

Abstract
Myeloid suppressor cells (MSCs) producing high levels of arginase I block T cell function by depleting l-arginine in cancer, chronic infections, and trauma patients. In cancer, MSCs infiltrating tumors and in circulation are an important mechanism for tumor evasion and impair the therapeutic potential of cancer immunotherapies. However, the mechanisms that induce arginase I in MSCs in cancer are unknown. Using the 3LL mouse lung carcinoma, we aimed to characterize these mechanisms. Arginase I expression was independent of T cell–produced cytokines. Instead, tumor-derived soluble factors resistant to proteases induced and maintained arginase I expression in MSCs. 3LL tumor cells constitutively express cyclooxygenase (COX)-1 and COX-2 and produce high levels of PGE2. Genetic and pharmacological inhibition of COX-2, but not COX-1, blocked arginase I induction in vitro and in vivo. Signaling through the PGE2 receptor E-prostanoid 4 expressed in MSCs induced arginase I. Furthermore, blocking arginase I expression using COX-2 inhibitors elicited a lymphocyte-mediated antitumor response. These results demonstrate a new pathway of prostaglandin-induced immune dysfunction and provide a novel mechanism that can help explain the cancer prevention effects of COX-2 inhibitors. Furthermore, an addition of arginase I represents a clinical approach to enhance the therapeutic potential of cancer immunotherapies.
T cell anergy is a common observation in patients and rodents with cancer. This tumor-induced phenomenon may help tumors evade the immune response and block the potential therapeutic benefit of immunotherapy. Of the several mechanisms described to explain anergy, the accumulation of myeloid suppressor cells (MSCs) in the tumor, spleen, and peripheral blood of tumor-bearing mice and cancer patients has gained considerable interest (1–6). Using the 3LL murine lung carcinoma, we recently demonstrated that l-arginine (l-Arg) depletion in the microenvironment by arginase I–producing MSCs inhibited T cell receptor CD3ζ expression and blocked T cell functions (5). However, the mechanisms that induce arginase I in MSCs in cancer are unclear.
In vitro models show that cytokines such as IL-4, IL-10, and IL-13 can induce the expression of arginase I in bone marrow and peritoneal macrophages through activation of nuclear transcription factor STAT6 (7, 8). Similarly, arginase I can also be induced in macrophages exposed to cAMP analogues, prostaglandin E2 (PGE2), LPS, hypoxia, and other cytokines including TGFβ (9). However, the role of these factors in the induction of arginase I in cancer has not been determined. Using the 3LL lung carcinoma model, we attempted to characterize the mechanism of arginase I induction in MSCs.
The results failed to demonstrate the presence of IL-4 or IL-13 in the tumor microenvironment or a role for T cell–produced cytokines in the induction of arginase I in MSCs. Instead, soluble factors produced by 3LL tumors were essential to induce and maintain arginase I production in MSCs. Prostanoid production by 3LL cells, including PGE2, induced arginase I expression in MSCs by signaling through the E-prostanoid (EP) 4 receptor. Genetic or pharmacological inhibition of cyclooxygenase (COX)-2 blocked arginase I expression and induced a T cell–mediated antitumor effect. This represents a novel mechanism for prostaglandin-induced immune dysfunction and may explain the cancer prevention effect of COX-2 inhibition.
RESULTS
Arginase I expression in tumor-infiltrating MSCs is dependent on tumor-derived factors
Increased arginase activity in cancer was thought to come from tumor cells metabolizing l-Arg to produce polyamines, which are needed to sustain rapid cell proliferation (10). However, our recent data showed that arginase I was produced by tumor-infiltrating MSCs (5). The mechanisms that induce arginase I in MSC-infiltrating tumors are not clear. In vitro models showed that arginase I can be induced in peritoneal and bone marrow macrophages by IL-4 and IL-13, which can be produced by some tumors, infiltrating T lymphocytes, or NKT cells (11–13). However, none of these cytokines was detected via protein or RNA assays in 3LL cells cultured in vitro or in single-cell suspensions of subcutaneous 3LL tumors (unpublished data). Furthermore, no significant differences in arginase I expression were observed in 3LL tumors excised from tumor-bearing SCID mice (C57BL/6-Prkdc scid), which lack functional T and B cells, and wild-type C57BL/6 mice (Fig. 1 A). In addition, MSC-infiltrating tumors isolated from wild-type and C57BL/6-Prkdc scid mice expressed similar levels of arginase I (Fig. 1 B), suggesting that T cells and T cell–produced cytokines were not responsible for arginase I induction in MSCs.
Figure 1.

Expression of arginase I in tumor-infiltrating MSCs is independent of lymphocytes. (A) Single-cell suspensions from individual subcutaneous 3LL tumors excised from tumor-bearing C57BL/6 and C57BL/6 Prkdc scid mice (n = 15 per group) were tested for arginase I expression via Western blot analysis. Representative results of 10 tumors are shown. (B) Arginase I expression was tested in freshly isolated MSCs infiltrating individual 3LL tumors from C57BL/6 and C57BL/6 Prkdc scid tumor-bearing mice (n = 15 per group). Representative results from 6 tumors are shown.
