

Abstract
The selectin family of adhesion molecules and their glycoconjugated ligands are essential for blood polymorphonuclear neutrophil (PMN) extravasation into inflammatory and infectious sites. However, E-selectin ligands on PMNs are not well characterized. We show here that CD44 immunopurified from G-CSF–differentiated 32D cells or from peripheral blood PMNs binds specifically to E-selectin. In contrast, CD44 extracted from bone marrow stromal or brain endothelial cell lines does not interact with E-selectin, suggesting cell-specific posttranslational modifications of CD44. PMN-derived CD44 binding activity is mediated by sialylated, α(1,3) fucosylated, N-linked glycans. CD44 enables slow leukocyte rolling on E-selectin expressed on inflamed endothelium in vivo and cooperates with P-selectin glycoprotein ligand–1 to recruit neutrophils into thioglycollate-induced peritonitis and staphylococcal enterotoxin A–injected skin pouch. CD44 extracted from human PMNs also binds to E-selectin. Moreover, we demonstrate that CD44 is hypofucosylated in PMNs from a patient with leukocyte adhesion deficiency type II, suggesting that it contributes to the syndrome. These findings thus suggest broader roles for CD44 in the innate immune response and uncover a potential new target for diseases in which selectins play a prominent role.
The selectins are vascular adhesion molecules interacting with sialylated and fucosylated glycan determinants, such as sialyl Lewis x, that decorate the terminal extensions of O- and / or N-linked carbohydrates (1). They play pivotal roles in cell–cell interactions among leukocytes, platelets, and endothelial cells (2). Several putative glycoprotein selectin ligands have been isolated from hematopoietic cells using in vitro affinity purification techniques (3–6), but the exact identity and contribution of physiological E-selectin ligands (ESLs) on PMNs is unknown. This knowledge gap, despite intense research effort, originates in part from the unavailability of inhibitory antibodies, due to the poor immunogenicity of highly glycosylated and conserved ESL epitopes (7). P-Selectin glycoprotein ligand-1 (PSGL-1) was identified as a ligand for all three selectins in vivo (8). PSGL-1−/− mice exhibit some deficits in E-selectin–mediated rolling but PSGL-1 appears dispensable for E-selectin–mediated PMN extravasation, suggesting a role for other cell surface ESLs (9, 10). An intriguing report has suggested that CD44 derived from primitive human hematopoietic stem cells, but not mature hematopoietic cells, could bind to E-selectin in vitro (6). The globular amino-terminal domain of CD44 contains hyaluronic acid (HA)–binding motifs and several potential glycosylation sites (see Fig. 1 A). CD44–HA interaction has been extensively characterized and shown to mediate a variety of physiological and pathological processes, including the recruitment of activated T cells in extralymphoid tissues (11), the recovery after noninfectious lung injury (12), and tumor metastasis (13). Here, we demonstrate that neutrophil CD44 is a physiological ESL.
Figure 1.

Neutrophil CD44 binds to E-selectin through N-linked glycans. (A) Structure of CD44 molecule. The globular amino-terminal domain of CD44 proteins contains several potential O- (circle) or N- (diamond) glycosylation sites (for review see reference 31). (B) Binding of E-selectin–IgM chimera to 32D cells differentiated for 4 d in media containing 10 ng/ml G-CSF. All 32D cells bind to E-selectin (left, open histogram) and binding is abrogated by treatment with EDTA (5 mM; gray-filled histograms) or sialidase treatment (right, open histogram). (C) Binding of E-selectin to CD44 immunopurified from the same 32D cells. (D) Contributions of O- and N-glycans in CD44 binding to E-selectin. Differentiated 32D cells were either treated with OSGE, sialidase, or vehicle before immunopurification of CD44. In the last three bars, ESLs were removed by sialidase and cells were allowed to recover for 36 h in the presence or absence of tunicamycin (15 μg/ml) to inhibit N-glycosylation. Data are average geometric mean values from at least three independent experiments. **, P ≤ 0.003. (E) PMN-derived CD44 is an ESL. PMNs were isolated from (i, iv, and vii) BM and (ii, v, and viii) blood of control mice, or (iii, vi, and ix) blood from FucT IV/VII−/− mice. Upper panels (i–iii) show CD44 expression on PMNs (gated on Gr-1hi). Middle panels (iv–vi) depict E-selectin binding on PMNs (Gr-1hi). Lower panels (vii–ix) show binding of E-selectin on immobilized CD44 extracted from purified PMNs (purity >95%). Gray-filled histograms represent isotype-matched control (CD44 staining) or EDTA treatment (E-selectin binding). (F and G) ESLs on wild-type and CD44−/− PMNs. Blood leukocytes were stained with E-selectin–IgM to determine ESL densities. (F) Histograms of E-selectin binding on Gr-1hi PMNs. (G) Geometric mean fluorescence of E-selectin binding on blood Gr-1hi PMNs. n = 12 mice per group. *, P = 0.02.
