

Abstract
The mechanisms through which regulatory T cells accumulate in lymphoid organs of tumor-bearing hosts remain elusive. Our experiments indicate that the accumulation of CD4+CD25+ regulatory T cells (T reg cells) expressing FoxP3 and exhibiting immunosuppressive function originates from the proliferation of naturally occurring CD25+ T cells and requires signaling through transforming growth factor (TGF)–β receptor II. During tumor progression, a subset of dendritic cells (DCs) exhibiting a myeloid immature phenotype is recruited to draining lymph nodes. This DC subset selectively promotes the proliferation of T reg cells in a TGF-β–dependent manner in mice and rats. Tumor cells are necessary and sufficient to convert DCs into regulatory cells that secrete bioactive TGF-β and stimulate T reg cell proliferation. In conclusion, tumor expansion can stimulate T reg cells via a specific DC subset.
CD4+CD25+ regulatory T cells (T reg cells) were initially defined as a subpopulation of suppressor T cells that mediate immune tolerance by suppressing autoreactive T cells (1, 2). They represent 1–3% of CD4+ T cells in humans and 5–10% of CD4+ T cells in rodents and express cell surface molecules associated with activated/memory cells (CD25, CD45Rblow, CD62L, CD103, CTLA4, and GITR), as well as the pathognomonic transcription factor protein Foxp3 (3). CD4+CD25+ T reg cells can recognize self-antigens more efficiently than other T cell subsets (4).
T reg cells require TCR ligation and IL-2 to become activated. Once they have been activated in an antigen-specific fashion, they can mediate immune suppression without antigen specificity (5–7). Under normal circumstances, T reg cells are anergic; i.e., they fail to proliferate or to produce IL-2 in response to conventional T cell stimuli. This anergy can be broken by the addition of high doses of exogenous IL-2, anti-CD28, or exposure to mature DCs (5–9). However, it has also been suggested that mature DCs can inhibit the immunosuppressive function of T reg cells (10). In vitro, anergy is the default state of naturally occurring T reg cells, to which they revert after withdrawal of the anergy-breaking stimulus (5). In vivo, however, T reg cell anergy has been controversial (11, 12).
In human neoplasia, T reg cells accumulate in tumors, draining LNs (DLNs), and the blood stream (13–17), and similar observations have been made in rodent models of cancer (18). However, the mechanisms leading to T reg cell accumulation in tumor-bearing hosts are poorly understood. We investigated the cellular and molecular requirements for T reg cell proliferation during tumor progression in rats bearing colon tumors (18) and in mice with melanoma (19). We found that a subset of DCs exposed to tumor cells acquired the capacity to secrete TGF-β and to stimulate the expansion of naturally occurring T reg cells in vivo.
Results
Proliferation of FOXP3+CD4+CD25+ T reg cells during tumor progression in mice and rats
CD4+CD25+ T cells increase in number in LNs draining syngeneic B16F10 melanoma cells. Both the percentage (Fig. 1 A) and the absolute numbers of CD4+CD25+ T cells significantly (P < 0.02) increased by day 15 in DLNs (9.3 ± 0.7 × 104 cells, n = 6) as compared with control LNs from tumor-free mice (TFM; 5.2 ± 0.6 × 104 cells, n = 6). Irrespective of the tumor status, ∼90% of such CD4+CD25+ LN cells expressed FOXP3, indicating that they are indeed T reg cells (Fig. 1 A). Accordingly, CD4+CD25+ splenic mouse T lymphocytes (which also expressed FOXP3; unpublished data) exhibited the functional characteristics of regulatory T cells (as opposed to those of activated effector T cells) because they inhibited the proliferation of alloreactive T cells (not depicted) and that of concanavalin A (ConA)–stimulated syngeneic T lymphocytes (Fig. 1 B). Next, we measured the incorporation of 5-bromo-2′-deoxyuridine (BrdU) into T reg cells residing in tumor beds and lymphoid organs of TFM versus tumor-bearing mice (TBM). In TBM, T reg cells proliferated in tumor beds and DLNs, but there was no increase in T reg cell proliferation in the spleen (Fig. 1 C). CD4+CD25+ T cells also accumulated in DLNs of BD-IX rats bearing advanced (28-d) PROb colon cancers (Fig. 1 D). These CD4+CD25+ cells also expressed FOXP3, as measured by RT-PCR (Fig. 1 D), and inhibited mixed lymphocyte reactions (not depicted) and ConA-induced T cell proliferation in vitro (Fig. 1 E). Proliferation measurements in vivo using BrdU again revealed that T reg cells proliferated in tumor beds, DLNs, and (as opposed to mice) in the spleen of tumor-bearing rats (TBR; Fig. 1 F). Thus, CD4+CD25+ cells accumulating in the tumors and in tumor DLNs from mice and rats do so by proliferation and share the biochemical and functional characteristics of T reg cells. We therefore assume that the natural history and functional role of such CD4+CD25+ cells is equivalent in both rodent species.
Figure 1.
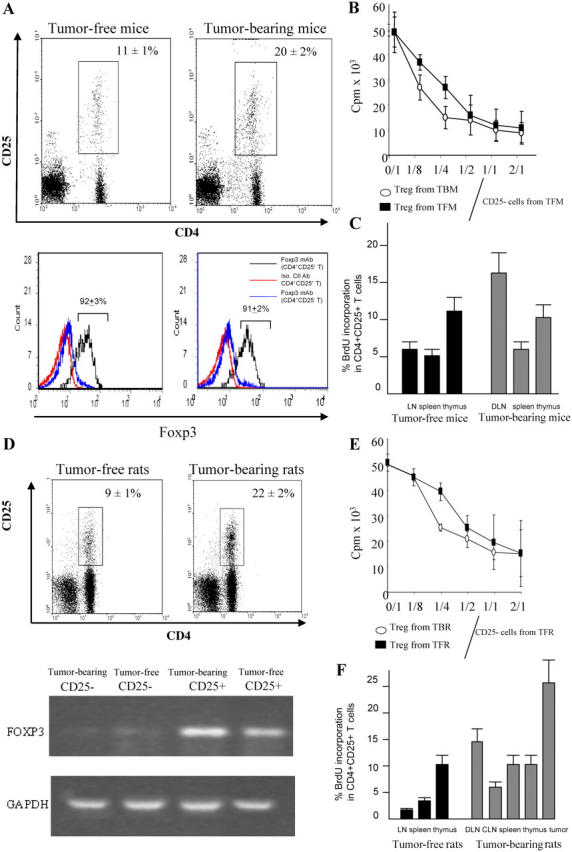
Accumulation and proliferation of T reg cells in lymphoid organs of tumor-bearing rodents. (A) CD4+CD25+ cells in DLNs from C57BL6 mice bearing B16F10 tumors. The expression of CD4 and CD25 was analyzed by flow cytometry on gated CD3+ T cells isolated from inguinal DLNs of 15-d-old B16F10 TBM or tumor-free controls. These cells were FACS purified and then subjected to intracellular staining for the detection of FOXP3, whereas background levels were determined with an isotype control (bottom). Percent values represent the mean ± SEM (n = 6 distinct animals). (B) Inhibitory effect of T reg cell splenocytes. 105 splenocytes harvested from a TFM were depleted in CD25+ cells and stimulated with 2 μg/ml of ConA. CD25+ T cells purified from TFM or TBM were added to the culture. Proliferation was determined after 3 d of culture using [3H]thymidine incorporation. Values represent the mean ± SEM of triplicates. (C) Proliferation of T reg cells in TBM. C57BL6 mice bearing B16F10 tumors (as in A) or TFM were injected with BrdU, and the frequency of BrdU+ cells was determined among CD3+CD4+CD25+ cells by flow cytometry. Values represent the mean ± SEM (n = 6). (D) Immunophenotyping of axillary DLNs from rats bearing 28-d-old PROb tumors or tumor free control LNs. Percent values represent the mean ± SEM (n = 6). RT-PCR analysis of FOXP3 expression in CD25+ and CD25− T cells from TBR and TFR. Data are representative of three experiments. (E) Antiproliferative action of T reg cells from rats. The experiment was analogous to that shown in B, with the difference that all cells were derived from BD-IX rats instead of C57BL/6 mice. (F) Proliferation of T reg cells in vivo in TBR. BD-IX rats bearing PROb tumors (as in D) or TFR were injected with BrdU, and the frequency of BrdU+ cells was determined among CD3+CD4+CD25+ cells as in C. Values represent the mean ± SEM (n = 6).