We then tested whether tumor-derived factors might be necessary to induce or sustain arginase I production in MSCs. Purified MSCs obtained from 3LL tumors cultured in vitro in standard tissue culture medium (RPMI 1640 which contains 1,000 μM arginine) lose arginase I expression after 24 h. However, if freshly isolated MSCs were cocultured in transwells with 3LL cells or 3LL supernatants, they maintained arginase I expression (Fig. 2 A) and arginase activity (not depicted). Furthermore, the reintroduction of 3LL tumor cells into the culture of MSCs that had lost arginase I induced the re-expression of arginase I within 48 h (Fig. 2 B). Similar results were obtained using peritoneal macrophages from normal mice cocultured with 3LL tumor cells or 3LL supernatants (Fig. 2 C).
Figure 2.

Arginase I expression in MSCs is induced by tumor-derived soluble factors. (A) MSCs (2 × 106) isolated from 3LL tumors were cultured alone or cocultured in transwells with 2 × 106 3LL cells or 3LL supernatants. Arginase I expression was tested via Western blot analysis at 24, 48, and 72 h. (B) MSCs (2 × 106) were cultured for 24 h in regular RPMI-1640, which results in the loss of arginase 1 expression. They were then cultured in RPMI-1640 alone or cocultured in transwells with 3LL cells (2 × 106). Arginase I expression was tested at 24, 48, or 72 h afterward via Western blot analysis. (C) Peritoneal macrophages (2 × 106) from normal mice were cocultured in transwells with 3LL cells (2 × 106) or 3LL supernatants. Arginase I expression was tested via Western blot analysis. Results shown are representative of 3 experiments.
We then tested whether 3LL cells produced cytokines that induce arginase I. An extended RNase protection assay using RNA isolated from cultured 3LL cells, and including IL-1a, IL-1b, IL-1Ra, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-11, IL-13, IL-15, MIF, LIF, SCF, G-CSF, macrophage CSF (mCSF), vascular endothelial growth factor (VEGF), GM-CSF, and IFN-γ only showed the expression of mCSF and VEGF (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20050715/DC1). However, culture of MSCs (Fig. S1 B) or peritoneal macrophages (not depicted) with increasing concentrations of mCSF or VEGF failed to induce arginase I. In addition, treatment of 3LL supernatants with various proteases—including V8 protease, chymotrypsin, trypsin, and proteinase K—only slightly decreased its ability to induce arginase I in MSCs or peritoneal macrophages (unpublished data), suggesting that cytokines were unlikely to be the inducers of arginase I.
Arginase I induction in MSCs by 3LL tumor cells is dependent on COX-2
COX-1 and COX-2 protein and messenger RNA (mRNA) were found to be constitutively expressed in 3LL cells cultured in vitro and freshly isolated 3LL tumors (Fig. 3, A and B). Consequently, PGE2 was found in the supernatants of 3LL cultures (Fig. 3 C). PGE2 is generated by the metabolism of PGH2 by three different PGE2 synthases (PGESs), one of which is expressed as a cytosolic form (cPGES) and two of which are microsomal-associated forms (mPGES-1 and -2). We found a high expression of mPGES-2 and cPGES but only a minimal expression of mPGES-1 in 3LL cells cultured in vitro and 3LL cells isolated from freshly excised tumors (Fig. 3 D).
Figure 3.

3LL cells and 3LL tumors express COX-1 and COX-2. 3LL cells cultured in vitro and single-cell suspension from subcutaneous tumors were tested for COX-1 and COX-2 protein (A) and mRNA (B). White lines indicate that intervening lanes have been spliced out. (C) Supernatants from 3LL cultured in vitro for 24 h were tested for PGE2 levels. (D) Cytoplasmic extracts from 3LL cells were also tested for PGES isoforms via Western blot analysis. Representative data are shown of experiments repeated three times.
PGE2 can induce arginase I in vitro in bone marrow–derived macrophages (14, 15). Consistent with these observations, the addition of PGE2 at concentrations found in 3LL supernatants (10 ng/ml) maintained arginase I expression in MSCs isolated from 3LL tumors (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050715/DC1). We then tested whether PGE2 produced by 3LL cells was responsible for arginase I induction in MSCs. The addition of 10 μg/ml of anti-PGE2 antibody, but not isotype control, partially prevented the up-regulation of arginase I by 3LL supernatants (Fig. 4 A). Furthermore, the addition of increasing concentrations of COX-2 inhibitor sc-58125 (16), but not COX-1 inhibitor FR122047 (17), into MSC-3LL cocultures completely blocked the induction of arginase I and arginase activity in MSCs (P < 0.0001) (Fig. 4, B and C). Similar results were obtained using other cell lines, including squamous cell carcinoma (SCC) VII (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20050715/DC1). To further confirm the role of COX-2 in the induction of arginase I we transiently (for 72 h) silenced COX-1 or COX-2 expression in 3LL cells by transfection with chemical designed small interfering RNA (siRNA) (Fig. 5 A). MSCs cocultured with COX-2–silenced 3LL cells did not express arginase I and had a lower arginase activity, similar to MSCs cultured in medium alone (Fig. 5, B and C). In contrast, MSCs cocultured with 3LL transfected with COX-1 siRNA or an irrelevant siRNA (negative control) expressed high levels of arginase I and had a higher arginase activity (P < 0.005) (Fig. 5, B and C). Experiments were repeated using peritoneal macrophages obtaining similar results (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20050715/DC1). These results support the hypothesis that prostanoids produced by 3LL tumor cells are responsible for the induction of arginase I in MSCs.
Figure 4.