Results and Discussion
To assess the ability of mature myeloid cell–derived CD44 to bind E-selectin, we induced the differentiation of the 32D cell line with G-CSF (Fig. 1, B and C). After 4 d of G-CSF treatment, the majority (68 ± 1%) of 32D cells exhibited a polymorphonuclear appearance. Differentiated 32D cells bound to soluble E-selectin, and binding was abrogated by chelation of divalent cations or by prior treatment of the cells with sialidase. Protein extracts from the same cells were incubated with immunomagnetic beads coated with anti-CD44. Immobilized CD44 bound to soluble E-selectin in a manner similar to intact cells; binding was eliminated with EDTA or by sialidase treatment before lysis (Fig. 1 C). No binding was observed when beads were coated with an isotype-matched antibody binding αMβ2 integrin, or with control rat IgG (unpublished data). Immunoblot analyses revealed that CD44 was the sole protein purified from anti-CD44–coated beads (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20042014/DC1). These results thus suggest that CD44 derived from mature myeloid cells interacts specifically with E-selectin. Further, we evaluated E-selectin binding specificity using CD44 extracted from a bone marrow stromal cell line (MS-5) and a brain endothelial cell line (bEnd.3), which express high levels of CD44 but do not bind to E-selectin. CD44 immunopurified from endothelial and stromal cells did not interact with E-selectin (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20042014/DC1), underscoring differential posttranslational modifications of CD44 between hematopoietic and nonhematopoietic cells.
To investigate the contribution of O-linked and N-linked carbohydrates in the formation of ESLs on myeloid cells, we treated G-CSF–differentiated 32D cells with O-sialoglycoprotein endopeptidase (OSGE) to remove O-glycans. OSGE affected neither ESLs on the cell surface, nor E-selectin binding to immunopurified CD44 (Fig. 1 D), whereas P-selectin ligands were completely cleaved by OSGE (unpublished data). To assess the contribution of N-linked carbohydrates, ESLs were removed with sialidase and differentiated 32D cells were then cultured in the presence or absence of tunicamycin. De novo synthesis of ESL determinants on CD44 was dramatically inhibited (>90%, P = 0.002) by incubation with tunicamycin and the addition of OSGE to tunicamycin-treated cells did not further reduce E-selectin binding (Fig. 1 D). Tunicamycin treatment, however, did not affect P-selectin binding (which is dependent on O-glycosylation; Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20042014/DC1). Thus, CD44 derived from mature myeloid cell binds to E-selectin through N-linked, but not O-linked, glycans.
We next assessed E-selectin binding on primary mouse PMNs. BM and peripheral blood (PB) PMNs (Gr-1hi) express high levels of CD44 but expression of ESLs is heterogeneous on BM PMNs whereas PB PMNs uniformly express high levels of ESLs (Fig. 1 E). E-Selectin binding to CD44 displayed a pattern similar to that of intact cells in that binding was lower on CD44 extracted from BM PMNs than on PB PMNs (Fig. 1 E), suggesting that the density of CD44 molecules that are appropriately decorated with E-selectin binding carbohydrates is greater on circulating PMNs than on maturing bone marrow PMNs. No binding was observed when beads were coated with anti-αM integrin or rat IgG (Fig. S2), or when CD44 was extracted from blood PMNs deficient in α(1,3) fucosyltransferase IV and VII (FucT IV/VII−/−; Fig. 1 E; reference 14). These results further validate the binding specificity of the assay and indicate that CD44 is a physiological target of leukocyte FucTs. Fluid-phase binding of E-selectin to peripheral blood PMNs, an assay largely PSGL-1–dependent (10), was significantly reduced in CD44−/− PMNs compared with wild-type control PMNs (Fig. 1, F and G). Taken together, these data clearly indicate that CD44 from PB neutrophils binds to E-selectin.