Subversion of immunosurveillance by endogenous and exogenous CD4+CD25+ T reg cells
We took advantage of an immunogenic colon carcinoma cell line (REGb) derived from PROb that underwent spontaneous regression when inoculated into immunocompetent BD-IX rats (18). However, when REGb-inoculated rats were adoptively transferred with T reg cells (but not with conventional CD25− T cells), they exhibited vigorous tumor growth. This subversion of immunosurveillance was obtained with T reg cells purified from tumor-free rats (TFR) or TBR (Fig. 2 A). Thus, T reg cells from both TBR and TFR were indistinguishable in their immunosuppressive activity (on a per-cell basis; Fig. 2 A), whereas they were more abundant in number in TBR (see above; Fig. 1). If T cells from rats immunized with live REGb tumor cells were cultured with DCs pulsed with the colon carcinoma cell line, they produced IFN-γ, and this IFN-γ production could be inhibited by addition of CD25+ T cells but not by addition of conventional CD25− T cells (Fig. 2 B). This corroborated that T reg cells were capable of inhibiting an antitumor immune response in vivo.
Figure 2.

Adoptive transfer of T reg cells into immunogenic tumor variants. (A) Adoptive transfer of T reg cells into rats bearing immunogenic REGb tumors promotes tumor outgrowth. 106 REGb cells were inoculated s.c. into the flank, and tumor kinetics were monitored. REGb cells were either injected alone or admixed with either 10 × 106 CD25+ or CD25− T cells derived from TFR or rats bearing 42-d-old PROb tumors. The results of a representative experiment containing five rats per group is shown, and statistical analyses were performed using Fisher's exact test at 95% confidence. Values represent the mean ± SEM. (B) CD25+ T cells abrogate antigen-driven IFN-γ secretion from effector T cells. 105 splenic T cells isolated from rats immunized against PROb cells were stimulated with 104 CD11c+ DCs purified from naive animals and admixed with lysates from 104 PROb cells. CD25−/+ T cells from tumor-bearing or naive animals were added to the DC/T cell co-culture. IFN-γ secretion in the culture supernatant was determined by ELISA after 48 h of co-culture. Values represent the mean ± SEM (n = 3). Asterisks indicate significant differences at a P < 0.05 confidence interval using Fisher's exact test.
Next, we determined whether the depletion of T reg cells stimulated tumor surveillance in vivo. When rats were inoculated with a progressive colon carcinoma cell line (PROb), they rapidly developed metastatic cancer. However, when T reg cells were depleted from PROb-inoculated rats using anti-CD25 mAb, tumor growth was attenuated (Fig. 3 A). These effects could be observed in a similar fashion using an ex vivo readout. Splenocytes recovered from animals bearing advanced PROb carcinomas failed to lyse PROb cells and failed to produce IFN-γ when co-cultured with DCs pulsed with PROb lysates. However, depletion of T reg cells from such splenocytes converted nonresponders to responders in both assays, tumor cell lysis, and IFN-γ secretion (Fig. 3, B and C). These data indicate that T reg cells were acting as endogenous immunosuppressors in vivo.
Figure 3.
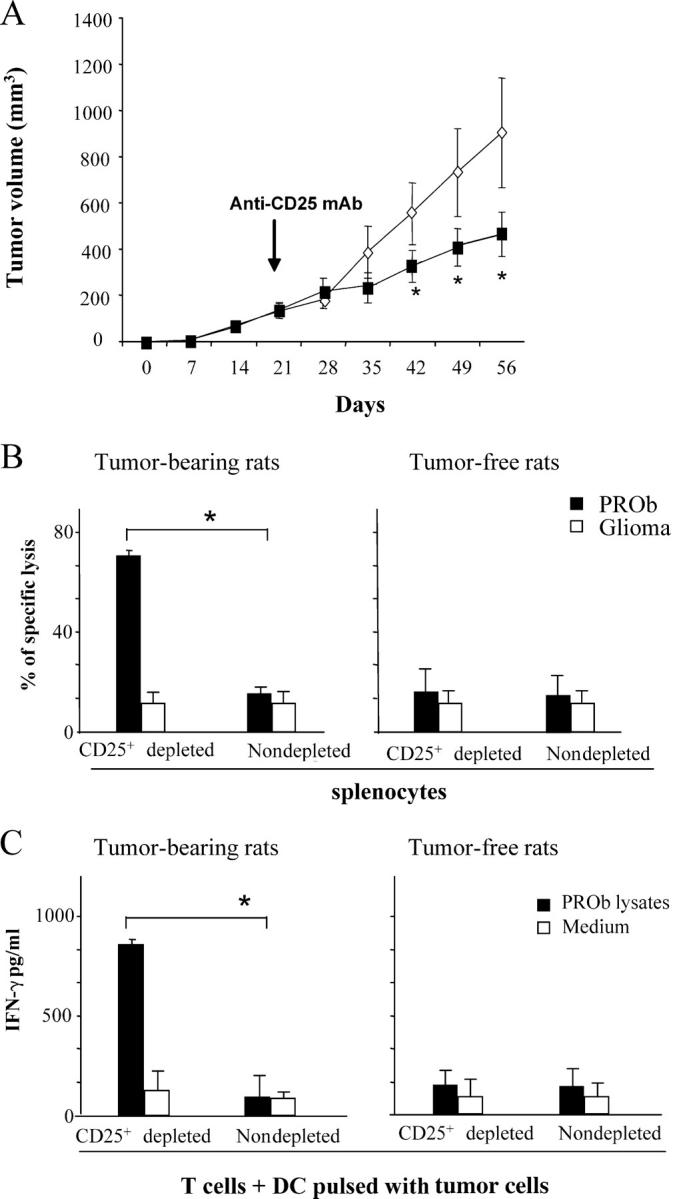
Depletion of T reg cells in nonimmunogenic tumors. (A) Depletion of CD25+ T reg cells promotes tumor regression in the PROb model. Rats bearing 21-d-old s.c. PROb tumors were injected with either 1 mg of anti–rat CD25 antibody (Clone OX-39; closed square) or control isotype (open diamond). The results of a representative experiment containing five rats per group is shown, and asterisks indicate that statistical analyses were performed using Fisher's exact test at 95% confidence. Values represent the mean ± SEM. (B and C) Depletion of CD25+ T reg cells from splenocytes derived from TBR restores tumor lysis and antigen–specific IFN-γ secretion. 104 PROb cells (or irrelevant glioma tumor cells) were cultured in the presence of 105 splenocytes depleted or not from CD25+ T reg cells and derived from TFR versus TBR at day 42. Cytotoxicity was determined with a crystal violet assay after 48 h of co-culture. (B) Increased tumor cell lysis on depletion of T reg cells. 105 T cells depleted or not from CD25+ T cells and derived from either TBR or naive rats were cultured in the presence of 104 CD11c+ DCs harvested from naive animals and pulsed with 104 PROb cell lysates. (C) Increased IFN-γ secretion on depletion of T reg cells. The accumulation of IFN-γ in the culture supernatant was determined by ELISA after 48 h of co-culture. Values represent the mean ± SEM (n = 3). Asterisks indicate significant differences (P < 0.05) using the Student's t test. This experiment has been repeated, yielding similar results.