Arginase I induction in MSCs by 3LL tumor cells is dependent on COX-2. (A) MSCs (2 × 106) were cultured in RPMI-1640 alone (NS or nonstimulated), 3LL supernatants, or 3LL supernatants containing 10 μg/ml anti-PGE2 or isotype control. Arginase I was tested 48 h later via Western blot analysis. (B, C) MSCs (2 × 106) were cultured in RPMI 1640 (NS) or cocultured in transwells with 3LL cells (2 × 106) in the presence of increasing concentrations (μM) of COX-1–specific (FR122047) and COX-2–specific (sc-58125) inhibitors. Arginase I expression was tested after 48 h of coculture via Western blot analysis, as was arginase activity via conversion of l-Arg to l-ornithine. *P < 0.0001 when comparing MSCs cocultured with 3LL with MSCs cocultured with 3LL in the presence of sc-58125 (20 μM).
Figure 5.
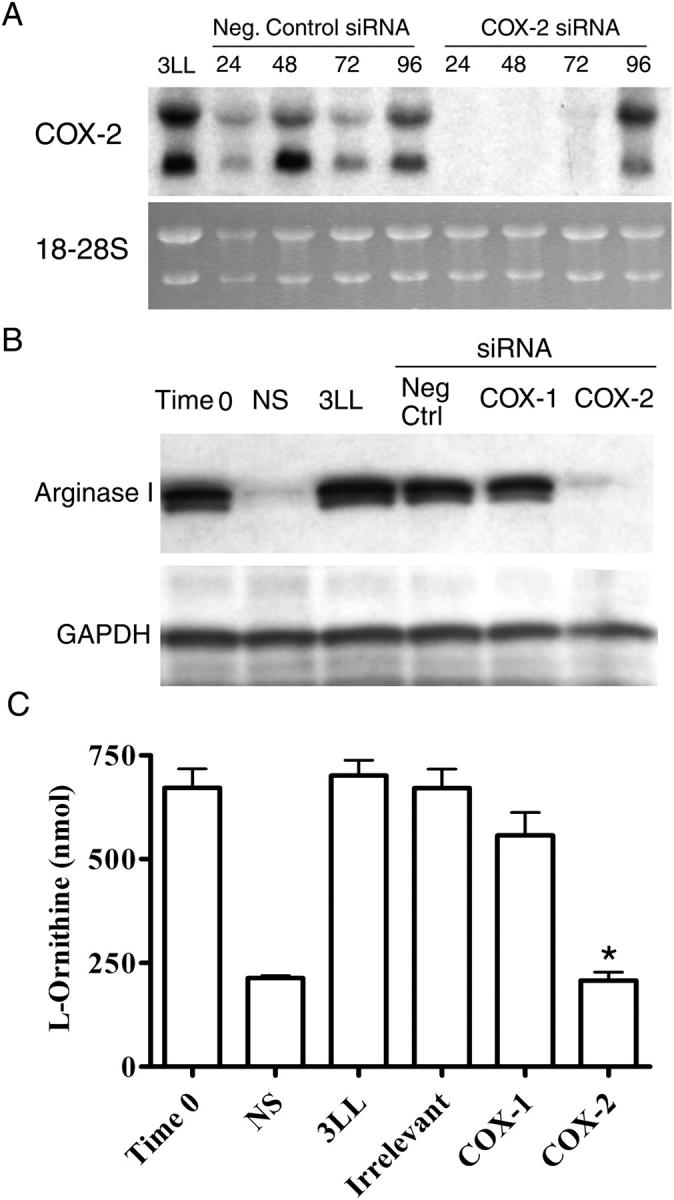
COX-2 silencing in 3LL tumor cells blocks arginase I induction. (A) 3LL cells were transfected using COX-2 siRNA or an irrelevant siRNA probe as described in Material and methods. COX-2 mRNA expression was determined via Northern blot analysis. (B, C) After 24 h of transfection, 3LL cells (2 × 106) were cocultured with freshly isolated MSCs (2 × 106) using transwells, and arginase I expression and activity were tested in MSCs after 48 h. All experiments were repeated 3 times. *P < 0.005 when comparing MSCs cocultured with 3LL with MSCs cocultured with 3LL transfected with COX-2 siRNA.
EP4 signaling induces arginase I expression
PGE2 can bind to different EP receptors (EP1, EP2, EP3, EP4) on target cells (18). The intracellular signaling pathways differ substantially and may in part explain the wide array of effects induced by PGE2. The EP1 receptor is coupled to intracellular calcium, while EP2 and EP4 receptors signal by stimulating adenylyl cyclase and cAMP. Signaling by EP3 receptor couples different signaling pathways including Gi, Gs, and calcium. MSCs strongly expressed EP2, EP3, and EP4, with low levels of EP1, whereas 3LL cells expressed EP2 and EP3 (Fig. 6 A). PGE2 analogues that differ in their affinity for EP receptors were then used to determine which EP receptor could induce arginase I in MSCs. Misoprostol at lower concentrations stimulates EP2, EP3, and EP4, whereas sulprostone and 17-phenyl-ω-trinor PGE2 are specific for EP1 and EP3. Butaprost is a selective agonist for EP2, whereas PGE1 alcohol is a selective agonist for EP3 and EP4. MSCs were stimulated for 24 h using 1 μM of each of the PGE2 agonists. Increased arginase activity and arginase I expression were induced by PGE1 alcohol and misoprostol, both being EP3 and EP4 agonists (Fig. 6, B and C). Sulprostone and 17-phenyl-ω-trinor, also EP3 agonists, did not induce arginase, which suggests that EP4 may be the receptor that preferentially signals the induction of arginase I. As described before, activation of EP4 induces increased cAMP levels. Similarly, MSCs cocultured with 3LL tumor cells had an increase in cAMP levels (unpublished data). Even though EP4 KO mice have been described previously, they were not available to the researchers to confirm this observation.