E-selectin closely cooperates with P-selectin in promoting PMN–endothelial interactions and PMN extravasation; mice lacking both endothelial selectins have much more severe defects than either singly deficient animals (15–18). E-Selectin–deficient mice display increased leukocyte rolling velocities, suggesting that it is critical in forming stronger adhesion bonds between endothelial cells and PMNs (19). However, leukocyte rolling velocities were not altered in PSGL-1−/− mice (10), suggesting that another leukocyte ESL mediates the slow rolling. We intercrossed CD44−/− with PSGL-1−/− mice to characterize the function of CD44 in E-selectin–mediated leukocyte-endothelial interactions. Double knockout (DKO) animals were viable, fertile, and displayed a significant increase in circulating leukocytes, including PMNs, monocytes and eosinophils (Table S1, available at http://www.jem.org/cgi/content/full/jem.20042014/DC1). We subjected age-matched male wild-type, CD44−/−, PSGL-1−/−, and DKO mice to intravital microscopic examination of TNF-α–treated cremaster muscle to explore the ability of CD44 to interact with E-selectin in vivo. Consistent with previous studies (9, 10) and with the notion that P-selectin mediates most rolling activity in inflamed venules, the rolling fraction was significantly reduced in PSGL-1−/− mice (Fig. 2 A and Table S2). Residual rolling activity in PSGL-1−/− mice was further reduced by ∼30% in mice lacking both PSGL-1 and CD44, although this did not reach the threshold statistical significance in a multigroup analysis of variance (Fig. 2 A). Although the average velocity of rolling leukocytes was similar between PSGL-1−/− and wild-type mice (Fig. 2, B and C and Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20042014/DC1), leukocyte rolling velocities were significantly increased in CD44−/− mice (P < 0.0001). Moreover, rolling velocities were further increased when both CD44 and PSGL-1 were absent (Fig. 2 B, P = 0.0002, comparison CD44−/− with DKO group). In keeping with the velocity analyses, the median transit times of rolling leukocytes were much shorter in CD44−/− than wild-type or PSGL-1−/− mice, and even more rapid in DKO mice (Fig. 2 D). Because CD44 is known to interact with HA, we treated wild-type mice with hyaluronidase at a dose previously shown to alter CD44-mediated lymphocytes recruitment (11). Hyaluronidase treatment neither influenced the leukocyte rolling flux nor affected leukocyte rolling velocities and transit times (Fig. 2 and Fig. S4). Thus, these data indicate that CD44 binding to E-selectin and not HA primarily controls the velocity of rolling, and that PSGL-1 can also contribute to this activity when CD44 is absent.
Figure 2.

Intravital microscopy of TNF-α–stimulated cremaster muscle venules. Leukocyte behavior in cremasteric venules was recorded between 150 and 210 min after TNF-α administration for off-line analyses. (A) Rolling flux fraction in wild-type (WT, n = 63 venules), WT treated with hyaluronidase (WT + Hase, n = 51), CD44−/− (n = 79), PSGL-1−/− (n = 50), and DKO (n= 59) mice. (B–D) The velocity of 2,610 rolling leukocytes from the five groups of mice was measured over 2 s (also see Fig. S4). (B) Mean rolling velocities. (C) Cumulative histograms of rolling velocities. (D) Cumulative histograms of transit times calculated per 100 μm of venule segment. Five-group comparisons were analyzed by one-way ANOVA with Bonferroni/Dunn post hoc test. **, P < 0.001 (1% risk level) compared with WT group or between indicated two groups.
To investigate whether CD44 binding to E-selectin can mediate the extravasation of PMNs into inflammatory sites, we injected mice with thioglycollate, a chemical that induces a severe peritoneal inflammation. In this model, PMN recruitment is initially (0–4 h) P-selectin dependent but subsequently (8 h) requires E-selectin expression (15, 17). Previous studies have revealed no significant defect in PMN recruitment 8 h after thioglycollate injection in PSGL-1−/− mice (9). Wild-type, CD44−/−, PSGL-1−/−, and DKO mice were thus treated with thioglycollate, and the number of PMNs in the peritoneal exudate was evaluated 8 h after injection. Although there was no significant reduction in PMN counts in CD44−/− and PSGL-1−/− mice, the recruitment of PMNs was significantly reduced (by 44%, P = 0.005) in DKO mice (Fig. 3 A). Because thioglycollate may not reproduce physiological inflammation, we also evaluated PMN extravasation elicited by the staphylococcal enterotoxin A (SEA) in a preformed air pouch model (20). In preliminary experiments using P- and E-selectin–deficient mice, we ascertained that SEA-mediated PMN recruitment was selectin dependent (unpublished data). We then instilled SEA in dorsal skin pouches of mice from the four genotypes to assess the contribution of CD44 and PSGL-1 in this model. We found a severe reduction in the extravasation of PMNs 6 h after SEA injection in DKO mice compared with wild-type controls (77% reduction, P = 0.006), whereas the numbers of extravasated PMNs were not significantly altered in either singly-deficient mice (Fig. 3 B). To exclude further the possibility that the reduced recruitment of PMNs observed in DKO mice was due to CD44 binding to HA, we repeated the thioglycollate-induced extravasation experiments in PSGL-1−/− mice treated with hyaluronidase (20 U i.v.) or vehicle. Hyaluronidase treatment did not significantly alter PMN recruitment (Fig. 3 C). Although we cannot completely rule out the possibility that extravascular HA may have remained available for PMN migration, the results from these two models strongly suggest that CD44 is an ESL that cooperates with PSGL-1 in PMN extravasation into inflamed sites.
Figure 3.

Neutrophil extravasation into inflammatory sites. (A) Thioglycollate-induced peritonitis. Extravasated PMNs were determined 8h after the i.p. injection of thioglycollate (n = 7). (B) SEA-induced inflammation model. Extravasated PMNs were quantified 6 h after instillation of SEA into preformed skin pouches (n = 6). Data are represented by box-and-whisker plots wherein each box represents an interquartile range (central 50%), the median is shown by the horizontal lines, and vertical lines show the full range of data points. Four-group comparisons were analyzed by one-way ANOVA with Bonferroni/Dunn post hoc test. *, P < 0.0083 (5% risk level) compared with WT group. (C) Hyaluronidase treatment does not affect PMN recruitment. PSGL-1−/− mice were injected i.v. with 20 U of hyaluronidase or PBS before i.p. injection of thioglycollate. Each circle represents the value of an individual PSGL-1−/− mouse. P = 0.24.
