells after in vivo treatment with anti-CD45RB mAb (16).
The mouse anti–human mAb A6 has a unique specificity and recognizes both the RO and RB isoforms of CD45 on human cells (17). It has been shown that in vitro depletion of A6+ cells from PBMCs dramatically decreased proliferation and cytotoxic activity of these cells in response to recall and alloantigens or anti-CD3 mAb stimulation (17). In the present study, we investigated the immunomodulatory properties of a chimeric A6 (chA6) mAb in which constant mouse regions of A6 mAb were substituted by human constant regions of human IgG1/κ isotype. Our results demonstrate that chA6 mAb is a potent immunomodulator that inhibits responses of both primary and preactivated T cells, selectively mediates apoptosis of CD4+CD45RO/RBbright T cells, and induces populations of CD4+ and CD8+ T reg cells in vitro. In addition, chA6 mAb mediates long-term survival of human pancreatic islet allograft in hu-PBL-NOD/SCID mice.
RESULTS
ChA6 mAb inhibits T cell proliferation
It has been shown that some mAbs that bind to the CD45RB isoform are capable of selectively inhibiting both mouse and human T cell responses (18–20). We investigated the effect of chA6 mAb, which specifically recognizes the CD45RO and CD45RB isoforms, on the proliferative responses of human CD4+ T cells following stimulation with anti-CD3 mAb, alloantigens, or tetanus toxoid (TT). ChA6 mAb inhibited the proliferation of CD4+ T cells activated with immobilized anti-CD3 mAb. The strongest inhibition was observed at anti-CD3 mAb concentrations of 0.01 μg/ml (81 ± 13%; n = 4; P < 0.05), whereas at concentrations of 0.1 μg/ml the inhibition was 45 ± 23% (P < 0.05). No significant inhibition (4 ± 0.5%) was observed at 1-μg/ml concentrations of anti-CD3 mAb. Comparable inhibitory effects were obtained when T cells were stimulated with anti-CD3 and anti-CD28 mAbs. The strongest inhibition (86 ± 12%; n = 4, P = 0.05) was observed at anti-CD3 mAb concentrations of 0.01 μg/ml, whereas inhibitions of 49 ± 24% and 24 ± 18% were obtained at anti-CD3 mAb concentrations of 0.1 μg/ml and 1 μg/ml, respectively (Fig. 1 A). The inhibitory effect of chA6 mAb was dose-dependent: the percentages of inhibition of T cells activated with anti-CD3 mAb (0.01 μg/ml) were 71 ± 10% (n = 2, P = 0.03), 66 ± 5% (n = 2, P = 0.01), and 42 ± 22% (n = 2, P = 0.03) at concentrations of 10, 1, and 0.1 μg/ml of chA6 mAb, respectively. Comparable dose-dependent inhibitory effects were observed on T cells activated with 0.1 μg/ml of anti-CD3 mAb. Under these conditions, inhibitions of 55 ± 12% (n = 3, P < 0.001), 35 ± 6% (n = 3, P < 0.01), 35 ± 12% (n = 3, p-value was not significant) were obtained at concentrations of 10, 1, and 0.1 μg/ml of chA6 mAb, respectively (Fig. 1 B). These data indicate that the strongest inhibition of proliferation is observed when T cells are activated with low concentrations of anti-CD3 mAb (0.01 μg/ml) and optimal doses of chA6 mAb (10 μg/ml). This inhibition is reproducible and specific, because it is not observed in the presence of a chimeric control mAb.
Figure 1.

ChA6 mAb inhibits polyclonal proliferation of CD4+T cells. (A) CD4+ T cells were stimulated with the indicated concentrations of coated anti-CD3 mAb in the presence or absence of chA6 or chimeric isotype control mAb (10 μg/ml) with or without soluble anti-CD28 mAb (1 μg/ml). Results from one representative donor out of four are shown. (B) Dose-dependent inhibition of proliferative polyclonal T cell response by chA6 mAb. CD4+ T cells were stimulated with 0.1 and 0.01 μg/ml of coated anti-CD3 mAb in the presence of the indicated concentrations of soluble chA6 mAb. Results from one representative donor out of three (0.1 μg/ml of anti-CD3 mAb) and out of two (0.01 μg/ml of anti-CD3 mAb) are shown. The percentages of inhibition of proliferation in the presence of chA6 mAb relative to control are indicated.
ChA6 mAb also inhibited proliferation of CD4+ T cells in primary (mean inhibition, 57 ± 17%; n = 14, P < 0.02) and secondary MLRs (Fig. 2 A). In the latter case, a mean inhibition of 30 ± 16% (n = 4, P < 0.05) was observed when CD4+ T cells, primed with allogeneic APC for 10 d without chA6 mAb, were restimulated with the original allogeneic cells in the presence of chA6 mAb (Fig. 2 A). As expected, chA6 mAb inhibited the proliferation of CD4+ T cells stimulated with allogeneic APC in a dose-dependent fashion: 41 ± 18% (n = 5, P = 0.04), 37 ± 18% (n = 5, P = 0.03), and 33 ± 9% (n = 5, p-value was not significant.) inhibition was obtained at concentrations of 10, 1, and 0.1 μg/ml of chA6 mAb, respectively (Fig. 2 A). No inhibition was observed when cells were cultured in the presence of a chimeric isotype control mAb.
Figure 2.

ChA6 mAb inhibits allogeneic and TT-specific proliferation of CD4+ T cells. (A) Primary MLR: CD4+ T cells were stimulated with allogeneic CD3-depleted cells with or without chA6 mAb (10 μg/ml). Results from one representative donor out of 14 are shown. Secondary MLR: CD4+ T cells were primed with allogeneic CD3-depleted cells for 10 d. Cells were collected and washed, and proliferative responses to the same allogenic cells used in the primary stimulation in the presence or absence of chA6 mAb (10 μg/ml) were tested. Results from one representative donor out of four are shown. Dose-dependent inhibition of primary MLR: CD4+ T cells were stimulated with allogeneic CD3-depleted cells in the presence of the indicated concentrations of chA6 or chimeric isotype control mAbs. Results from one representative donor out of five are shown. R, responder; S, stimulator. (B) TT response: total PBMCs were stimulated with TT with or without chA6 mAb (10 μg/ml). Results from one representative donor out of 16 are shown. The percentages of inhibition of proliferation in the presence of chA6 mAb relative to control are indicated.
Finally, chA6 mAb also inhibited TT-specific memory T cell responses. A mean inhibition of 47 ± 24% (n = 16, P = 0.02) of the proliferative responses of PBMCs stimulated with TT was observed (Fig. 2 B). Taken together, these data indicate that chA6 mAb inhibits primary, secondary, and memory T cell proliferative responses.
ChA6 mAb induces apoptosis in A6brightCD4+ T cells
To determine whether the inhibition of proliferation was caused by depletion of responder T cells, the ability of chA6 mAb to induce T cell apoptosis was investigated. Overnight incubation of CD4+ T cells with chA6 mAb in the presence or absence of anti-CD3 and anti-CD28 mAb resulted in increased percentages of early apoptotic (annexin V+/PI−) cells. The percentages of annexin V+/PI− cells were significantly higher in cultures incubated with 10 μg/ml or 1 μg/ml of chA6 mAb (38 ± 13% and 30 ± 11%, respectively; n = 20, P ≤ 0.005) than in cultures incubated with an isotype control mAb (20 ± 9%) (Fig. 3 A). The mean value of ED50 for the induction of apoptosis was 20.6 ± 8.7 μg/ml (Fig. 3 B). Apoptosis induced by chA6 mAb was not significantly enhanced when CD4+ T cells were activated with anti-CD3 and anti-CD28 mAbs (Fig. 3 A). Double staining of CD4+ T cells with annexin V–FITC and A6-PE mAb revealed that apoptosis was induced mainly in CD4+A6bright cells (Fig. 3 C), which represent the CD45RO/RBbright cells (not depicted; reference 17). These results indicate that chA6 mAb induces apoptosis in CD4+ T cells in a dose-dependent manner, which does not require T cell activation, and selectively depletes CD4+CD45RO/RBbright T cells, which represent the CD4+ effector/memory T cell population.
Figure 3.