Figure 6.

EP4 activation induces arginase I in MSCs. (A) 3LL cells kept in culture and MSCs isolated from 3LL tumors were tested via Western blot analysis for EP receptor expression. (B, C) MSCs (2 × 106) were cultured in the presence of 1 μM EP analogues for 24 h, after which arginase activity and arginase I expression were tested. White lines indicate that intervening lanes have been spliced out.
COX-2 inhibition blocks the induction of arginase I in vivo and has an antitumor effect
To test the role of COX-2 in the induction of arginase I in vivo, tumor-bearing mice were injected with COX-2 inhibitor sc-58125 (5 mg/kg) or vehicle control (DMSO) every other day in the contralateral side from the tumor, starting on the day of tumor injection (1 × 106 3LL cells). After 18 d, tumors were excised and tested for arginase I expression. COX-2 inhibitor sc-58125 completely blocked the induction of arginase I in the tumor (Fig. 7 A) and also decreased tumor growth (P < 0.01) (Fig. 7, B and C). The antitumor effect induced by the COX-2 inhibitor in tumor-bearing C57BL/6 mice was dependent on the presence of competent lymphocytes, as demonstrated by the loss of the antitumor effect in C57BL/6 Prkdc scid mice (Fig. 7 C). In addition, experiments performed in CD4−/− and CD8−/− mice suggest that both CD4+ and CD8+ T cells contribute to the antitumor effect induced by inhibition of COX-2 (Fig. 7 D). Furthermore, the antitumor effect induced by sc-58125 did not appear to be mediated by an anti-angiogenic response, because mice receiving sc-58125 or vehicle showed no major differences in the expression of pro-angiogenic factor VEGF and the metastasis marker E-cadherin (Fig. 7 E).
Figure 7.

COX-2 inhibitor sc-58125 blocks arginase I induction in vivo. (A) Mice (n = 10) were injected subcutaneously with 3LL cells (1 × 106) in the right flank and received subcutaneous injections of the COX-2 inhibitor sc-58125 (5 mg/kg) or DMSO (dilution vehicle) every other day for 18 d in the left flank. Tumor was excised and tumor single-cell suspensions were tested for arginase I and COX-2 via Western blot analysis. (B–D) Tumor volume was determined in tumor-bearing C57BL/6-Prkdc scid (n = 8), CD4−/− (n = 8), and CD8 −/− (n = 8) mice receiving sc-58125 or DMSO. (E) Single-cell suspensions from tumors were lysed and tested for VEGF and E-cadherin expression. One representative sample from eight different tumors is shown.
Arginase I is induced by tumor-derived COX-2
COX-2 and prostanoid production in the tumor could come from the 3LL cells or from other host tissues such as macrophages or endothelial cells. To determine the relative contribution of tumor-derived COX-2 and host-derived COX-2 in the induction of arginase I, we injected COX-2 knockout mice with 3LL tumor cells and measured arginase I expression in MSCs. Arginase I was equally induced in both wild-type and COX-2 knockout mice (Fig. 8), suggesting that tumor-derived COX-2 and PGE2 produced by the tumor cells is more important in arginase I induction than endogenous expression of COX-2 and PGE2 by host tissues. Furthermore, no major changes in COX-2 expression were observed in tumors excised from wild-type and COX-2 knockout mice, as previously described (19).
Figure 8.

Arginase I is induced by tumor-derived COX-2. Arginase was tested in tumors harvested 18 d after tumor injection from COX-2 knockout mice. One representative sample of two different tumors is shown.
Discussion
Lung cancer remains the major cause of cancer-related deaths in the United States (20). Although immunological-based therapies have shown some success in other malignancies, lung cancer has been largely unresponsive (21, 22). This might be explained in part by the highly immunosuppressive microenvironment of lung tumors, which could induce a state of immune tolerance, blocking the efficacy of cancer immunotherapies. We recently described the presence of MSCs producing arginase I in murine and human lung carcinoma, which induced major alterations in the T cell receptor expression and T cell function (5). However, the mechanisms leading to arginase I induction in MSCs were unknown.
Even though IL-4 and IL-13 induce arginase I in murine peritoneal and bone marrow–derived macrophages, our studies failed to show a role for these cytokines (Fig. S1) or their receptors (not depicted) in the induction of arginase I by 3LL lung carcinoma. Instead, the data demonstrate that prostanoids produced by tumor cells induce and sustain arginase I production in MSC-infiltrating tumors as well as peritoneal macrophages in vitro. The data also suggest that the most likely prostanoid mediating this process is PGE2. PGE2 is generated by the metabolism of PGH2 by three different PGESs: one cytosolic form (cPGES) and two microsomal-associated forms (mPGES-1 and -2). mPGES-1 preferentially metabolizes PGH2 derived from COX-2, while mPGES-2 has similar affinity for PGH2 derived from COX-1 and COX-2 and is up-regulated in several carcinomas (23). 3LL cells expressed mPGES-2 and cPGES, but not mPGES-1. However, only the inhibition of COX-2, but not COX-1 (by pharmacological or genetic methods) inhibited arginase I induction in MSCs by the tumor cells in vitro and in vivo.