Immature myeloid DCs (IMDCs) promote T reg cell proliferation in vitro and in vivo
We next characterized the APCs residing in lymphoid organs that could promote the proliferation of T reg cells isolated from tumor bearing hosts in vitro. Tumor cells that lacked MHC class II expression (even after in vitro stimulation with TNF-α and IFN-γ; unpublished data) could not directly induce the proliferation of T reg cells in mixed tumor/lymphocyte cultures (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050463/DC1). Extensive immunophenotyping of DLNs revealed that, beyond T reg cells, IMDCs accumulated in DLNs during tumor progression. Thus, CD11c+ cells coexpressing CD11b, low levels of MHC class II, and CD80 and CD86 co-stimulatory molecules selectively accumulated in the spleen of TBR (12.8 ± 1.6 vs. 3.6 ± 0.6 × 106 cells in TBR versus TFR; P < 0.05) and in the DLNs of TBM (86 ± 13 vs. 14 ± 3 × 103 in TBM vs. TFM, respectively; P < 0.001; Table I and Fig. S2). In BD-IX rats, these CD11c+CD11b+ cells coexpressed the DC lineage marker CD103 but not the macrophage marker CD163. 80% among purified CD11b+ splenocytes expressed CD11c and were therefore considered bona fide IMDCs.
Table I.
Phenotype of mice and rat IMDCs in lymphoid organs
| Percentage of IMDCs coexpressing indicated markers
|
||||||
|---|---|---|---|---|---|---|
| Number of IMDCs | CD11b | CD11c | MHC II | CD80 | CD86 | |
| % | % | % | % | % | ||
| Mouse DLN (CD11c+ cells) | ||||||
| Tumor bearing | 86 ± 13 × 103 | 87 ± 8 | >90 | 80 ± 6 | 26 ± 2a | 29 ± 8a |
| Tumor free | 14 ± 3 × 103 | 84 ± 7 | >90 | 90 ± 8 | 52 ± 4 | 61 ± 6 |
| Rat spleen (CD11b+ cells) | ||||||
| Tumor bearing | 12.8 ± 1.6 × 106 | >90 | 45 ± 6 | 21 ± 3 | 2 ± 1 | 5 ± 1 |
| Tumor free | 3.6 ± 0.6 × 106 | >90 | 52 ± 8 | 52 ± 4a | 19 ± 2a | 10 ± 1a |
Flow cytometry analysis of DC phenotypes in the LNs (mice, CD11c+ cells) and in the spleen (rats, CD11b+ cells) of tumor-free and tumor-bearing animals. Mean ± SD of three determinations.
P < 0.05 using the Student's t test.
In a further series of experiments, we harvested IMDCs from the spleen of TBR and TFR and evaluated their capacity to induce the proliferation of T reg cells. Splenic IMDCs from TBR induced the proliferation of T reg cells derived from both TFR or TBR in vitro (Fig. 4 A). No such effect was observed for any of the other APCs isolated from the spleen (Fig. S1). Moreover, splenic IMDCs from TBR selectively stimulated CD25+ (but not CD25−; unpublished data) T cells. Of note, splenic IMDCs purified from TFR failed to stimulate T reg cell proliferation in vitro (Fig. 4 A). The DC-mediated T reg cell proliferation did not revert the suppressive functions of T reg cells (Fig. 4 B). Next, we monitored the proliferation of ex vivo purified T reg cells in the presence of T cell–deprived splenocytes or of T cell–deprived splenocytes that were also depleted from CD11b+ or MHC class II+ cells. T reg cells only proliferated strongly after stimulation with splenocytes purified from TBR and not from tumor-free littermates. The proliferation of T reg cells elicited by non–T splenic cells was abolished on depletion of CD11b+ or MHC class II+ cells. Again, both T reg cells from TFR or TBR proliferated when co-cultured with IMDCs from TBR (Fig. 4 C). Collectively, these data indicate that T reg cells proliferate on stimulation with IMDCs in a MHC class II–restricted fashion, but only when such IMDCs are derived from tumor-bearing hosts. These data, which were obtained in the rat model (Fig. 4, A–C), could be completely reproduced in C57BL/6 mice carrying B16F10 melanomas. CD11c+ cells purified from tumor DLNs (but not from TFM) elicited the proliferation of T reg cells but not that of CD4+CD25− T cells (Fig. 4 D).
Figure 4.

DCs from tumor bearers promote T reg cell proliferation in vitro. (A) Increased T reg cell proliferation induced by DCs from TBR. 105 CD25+ T cells and CD11b+ DCs were isolated from the spleen of either TFR or TBR and co-cultured alone or together at a 1:1 ratio for 5 d before pulsing with [3H]thymidine. (B) T reg cells maintain their inhibitory potential after stimulation by DCs from TBR. Spleen cells from a BD-IX rat were cultured for 5 d with mitomycin C–treated spleen cells from a Wistar rat, and BrdU was added during the last 12 h of the mixed lymphocyte culture before measuring BrdU incorporation. CD25+ T cells were isolated from tumor-bearing BD-IX rats and added at a ratio of 1:1, either immediately (+CD25+) or after a 3-d mixed culture with CD11b+ cells from TBR (+CD25+ expanded). (C) Requirement of class II and CD11b cells for T reg cell proliferation. 105 TCR+ cell-depleted splenocytes from TBR or TFR were further depleted from MHC class II+ cells or CD11b+ cells and co-cultured in the presence of 105 splenic CD25+ T cells from TBR (left) or TFR (right) for 5 d before pulsing with [3H]thymidine. Values represent the mean ± SEM of triplicate wells. The asterisks indicate statistical significance (P = 0.01) at 95% confidence using Fisher's exact test. (D) 105 CD11c− cells were isolated from the DLNs of TFM and TBM and co-cultured for 5 d with 105 spleen CD25− T cells from TBM before measuring [3H]thymidine incorporation. Values represent means ± SEM of triplicate wells. The asterisk indicates statistical significance (P = 0.005) at 95% confidence using Fisher's exact test.
To recapitulate the in vitro data mentioned in Fig. 4 in an in vivo system, we inoculated splenic IMDCs from TBR or controls into the footpad of TFR. 4 d later, we observed a twofold increase in the BrdU incorporation into T reg cells in the ipsi- versus contra-lateral LNs (Fig. 5 A) but not into other T cell subsets (not depicted), provided that IMDCs were purified from TBR. Similarly, IMDCs from TBR stimulated the proliferation of carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled T reg cells in vivo after adoptive transfer of both populations by i.v. injection into TFR. In contrast, IMDCs from TFR failed to stimulate the division of coadministered T reg cells in vivo (Fig. 5 B). Collectively, these data support the contention that IMDCs drive T reg cell proliferation associated with tumor progression.