Dose-dependent apoptosis induced in CD4+A6bright T cells by chA6 mAb. (A) CD4+ T cells were incubated with the indicated concentrations of chA6 or isotype control mAbs. Cells were cultured overnight without (unstimulated) or with (stimulated) coated anti-CD3 (0.1 μg/ml) and soluble anti-CD28 (1 μg/ml) mAb and were analyzed for apoptosis. The percentages of early apoptotic (annexin V+/PI−) cells and mean ± SE are shown. P values were calculated by t test: *P, effect of chA6 mAb on total CD4+ T cells; $ P, effect on annexin V–depleted CD4+ T cells (*P or $ P ≤ 0.05; **P ≤ 0.005). (B) PBMCs of three healthy donors were cultured overnight in the presence of the indicated concentrations of chA6 mAb (filled symbols) or an IgG1 isotype control antibody (open symbols) and analyzed for apoptosis. Curve fitting and ED50 value calculations were performed. (C) Total or annexin V–depleted CD4+ T cells were incubated with the indicated concentrations of chA6 mAb. Cells were cultured overnight without or with (stimulated) coated anti-CD3 (0.1 μg/ml) and soluble anti-CD28 (1 μg/ml) mAbs and were stained with annexin V FITC mAb and chA6 mAb followed by anti-human IgG1PE mAb and analyzed by flow cytometry. Percentages of positive cells, set according to the isotype-matched controls (not shown), are shown in the top corner of the quadrant. Results from one representative out of 10 different donors tested are shown. (D) CD4+ T cells were incubated with the indicated concentrations of anti-CD45RO (UCLH-1), anti-CD45RA (HI-100), chA6, or isotype control mAbs. Cells were stimulated with coated anti-CD3 (0.1 μg/ml) and soluble anti-CD28 (1 μg/ml) mAbs, cultured overnight, and analyzed for apoptosis. The percentages of early-apoptotic (annexinV+/PI−) cells are shown. (E) Total or annexin V–depleted CD4+ T cells were stimulated with allogeneic CD3-depleted cells with or without chA6 mAb (10 μg/ml). Results from one representative donor out of four are shown. The percentages of inhibition of proliferation in the presence of chA6 mAb relative to control are presented.
Cross-linking of CD45RO or CD45RA isoforms by specific mAb did not induce apoptosis on human CD4+ T cells (Fig. 3 D), indicating the specific effect of the cross-link of CD45RO/RB isoform by chimeric A6 mAb. ChA6 mAb failed to induce apoptosis of CD8+ T cells and of non–T cells at concentrations up to 10 μg/ml, indicating a specific effect on CD4+ T cells (unpublished data).
To verify whether the apoptosis mediated by chA6 mAb was targeting preexisting CD4+A6bright responding T cells, we examined the effect of chA6 mAb on cells preincubated with chA6 mAb and depleted of annexin V+ cells. As expected, depletion of annexin V+ cells resulted in a reduced percentage of CD4+A6bright T cells, whereas the proportion of CD4+A6low T cells increased (unpublished data). Annexin V–depleted CD4+ T cells reexpressed the A6 epitope on the cell surface (not depicted) and subsequently became susceptible to apoptosis induced by chA6 mAb (Fig. 3, A and C). Together, these data show that ligation of CD45RB/RO isoforms by chA6 mAb leads to the death of preexisting and de novo induced CD4+A6bright memory T cells. The observation that chA6 mAb inhibited primary allogeneic proliferative responses of freshly isolated CD4+ T cells (mean value of inhibition, 46 ± 22%; n = 4, P < 0.005) and annexin V–depleted CD4+ T cells (mean value of inhibition, 53 ± 21%; n = 4, P < 0.05) in a comparable fashion (Fig. 3 E) indicates that the immunosuppressive effect of chA6 mAb is caused by the induction of apoptosis of preexisting CD4+A6bright T cells and of newly activated effector cells, which expressed the A6 epitope at high levels.
ChA6 mAb induces apoptosis through the intrinsic pathway
We investigated the mechanism involved in the apoptosis induced by chA6 mAb by analyzing the expression and activation of several caspases, including caspase-3, one of the key molecules involved in apoptosis. The p17 active subunit of caspase-3 was expressed in CD4+ T cells cultured with chA6 alone, indicating that ligation of CD45RO/RB results in activation of the caspase cascade and induction of cell death in unstimulated CD4+ T cells. As expected, the p17 subunit was expressed in CD4+ T cells activated with anti-CD3 and anti-CD28 mAbs in the presence or absence of chA6 mAb (Fig. 4 A). Next we investigated the processing and expression of caspase-8 and caspase-9 in CD4+ T cells treated with chA6 mAb to determine whether chA6 mAb induces apoptosis through the activation of the death receptors CD95 and TNF-R, which requires caspase-8 (21), or by direct activation of the intrinsic apoptotic pathway, which requires activation of caspase-9 (22). As shown in Fig. 4 A, the full-length protein (p57) and the cleavage products (p43/p41) of caspase-8 were detected in all conditions tested, whereas the p18 active subunit of caspase-8 was not detected. Conversely, both the full-length protein (p46) and the cleaved active forms (p37/35) of caspase-9 were detected in CD4+ T cell cultured with chA6 mAb.
Figure 4.

ChA6 mAb induce apoptosis in CD4+ T cells through the activation of the intrinsic pathway. (A) CD4+ T cells were incubated with the indicated concentrations of chA6 mAb. Cells were cultured overnight with or without coated anti-CD3 (0.1 μg/ml) and soluble anti-CD28 (1 μg/ml) mAbs. Western blot tests with anti-caspase-3, anti-caspase-8, and anti-caspase-9 mAb were performed. As positive control, Hela cells were treated with staurosporine, 5 μM. Amounts of loaded proteins have been controlled for homogeneity by probing membranes with an anti- β-actin mAb. Weak expression of the cleavage products of caspase-9 in CD4+ T cells cultured in medium is considered background apoptosis. Results from one representative donor out of three are shown. (B) CD4+ T cells were incubated with chA6 mAb (5 μg/ml) in the presence of 15 μg/ml of cross-linking goat anti–human IgG (F(ab′)2). Mitochondrial-based death pathways were assessed using 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3)) accumulation that reflects changes in Δψm in the mitochondria. Results from one representative donor out of three are shown.
One of the first events required for induction of apoptosis via caspase-9 is perturbation of the mitochondria that results in the release of cytochrome c and proapoptotic factors and ultimately in caspase-9 activation (23). The mitochondrial accumulation of DiOC6(3) was used to assess the value of change in the mitochondria transmembrane potential (Δψm) (24) in CD4+ T cells treated with chA6 mAb. No Δψm was observed in medium or isotype control mAb-treated CD4+ T cells (Fig. 5 B), whereas Δψm was significantly reduced in CD4+ T cells incubated with chA6 mAb. Together, these results indicate that chA6 mAb–induced apoptosis of CD4+ T cells is caused by triggering of the intrinsic pathway and is independent from CD95 and TNF-R receptor/ligation.
Figure 5.

ChA6 mAb induced TT-specific CD4+ T reg cells. (A) Total PBMCs were stimulated with TT with or without chA6 mAb (10 μg/ml). TT-specific cell lines were rechallenged with TT-pulsed and nonpulsed autologous irradiated monocytes in the absence of chA6 mAb. Results from one representative donor out of 18 are shown. The percentage of inhibition of proliferation in the presence of chA6 mAb relative to control is presented. Alternative: after 48 h culture, IFN-γ was quantified in culture supernatants by ELISA. Results from one representative donor out of 10 are shown. The percentage of inhibition of IFN-γ release in the presence of chA6 mAb relative to control is shown. Iono, ionomycin; TPA, 12-0-tetradecanoylphorbol-13-acetate. (B). TT-specific cell lines were cocultured with increased amounts of TT-specific T cell lines generated in the presence of chA6 mAb (TT/chA6) and stimulated with TT-pulsed and nonpulsed autologous irradiated monocytes in the absence of chA6 mAb. After 48 h of culture, IFN-γ was quantified in culture supernatants by ELISA. Results from one representative donor out of four are shown. The percentages of inhibition of proliferation in the presence of chA6 mAb relative to control are presented.