Increasing evidence supports the multifaceted effect of tumor-produced prostanoids on cancer progression. PGE2 not only enhances tumorigenesis by conferring a metastatic phenotype, increasing resistance to apoptosis and stimulating angiogenesis, it also impairs the host immune response. PGE2 has been shown to decrease IL-12 and increase IL-10 production in dendritic cells and macrophages (24–26). PGE2 may also influence a wide range of T cell functions, including inhibition of T lymphocyte activation and proliferation (27), promoting the development of a Th2 response and inhibiting the production of the Th1 cytokines IL-2 and interferon γ (28). PGE2 produced by macrophages may also decrease proliferation and inhibit T cell cytotoxic responses (29, 30). However, macrophage-derived PGE2 is not playing a role in the induction of arginase I, because the injection of 3LL in COX-2 knockout mice was similar to wild-type mice bearing tumors. The multiplicity of effects caused by PGE2 may be explained in part by the various receptors it engages. PGE2 interacts with different isoforms of transmembrane G protein-coupled receptors. Four main PGE2 EP receptor subtypes have been identified (EP1, EP2, EP3, and EP4), encoded by distinct genes, which use alternate and in some cases opposing intracellular signaling pathways (31). Our data suggest that signaling through EP4 may be responsible for arginase I induction in MSCs. The distinguishing feature of EP2 and EP4 receptors is their ability to stimulate intracellular cAMP formation. MSCs cocultured with 3LL had increased intracytoplasmic cAMP levels (unpublished data); however, EP4 can also induce other signal transduction pathways in a cAMP-independent manner (32, 33). Further evidence of the potential involvement of EP4 receptor in other tumors such as colon cancer has come from the use of an EP4-selective antagonist resulting in a decrease in the number of aberrant crypt foci and polyps (34).
Our data show that prostanoids produced by COX-2 metabolism in 3LL tumor cells induces arginase I expression, a potent mechanism of tumor evasion found in various murine tumor models in the lung and colon (2, 5). This observation is not limited to 3LL cells, because we have also found a high expression of COX-2 and the ability to induce arginase I expression in MSCs in SCC VII head and neck tumors (Fig. S3), three different renal cell carcinoma lines, and colon carcinoma MCA-38 (not depicted). Furthermore, increased COX-2 expression and PGE2 production has been reported in human colon carcinomas, breast cancer (35), renal cell carcinoma (36), and lung carcinoma (37–39), which has led to the testing of COX-2 inhibitors as a means for cancer prevention. The observations presented here provide a new explanation for the antitumor effects seen by COX 2 inhibitors. Aspirin, sulindac, and NS-398 decreased lung cancer incidence in a dose–response manner in mice exposed to tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by inhibition of COX-2 expression and induction of apoptosis (40). Indomethacin inhibited the accumulation of tumor cells in mouse lungs and subsequent growth of lung metastases (41). Celecoxib, a selective COX-2 inhibitor, dose-dependently inhibited primary tumor growth as well as the number and size of lung metastases in 3LL cells and human colon carcinoma HT29 cells (42). Meloxicam, a preferential COX-2 inhibitor, inhibited PGE2 production and the growth of non–small cell lung cancer cell lines (43). All of these studies strongly support the notion that blocking COX-2 inhibits tumor formation. It is therefore possible that one of the pathways for tumor prevention induced by the use of COX-2 inhibitors may be mediated by the inhibition of arginase I in MSCs.
An increased arginase activity has been described in patients with different types of tumors and can be produced by certain tumors or MSCs (5, 44, 45). However, the mechanisms by which arginase leads to tumor growth have been poorly understood. Macrophages expressing arginase I increased the production of polyamines, which increases tumor proliferation (10). In addition, recent data published by us and others show that infiltrating MSCs are the primary producers of arginase I and are potent inhibitors of TCR expression and antigen-specific T cell responses in vivo (2, 5, 46). The depletion of l-Arg by MSCs blocks CD3ζ expression (5) and T cell receptor signaling, and also increases the production of reactive oxygen species, which induce T cell apoptosis (46). Therefore, the increase in arginase I expression may not only facilitate tumor growth by producing more polyamines, but may also facilitate tumor escape by blocking the immune response. In contrast, low levels of l-Arg can also affect tumor growth in vitro by inducing tumor cell proliferation arrest (47, 48). However, the mechanisms by which low levels of l-Arg affect tumor growth in vivo are unknown. We have found that the injection of exogenous l-Arg without an arginase inhibitor in tumor-bearing mice potentiated tumor growth (unpublished data).