Because CD44 is expressed on both leukocyte and endothelial cells, we wished to clarify further whether CD44 deficiency on PMNs was sufficient to account for impaired PMN extravasation. We generated chimeric mice by transplantation of a mixture of PSGL-1−/−CD44+/+ and PSGL-1−/−CD44−/− BM-nucleated cells into lethally irradiated wild-type recipients. 6 wk after transplantation, >96% Gr-1hi leukocytes did not express PSGL-1. Peritoneal inflammation was then induced by thioglycollate for 8 h, and the ratios of PSGL-1−/−CD44+/+ PMNs over DKO PMNs in blood and peritoneal exudates were assessed by FACS (Fig. 4 A). In this competitive setting, we found that PSGL-1−/−CD44+/+ PMNs were preferentially recruited in the peritoneum (an approximate twofold increase) over those that did not express PSGL-1 and CD44 (Fig. 4 B), suggesting that neutrophil rather than endothelial CD44 plays a critical role for migration into inflammatory sites.
Figure 4.

Competitive recruitment of neutrophils into inflammatory sites. Wild-type recipient mice were lethally irradiated and transplanted with mixture of BM cells from PSGL-1−/−CD44+/+ and PSGL-1−/−CD44−/− mice. 6 wk after transplantation, PMNs were elicited by thioglycollate for 8 h. Blood and peritoneal exudates were analyzed by FACS. (A) Representative dot plots for CD44 and PSGL-1 in Gr-1hi cells from blood and peritoneum. (B) Ratios PSGL-1−/−CD44+/+ over PSGL-1−/−CD44−/− Gr-1hi cells. Bars show mean values. n = 4, *, P < 0.01.
Our data suggest that ESLs may have specialized functions. It is interesting to speculate that this may be controlled by the spatial distribution on the cell surface. For example, PSGL-1 is primarily a tethering molecule localized on the tip of microvilli (21), whereas we show here that CD44, a receptor located on the cell body (22, 23), primarily controls rolling velocity. It is notable that β2 integrins, which can also mediate slow rolling (24), are located on the cell body. Thus, our data are consistent with the notion that adhesion receptors located on microvilli may determine tethering efficiency but not rolling velocity (23, 25). As suggested by the partial defect in leukocyte rolling and recruitment of DKO mice, other functional ESLs exist on PMNs. The various ESLs may exert distinct functions in E-selectin–dependent adhesive and migratory activities. Further studies are needed to ascertain in vivo functions of major candidate ESL glycoproteins, including ESL-1 (expressed on mouse microvilli of myeloid cells; 7) and L-selectin (candidate ESL on human PMNs; 22, 26).
To investigate whether human neutrophil CD44 was a functional ESL, we purified peripheral blood PMNs from healthy donors and extracted PMN-derived human CD44 from cell lysates for the E-selectin binding assay. To control for binding specificity, we used sialidase-treated PMNs from the same donors and PMNs from a patient with leukocyte adhesion deficiency type II (LADII), characterized by a complete deficit in functional selectin ligands due to G588 nucleotide deletion in the GDP–fucose transporter gene (27). As shown in Fig. 5 A, healthy PMN-derived CD44 immobilized on beads bound to soluble E-selectin, and binding was abrogated when cells were treated with sialidase before the preparation of the lysates. In contrast, CD44 extracted from LADII PMNs did not appreciably bind to E-selectin (Fig. 5 B), despite normal expression of CD44 (Fig. 5, insets). Interestingly, a short incubation of LADII PMNs with recombinant FucTVI enabled CD44 to bind normally to E-selectin, indicating that CD44 on these cells possesses terminal N-glycans that can be modified by extracellular FucT activity (Fig. 5 C). The rescue of selectin ligand activity by treatment with FucTVI may represent an effective therapy to prevent infectious episodes associated with this syndrome.
Figure 5.