ChA6 mAb modulates antigen-specific CD4+ T cell responses
Although apoptosis of CD4+ T cells may contribute to the antiproliferative effects of chA6 mAb, chA6 mAb inhibited both polyclonal and alloantigen-induced proliferation of T cells at concentrations of 0.1 μg/ml, which failed to induce significant apoptosis in CD4+ T cells (Figs. 1–3).
To determine further whether chA6 mAb, in addition to its apoptotic effect on T effector cells, also has immunomodulatory effects, induction of antigen-specific anergic T reg cells was investigated. Total PBMCs were activated with TT in the presence (TT/chA6) or absence (TT) of chA6 mAb. After two rounds of stimulation under the same conditions, CD4+ T cell lines were rechallenged with TT in the absence of chA6 mAb. Results shown in Fig. 5 A demonstrate that chA6 mAb induced a profound state of unresponsiveness in TT-specific CD4+ T cells. Both proliferation (mean value of inhibition, 72 ± 28%; n = 18, P = 0.03) and IFN-γ production (mean value of inhibition, 91 ± 10%; n = 10, P = 0.027) were strongly inhibited. The unresponsiveness was dependent on TCR activation because TT and TT/chA6 cell lines produced comparable levels of IFN-γ in response to activation by 12-0-tetradecanoylphorbol-13-acetate and ionomycin (n = 3, P > 0.1) (Fig. 5 A).
To determine whether anergic TT/chA6 cell lines contain T reg 1 cells, TT-specific effector CD4+ T cell lines generated as described previously were rechallenged with TT and autologous APC in the presence of increasing numbers of anergic TT/chA6 cells. Results shown in Fig. 5 B demonstrate that anergic TT/chA6 cells suppressed the IFN-γ production of effector TT-specific CD4+ T cells in a dose-dependent manner (mean value of inhibition, 41 ± 15% at 1:2 ratio, P = 0.03; 33 ± 11% at 1:1 ratio, P = 0.02; and 21 ± 12% at 1:0.5 ratio in four donors tested).
TT/chA6 cell lines and control TT cell lines contained comparable percentages of CD4+ T cells expressing CD25, but the percentages of CD4+ T cells expressing CD69 was reduced (n = 10, P = 0.03; Fig. 6 A). This reduction resulted in lower percentages of CD25+CD69+ T cells in anergic TT/chA6 cell lines as compared with TT control cell lines. CTLA-4, GITR, and FOXP3 mRNA expression was similar in the anergized and nonanergized TT cell lines (unpublished data). From these data it can be concluded that TT-specific suppressor T cell lines generated in the presence of chA6 mAb did not contain higher proportion of CD4+ CD25+ T reg cells.
Figure 6.

ChA6 mAb induced TT-specific CD4+ T reg 1 cells. (A) TT-specific cell lines were stained with the indicated surface molecules and analyzed by flow cytometry with gating on live CD4+ T cells. Thin lines represent staining with the appropriate control mAbs. Results from one representative donor out of 10 are shown. (B) TT and TT/chA6 cell lines were stimulated with TT-pulsed autologous irradiated monocytes in the absence of chA6 mAb. Supernatants were collected after 72 h, and levels of IL-10 and TGF-β were determined by ELISA. (C) TT cell lines were cocultured with TT/chA6 cell lines (1:1 ratio) and stimulated with TT-pulsed and nonpulsed autologous irradiated monocytes in the presence of 10 μg/ml of anti–IL-10R mAb. After 48 h culture, IFN-γ was quantified in culture supernatants by ELISA. Results from one representative donor out of two are shown. The percentage of inhibition of IFN-γ release in the presence of chA6 mAb relative to control is presented.
The overall number of cytokines produced by anergized TT/chA6 cell lines was low. After antigen-specific activation, these anergized T cells did not produce IL-2 and IL-4 and secreted significantly lower amounts of IFN-γ than did nonanergized control cells (Fig. 5 A and not depicted). However, the amounts of IL-10 were significantly higher than those produced by nonanergized control cells, 277 ± 54 pg/ml versus 147 ± 60 pg/ml (n = 4, P = 0.004), whereas the levels of TGF-β were not significantly different (Fig. 6 B). Thus, TT-specific T cell lines generated in the presence of chA6 mAb display a profile of cytokine production similar to that of T reg 1 cells (25).
Reversal of suppression of both proliferation (not depicted) and IFN-γ production (Fig. 6 C) was observed when a neutralizing anti–IL-10R mAb was added to the cultures, demonstrating that suppression by TT/chA6 cells is mediated by IL-10. Together, these data indicate that repetitive stimulation of CD4+ T cells with TT in the presence of chA6 mAb leads to the induction of T reg cells that are phenotypically and functionally equivalent to T reg 1 cells.
ChA6 mAb modulates antigen-specific CD8+ T cell responses
To test whether chA6 mAb could also modulate antigen-specific CD8+ T cells, PBMCs from HLA-A*0201 individuals were stimulated in a mixed lymphocyte-peptide (MLP)–specific reaction with an immunodominant influenza A matrix protein-derived peptide (MP.58-66) in the presence (MLPchA6) or in the absence (MLP) of chA6 mAb. After two rounds of stimulation, MLP cultures were rechallenged with MP.56-88 in the absence of chA6 mAb. MLPchA6 cells were far less responsive to antigen stimulation than were their MLP counter parts, as demonstrated by the reduced production of IFN-γ in response to MP.58-66 (mean value of inhibition, 59 ± 30%; n = 16, P = 0.02; Fig. 7 A). Furthermore, CD8+ T cells generated in the presence of chA6 mAb displayed lower antigen-specific cytotoxic activity than did control CD8+ T cells (unpublished data). MP.58-66–specific CD8+ T effector cells can be monitored by staining with a mAb recognizing TCR Vβ17, the dominant Vβ chain used by these cells (26). The percentages of MP.58-66–specific Vβ17+CD8+ T cells in chA6-anergized and control cultures were comparable (Fig. 7 B), indicating that MP.58-66–specific CD8+ T cells were not deleted during stimulation in the presence of chA6 mAb but instead became functionally inactivated. We next investigated whether MP.58-66–specific CD8+ T cells generated in the presence of chA6 mAb have suppressive activity. MP.58-66–specific effector CD8+ T cells were rechallenged with APC, pulsed with MP.58-66, in the presence of increasing number of MLPchA6 cells. MLPchA6 cells inhibited IFN-γ production by MLP-specific effector CD8+ T cells in a dose-dependent fashion (mean value of inhibition, 42 ± 19% at 1:2 ratio; 23 ± 9% at 1:1 ratio; and 15 ± 8% at 1:0.5 ratio in three donors tested; Fig. 7 C).
Figure 7.

ChA6 mAb induced MP.58-66–specific CD8+ T reg cells. (A) Enriched CD8+ T cells were stimulated with 10 μg/ml peptide MP.58-66, in the absence (MLP) or presence of 10 μg/ml chA6 mAb (MLPchA6). After two rounds of stimulation, MLP and MLPchA6 cells were rechallenged with T2 cells previously pulsed with the MP.58-66 peptide. After 24 h culture, IFN-γ was quantified in culture supernatants by ELISA. Results from one representative donor out of 16 are shown. The percentage of inhibition of IFN-γ release in the presence of chA6 mAb relative to control is presented. (B) MLP and MLPchA6 cells were stained with TCR-Vβ17–specific mAb. Results from one representative out of eight donors tested are shown. (C) After two rounds of stimulation, MLP cells were cocultured with increased amounts of MLPchA6 cells with MP.56-88 pulsed and nonpulsed T2 cells in the absence of chA6 mAb. After 24 h culture, IFN-γ was quantified in culture supernatants by ELISA. Results from one representative out of three donors tested are shown. The percentage of inhibition of IFN-γ release in the presence of chA6 mAb relative to control is presented. (D) MP.58-66–specific T cell lines were stained with the indicated surface molecules and analyzed by flow cytometry. Percentages of positive cells, set according to the isotype controls, are shown in the relative quadrant. Results from one representative donor out of four donors tested are shown. (E) MLP cells were cocultured with MLPchA6 cells (1:1 ratio) and stimulated with MP.56-88 pulsed and nonpulsed APC in the presence of 10 μg/ml anti-IL-10R and 10 μg/ml anti-TGF-β mAb. After 24 h of culture, IFN-γ was quantified in culture supernatants by ELISA. Results from one representative out of two donors tested are shown. The percentages of inhibition of IFN-γ release in the presence of chA6 mAb relative to control are presented.