The data found here may also open new approaches to enhance the therapeutic efficacy of immunotherapy. Previous data have shown that inhibition of arginase I in vivo with the arginase inhibitor Nor-NOHA prevented tumor growth through an immunomediated mechanism (5). Here, we described that blocking COX-2 can achieve a similar response. Furthermore, De Santo et al. (49) have also shown that arginase inhibition using nitro-aspirin renders an anergic mouse, responsive a tumor vaccine resulting in a therapeutic antitumor response. In addition, inhibition of arginase can boost T cell responses in human prostate tumors (50)
The importance of arginase-producing MSCs is not limited to cancer. The association of increased arginase activity and T cell dysfunction was initially described in liver transplantation and trauma patients. This process was reversed by the enteral or parenteral supplementation of l-Arg (51). Arginase production has also been described in models of chronic infections by Helicobacter pylori (37, 52) and leishmaniasis (53, 54). In cancer patients (44, 45) the increased arginase activity was thought to come from tumor cells metabolizing l-Arg to produce polyamines to sustain rapid proliferation (10). However, as shown by Rodriguez et al. (5) in mice and more recently by Zea et al. (36) in patients with renal cell carcinoma, as well as Bronte et al. in prostate carcinoma (50), arginase comes from MSCs circulating in peripheral blood and infiltrating the tumors, respectively. However, Munder et al. (55) have suggested that arginase I is also expressed in the granulocytes of healthy individuals. We are currently studying whether arginase I is differentially regulated in the granulocytes of normal individuals and cancer patients.
Finally, the therapeutic promise of immunotherapy of cancer using cytokines, activated T cells, or cancer vaccines has been eclipsed by the presence of evasion mechanisms in the tumor. Inhibition of the immune response by limiting amino acid availability has now been described for tryptophan and l-Arg. However, it is possible that blocking arginase I through the careful use of COX-2 inhibitors and monitoring the expression of arginase I will allow us to better use the different immunotherapies at the time when tumor-induced tolerance mediated by arginase I is blocked. However, it is important to consider that the mechanisms of evasion may differ between the different types of tumors and will need to be determined before the initiation of treatment.
Materials and Methods
Cells and animals.
Lewis lung carcinoma (3LL) cells, a murine lung carcinoma cell line (American Type Culture Collection), and SCC VII, a squamous cell carcinoma cell line (provided by S. Strom, University of Maryland, Baltimore, MD), were maintained in RPMI 1640 (BioWhittaker) supplemented with 10% fetal calf serum (Hyclone), 25 mM Hepes (Gibco-Invitrogen), 4 mM l-glutamine (BioWhittaker), and 100 U/ml of penicillin/streptomycin (Gibco-Invitrogen). 6-wk-old female C57BL/6 mice, C3H/HeJ (Harlan) mice, C57BL/6 Prkdc scid mice, C57BL/6-cd4 tm1Knw (CD4 KO) mice, C57BL/6-Cd8a tm1Mak (CD8 KO) mice (The Jackson Laboratory), and C57BL/6 COX-2 knockout mice (Taconic) were injected subcutaneously with 1 × 106 3LL cells. Tumors were harvested at different time points as indicated in Results. All experiments using animals were approved by the LSU-IACUC and were performed following LSU animal care facility guidelines.
Antibodies.
Antibodies used included: COX-1 (H-62), COX-2 (M-19) (Santa Cruz Biotechnology, Inc.), arginase I (Transduction-Becton Dickinson), cPGES, mPGES-1, mPGES-2, and EP1, EP2, EP3, and EP4 (Cayman Chemical and Santa Cruz Biotechnology, Inc.). Positive controls included murine kidney (EP1) and RAW 264.7 cells stimulated with 100 ng/ml LPS (EP2, EP3, and EP4). VEGF was obtained from Santa Cruz Biotechnology, Inc., and E-cadherin was obtained from Transduction-Becton Dickinson.
Reagents.
Proteases including V8 protease, chymotrypsin, trypsin, and proteinase K (Sigma-Aldrich) were used to determine the protein nature of the 3LL cells factor that induced arginase I. Specific inhibitors of COX-1 (FR122047) (Calbiochem) (17) and COX-2 (sc-58125) (Calbiochem) (16) were used in in vitro experiments or were injected subcutaneously in mice every other day. Control mice were injected with DMSO. PGE2 agonists included misoprostol, sulprostone, 17-phenyl-ω-trinor PGE2, butaprost, and PGE1 alcohol (Cayman Chemical).
siRNA probes.
The constitutive expression of COX-1 and COX-2 in 3LL cells was silenced with chemically synthesized siRNAs. The transfection experiments were standardized using siRNA for GAPDH (Ambion) based on the number of cells to be transfected (1 × 105), the volume of transfection agent (Lipofectamine 2000, Invitrogen) and quantity of siRNA (200 nM). Chemically synthesized siRNA sequences included: COX-1, sense strand siRNA 1, 5′-GGGAAGAAACAGUUACCAGtt-3′, antisense strand siRNA, 5′-CUGGUAACUGUUUCUUCCCtt-3′; COX-2, sense strand siRNA, 5′-GGAUUUGACCAGUAUAAGUtt-3′, antisense strand siRNA, 5′-ACUUAUACUGGUCAAAUCCtg-3′. The negative control siRNA (Ambion) had no homology to known sequences from mice, rats, or humans. The expression of the targeted gene was tested after 24, 48, and 72 h via Northern blot analysis. Cocultures using transwells of the siRNA-transfected 3LL cells and MSCs were established 24 h after transfection. MSCs were harvested after 48 h and tested for arginase I expression and arginase activity. 3LL cells were also collected to reconfirm the silencing of COX enzymes. Controls included cocultures of MSCs with wild-type 3LL cells as positive control and 3LL cells transfected with GAPDH siRNA or the negative control siRNA purchased from Ambion.
Cell subset isolation from 3LL tumors.