Figure 5.
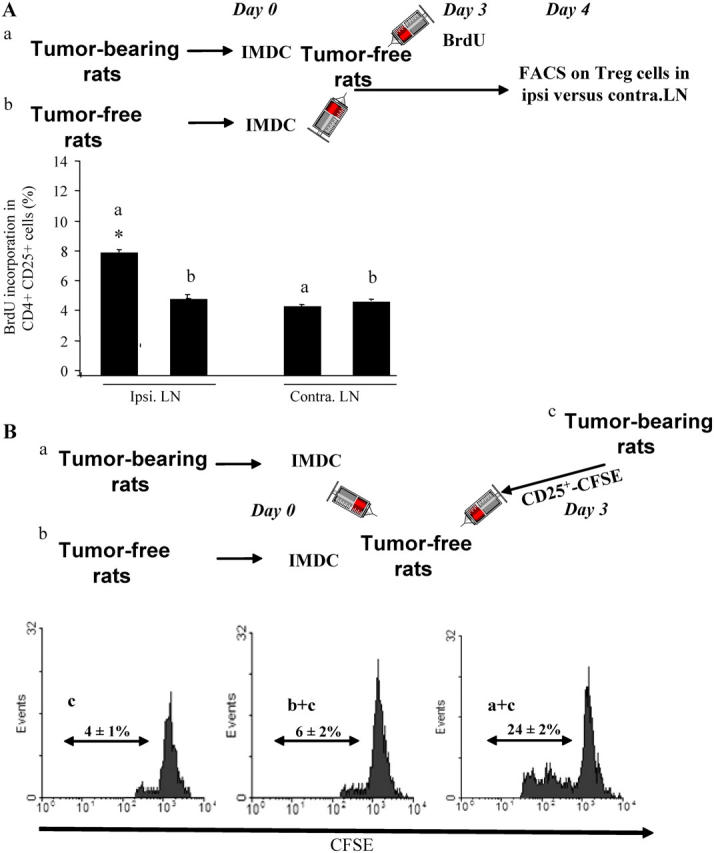
DCs from tumor bearers promote T reg cell proliferation in vivo. (A) Measurement of T reg cell proliferation elicited by DCs using BrdU. 2 × 106 CD11b+ DCs isolated from the spleen of TBR or TFR were injected into the left rear foot pad of TFR. 3 d later, BrdU was injected i.p. The percentages of BrdU+ T reg cells in the ipsi- versus contra-lateral LNs were determined by flow cytometry. The mean ± SEM of three rats per group are shown. The asterisk represent statistical significance (P = 0.02) at 95% confidence using the Mann-Whitney U test. (B) Measurement of T reg cell proliferation elicited by DCs using CFSE. 5 × 106 CFSE-labeled T reg cells isolated from the spleen of TBR were injected i.v. into TFR either alone (control) or in combination with 2 × 106 CD11b+ DC cells isolated from the spleen of TFR or TBR. 3 d later, CFSE dilution was studied in splenic CD4+ lymphocytes by flow cytometry. Percentages indicate the rate of proliferating cells. One representative out of two independent experiments is shown.
IMDC-induced T reg cell proliferation is TGF-β dependent
In autoimmune disease models, the proliferation of T reg cells requires signaling through TGF-βRII (20, 21). Hence, we addressed the question as to whether tumor-dependent T reg cell proliferation would involve TGF-βRII. We took advantage of a model of C57BL/6 dominant-negative TGF-β receptor II–signaling mice bearing transgenic T cells (DN TGF-βRII mice). Although nontransgenic C57BL/6 controls exhibited T reg cell accumulation (Fig. 6 A) and proliferation (Fig. 6 B) in DLNs of B16F10 melanomas, no such effect was found in tumor-bearing DN TGF-βRII mice. Moreover, B16F10 melanoma growth was reduced in DN TGF-βRII mice (Fig. 6 C), suggesting a more efficient tumor surveillance. Even at later stages of tumor development, DN TGF-βRII mice failed to manifest any sign of T reg cell accumulation in DLNs, indicating that the failure of T reg cell proliferation/accumulation was not a simple consequence of reduced tumor growth.
Figure 6.
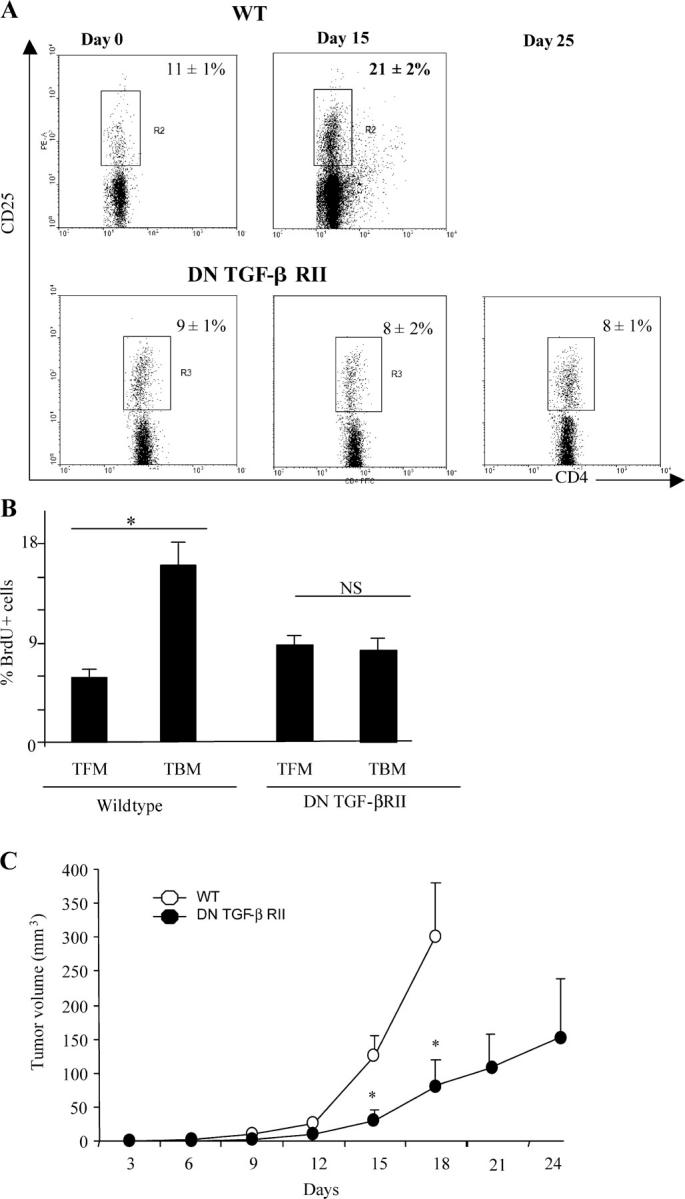
TGF-β signaling is critical for T reg cell proliferation and tumor escape. (A) Flow cytometry analyses of CD3+CD4+CD25+ cells isolated from the inguinal LNs of WT and DN TGF-βRII mice. The animals were either tumor free (0), or bore 15- or 25-d-old tumors. Data from one representative animal in each group is shown. Percentages of CD25 expression among CD3+CD4+ cells are shown. Values represent the mean ± SEM (n = 6). (B) BrdU incorporation into CD3+CD4+CD25+-gated T cells isolated from the inguinal LNs of WT and DN TGF-βRII mice. Animals were either tumor free or bore 15-d-old tumors, as indicated by the symbols, and the frequency of BrdU+ cells was determined. Values represent the mean ± SEM (n = 6). One representative experiment out of two is shown. The asterisk indicates statistical significance (P < 0.05) using the Student's t test. (C) Influence of the DN TGF-βRII transgene on PROb tumor growth. WT (open circles) and transgenic DN TGF-βRII mice (closed circles) were challenged s.c. with B16F10 cells, and tumor growth was monitored biweekly. One representative out of two experiments including six mice per group following the mean tumor volume ± SEM is depicted. The asterisk indicates statistical significance (P = 0.001) at 95% confidence using the Student's t test.