The percentages of MP.58-66–specific CD8+ T cells expressing CD25 were reduced in MLPchA6 cultures as compared with MLP cultures (n = 4, P = 0.01) (Fig. 7 D), indicating that CD8+CD25+ T reg cells were not responsible for the reduced IFN-γ production by MLPchA6 cells. In addition, the reduced percentage of MP.58-66–specific CD8+ T cells expressing CD69 in MLPchA6 cultures supports the conclusion that antigen-specific CD8+ T cells generated with chA6 mAb remain functionally inactivated. Both MLP and MLPchA6 cultures expressed comparable levels of CD28, excluding the possibility that MP.58-66–specific CD8+ T reg cells generated in the presence of chA6 mAb contained CD8+CD28− suppressor T cells.
The overall cytokine levels (IL-2, IL-4, IL-10, and TGF-β) produced after antigen-specific stimulation by MP.58-66–specific CD8+ T cell lines was below the detection level (unpublished data). However, the suppression mediated by anergic MLPchA6 cells was partially reversed by neutralizing anti-TGF-β and anti–IL-10R mAbs (Fig. 7 E), suggesting that chA6 mAb induces antigen-specific CD8+ T reg cells that have a mode of action similar to that of CD4+ T reg 1 cells.
ChA6 mAb prolongs human islet allograft survival in NOD/SCID mice
To determine whether chA6 mAb also exert immunomodulatory effects in vivo, we established a modified model of human islet transplantation in NOD/SCID mice. Human islets were transplanted under the kidney capsule of NOD/SCID mice rendered diabetic by a single injection of streptozotocin. NOD/SCID recipient mice were injected intraperitoneally with freshly isolated allogeneic PBMCs. Hu-PBL-NOD/SCID recipient mice were treated with chA6 mAb at 1 mg/kg subcutaneously at days 0, 3, and 5 after transplantation. Normal NOD/SCID mice transplanted with human islets remained normoglycemic up to 100 d after transplantation, whereas the mean rejection time of hu-PBL-NOD/SCID mice transplanted with human islets was 35 ± 13 d. The short treatment of transplanted hu-PBL-NOD/SCID mice with chA6 mAb significantly prolonged the survival of human islets (survival rate of >70% at day 60 and of 50% at day 100 after transplantation; Fig. 8 A). Comparison of the in vivo effect of chA6 mAb with sirolimus and with a combined immunosuppressive therapy (sirolimus, tacrolimus, and anti-IL2Rα mAb anti-TAC) defined as the Edmonton protocol (27) clearly demonstrated that a short treatment with chA6 mAb is significantly more effective that monotherapy with sirolimus but less powerful than the Edmonton protocol in preventing allograft rejection in hu-PBL-NOD/SCID mice. (Fig. 8 A). Histological analyses of human islet grafts performed 100 d after transplantation showed a massive infiltration of human CD3+, CD4+, and CD8+ T cells (Fig. 8 B and not depicted) in control mice. In contrast, significantly lower numbers of infiltrating cells were observed in mice treated with chA6 mAb (Fig. 8 B and not depicted). The staining for insulin was similar in hu-PBL-NOD/SCID–recipient mice treated with chA6 mAb and in transplanted mice not injected with PBMCs, demonstrating the graft function. Collectively, these data indicate that a short treatment with chA6 mAb prolongs human islet allograft survival in vivo.
Figure 8.
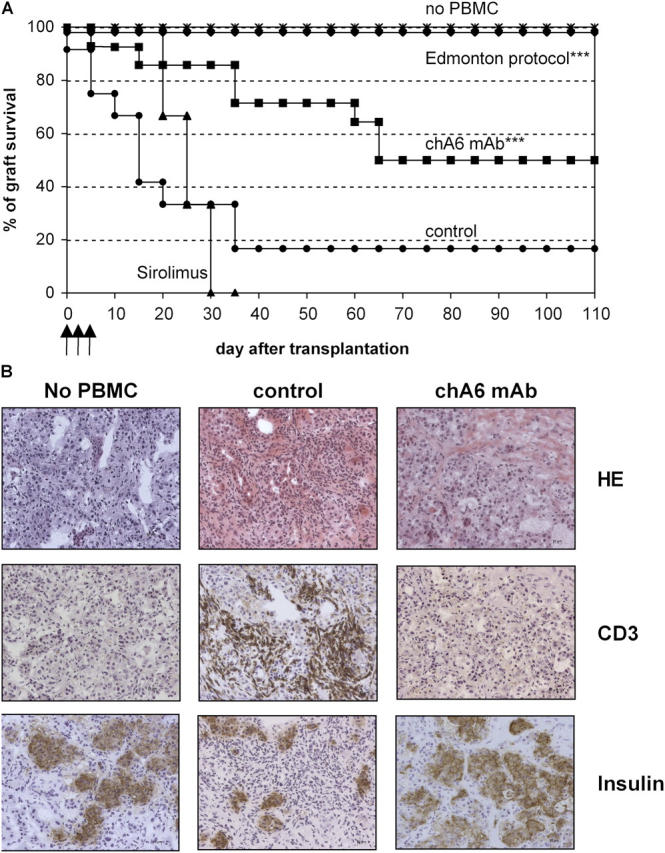
ChA6 mAb treatment prolongs human islet allograft survival in hu-PBL-NOD/SCID recipient mice. (A) Diabetic NOD/SCID mice were transplanted under the kidney capsule with human islets. Mice were injected intraperitoneally with 50 × 106 allogeneic human PBMCs. Normal NOD/SCID mice (*, n = 8) were used as control of human islets function. Mice were treated with vehicle (•, n = 12); with the Edmonton protocol (♦, n = 4); chA6 mAb at days 0, 3, and 5 after transplantation (▪, n = 14); or sirolimus (▴, n = 3). Glycemia levels monitored graft survival. Asterisks indicate statistical analysis in which treated mice were compared with control mice (*** ≤ 0.0001). (B) Kidney bearing the human-islet graft from control, chA6 mAb–treated, or normal NOD/SCID mice at 100 d after transplantation were snap-frozen, and 5-μm-thick sections were stained with hematoxylin and eosin (HE). Alternatively, sections were stained for the expression of CD3 and insulin.
DISCUSSION
In the present study, we analyzed the immunomodulatory effects of a chimeric A6 mAb that has unique specificity and recognizes both the RB and RO isoforms of CD45 on human cells (17). We demonstrated that chA6 mAb suppresses T cell responses in vitro through several mechanisms: inhibition of proliferation of primary, activated, and memory T cells; induction of apoptosis in effector/memory CD4+ CD45RO/RBbright T cells; and generation of antigen-specific T reg cells in both the CD4+ and CD8+ T cell subsets. Moreover, administration of chA6 mAb prolongs human islet allograft survival in hu-PBL-NOD/SCID mice.
Several studies demonstrated that CD45 RO- and RB-specific mAbs inhibit proliferative primary responses of T cells in humans (13, 18, 19) and mice (20). Here, we show that chA6 mAb inhibits not only primary polyclonal and alloantigen-specific T cell responses but also secondary and memory responses, indicating that chA6 mAb has a broad and powerful suppressive effect on T cell proliferation.