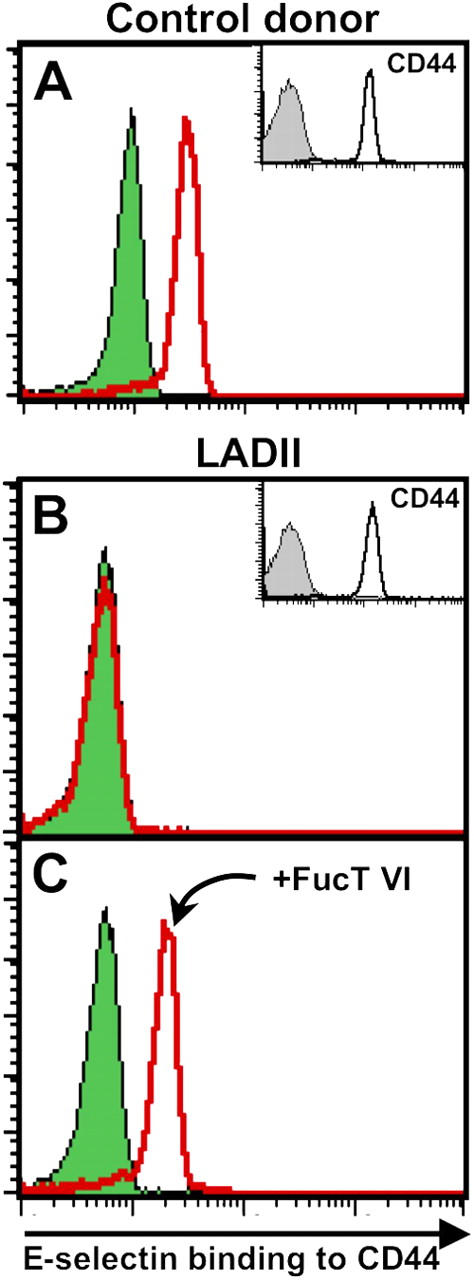
Human neutrophil CD44 binds to E-selectin. PMNs were purified from healthy donors or from a patient with LADII. PMNs were treated with sialidase or incubated with vehicle. (A) CD44 derived from healthy PMNs binds to E-selectin (empty red histogram), and binding is abrogated by sialidase (green-filled histogram). (B) In contrast, there is no difference in E-selectin binding to immunopurified CD44 between sialidase-treated and sham-treated LADII PMNs. (C) Incubation of LADII PMNs with recombinant FucTVI (20 mU/ml, 40 min at 37°C) and GDP-fucose (1 mM) restores the ability of human CD44 to bind to E-selectin.
In summary, these data clearly indicate that CD44 is a physiological ESL on mature human and mouse myeloid cells. CD44 is widely expressed in multiple cell types but only hematopoietic cells bind to E-selectin, indicating that the affinity for E-selectin is regulated by cell-specific posttranslational modifications. Selective binding to E-selectin (myeloid cells) or HA (activated T cells) endows CD44 with pivotal functions at the nexus of innate and adaptative immunity.
Materials and Methods
Cell lines and E-selectin/IgM chimeric protein
The 32D mouse myeloid progenitor cell line (American Type Culture Collection) was cultured in RPMI containing 10% FBS (Hyclone) and 10% Wehi-3B (a murine IL-3–secreting cell line) conditioned medium. Differentiation of 32D cells was induced with 10 ng/ml recombinant human G-CSF (R&D Systems) for 4 d. Mouse BM-derived stromal cell line MS-5 was cultured in αMEM containing 10% FBS, and mouse endothelial cell line bEnd.3 (American Type Culture Collection) was cultured in DMEM containing 10% FBS.
Murine E-selectin/IgM chimera was produced by the transfection of 293T cells with E-selectin/IgM DNA vector using Lipofectamine 2000 (Invitrogen). Saturating concentrations of culture supernatants were used for the fluid-phase selectin binding assay.
Glycan analyses
32D cells or purified PMNs were treated with 150 mU sialidase from Arthrobacter ureafaciens (Roche), or with 20 μg OSGE (partially purified by membrane diafiltration and isoelectric point precipitation from Pasteurella haemolytica; Cedarlane) for 1 h at 37°C in HBSS media containing 20 mM Hepes and 0.1% human serum albumin (Baxter). Both treatments completely eliminated P-selectin ligands. Inhibition of N-glycosylation was achieved by culture of sialidase-treated 32D cells in the presence or absence of 15 μg/ml tunicamycin (Calbiochem) for 36 h.
Preparation of murine and human neutrophils
Age-matched (6–12 wk) C57BL/6 (NCI, Frederick Cancer Research and Developmental Center) and FucT IV/VII−/− mice (14; provided by John B. Lowe, University of Michigan Medical School, Ann Arbor, MI) were used as BM and blood donors. In some experiments, lymphopenic NOD/SCID mice were used as blood donor to obtain greater PMN purity. Expression of ESLs and binding of E-selectin to CD44 were similar between C57BL/6- and NOD/SCID-derived PMNs. BM cells were harvested by flushing femora in RPMI using 21-gauge needle and blood was harvested by retroorbital sampling using heparinized capillary tubes. For blood PMNs purification, RBCs were depleted by blood sedimentation in 0.1% methylcellulose for 30 min at room temperature. Cells remaining in the supernatant were washed and resuspended in PBS containing 0.5% BSA and 2 mM EDTA, and PMNs separated by overlaying on a Percoll gradient (65% in HBSS). Contaminating RBCs were lysed with 0.8%NH4Cl.
Human neutrophils were purified from human venous blood as described previously (27). Human blood samples were obtained in accordance with protocols approved by the Internal Review Board of Mount Sinai.