We next investigated the capacity of IMDCs to actively produce TGF-β ex vivo. FACS-purified IMDCs from TBR or TBM (but not from tumor-free controls) revealed positive intracellular TGF-β staining (Fig. 7). Moreover, IMDCs purified from TBR or TBM (but not from tumor-free controls) spontaneously produced biologically active TGF-β within 48 h (Fig. 7 B), in sharp contrast with B16F10 and PROb tumor cells that only produced latent, inactive TGF-β (not depicted). Because tumor cells did not produce biologically active TGF-β, we investigated the possibility that they would license IMDCs to produce TGF-β. Incubation of IMDCs from TFM with culture supernatants from B16F10 or PROb tumor cells (but not from NIH 3T3 fibroblasts) induced TGF-β production by IMDCs within 24 h, as shown by intracellular staining of TGF-β (Fig. 7 C). We ruled out that intracellular TGF-β labeling observed in IMDCs resulted from the endocytosis of this cytokine because addition of exogenous TGF-β failed to yield a positive staining (Fig. 7 C). It is noteworthy that though LPS induced IMDCs to produce IL-10 and IL-12 and to up-regulate CD80 and CD86 co-stimulatory molecules, the tumor supernatants could only promote the secretion of TGF-β (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050463/DC1). Collectively, these results indicate that tumor cells produce soluble factors that cause DCs to secrete bioactive TGF-β.
Figure 7.

Tumor cells license IMDCs to produce TGF-β. (A) Intracellular staining of IMDCs from tumor-free or tumor-bearing rats and mice. CD11b+ DCs isolated from the spleen of TBR and TFR or CD11c+ IMDCs from DLNs of TFM or TBM were permeabilized and stained with a polyclonal anti–TGF-β antibody. Shaded histogram, anti–TGF-β; bold line histogram, isotype control. Note that no positive staining was obtained with nonpermeabilized cells (not depicted). (B) TGF-β secretion by IMDCs. IMDCs purified as in A were cultured for 48 h in serum-free medium and TGF-β1 levels in the supernatants were assessed by ELISA. Values represent the mean ± SEM (n = 3). (C) Induction of TGF-β production in mouse splenic IMDCs by tumor supernatants. CD11c+ DCs from inguinal LNs of TFM were cultured for 24 h in the absence or presence of B16F10 or NIH 3T3 culture supernatants or 10 ng/ml TGF-β1. Shaded histogram, test tube; open histogram, isotype control. At 24 h, TGF-β–expressing cells were identified by FACS analysis after permeabilization. The percentages of TGF-β expression cells are given. Data representative of two independent experiments are shown.
TGF-β acts as a co-stimulator of T reg cells
We investigated the contribution of TGF-β to the proliferation of T reg cells elicited by IMDCs from tumor-bearing rodents. Anti–TGF-β antibody significantly (P = 0.03) decreased the T reg cell proliferation induced by splenic IMDCs isolated from TBR (Fig. 8 A). Furthermore, IMDCs derived from TFR acquired the capacity to elicit CD25+ T reg cells (but not CD25−) T cell proliferation in the presence of exogenous TGF-β (Fig. 8 B and not depicted). These data indicate that it is truly TGF-β production that marks the difference between IMDCs capable of stimulating T reg cells. In a further set of experiments, we investigated whether TGF-β alone could provide a co-stimulatory signal for T reg cell proliferation. T reg cells proliferated in response to a combination of CD3 cross-linking and TGF-β but exhibited minor or absent proliferative responses to either CD3 cross-linking or TGF-β alone (Fig. 8 C). In contrast, TGF-β decreased the anti-CD3 mAb–driven proliferation of CD4+CD25− conventional T cells (unpublished data), underscoring the specific role of TGF-β on T reg cells.
Figure 8.

IMDCs stimulate T reg cell proliferation in a TGF-β–dependent fashion. (A) Neutralization of TGF-β curtails IMDC-driven T reg cell proliferation. 105 IMDCs were cultured for 5 d with T reg cells at a 1:1 ratio along with 1 ng/ml blocking anti–TGF-β antibody or an isotype-matched control before pulsing with [3H]thymidine. Both IMDCs and T reg cells were isolated from the spleen of TBR. The inset shows a similar experiment in which CD25+ splenic cells were labeled with CFSE, and CFSE expression in T cells was monitored by flow cytometry at day 5 (one representative of three experiments is shown). Values represent the mean ± SEM. The asterisk indicates statistical significance (P = 0.03). (B) Exogenous TGF-β cooperates with IMDCs from TFM to drive the proliferation of T reg cells. 105 splenic T reg cells from TBR were cultured in FBS-free medium supplemented with 200 UI/ml rIL-2 along with CD11b+ splenic DCs from tumor-free rats at a 1:1 ratio (left) in the presence of 1 ng/ml TGF-β1 before pulsing with [3H]thymidine. One representative out of five experiments is shown. Means and SEM of cpm of triplicate wells are shown. In all proliferation assays, CD11b+ DCs alone did not incorporate [3H]thymidine above background levels (not depicted). The asterisk indicates statistical significance (P < 0.05) using Fisher's exact test. (C) Stimulation of T reg cell proliferation by anti-CD3 and TGF-β1. T reg cells were stimulated with immobilized anti-CD3 and/or TGF-β1 (as in B). (D and E) In vitro cooperation between tumor cell supernatants and IMDCs to drive TGF-β–dependent T reg cell proliferation in mice (D) and in rats (E). 2 × 104 IMDCs isolated from tumor-free rodents and incubated in tumor cell supernatants for 24 h were co-cultured with 105 T reg cells from tumor-bearing rodents with anti–TGF-β1 blocking antibodies for 5 d. Proliferation of T reg cells was monitored in triplicate wells by measuring [3H]thymidine incorporation. One representative experiment out of three is depicted. The asterisk indicates statistical significance (P < 0.05) using Fisher's exact test. It is noteworthy that supernatants of NIH 3T3 did not license DCs for T reg cell proliferation (not depicted).
Finally, we attempted to recapitulate the entire pathway delineated in this paper in vitro while comparing the mouse and rat systems. We found that incubation of IMDCs isolated from tumor-free rodents with tumor cell supernatants licensed IMDCs to induce T reg cell proliferation. This effect was suppressed by the addition of a neutralizing anti–TGF-β antibody. Very similar data were obtained in C57BL/6 mice (Fig. 8 D) and BD-IX rats (Fig. 8 E). Again, in this system, DC-mediated T reg cell proliferation did not revert the suppressive functions of T reg cells (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20050463/DC1). Thus, in both rodent systems, TGF-β appears to be the determining factor that drives T reg cell proliferation by IMDCs exposed to tumor products.