Induction of apoptosis in human T cells (28, 29) and murine thymocytes by ligation of CD45 has been described (30). It has been shown that cell death induced by cross-linking of CD45 in human T and B cells resembles cell death induced by CD95 (29), indicating that in human cells ligation of CD45 induces apoptosis via the extrinsic pathway. On the other hand, apoptosis of murine T lymphocytes induced by CD45 cross-linking resulted in a rapid increase in Δψm that was not inhibited by caspase inhibitors (30), indicating the use of the intrinsic apoptotic pathway. Similarly, anti-CD45RB mAb induced a rapid elimination of both murine CD4+ and CD8+ T cells in vitro caused by mitochondrial-dependent cell death mechanisms (16). Interestingly, the apoptotic effects induced by CD45 ligation in murine T lymphocytes was independent of the PTPase activity of the CD45 molecules, indicating a crucial role of the extracellular domain of the CD45 (28). Here, we show that CD45RB/RO ligation induces selective cell death in human CD4+ T cells through a CD95-independent mechanism. This effect is specific for the chA6 mAb, because it was not observed with anti-CD45RA and anti-CD45RO mAbs. Apoptosis induced by chA6 mAb is mediated via the intrinsic pathway, as demonstrated by the presence of caspase-9– and -3–activated subunits and by the reduction in mitochondrial transmembrane potential which occurs 2 h after CD45RB/RO ligation, a time at which up-regulation of CD95 on T cells has not yet occurred.
Our findings that ligation of CD45RO/RB isoforms by chA6 mAb results in a selective depletion of preexisting and recently activated CD4+CD45RO/RBbright T cells, which represent the memory/effector T cells compartment (17), is consistent with results obtained in vivo. Treatment with anti-CD45RB mAb in mice or with a pan–anti-CD45 mAb in rats resulted in a reduction of the number of peripheral T cells and ultimately in tolerance (11, 31, 32). In murine models the selective elimination of CD45RBhigh cells by anti-CD45RB mAb treatment promoted the survival of a T reg cell subset within the CD45RBlow population that was able to inhibit allograft rejection (10–12). Similarly, in our study depletion of preexisting and newly activated CD4+ CD45RO/RBbright human T cells mediated by chA6 mAb results in an increased percentage of CD4+A6low T cells (unpublished data), which may reset the T cell repertoire and permit the induction of T reg cells. The A6+ population does contain memory T cells, because depletion of the A6+ cell subset from PBMCs of TT- or hepatitis B–sensitized individuals by murine A6 mAb resulted in considerably reduced responses to recall antigens (17).
ChA6 mAb selectively eliminates human CD4+ memory T cells, but the proportion of MP.58-66–specific CD8+ T cells generated with chA6 mAb was comparable to that observed in controls, indicating that the CD8+ T cell population is unaffected. This finding is consistent with previous observations that showed that murine A6 mAb did not alter specific target-cell lysis mediated by cytotoxic T cells (17). The molecular mechanism underlying this differential apoptotic effect of chA6 mAb in CD4+ and CD8+ T cells remains to be defined.
In addition to apoptosis, modulation of antigen-specific T cell responses by chA6 mAb, with the induction of T reg 1 cells, is an important mode of action for this mAb. ChA6 mAb induces antigen-specific CD4+ T reg cells that do not acquire the CD4+CD25+ T reg cell phenotype and do not express FOXP3 (unpublished data), which is now recognized as a critical factor in the differentiation and function of mouse (33, 34) and human (35) CD4+CD25+ T reg cells. ChA6 mAb induces T reg cells that display a T reg 1 cell phenotype and function. So far, T reg 1 cells have been generated from naive CD4+ T cells in vitro either by using exogenous IL-10 and IFN-α (36) or vitamin D3 and dexamethasone (37) or by repetitive stimulation with immature DC (38). Here, we induced TT-specific T reg 1 cells from the memory CD4+ CD45RO/RBlow T cell compartment. Because chA6 mAb depletes CD4+CD45RO/RBbright T cells, which represent the effector/memory compartment, we suggest that chA6 mAb modulates central/memory cells, which are a part of the CD4+CD45RO/RBlow T population, leading to the generation of antigen-specific T reg 1 cells.
Interestingly, chA6 mAb induces not only antigen-specific CD4+ T reg 1 cells but also antigen-specific CD8+ T reg cells. Studies in human CD8+ T reg cells are still limited, probably because of their poor proliferative capacity in vitro. However, suppressor CD8+CD28− T cell lines can be generated in vitro (39) and have been isolated from peripheral blood of successfully transplanted patients (40). Antigen-specific CD8+ T cells generated with chA6 mAb express similar levels of CD28 and lower levels of CD25+ than do control cells, indicating that chA6 mAb does not induce CD8+ CD28− T reg cells and has no effect on CD8+CD25+ T reg cells. ChA6-induced CD8+ T reg cells share many similarities with the CD8+ T reg cells generated by plasmacytoid dendritic cells (DC2) (41) or by IL-10–treated DC (42). CD8+ T reg cells induced by these three different modalities are anergic and suppress T cell responses. However, CD8+ T reg cells induced by DC2 did not suppress secondary responses of activated effector T cells (41), whereas chA6-induced CD8+ T reg cells are able to suppress proliferation of activated T cells of the same specificity. Interestingly, CD8+ T reg cells induced by IL-10–treated DCs did not secrete IL-10 (43). Similarly, we were unable to detect IL-10 production by chA6-induced CD8+ T reg cells (unpublished data). These findings suggest that chA6 mAb induces antigen-specific CD8+ T reg cells that have phenotypical and functional properties similar to those of IL-10–induced CD8+ suppressor T cells.
To test the immunomodulatory effects of chA6 mAb in vivo, we modified the model for human islet allograft rejection described by Shiroki et al. (44). In our model, injection of freshly isolated allogeneic PBMCs at the time of the human islet transplantation in NOD/SCID mice resulted in the rejection of the graft. Interestingly, three injections of chA6 mAb resulted in long-term survival of islet allograft in transplanted hu-PBL-NOD/SCID mice. This prolonged survival was accompanied by a reduced infiltration of human lymphocytes. Similar to the effect observed in mouse islet allografts with anti-CD45RB mAb treatment, three injections of chA6 mAb induced long-term engraftment in 50% of the hu-PBL-NOD/SCID–recipient mice. This in vivo protective effect of chA6 mAb was opposed to the inability of sirolimus to prolong graft survival in this model. Treatment for 30 d with the Edmonton protocol resulted in a higher incidence of graft survival. These data suggest that chA6 mAb administration early after transplantation may induce long-term tolerance in recipient mice, possibly through the apoptosis of activated CD4+ T cells and the induction of T reg 1 cells.
The mechanism by which chA6 mAb induces T reg 1 cells remains unclear and may involve both direct and indirect effects on T cells. ChA6 mAb modulates T cell responses at nonapoptotic concentrations and increases the calcium influx in T cells (unpublished data), indicating that it can directly modulate T cell activation. In addition, it has been demonstrated that CD45 suppresses JAK kinases and negatively regulates cytokine receptor signaling including those of IL-3, IL-4, and IFN-α (45). Therefore, ligation of CD45RB/RO by chA6 mAb may also directly interfere with signaling through cytokine receptors and modulation of cytokine responses by T cells, allowing the induction of T reg cells. Alternatively, chA6 mAb may act indirectly on antigen-specific CD4+ and CD8+ T cells through modulation of the APC that express the CD45RO/RB isoforms.
Different mechanisms, which are not mutually exclusive, have been associated with tolerance induction: deleting mechanisms in which either allo- or autoreactive T cells are eliminated and nondeleting mechanisms including anergy (46), immune deviation, and active immunosuppression mediated by T reg cells. Here we describe a new chimeric mAb, which selectively depletes memory/effector CD4+CD45RO/RBbright T cells, induces CD4+ T reg 1 cells and CD8+ T reg cells, and prevents human islet allograft rejection in hu-PBL-NOD/SCID mice. Therefore, it can be hypothesized that chA6 mAb may induce immunological tolerance in vivo by inducing both clonal deletion and active immunosuppression.
MATERIALS AND METHODS
Generation of chimeric A6 antibody.
ChA6 was generated by linking the variable regions of mAb A6 (17), cloned by RT-PCR, with human γ1 heavy chain– and human κ light chain–constant regions. After transfection into SP2/0 cells and selection of clones using G418 and methotrexate, the antibody was purified by affinity chromatography on goat anti–human IgG followed by size exclusion chromatography. Endotoxin was removed using ActiClean Etox (Sterogene Bioseparations, Inc.). Final endotoxin levels were below 30 pg/mg protein.
Mixed lymphocyte cultures and proliferation assay.