Flow cytometry and E-selectin binding assay
Cells were stained by incubation with 10 μg/ml of a biotin-labeled anti-CD44 antibody (clone IM7; BD Biosciences) or control antibody followed by incubation with CY5-conjugated streptavidin (Jackson ImmunoResearch Laboratories), and washed in PBS before analysis by flow cytometry. For the fluid-phase selectin binding assay, cells or CD44-coated beads were incubated with E-selectin/IgM chimera followed by incubation with Cy5-conjugated anti–human IgM antibody (1:50 dilution; Jackson ImmunoResearch Laboratories) as described previously (28). Control samples were stained in the presence of 5 mM EDTA. A similar procedure was followed for staining with the P-selectin/IgM chimera. For staining of murine PMNs, 50 μl of blood or 105 BM mononuclear cells were incubated with FITC-conjugated anti–Gr-1 antibody (BD Biosciences) and then labeled with the E-selectin chimera as described above. PMNs were gated on the basis of high Gr-1 expression. All incubations were performed at 6°C for 15 min. Samples were analyzed using a FACSCalibur flow cytometer and the CellQuest software (Becton Dickinson).
Preparation of immunomagnetic beads for E-selectin binding analyses
Lysates were prepared by incubation of cells in lysis buffer (50 mM Tris-HCl, pH 7.5, 1% Triton X-100, 150 mM NaCl, and 1 mM CaCl2 plus 0.1 M PMSF and protease inhibitor cocktail; Sigma-Aldrich) for 30 min on ice and cell debris were removed by centrifugation at 14,000 rpm for 10 min at 4°C. Anti–rat IgG-coated beads (106 M-450 Dynabeads; Dynal) were incubated with 2.5 μg anti-CD44 (IM7), anti-αM integrin (M1/70 from American Type Culture Collection, purified from hybridoma supernatants) or rat IgG (Sigma-Aldrich) for 4 h at 4°C under rotation. Beads were then washed twice in cold lysis buffer and incubated overnight with the appropriate cell lysates (2 × 106 cells/106 beads) at 4°C under rotation. Beads were washed once in cold lysis buffer and once in cold RPMI containing 5% FBS and 0.05% NaN3 before fluid-phase E-selectin binding assay.
In vivo models of leukocyte recruitment
Leukocyte-endothelial interactions were assessed using intravital microscopy of the cremaster muscle as described previously (29). More detailed information is available at http://www.jem.org/cgi/content/full/jem.20042014/DC1. PMN recruitment in the thioglycollate-induced peritonitis model was assessed as described (15, 17). In some experiments, mice were injected i.v. with 20 U of hyaluronidase (Sigma-Aldrich) or control PBS before injection of thioglycollate. SEA-induced inflammation in air pouch was performed as described elsewhere (20). In brief, air pouches were raised on the dorsum by s.c. injections of 2.5 ml sterile air on days 0 and 3 and the pouch was allowed to form until day 6. On day 6, 10 μg of SEA (endotoxin <1 EU/mg; Toxin Technology) in 1 ml PBS was injected in the pouch. Mice were killed 6 h after SEA instillation, and the pouches were washed with 5 ml of PBS containing 1% BSA, 0.5 mM EDTA, and 10 U/ml heparin. Total cell numbers in the peritoneal and air pouch exudates were determined by hemocytometer. Differential leukocyte counts were determined from Wright-stained cytospin preparations. All animal experimental procedures were approved by the Animal Care and Use Committee of Mount Sinai.
Generation of PSGL-1/CD44 double-deficient mice
PSGL-1−/− mice were generated by gene targeting (9) and provided by Bruce Furie (Harvard Medical School, Boston, MA). CD44−/− mice were also generated by gene targeting (30) and purchased from The Jackson Laboratory. PSGL-1−/− mice were intercrossed with CD44−/− mice to generate double-heterozygous animals. These doubly heterozygous mice were then bred to yield wild-type control, CD44−/−, PSGL-1−/−, and double-deficient mice. The genotypes were determined by PCR using primers and conditions detailed in supplemental materials. Separate colonies of each genotype were then expanded to yield age- and sex-matched experimental mice with similar mixed background.
Generation of chimeric mice and competitive recruitment assay
Wild-type–recipient mice were lethally irradiated (12 Gy, split dose) and transplanted with 1.5 × 106 PSGL-1−/− BM-nucleated cells and 1.5 × 106 PSGL-1/CD44 double deficient (DKO) BM-nucleated cells. 6 wk after transplantation, engraftment was confirmed by FACS. Leukocytes from thioglycollate-induced exudates were stained with FITC-conjugated anti–Gr-1 antibody (BD Biosciences), PE-conjugated anti-CD44 antibody (KM201; Southern Biotechnologies Associates), and biotin–anti–PSGL-1 antibody (clone 4RA10; gift of Dr. Dietmar Vestweber, The Institute of Cell Biology, University of Münster, Münster, Germany), followed by Cy5-conjugated streptavidin (Jackson ImmunoResearch Laboratories).