Discussion
The data exposed in this article suggest the existence of a linear pathway that links tumor progression to immune suppression. Tumor cells produce factors that stimulate local IMDCs to produce TGF-β. TGF-β then acts as a co-stimulator to drive the proliferation of FOXP3+CD4+CD25+ T reg cells in a TGF-βRII–dependent fashion, and T reg cells in turn mediate immunosuppression. This pathway has been unraveled in two distinct rodent species, enhancing the likelihood (but by no means proving) that it also applies to the human system.
During tumor progression, T reg cells accumulate in tumors and secondary lymphoid organs of humans (13–18). It has been suggested that chemokines produced by tumor cells or tumor-infiltrating macrophages recruit T reg cells into the tumor bed (17). On theoretical grounds, T reg cell accumulation could also result from the priming and differentiation of naive CD4+CD25− T lymphocytes into T reg cells, a phenomenon well established in vitro (22–24) and on homeostatic proliferation in vivo (25). Here, we formally demonstrated that the T reg cell accumulation in tumor-draining lymphoid organs largely resulted from their local proliferation (as opposed to reduced apoptosis or nonproliferative differentiation). These results are in line with the seminal work of North and Awwad, who highlighted the expansion of cycling CD4+ suppressor cells in tumors and their role in tumor-induced immune tolerance (26).
T reg cell proliferation may be related to IL-2 (11, 27) and MHC class II peptide complexes (28, 29), as well as TGF-β (20, 21). We retain that it is unlikely that IL-2 drives T reg cell proliferation stimulated by IMDCs during tumor growth. Indeed, to date, DCs have solely been reported to produce IL-2 after LPS stimulation, bacterial infection, or CD40 ligation (27), conditions that are unlikely to be met in tumor-bearing hosts. Reportedly, immunization with MHC class II epitopes in TCR transgenic mice promoted the expansion of peptide-specific T reg cells in vivo (28). Similarly, MHC class II peptide–pulsed mature DCs could elicit peptide-specific T reg cells capable of preventing the onset of diabetes (9, 29). Our data suggest that IMDC-mediated T reg cell expansion occurring in tumor-bearing hosts is MHC class II restricted because T reg cell proliferation induced by IMDCs from TBR was abolished by the depletion of MHC class II+ cells (Fig. 4 C). Moreover, T reg cell proliferation could be elicited by a combination of anti-CD3 mAb (which mimics the MHC class II peptide complex) and TGF-β co-stimulation (Fig. 8 C). Peng et al. (20) first unraveled the role of TFG-β in inducing T reg cell proliferation for the control of diabetes. Working in a model of dextran sulfate–induced autoimmune colitis, Huber et al. (21) confirmed the requirement for TGF-βRII signaling in T reg cells for their proliferation in vivo. However, the source of TGF-β was either transfected pancreatic islet β cells (20) or not identified (21). Here, we demonstrate that a specific subset of DC-infiltrated lymphoid organs during tumor progression provides a source of TGF-β (Fig. 7).
The role of TGF-β in the regulation of T reg cell proliferation in tumor-bearing hosts has not been reported so far. An immunosuppressive effect and a tumor-enhancing role of TGF-β has been demonstrated in the model of syngeneic mice bearing T cells defective in TGF-βRII signaling (30), but it was assumed that TGF-β produced by tumor cells exerted a direct suppressive effect on the antitumor-specific T cells (30). We provide compelling evidence indicating that TGF-β produced by tumor cell–licensed IMDCs was critical for the proliferation and accumulation of T reg cells. Indeed, IMDCs isolated from tumor bearers, but not from TFR or TFM, elicited T reg cell proliferation (Fig. 4) in a TGF-β–dependent fashion (Fig. 8). Moreover, IMDCs from TFM were converted to a T reg cell-stimulatory phenotype when licensed by tumor cells (Figs. 7 and 8).
Our experiments using positive (Fig. 4, A and B) or negative selection (Fig. 4 C) of either CD11c+/CD11b+ cells or MHC class II+ cells suggest that IMDCs are essential for the proliferation of T reg cells in vitro. This particular DC population was CD11c+/I-Ab+ in mice and OX42+/OX17+ in rats, coexpressed CD11b, and exhibited low levels of co-stimulatory molecules. Several arguments support the relevance of these IMDC for the expansion of T reg cells in tumor-bearing animals. First, both IMDCs and T reg cells resided and expanded in tumor DLNs during tumor progression (Fig. 1 and Table I). IMDCs expanded by fivefold and T reg cells by twofold by day 15 or 28 in DLNs of animals with B16F10 and PROb tumors, respectively. Second, IMDCs isolated from tumor bearers induced T reg cell proliferation when adoptively transferred into tumor-free hosts (Fig. 5). Third, the IMDC-mediated T reg cell priming depended on both MHC class II and TGF-β molecules (Fig. 8).
Mature DCs expressing high levels of MHC class II and CD80 (9), as well as immature DCs (31), have been reported to be mitogenic for T reg cells. In the two rodent tumor cells that we investigated in this study, we failed to detect significant (P > 0.10) numbers of completely mature DCs in tumor DLNs (unpublished data), suggesting that IMDCs are indeed the preponderant T reg cell stimulators. In line with this consideration, immature DCs were shown to produce TGF-β more actively than mature DCs (32). Moreover, the phenotype of IMDCs is clearly different from that of indoleamine 2,3-dioxygenase–expressing plasmacytoid DCs that also may exert immunosuppressive functions (33).
Tumor cells licensed DCs to promote T reg cell proliferation via TGF-β (Figs. 7 and 8). This licensing effect involved soluble factors produced by B16F10 and PROb tumor cells. In this context, it appears intriguing that tumors reportedly produce a variety of distinct cytokines, including vascular endothelial growth factor, macrophage CSF, granulocyte-macrophage CSF, IL-6 and IL-10, capable of blocking DC differentiation, for instance through activation of STAT-3 and inactivation of the NF-κB signaling pathway (34, 35). Whether such soluble factors could also induce TGF-β expression and secretion by DCs remains to be assessed.
Irrespective of these questions, the data outlined in this paper delineate a novel pathway linking tumor expansion to immunosuppression. Several elements among this pathway are new (such as IMDCs stimulating T reg cell proliferation) or are newly placed (such as TGF-β as a co-stimulatory factor for T reg cells). Future research will address the question as to whether interruption of this cascade may have positive consequences for the clinical management of human cancer.
Material and Methods
Animals and cell lines.
BD-IX strain inbred rats were bred at the University of Burgundy animal facilities. The tumor cell line PROb (available from the European Collection of Cell Culture under accession no. DHD-K12-TRb) was established from a BD-IX rat colon carcinoma (36). PROb cells were cultured in Ham F10 medium supplemented with 10% FBS. PROb tumors were induced by s.c. injection of 106 tumor cells in the thoracic wall of 10–12-wk-old BD-IX rats. C57BL6 WT mice were purchased from Charles River Laboratories. C57BL6 DN TGF-βRII mice, with an impaired TGF-β signaling pathway in T cells by overexpressing a truncated version of the TGF-β type II receptor under control of a T cell–specific promoter (37), were a gift from P. Lucas (National Institutes of Health, Bethesda, MD). B16F10 cells, obtained from the American Type Culture Collection, were cultured in RPMI 1640 with 10% FBS and injected s.c. in the hind leg in C57BL6 mice. All cell lines were monitored routinely and found to be free of Mycoplasma infection. Animal use and handling were approved by the local veterinary committee and were performed according to the French laws for animal experimentation.