Human peripheral blood was obtained from healthy donors in accordance with local ethical committee standards. PBMCs were separated by density-gradient centrifugation over Lymphoprep (Amersham Biosciences). CD4+ T cells enriched by selective depletion of CD8, CD14, and CD19 cells (Oxoid Linited) were used as responder, and CD3-depleted and irradiated (6,000 rad) cells were used as stimulators. CD4+ T cells (105/well) were cultured in RPMI 1640 medium (GIBCO BRL) supplemented with 100 U/ml penicillin/streptomycin (Bristol-Myers Squibb), 10% heat-inactivated FCS (Mascia Brunelli S.p.A.) in 96-well plates (Costar) for 5 d with CD3-depleted cells (105/well) with or without chA6 or chimeric isotype control mAbs (10 μg/ml). To evaluate secondary responses, primary cultures were carried on for 10 d; then cells were collected, washed, and plated in 96-well plates with newly prepared stimulator cells with or without of chA6 mAb (10 μg/ml) for 2 d.
To analyze proliferation in response to polyclonal activation, CD4+ T cells (105/well) were cultured in 96-well plates precoated with anti-CD3 mAb (OKT3, Janssen Cilag, Ltd.) for 3 d in the presence or absence of chA6 or chimeric isotype control mAbs (10 μg/ml) with or without soluble anti-CD28 mAb (1 μg/ml) (BD Biosciences). To measure cell proliferation, cultures were pulsed with [3H]thymidine during the last 16 h.
Apoptosis.
After overnight incubation with chA6, anti-CD45RA (HI100, BD Biosciences), anti-CD45RO (UCLH-1, BD Biosciences), or isotype control, mAb cells were stained with annexin V–FITC (BD Biosciences) and propidium iodide (50 μg/ml, Sigma-Aldrich). Alternatively, cells were stained with annexin V–FITC and anti–human IgG1 PE mAb (Sigma-Aldrich). Analyses were performed using FACScan flow cytometer using CellQuest software (BD Biosciences). To calculate the ED50 value, CD4+ T cells were stained, after overnight culture, with annexin V–FITC and CD2-PE (BD Biosciences). Curve fitting and ED50 value calculations were performed using Origin v7.5 SR2 software (OriginLab Corporation). To evaluate primary proliferative responses on annexin V+ cell depletion, CD4+ T cells were incubated with chA6 mAb (10 μg/ml) for 4 h at 37°C and depleted using a dead cell removal kit (Miltenyi Biotec). Totally depleted or annexin V–depleted CD4+ T cells were used as responder cells, and irradiated CD3-depleted cells were used as stimulators.
Western blot assay.
Treated cells were resuspended in lysis buffer containing 10 mM Tris-HCl, pH 7.2, 100 mM NaCl, 1 mM EDTA, 1% deoxycholate acid, 1% Triton X-100, 0.1% SDS, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml peptstatin, and 0.23 U/ml aprotinin. The supernatants were then centrifuged at 14,000 revolutions/min for 10 min at 4°C to remove lysosome and mitochondria. 40 μg of protein were separated on SDS-PAGE and transferred to a nitrocellulose membrane (Amersham Biosciences). Blots were blocked 30 min at 37°C in PBS containing 10% nonfat dried milk and incubated at 4°C with anti–caspase-3 (anti-CCP32), anti–caspase-8 (1C12, Cell Signaling Technology), and anti-caspase-9 (Asp 315, Cell Signaling Technology) mAbs. Detection was achieved with the appropriate secondary antibodies coupled to horseradish peroxidase followed by enhanced chemiluminescence Western blotting (Amersham Biosciences).
Mitochondrial permeability assay.
CD4+ T cells were incubated for 2 h with 5 μg/ml chA6 or isotype control mAbs in the presence of 15 μg/ml of cross-linking goat anti–human IgG (F[ab′]2). Cells were washed and resuspended in culture medium without FCS containing 20 nM of 3,3′-dihexyloxacarbocyanine iodide (DiOC6[3]) (Molecular Probes). After incubation for 20 min, cells were washed, resuspended in PBS, and analyzed on a FACSCalibur (BD Biosciences). The mitochondrial accumulation of DiOC6(3) was used to assess Δψm in the mitochondria.
Antigen-specific responses.
For in vitro induction of effector cells specific for TT, PBMCs depleted of CD8+ and CD19+ cells (2 × 106 cells/well) were cultured in 24-well plates with 5 μg/ml TT (Calbiochem) with or without chA6 mAb (10 μg/ml) in X-VIVO 15 (Cambrex BioScience Inc.) supplemented with 100 U/ml penicillin/streptomycin, 5% heat-inactivated human serum (Cambrex BioScience, Inc.). After 10 d, the bulk populations were restimulated with autologous irradiated (6,000 rad) CD3-depleted cells pulsed with TT with or without chA6 mAb (10 μg/ml). The responder populations were analyzed after the second stimulation for proliferation: 104 T cell lines were stimulated with 5 × 104 autologous irradiated monocytes with or without TT, in the absence of chA6 mAb for 2 d and pulsed with [3H]thymidine during the last 16 h. For suppressor assay, TT-specific T cell lines generated without chA6 mAb were cocultured with increased amounts of TT-specific T cell lines generated in the presence of chA6 mAb with TT-pulsed and nonpulsed autologous irradiated monocytes in the absence of chA6 mAb. After 48 h of culture, IFN-γ was quantified in culture supernatants by two-site ELISA.
For in vitro induction of effector cells specific for influenza peptide MP.58-66, CD8-enriched T cells from HLA-A*0201+ healthy donors were seeded (2 × 106/well) in 24-well plates with 10 μg/ml peptide MP.58-66, with or without chA6 mAb (10 μg/ml). After 10 d, responder CD8+ T cells were restimulated with autologous irradiated monocytes pulsed with the MP.58-66 peptide. The responder populations were analyzed after the second stimulation for IFN-γ release against peptide-pulsed and nonpulsed T2 cells, which are HLA-A*0201+ APC 8 × 103 T cell lines were stimulated with 2 × 104 irradiated (10,000 rad) T2 cell lines. At the end of each round of stimulation with the MP.58-66 peptide, cells (0.5–1 × 106) were stained with T cell receptor Vβ17-specific mAb.
FACS analysis.
Anti-CD4, -CD8, -CD25, and -CD28 mAbs directly coupled with FITC and PE were purchased from BD Bioscience. Cells were incubated with the indicated mAbs for 20 min at 4°C in PBS plus 2% FCS, washed twice, and analyzed by FACS using CELLQuest software (BD Biosciences).
Cytokine secretion.
TT-specific CD4+ T cells (5 × 103) were stimulated with 2 × 104 autologous irradiated monocytes with or without 5 μg/ml TT. Supernatants were collected after 24 h for IL-2, after 48 h for IL-4 and IFN-γ, and after 72 h for IL-10 and TGF-β. Levels of IL-2, IL-4, IL-10, and IFN-γ were determined by capture ELISA according to the manufacturer's instructions (BD Bioscience). Levels of TGF-β in acidified supernatants were determined by capture ELISA according to the manufacturer's instructions (R&D Systems). The limits of detection were as follows: IL-2: 20 pg/ml; IL-4: 20 pg/ml; IL-10: 20 pg/ml; IFN-γ: 60 pg/ml; TGF-β: 60 pg/ml.
Mice.
NOD/SCID female mice were purchased from Charles River Laboratories. All mice were kept under specific-pathogen–free conditions. All animal care procedures were performed according to protocols approved by the HSR Institutional Animal Care and Use Committee, IACUC 156. Blood glucose level was quantified using Glucometer Elite system (Bayer AG). Diabetes was induced in NOD/SCID mice by intravenous injection of streptozotocin (Sigma-Aldrich) at 180 mg/kg. A diagnosis of diabetes was made after two consecutive glucose measurements >350 mg/dl.
Islet preparations and transplantation.
Pancreases were obtained from cadaveric multiorgan donors through the North Italian Transplant Organization. Islets were isolated according to a modification of the automated method (47). The purified islets were cultured in M199 medium (Seromed Biochrom KG) supplemented with 10% FCS, 1% l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin and incubated at 30°C in 5% CO2 and 95% humidified air. Mice were anesthetized with an intraperitoneal injection of avertin, and aliquots of 1,500 IE of human islets were transplanted under the kidney capsule of recipient diabetic NOD/SCID mice as previously described (48). After transplantation, recipient NOD/SCID mice were injected intraperitoneally with 50 × 106 freshly isolated allogeneic PBMCs.