Tumors were removed from mice under sterile conditions 14 d after tumor injection. Tumors were digested with trypsin-EDTA (Invitrogen) for 3 h, and the single cell suspension was passed through a 40-μm cell strainer (Becton Dickinson-Falcon). To isolate tumor-associated MSCs, excised tumors were stained for CD11b, CD16/CD32, and class II and separated with anti-FITC immunomagnetic beads (Miltenyi Biotec) as described previously (5). Purity ranged between 95 and 99%.
Peritoneal macrophages purification.
6-wk-old female C57BL/6 mice were used to isolate peritoneal macrophages as previously described (56). After 2 d of culture in RPMI with 4% fetal bovine serum, unattached cells were washed off and attached cells were used in cocultures.
PGE2 and PGE2 metabolites.
PGE2 and its metabolite levels were tested in supernatants and cytoplasmic extracts by EIA using kits from Cayman Chemicals, following the vendor's recommendations.
Ribonuclease protection assay.
Five micrograms of RNA isolated using TRIzol (Invitrogen) were tested for several cytokine expressions using ribonuclease protection assay (BD PharMingen) following the vendor's recommendations. In brief, RNA were mixed with the templates (BD Biosciences) and incubated at 90°C allowing the temperature to decrease slowing to 56°C. Samples were then treated with RNase followed by proteinase K and extracted using phenol-chloroform and precipitated using ethanol. Samples were separated in polyacrylamide gel containing 8 M urea, dried, and exposed to Kodak Biomax-MR films (Eastman Kodak).
Northern blot analysis.
Two million cells were used for RNA extraction using lysis with TRIzol (Invitrogen) following the manufacturer's specifications. Five micrograms of total RNA from each sample were electrophoresed under denaturing conditions, blotted onto nytran membranes (Schleicher & Schuell), and cross-linked by UV irradiation. Membranes were prehybridized at 42°C in ULTRAhyb buffer (Ambion) and hybridized overnight with 106 cpm/ml of 32P-labeled probe. Probes for detection of arginase I, COX-1, COX-2, and GAPDH (CLONTECH Laboratories, Inc.) mRNA were labeled by random priming using a RediPrime Kit (GE Healthcare) and [α-32P] dCTP (3,000 Ci/mmol; NEN Life Science Products). Membranes were washed and subjected to autoradiography at −70°C using Kodak Biomax-MR films (Eastman Kodak) and intensifying screens.
Western blot analysis.
Cell extracts were obtained as previously described (56). The expression of arginase I, COX-1, COX-2, EP receptors, PGES, E-cadherin, VEGF, and GAPDH were detected by immunoblotting using 30 μg of cell extracts. Cytoplasmic extracts were electrophoresed in 10 or 8% Tris-Glycine gels (Novex), transferred to PVDF membranes, and immunoblotted with the appropriate antibodies. The reactions were detected using an electrochemiluminescence kit (GE Healthcare).
Arginase activity assay.
Cell lysates (2 μg) were tested for arginase activity by measuring the production of l-ornithine and urea from l-Arg (56). In brief, cell lysates were added to 25 μl of Tris-HCl (50 mM; pH 7.5) containing 10 mM MnCl2. This mixture was heated at 55–60°C for 10 min to activate arginase. Then, a solution containing 150 μl carbonate buffer (100 mM) (Sigma-Aldrich) and 50 μl l-Arg (100 mM) was added and incubated at 37°C for 20 min. The hydrolysis reaction from l-Arg to l-ornithine was identified via colorimetric assay after the addition of ninhydrin solution and incubation at 95°C for 1 h. In addition, the hydrolysis reaction from l-Arg to urea was detected with diacetyl monoxime (Sigma) and incubation at 95°C for 10 min.
Statistical analysis.
Comparison of values for arginase activity, PGE2 levels, and tumor volume was performed via one-way analysis of variance using the Graph Pad statistical program (Graph Pad).
Online supplemental material.
Fig. S1 shows that the only cytokines found in the 3LL tumor cells were mCSF and VEGF (A). However, increasing concentrations of mCSF and VEGF did not maintain arginase I expression in MSCs (B). Fig. S2 shows that addition of PGE2 (10 ng/ml) maintains arginase I expression in MSCs. Fig. S3 shows that inhibition of COX-2 in SCC VII tumor cells (sc-58125, 20 μM) blocks arginase I induction in MSCs. Fig. S4 shows that inhibition of COX-2 activity and expression blocks arginase I induction in peritoneal macrophages. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050715/DC1.
Acknowledgments
This work was supported by National Institutes of Health National Cancer Institute grants RO1 CA 82689, RO1 CA 107974, and RO1 CA 88885 (to A.C. Ochoa), Seed Grant Louisiana Cancer Consortium (to A.C. Ochoa), and grant R01-10655914-NIGMS (to J.B. Ochoa).
The authors have no conflicting financial interests.
Abbreviations used: cAMP, cyclic adenosine monophosphate; COX, cyclooxygenase; EP, E-prostanoid; l-Arg, l-arginine; mCSF, macrophage CSF; mRNA, messenger RNA; MSC, myeloid suppressor cell; PGE2, prostaglandin E2; PGES, prostaglandin E2 synthase; SCC, squamous cell carcinoma; siRNA, small interfering RNA; VEGF, vascular endothelial growth factor.