Antibodies.
Mouse mAbs against rat CD4 (W3/25), CD25 (OX-39), CD11b (OX-42, Alexa 647 conjugated), CD11c (8A2), CD68 (ED1, FITC conjugated), CD80 (3H5), CD86 (24F, FITC conjugated), CD163 (ED2, FITC conjugated), MHC class II (OX-17, FITC conjugated), Ki67, BrdU (BU1/75), CD103 (αE2 chain of integrins, OX-62), and isotype control mAbs were purchased from Serotec. Rat mAbs against mouse CD3–Cy-Chrome (17A2), CD4–FITC (RM4-5), CD25 (PC61), CD11c–FITC (HL3), CD11b–APC (M1/70), CD80–FITC (16-10A1), CD86 (24F), and control isotypes were obtained from BD Biosciences. Antibodies against TGF-β1 and recombinant TGF-β1 were obtained from R&D Systems, and FITC–secondary antibodies were purchased from ABCAM.
Isolation of T lymphocytes, macrophages, and DCs.
Rat cell suspensions containing ≥95% T cells after FACScan analysis were obtained from spleen and axillary LNs as previously described (18). The purification of CD25+ T cells was performed using an anti–rat CD25 mAb and anti–mouse IgG1-coated magnetic beads according to the manufacturer's instructions (Miltenyi Biotec). Approximately 90% of the positively selected cells expressed both CD4 and CD25 after a subsequent flow cytometry analysis. Rat splenic macrophages were prepared according to their fast attachment to a plastic surface. Approximately 90% of the adherent cells were mature macrophages according to their labeling with anti-CD163 antibody. Rat splenic CD11b cells were isolated using anti-CD11b mAb and anti–mouse IgG1-coated magnetic beads from Miltenyi Biotec. Approximately 95% of the positively selected cells expressed CD11b, as determined by flow cytometry analysis. For mice, splenic CD4+CD25+ regulatory T cells were isolated using the CD4+CD25+ regulatory T cells isolation kit from Miltenyi Biotec. Mouse DCs were isolated from inguinal LNs using CD11c magnetic beads (Miltenyi Biotec). Cytometry analyses were performed on FACScan or LSRII (Becton Dickinson) using WinMDI software.
Semiquantitative RT-PCR and mouse Foxp3 immunolabeling.
Total RNA was extracted from freshly purified T cell subsets using Trizol (Sigma-Aldrich) according to the manufacturer's instructions. cDNA synthesis was obtained with a cDNA synthesis kit (Roche Diagnostic). For semiquantitative RT-PCR, the following primers were used: rat FOXP3 (5′-GCACAAGTGCTTTGTGCGAGT-3′ and 5′-TGTCTGTGGTTGCAGACGTTGT-3′ with an annealing temperature of 62°C for 30 cycles with an expected size 572 bp) and GAPDH (5′-TAAAGGGCATCCTGGGCTACACT-3′ and 5′-TTACTCCTTGGAGGCCATGTAGG-3′ with an annealing temperature of 62°C for 30 cycles with an expected size of 200 bp). Samples were normalized using GADPH expression and run using 1 μg of total mRNA.
Proliferation of CFSE-labeled CD25+ and CD25− T cells.
After MACS purification, CD4+CD25+ and CD4+CD25− T cells were incubated with 5 μM CFSE (Molecular Probes) for 10 min at 37°C for in vivo studies. Afterward, cells were washed three times in complete medium.
BrdU staining in vitro and in vivo.
Proliferation of T cells was determined in vitro using a BrdU proliferation ELISA kit (Roche) according to the manufacturer's instructions. To investigate cell proliferation in vivo, tumor-bearing or tumor-free animals received an i.p. injection of 50 mg BrdU/kg of body weight. Animals were killed 18 h after the last injection. CD25+ T cells were isolated from the thymus, spleen, tumor-draining (axillary), and non–tumor-draining (popliteal) LNs by MACS as described above. The same procedure was applied for isolating tumor-infiltrating CD25+ T cells.
Determination of TGF-β1 concentrations.
IMDCs purified from rat spleen by MACS were cultured for 24 h at a concentration of 106 cells/ml in serum-free RPMI medium with 200 μg/ml BSA. TGF-β1 concentration was determined in the supernatant using an ELISA kit (R&D Systems) after conversion of the latent TGF-β1 in its active form through acidification according to the manufacturer's instructions.
Mixed leukocyte reaction.
105 BD-IX rat T lymphocytes were co-cultured for 5 d with 105 total spleen cells from a Wistar rat pretreated with 50 μg/ml mitomycin-C for 2 h. Cells were co-cultured for 5 d. 1 μCi/well of [3H]thymidine (Isotopchim) was added 24 h before the end of the culture. Cells were harvested by filtration onto glass fiber filters. The amount of [3H]thymidine incorporated in the cells was determined by liquid scintillation counting (Packard Biosciences).
Statistics.
Statistical significance (P < 0.05) was determined by the two-tailed Student's t test or by Fisher's exact test.
Online supplemental material.
Fig. S1 shows that splenic B cells, macrophages, or tumor cells from TBR did not induce CD4+CD25+ proliferation. Fig. S2 depicts a phenotype of mouse DCs in lymphoid organs. Fig. S3 shows that tumor supernatants induced TGF-β production by IMDCs. Fig. S4 shows that T reg cells expanded by DCs licensed by tumor cell supernatants maintained their suppressive functions and are able to inhibit mixed lymphocyte reactions. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050463/DC1.
Acknowledgments
We are grateful to Monique Moutet and Annie Fromentin for their technical assistance.
This work was supported by grants from the Ligue Labelisée contre le Cancer, the Ligue contre le Cancer (Comités national, Saône-et-Loire, and Nievre), and AlloStem (503319).
The authors have no conflicting financial interests.
Abbreviations used: BrdU, 5-bromo-2′-deoxyuridine; CFSE, carboxyfluorescein diacetate succinimidyl ester; ConA, concanavalin A; DLN, draining LN; DN TGF-βRII mice, dominant-negative TGF-β receptor II–signaling mice bearing transgenic T cells; IMDC, immature myeloid DC; TBM, tumor-bearing mice; TBR, tumor-bearing rats; TFM, tumor-free mouse/mice; TFR, tumor-free rats; T reg cells, CD4+CD25+ regulatory T cells.