Treatments of transplanted mice.
Hu-PBL-NOD/SCID transplanted mice were treated at days 0, 3, and 5 with chA6 mAb 1 mg/kg subcutaneously; or with the Edmonton protocol: anti-IL-2R mAbs (Zenapax, Roche), 200 μg/mouse, administered intraperitoneally at day 0 and at day 4 after transplantation; sirolimus (Rapamune, Wyeth-Ayerst Research) diluted in peanut oil (Sigma-Aldrich) and administered once daily at 1 mg/kg by gavage from days 0 to 30 after transplantation, and tacrolimus (Prograf, Fujisawa) diluted in saline solution and administered once daily at 0.3 mg/kg intraperitoneally from days 0 to 30 after transplantation; or with sirolimus alone once daily at 1 mg/kg by gavage from days 0 to 30 after transplantation. Control mice were treated with saline solution or with IgG1-purified mAb (Vinci-Biochem).
Histological analysis.
Kidneys containing human islet grafts were snap-frozen in Tissue Tek (Miles Laboratories, Inc.) and stored at −70°C. Frozen sections, 5 μm thick, were stained with biotinylated mAb against human insulin followed by streptavidin-peroxidase conjugate. Diaminobenzidine (DakoCytomation) was used as chromogen, and hematoxylin as was used as counterstain. Lymphocyte infiltration of the grafts was evaluated on hematoxylin and eosin–stained frozen sections.
Statistical analysis.
All analyses for statistically significant differences were performed with the Student's t test or nonparametric Wilcoxon test. p-values <0.05 were considered significant.
Acknowledgments
The authors thank G. Aversa for valuable scientific input, F. Bertuzzi and E. Bonifacio for providing human islets preparation; F. Bottazzoli, B. Migliavacca, A. Miluzio, M. Bongers, S. Gronalt, and A. Hren for the excellent technical assistance provided; and F. Sanvito, and S. Olivieri for immunohistochemical analysis.
This study is partially supported by a Novartis grant to the San Raffaele Park Raf and by grant no. JT-01 from the Juvenile Diabetes Research Foundation and Telethon Foundation (to M.-G. Roncarolo).
The Novartis Institutes for Biomedical Research (NIBR) are developing a modified version of the monoclonal antibody described in this paper. F. Kolbinger, J.M. Carballido, U. Korthäuer, and J.E. de Vries are employed by NIBR and receive stock options, or restricted stocks (J.E. de Vries). The authors have no other conflicting financial interests.
Abbreviations used: Δψm, value of change in mitochondria transmembrane potential; chA6, chimeric A6; MLP, mixed lymphocyte-peptide; MP, matrix protein-derived peptide; PTPase, protein tyrosine phosphatase; T reg, regulatory T; TT, tetanus toxoid.
References
- 1.Sasaki, T., J. Sasaki-Irie, and J.M. Penninger. 2001. New insights into the transmembrane protein tyrosine phosphatase CD45. Int. J. Biochem. Cell Biol. 33:1041–1046. [DOI] [PubMed] [Google Scholar]
- 2.Byth, K.F., L.A. Conroy, S. Howlett, A.J. Smith, J. May, D.R. Alexander, and N. Holmes. 1996. CD45-null transgenic mice reveal a positive regulatory role for CD45 in early thymocyte development, in the selection of CD4+CD8+ thymocytes, and B cell maturation. J. Exp. Med. 183:1707–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothstein, D.M., A. Yamada, S.F. Schlossman, and C. Morimoto. 1991. Cyclic regulation of CD45 isoform expression in a long term human CD4+CD45RA+ T cell line. J. Immunol. 146:1175–1183. [PubMed] [Google Scholar]
- 4.Bottomly, K., M. Luqman, L. Greenbaum, S. Carding, J. West, T. Pasqualini, and D.B. Murphy. 1989. A monoclonal antibody to murine CD45R distinguishes CD4 T cell populations that produce different cytokines. Eur. J. Immunol. 19:617–623. [DOI] [PubMed] [Google Scholar]
- 5.Lee, W.T., X.M. Yin, and E.S. Vitetta. 1990. Functional and ontogenetic analysis of murine CD45Rhi and CD45Rlo CD4+ T cells. J. Immunol. 144:3288–3295. [PubMed] [Google Scholar]
- 6.Powrie, F., R. Correa-Oliveira, S. Mauze, and R.L. Coffman. 1994. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J. Exp. Med. 179:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fowell, D., and D. Mason. 1993. Evidence that the T cell repertoire of normal rats contains cells with the potential to cause diabetes. Characterization of the CD4+ T cell subset that inhibits this autoimmune potential. J. Exp. Med. 177:627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powrie, F., M.W. Leach, S. Mauze, S. Menon, L.B. Caddle, and R.L. Coffman. 1994. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1:553–562. [DOI] [PubMed] [Google Scholar]
- 9.Davies, J.D., E. O'Connor, D. Hall, T. Krahl, J. Trotter, and N. Sarvetnick. 1999. CD4+ CD45RB low-density cells from untreated mice prevent acute allograft rejection. J. Immunol. 163:5353–5357. [PubMed] [Google Scholar]
- 10.Auersvald, L.A., D.M. Rothstein, S.C. Oliveira, C.Q. Khuong, H. Onodera, A.I. Lazarovits, and G.P. Basadonna. 1997. Indefinite islet allograft survival in mice after a short course of treatment with anti-CD45 monoclonal antibodies. Transplantation. 63:1355–1358. [DOI] [PubMed] [Google Scholar]
- 11.Basadonna, G.P., L. Auersvald, C.Q. Khuong, X.X. Zheng, N. Kashio, D. Zekzer, M. Minozzo, H. Qian, L. Visser, A. Diepstra, et al. 1998. Antibody-mediated targeting of CD45 isoforms: a novel immunotherapeutic strategy. Proc. Natl. Acad. Sci. USA. 95:3821–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fecteau, S., G.P. Basadonna, A. Freitas, C. Ariyan, M.H. Sayegh, and D.M. Rothstein. 2001. CTLA-4 up-regulation plays a role in tolerance mediated by CD45. Nat. Immunol. 2:58–63. [DOI] [PubMed] [Google Scholar]
- 13.Lazarovits, A.I., S. Poppema, Z. Zhang, M. Khandaker, C.E. Le Feuvre, S.K. Singhal, B.M. Garcia, N. Ogasa, A.M. Jevnikar, M.H. White, et al. 1996. Prevention and reversal of renal allograft rejection by antibody against CD45RB. Nature. 380:717–720. [DOI] [PubMed] [Google Scholar]
- 14.Sho, M., H. Harada, D.M. Rothstein, and M.H. Sayegh. 2003. CD45RB-targeting strategies for promoting long-term allograft survival and preventing chronic allograft vasculopathy. Transplantation. 75:1142–1146. [DOI] [PubMed] [Google Scholar]
- 15.Auersvald, L.A., D.M. Rothstein, S.C. Oliveira, C.Q. Khuong, and G.P. Basadonna. 1997. Anti-CD45RB treatment prolongs islet allograft survival in mice. Transplant. Proc. 29:771. [DOI] [PubMed] [Google Scholar]
- 16.Luke, P.P., J.P. Deng, C.A. O'Brien, M. Everest, A.V. Hall, S. Chakrabarti, P.J. O'Connell, R. Zhong, and A.M. Jevnikar. 2003. Alteration in CD45RBhi/CD45RBlo T-cell ratio following CD45RB monoclonal-antibody therapy occurs by selective deletion of CD45RBhi effector cells. Transplantation. 76:400–409. [DOI] [PubMed] [Google Scholar]
- 17.Aversa, G., J.A. Waugh, and B.M. Hall. 1994. A monoclonal antibody (A6) recognizing a unique epitope restricted to CD45RO and RB isoforms of the leukocyte common antigen family identifies functional T cell subsets. Cell. Immunol. 158:314–328. [DOI] [PubMed] [Google Scholar]