Online supplemental material
Online supplemental materials contain detailed information about genotyping of mutant mice, intravital microscopy studies, and statistical analyses. Figs. S1 and S2 further evaluate the specificity of E-selectin binding to CD44-coated beads. Fig. S3 shows the effect of tunicamycin treatment on O-glycosylation. Fig. S4 shows the leukocyte rolling velocity distributions in TNF-α–treated cremaster muscle venules. Table S1 shows peripheral blood counts and Table S2 details venular hemodynamic parameters of intravital microscopy experiments. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20042014/DC1.
Acknowledgments
We thank B. Furie for providing PSGL-1−/− mice, J. B. Lowe for FucT IV/VII−/− mice, Dietmar Vestweber for the 4RA10 mAb, and C. Cunningham-Rundles (Mount Sinai School of Medicine) for procuring blood samples of the LADII patient. We also thank Elaine Chiang for reviewing the manuscript.
Supported by a Fellowship from National Blood Foundation (A. Hidalgo), a training grant from the National Institutes of Health T32 DK07792 (J. Chang), and R01 grants DK56638 and HL69438 (P.S. Frenette).
The authors have no conflicting financial interests.
Abbreviations used: DKO, double knockout; ESL, E-selectin ligand; FucT, fucosyltransferase; HA, hyaluronic acid; LADII, leukocyte adhesion deficiency type II; OSGE, O-sialoglycoprotein endopeptidase; PB, peripheral blood; PMN, polymorphonuclear neutrophil; PSGL1; P-selectin glycoprotein ligand-1; SEA, staphylococcal enterotoxin A.
Y. Katayama, A. Hidalgo, and J. Chang contributed equally to this work.
Y. Katayama's present address is Department of Hematology, Oncology and Respiratory Medicine, Okayama University Graduate School of Medicine and Dentistry, 2-5-1 Shikata-cho, Okayama 700-8558, Japan.
References
- 1.Lowe, J.B. 2003. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr. Opin. Cell Biol. 15:531–538. [DOI] [PubMed] [Google Scholar]
- 2.Ley, K. 2003. The role of selectins in inflammation and disease. Trends Mol. Med. 9:263–268. [DOI] [PubMed] [Google Scholar]
- 3.Steegmaier, M., A. Levinovitz, S. Isenmann, E. Borges, M. Lenter, H.P. Kocher, B. Kleuser, and D. Vestweber. 1995. The E-selectin-ligand ESL-1 is a variant of a receptor for fibroblast growth factor. Nature. 373:615–620. [DOI] [PubMed] [Google Scholar]
- 4.Jones, W.M., G.M. Watts, M.K. Robinson, D. Vestweber, and M.A. Jutila. 1997. Comparison of E-selectin-binding glycoprotein ligands on human lymphocytes, neutrophils, and bovine gamma delta T cells. J. Immunol. 159:3574–3583. [PubMed] [Google Scholar]
- 5.Montoya, M.C., K. Holtmann, K.R. Snapp, E. Borges, F. Sanchez-Madrid, F.W. Luscinskas, G. Kansas, D. Vestweber, and M.O. de Landazuri. 1999. Memory B lymphocytes from secondary lymphoid organs interact with E-selectin through a novel glycoprotein ligand. J. Clin. Invest. 103:1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dimitroff, C.J., J.Y. Lee, S. Rafii, R.C. Fuhlbrigge, and R. Sackstein. 2001. CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J. Cell Biol. 153:1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vestweber, D., and J.E. Blanks. 1999. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 79:181–213. [DOI] [PubMed] [Google Scholar]
- 8.McEver, R.P. 2002. Selectins: lectins that initiate cell adhesion under flow. Curr. Opin. Cell Biol. 14:581–586. [DOI] [PubMed] [Google Scholar]
- 9.Yang, J., T. Hirata, K. Croce, G. Merrill-Skoloff, B. Tchernychev, E. Williams, R. Flaumenhaft, B.C. Furie, and B. Furie. 1999. Targeted gene disruption demonstrates that P-selectin glycoprotein ligand 1 (PSGL-1) is required for P-selectin-mediated but not E-selectin-mediated neutrophil rolling and migration. J. Exp. Med. 190:1769–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia, L., M. Sperandio, T. Yago, J.M. McDaniel, R.D. Cummings, S. Pearson-White, K. Ley, and R.P. McEver. 2002. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J. Clin. Invest. 109:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeGrendele, H.C., P. Estess, and M.H. Siegelman. 1997. Requirement for CD44 in activated T cell extravasation into an inflammatory site. Science. 278:672–675. [DOI] [PubMed] [Google Scholar]
- 12.Teder, P., R.W. Vandivier, D. Jiang, J. Liang, L. Cohn, E. Pure, P.M. Henson, and P.W. Noble. 2002. Resolution of lung inflammation by CD44. Science. 296:155–158. [DOI] [PubMed] [Google Scholar]