B. Chauffert and L. Zitvogel contributed equally to this work.
References
- 1.Sakaguchi, S. 2000. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 101:455–458. [DOI] [PubMed] [Google Scholar]
- 2.Shevach, E.M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 3.Brunkow, M.E., E.W. Jeffery, K.A. Hjerrild, B. Paeper, L.B. Clark, S.A. Yasayko, J.E. Wilkinson, D. Galas, S.F. Ziegler, and F. Ramsdell. 2001. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27:68–73. [DOI] [PubMed] [Google Scholar]
- 4.Sakaguchi, S. 2004. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22:531–562. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi, T., Y. Kuniyasu, M. Toda, N. Sakaguchi, M. Itoh, M. Iwata, J. Shimizu, and S. Sakaguchi. 1998. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10:1969–1980. [DOI] [PubMed] [Google Scholar]
- 6.Thornton, A.M., and E.M. Shevach. 1998. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin-2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thornton, C.A., J.W. Upham, M.E. Wikstrom, B.J. Holt, G.P. White, M.J. Sharp, P.D. Sly, and P.G. Holt. 2004. Functional maturation of CD4+CD25+CTLA4+CD45RA+ T regulatory cells in human neonatal T cell responses to environmental antigens/allergens. J. Immunol. 173:3084–3092. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi, T., T. Tagami, S. Yamazaki, T. Uede, J. Shimizu, N. Sakaguchi, T.W. Mak, and S. Sakaguchi. 2000. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J. Exp. Med. 192:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamazaki, S., T. Iyoda, K. Tarbell, K. Olson, K. Velinzon, K. Inaba, and R.M. Steinman. 2003. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 198:235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasare, C., and R. Medzhitov. 2003. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 299:1033–1036. [DOI] [PubMed] [Google Scholar]
- 11.Almeida, A.R., N. Legrand, M. Papiernik, and A.A. Freitas. 2002. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 169:4850–4860. [DOI] [PubMed] [Google Scholar]
- 12.Gavin, M.A., S.R. Clarke, E. Negrou, A. Gallegos, and A. Rudensky. 2001. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat. Immunol. 3:33–41. [DOI] [PubMed] [Google Scholar]
- 13.Liyanage, U.K., T.T. Moore, H.G. Joo, Y. Tanaka, V. Herrmann, G. Doherty, J.A. Drebin, S.M. Strasberg, T.J. Eberlein, P.S. Goedegebuure, and D.C. Linehan. 2002. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 169:2756–2761. [DOI] [PubMed] [Google Scholar]
- 14.Wolf, A.M., D. Wolf, M. Steurer, G. Gastl, E. Gunsilius, and B. Grubeck-Loebenstein. 2003. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin. Cancer Res. 9:606–612. [PubMed] [Google Scholar]
- 15.Sasada, T., M. Kimura, Y. Yoshida, M. Kanai, and A. Takabayashi. 2003. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer. 98:1089–1099. [DOI] [PubMed] [Google Scholar]
- 16.Woo, E.Y., C.S. Chu, T.J. Goletz, K. Schlienger, H. Yeh, G. Coukos, S.C. Rubin, L.R. Kaiser, and C.H. June. 2001. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 61:4766–4772. [PubMed] [Google Scholar]
- 17.Curiel, T.J., G. Coukos, L. Zou, X. Alvarez, P. Cheng, P. Mottram, M. Evdemon-Hogan, J.R. Conejo-Garcia, L. Zhang, M. Burow, et al. 2004. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10:942–949. [DOI] [PubMed] [Google Scholar]
- 18.Ghiringhelli, F., N. Larmonier, E. Schmitt, A. Parcellier, D. Cathelin, C. Garrido, B. Chauffert, E. Solary, B. Bonnotte, and F. Martin. 2004. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 34:336–344. [DOI] [PubMed] [Google Scholar]
- 19.Turk, M.J., J.A. Guevara-Patino, G.A. Rizzuto, M.E. Engelhorn, S. Sakaguchi, and A.N. Houghton. 2004. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J. Exp. Med. 200:771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng, Y., Y. Laouar, M.O. Li, E.A. Green, and R.A. Flavell. 2004. TGF-beta regulates in vivo expansion of Foxp3-expressing CD4+ CD25+ regulatory T cells responsible for protection against diabetes. Proc. Natl. Acad. Sci. USA. 101:4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huber, S., C. Schramm, H.A. Lehr, A. Mann, S. Schmitt, C. Becker, M. Protschka, P.R. Galle, M.F. Neurath, and M. Blessing. 2004. Cutting edge: TGF-beta signaling is required for the in vivo expansion and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J. Immunol. 173:6526–6531. [DOI] [PubMed] [Google Scholar]
- 22.Chen, W., W. Jin, N. Hardegen, K.J. Lei, L. Li, N. Marinos, G. McGrady, and S.M. Wahl. 2003. Conversion of peripheral CD4+ CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 198:1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker, M.R., D.J. Kasprowicz, V.H. Gersuk, A. Benard, M. Van Landeghen, J.H. Buckner, and S.F. Ziegler. 2003. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25− T cells. J. Clin. Invest. 112:1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fantini, M.C., C. Becker, G. Monteleone, F. Pallone, P.R. Galle, and M.F. Neurath. 2004. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 172:5149–5153. [DOI] [PubMed] [Google Scholar]
- 25.Curotto de Lafaille, M.A., A.C. Lino, N. Kutchukhidze, and J.J. Lafaille. 2004. CD25− T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J. Immunol. 173:7259–7268. [DOI] [PubMed] [Google Scholar]
- 26.North, R.J., and M. Awwad. 1990. Elimination of cycling CD4+ suppressor T cells with an anti-mitotic drug releases non cycling CD8+ T cells to cause regression of an advanced lymphoma. Immunology. 71:90–95. [PMC free article] [PubMed] [Google Scholar]
- 27.Granucci, F., I. Zanoni, S. Feau, and P. Ricciardi-Castagnoli. 2003. Dendritic cell regulation of immune responses: a new role for interleukin 2 at the intersection of innate and adaptive immunity. EMBO J. 22:2546–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker, L.S., A. Chodos, M. Eggena, H. Dooms, and A.K. Abbas. 2003. Antigen-dependent proliferation of CD4+CD25+ regulatory T cells in vivo. J. Exp. Med. 198:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarbell, K.V., S. Yamazaki, K. Olson, P. Toy, and R.M. Steinman. 2004. CD25+CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 199:1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorelik, L., and R.A. Flavell. 2001. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat. Med. 7:1118–1121. [DOI] [PubMed] [Google Scholar]
- 31.Mahnke, K., Y. Qian, J. Knop, and A.H. Enk. 2003. Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood. 101:4862–4869. [DOI] [PubMed] [Google Scholar]
- 32.Morelli, A.E., A.F. Zahorchak, A.T. Larregina, B.L. Colvin, A.J. Logar, T. Takayama, L.D. Falo, and A.W. Thomson. 2001. Cytokine production by mouse myeloid dendritic cells in relation to differentiation and terminal maturation induced by lipopolysaccharide or CD40 ligation. Blood. 98:1512–1523. [DOI] [PubMed] [Google Scholar]
- 33.Munn, D.H., M.D. Sharma, D. Hou, B. Baban, J.R. Lee, S.J. Antonia, J.L. Messina, P. Chandler, P.A. Koni, and A.L. Mellor. 2004. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Invest. 114:280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gabrilovich, D. 2004. Mechanisms and functional significance of tumour-induced dendritic cell defects. Nat. Rev. Immunol. 4:941–952. [DOI] [PubMed] [Google Scholar]
- 35.Wang, T., G. Niu, M. Kortylewski, L. Burdelya, K. Shain, S. Zhang, R. Bhattacharya, D. Gabrilovich, R. Heller, D. Coppola, et al. 2004. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10:48–54. [DOI] [PubMed] [Google Scholar]
- 36.Caignard, A., H. Pelletier, and F. Martin. 1988. Specificity of the immune response leading to protection or enhancement by regressive and progressive variants of a rat colon carcinoma. Int. J. Cancer. 42:883–886. [DOI] [PubMed] [Google Scholar]
- 37.Lucas, P.J., S.J. Kim, S.J. Melby, and R.E. Gress. 2000. Disruption of T cell homeostasis in mice expressing a T cell–specific dominant negative transforming growth factor βII receptor. J. Exp. Med. 191:1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]