- 18.Nishikawa, M., T. Mukuta, G. Arreaza, E. Resetkova, S. Poppema, H. Tamai, R. Volpe, and A.I. Lazarovits. 1995. Effects of monoclonal antibody against CD45RB on peripheral blood mononuclear cell proliferation and on HLA-DR and adhesion molecule expression on thyrocytes of patients with autoimmune thyroid disease. Thyroid. 5:265–272. [DOI] [PubMed] [Google Scholar]
- 19.Shanafelt, M.C., H. Yssel, C. Soderberg, L. Steinman, D.C. Adelman, G. Peltz, and R. Lahesmaa. 1996. CD45 isoforms on human CD4+ T-cell subsets. J. Allergy Clin. Immunol. 98:433–440. [DOI] [PubMed] [Google Scholar]
- 20.Min, W.P., D. Zhou, T.E. Ichim, G.H. Strejan, X. Xia, J. Yang, X. Huang, B. Garcia, D. White, P. Dutartre, et al. 2003. Inhibitory feedback loop between tolerogenic dendritic cells and regulatory T cells in transplant tolerance. J. Immunol. 170:1304–1312. [DOI] [PubMed] [Google Scholar]
- 21.Varfolomeev, E.E., M. Schuchmann, V. Luria, N. Chiannilkulchai, J.S. Beckmann, I.L. Mett, D. Rebrikov, V.M. Brodianski, O.C. Kemper, O. Kollet, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 9:267–276. [DOI] [PubMed] [Google Scholar]
- 22.Kuida, K., T.F. Haydar, C.Y. Kuan, Y. Gu, C. Taya, H. Karasuyama, M.S. Su, P. Rakic, and R.A. Flavell. 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 94:325–337. [DOI] [PubMed] [Google Scholar]
- 23.Bernardi, P., L. Scorrano, R. Colonna, V. Petronilli, and F. Di Lisa. 1999. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem. 264:687–701. [DOI] [PubMed] [Google Scholar]
- 24.Kroemer, G., B. Dallaporta, and M. Resche-Rigon. 1998. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60:619–642. [DOI] [PubMed] [Google Scholar]
- 25.Roncarolo, M.G., R. Bacchetta, C. Bordignon, S. Narula, and M.K. Levings. 2001. Type 1 T regulatory cells. Immunol. Rev. 182:68–79. [DOI] [PubMed] [Google Scholar]
- 26.Lehner, P.J., E.C. Wang, P.A. Moss, S. Williams, K. Platt, S.M. Friedman, J.I. Bell, and L.K. Borysiewicz. 1995. Human HLA-A0201-restricted cytotoxic T lymphocyte recognition of influenza A is dominated by T cells bearing the V beta 17 gene segment. J. Exp. Med. 181:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shapiro, A.M., J.R. Lakey, E.A. Ryan, G.S. Korbutt, E. Toth, G.L. Warnock, N.M. Kneteman, and R.V. Rajotte. 2000. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 343:230–238. [DOI] [PubMed] [Google Scholar]
- 28.Fortin, M., A.M. Steff, J. Felberg, I. Ding, B. Schraven, P. Johnson, and P. Hugo. 2002. Apoptosis mediated through CD45 is independent of its phosphatase activity and association with leukocyte phosphatase-associated phosphoprotein. J. Immunol. 168:6084–6089. [DOI] [PubMed] [Google Scholar]
- 29.Klaus, S.J., S.P. Sidorenko, and E.A. Clark. 1996. CD45 ligation induces programmed cell death in T and B lymphocytes. J. Immunol. 156:2743–2753. [PubMed] [Google Scholar]
- 30.Lesage, S., A.M. Steff, F. Philippoussis, M. Page, S. Trop, V. Mateo, and P. Hugo. 1997. CD4+ CD8+ thymocytes are preferentially induced to die following CD45 cross-linking, through a novel apoptotic pathway. J. Immunol. 159:4762–4771. [PubMed] [Google Scholar]
- 31.Dahlke, M.H., O.S. Lauth, M.D. Jager, T. Roeseler, K. Timrott, S. Jackobs, M. Neipp, K. Wonigeit, and H.J. Schlitt. 2002. In vivo depletion of hematopoietic stem cells in the rat by an anti-CD45 (RT7) antibody. Blood. 99:3566–3572. [DOI] [PubMed] [Google Scholar]
- 32.Ko, S., M.D. Jager, T.Y. Tsui, A. Deiwick, A. Dinkel, F. Rohde, M.H. Dahlke, O. Lauth, K. Wonigeit, and H.J. Schlitt. 2001. Long-term allograft acceptance induced by single dose anti-leukocyte common antigen (RT7) antibody in the rat. Transplantation. 71:1124–1131. [DOI] [PubMed] [Google Scholar]
- 33.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 34.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 35.Walker, M.R., D.J. Kasprowicz, V.H. Gersuk, A. Benard, M. Van Landeghen, J.H. Buckner, and S.F. Ziegler. 2003. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+ CD25− T cells. J. Clin. Invest. 112:1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levings, M.K., R. Sangregorio, F. Galbiati, S. Squadrone, R. de Waal Malefyt, and M.G. Roncarolo. 2001. IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J. Immunol. 166:5530–5539. [DOI] [PubMed] [Google Scholar]
- 37.Barrat, F.J., D.J. Cua, A. Boonstra, D.F. Richards, C. Crain, H.F. Savelkoul, R. de Waal-Malefyt, R.L. Coffman, C.M. Hawrylowicz, and A. O'Garra. 2002. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 195:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonuleit, H., E. Schmitt, G. Schuler, J. Knop, and A.H. Enk. 2000. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J. Exp. Med. 192:1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu, Z., S. Tugulea, R. Cortesini, and N. Suciu-Foca. 1998. Specific suppression of T helper alloreactivity by allo-MHC class I-restricted CD8+CD28− T cells. Int. Immunol. 10:775–783. [DOI] [PubMed] [Google Scholar]
- 40.Ciubotariu, R., R. Vasilescu, E. Ho, P. Cinti, C. Cancedda, L. Poli, M. Late, Z. Liu, P. Berloco, R. Cortesini, and N. Suciu-Foca Cortesini. 2001. Detection of T suppressor cells in patients with organ allografts. Hum. Immunol. 62:15–20. [DOI] [PubMed] [Google Scholar]
- 41.Gilliet, M., and Y.J. Liu. 2002. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J. Exp. Med. 195:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinbrink, K., H. Jonuleit, G. Muller, G. Schuler, J. Knop, and A.H. Enk. 1999. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood. 93:1634–1642. [PubMed] [Google Scholar]
- 43.Steinbrink, K., E. Graulich, S. Kubsch, J. Knop, and A.H. Enk. 2002. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood. 99:2468–2476. [DOI] [PubMed] [Google Scholar]
- 44.Shiroki, R., N.J. Poindexter, E.S. Woodle, M.S. Hussain, T. Mohanakumar, and D.W. Scharp. 1994. Human peripheral blood lymphocyte reconstituted severe combined immunodeficient (hu-PBL-SCID) mice. A model for human islet allograft rejection. Transplantation. 57:1555–1562. [PubMed] [Google Scholar]
- 45.Irie-Sasaki, J., T. Sasaki, W. Matsumoto, A. Opavsky, M. Cheng, G. Welstead, E. Griffiths, C. Krawczyk, C.D. Richardson, K. Aitken, et al. 2001. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature. 409:349–354. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz, R.H. 1997. T cell clonal anergy. Curr. Opin. Immunol. 9:351–357. [DOI] [PubMed] [Google Scholar]
- 47.Bertuzzi, F., A.M. Davalli, R. Nano, C. Socci, F. Codazzi, R. Fesce, V. Di Carlo, G. Pozza, and F. Grohovaz. 1999. Mechanisms of coordination of Ca2+ signals in pancreatic islet cells. Diabetes. 48:1971–1978. [DOI] [PubMed] [Google Scholar]
- 48.Davalli, A.M., L. Scaglia, D.H. Zangen, J. Hollister, S. Bonner-Weir, and G.C. Weir. 1996. Vulnerability of islets in the immediate posttransplantation period. Dynamic changes in structure and function. Diabetes. 45:1161–1167. [DOI] [PubMed] [Google Scholar]