- 13.Gunthert, U., M. Hofmann, W. Rudy, S. Reber, M. Zoller, I. Haussmann, S. Matzku, A. Wenzel, H. Ponta, and P. Herrlich. 1991. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 65:13–24. [DOI] [PubMed] [Google Scholar]
- 14.Homeister, J.W., A.D. Thall, B. Petryniak, P. Maly, C.E. Rogers, P.L. Smith, R.J. Kelly, K.M. Gersten, S.W. Askari, G. Cheng, et al. 2001. The alpha(1,3)fucosyltransferases FucT-IV and FucT-VII exert collaborative control over selectin-dependent leukocyte recruitment and lymphocyte homing. Immunity. 15:115–126. [DOI] [PubMed] [Google Scholar]
- 15.Mayadas, T.N., R.C. Johnson, H. Rayburn, R.O. Hynes, and D.D. Wagner. 1993. Leukocyte rolling and extravasation are severely compromised in P-selectin deficient mice. Cell. 74:541–554. [DOI] [PubMed] [Google Scholar]
- 16.Labow, M.A., C.R. Norton, J.M. Rumberger, K.M. Lombard-Gillooly, D.J. Shuster, J. Hubbard, R. Bertko, P.A. Knaack, R.W. Terry, M.L. Harbison, et al. 1994. Characterization of E-selectin-deficient mice: demonstration of overlapping function of the endothelial selectins. Immunity. 1:709–720. [DOI] [PubMed] [Google Scholar]
- 17.Frenette, P.S., T.N. Mayadas, R. H., R.O. Hynes, and D.D. Wagner. 1996. Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell. 84:563-574. [DOI] [PubMed] [Google Scholar]
- 18.Bullard, D.C., E.J. Kunkel, H. Kubo, M.J. Hicks, I. Lorenzo, N.A. Doyle, C.M. Doerschuk, K. Ley, and A.L. Beaudet. 1996. Infectious susceptibility and severe deficiency of leukocyte rolling and recruitment in E-selectin and P-selectin double mutant mice. J. Exp. Med. 183:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunkel, E.J., and K. Ley. 1996. Distinct phenotype of E-selectin-deficient mice. E-selectin is required for slow leukocyte rolling in vivo. Circ. Res. 79:1196–1204. [DOI] [PubMed] [Google Scholar]
- 20.Diener, K., P. Tessier, J. Fraser, F. Kontgen, and S.R. McColl. 1998. Induction of acute inflammation in vivo by staphylococcal superantigens I: leukocyte recruitment occurs independently of T lymphocytes and major histocompatibility complex class II molecules. Lab. Invest. 78:647–656. [PubMed] [Google Scholar]
- 21.Moore, K.L., K.D. Patel, R.E. Bruehl, F. Li, D.A. Johnson, H.S. Lichenstein, R.D. Cummings, D.F. Bainton, and R.P. McEver. 1995. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 128:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picker, L.J., R.A. Warnock, A.R. Burns, C.M. Doerschuk, E.L. Berg, and E.C. Butcher. 1991. The neutrophil selectin LECAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140. Cell. 66:921–933. [DOI] [PubMed] [Google Scholar]
- 23.von Andrian, U.H., S.R. Hasslen, R.D. Nelson, S.L. Erlandsen, and E.C. Butcher. 1995. A central role for microvillous receptor presentation in leukocyte adhesion under flow. Cell. 82:989–999. [DOI] [PubMed] [Google Scholar]
- 24.Dunne, J.L., C.M. Ballantyne, A.L. Beaudet, and K. Ley. 2002. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 99:336–341. [DOI] [PubMed] [Google Scholar]
- 25.Stein, J.V., G. Cheng, B.M. Stockton, B.P. Fors, E.C. Butcher, and U.H. von Andrian. 1999. L-selectin-mediated leukocyte adhesion in vivo: microvillous distribution determines tethering efficiency, but not rolling velocity. J. Exp. Med. 189:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green, C.E., D.N. Pearson, R.T. Camphausen, D.E. Staunton, and S.I. Simon. 2004. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J. Immunol. 172:7780–7790. [DOI] [PubMed] [Google Scholar]
- 27.Hidalgo, A., S. Ma, A.J. Peired, L.A. Weiss, C. Cunningham-Rundles, and P.S. Frenette. 2003. Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood. 101:1705–1712. [DOI] [PubMed] [Google Scholar]
- 28.Hidalgo, A., L.A. Weiss, and P.S. Frenette. 2002. Functional selectin ligands mediating human CD34(+) cell interactions with bone marrow endothelium are enhanced postnatally. J. Clin. Invest. 110:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turhan, A., L.A. Weiss, N. Mohandas, B.S. Coller, and P.S. Frenette. 2002. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc. Natl. Acad. Sci. USA. 99:3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Protin, U., T. Schweighoffer, W. Jochum, and F. Hilberg. 1999. CD44-deficient mice develop normally with changes in subpopulations and recirculation of lymphocyte subsets. J. Immunol. 163:4917–4923. [PubMed] [Google Scholar]
- 31.Ponta, H., L. Sherman, and P.A. Herrlich. 2003. CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 4:33–45. [DOI] [PubMed] [Google Scholar]