References
- 1.Gabrilovich, D.I., M.P. Velders, E.M. Sotomayor, and W.M. Kast. 2001. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J. Immunol. 166:5398–5406. [DOI] [PubMed] [Google Scholar]
- 2.Bronte, V., P. Serafini, C. De Santo, I. Marigo, V. Tosello, A. Mazzoni, D.M. Segal, C. Staib, M. Lowel, G. Sutter, et al. 2003. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J. Immunol. 170:270–278. [DOI] [PubMed] [Google Scholar]
- 3.Liu, Y., J.A. Van Ginderachter, L. Brys, P. De Baetselier, G. Raes, and A.B. Geldhof. 2003. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J. Immunol. 170:5064–5074. [DOI] [PubMed] [Google Scholar]
- 4.Mazzoni, A., V. Bronte, A. Visintin, J.H. Spitzer, E. Apolloni, P. Serafini, P. Zanovello, and D.M. Segal. 2002. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J. Immunol. 168:689–695. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez, P.C., D.G. Quiceno, J. Zabaleta, B. Ortiz, A.H. Zea, M.B. Piazuelo, A. Delgado, P. Correa, J. Brayer, E.M. Sotomayor, et al. 2004. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 64:5839–5849. [DOI] [PubMed] [Google Scholar]
- 6.Zarour, H., C. De Smet, F. Lehmann, M. Marchand, B. Lethe, P. Romero, T. Boon, and J.C. Renauld. 1996. The majority of autologous cytolytic T-lymphocyte clones derived from peripheral blood lymphocytes of a melanoma patient recognize an antigenic peptide derived from gene Pmel17/gp100. J. Invest. Dermatol. 107:63–67. [DOI] [PubMed] [Google Scholar]
- 7.Pauleau, A.L., R. Rutschman, R. Lang, A. Pernis, S.S. Watowich, and P.J. Murray. 2004. Enhancer-mediated control of macrophage-specific arginase I expression. J. Immunol. 172:7565–7573. [DOI] [PubMed] [Google Scholar]
- 8.Rutschman, R., R. Lang, M. Hesse, J.N. Ihle, T.A. Wynn, and P.J. Murray. 2001. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J. Immunol. 166:2173–2177. [DOI] [PubMed] [Google Scholar]
- 9.Morris, S.M., Jr. 2004. Recent advances in arginine metabolism. Curr. Opin. Clin. Nutr. Metab. Care. 7:45–51. [DOI] [PubMed] [Google Scholar]
- 10.Chang, C.I., J.C. Liao, and L. Kuo. 2001. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 61:1100–1106. [PubMed] [Google Scholar]
- 11.Munder, M., K. Eichmann, and M. Modolell. 1998. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J. Immunol. 160:5347–5354. [PubMed] [Google Scholar]
- 12.Munder, M., K. Eichmann, J.M. Moran, F. Centeno, G. Soler, and M. Modolell. 1999. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J. Immunol. 163:3771–3777. [PubMed] [Google Scholar]
- 13.Terabe, M., S. Matsui, N. Noben-Trauth, H. Chen, C. Watson, D.D. Donaldson, D.P. Carbone, W.E. Paul, and J.A. Berzofsky. 2000. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 1:515–520. [DOI] [PubMed] [Google Scholar]
- 14.Corraliza, I.M., G. Soler, K. Eichmann, and M. Modolell. 1995. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem. Biophys. Res. Commun. 206:667–673. [DOI] [PubMed] [Google Scholar]
- 15.Modolell, M., I.M. Corraliza, F. Link, G. Soler, and K. Eichmann. 1995. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur. J. Immunol. 25:1101–1104. [DOI] [PubMed] [Google Scholar]
- 16.Sheng, H., J. Shao, S.C. Kirkland, P. Isakson, R.J. Coffey, J. Morrow, R.D. Beauchamp, and R.N. DuBois. 1997. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J. Clin. Invest. 99:2254–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochi, T., and T. Goto. 2002. Differential effect of FR122047, a selective cyclo-oxygenase-1 inhibitor, in rat chronic models of arthritis. Br. J. Pharmacol. 135:782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narumiya, S., Y. Sugimoto, and F. Ushikubi. 1999. Prostanoid receptors: structures, properties, and functions. Physiol. Rev. 79:1193–1226. [DOI] [PubMed] [Google Scholar]
- 19.Williams, C.S., M. Tsujii, J. Reese, S.K. Dey, and R.N. DuBois. 2000. Host cyclooxygenase-2 modulates carcinoma growth. J. Clin. Invest. 105:1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenlee, R.T., T. Murray, S. Bolden, and P.A. Wingo. 2000. Cancer statistics, 2000. CA Cancer J. Clin. 50:7–33. [DOI] [PubMed] [Google Scholar]
- 21.Carney, D.N., and H.H. Hansen. 2000. Non-small-cell lung cancer–stalemate or progress? N. Engl. J. Med. 343:1261–1262. [DOI] [PubMed] [Google Scholar]
- 22.Carney, D.N. 2002. Lung cancer–time to move on from chemotherapy. N. Engl. J. Med. 346:126–128. [DOI] [PubMed] [Google Scholar]
- 23.Murakami, M., H. Naraba, T. Tanioka, N. Semmyo, Y. Nakatani, F. Kojima, T. Ikeda, M. Fueki, A. Ueno, S. Oh, and I. Kudo. 2000. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 275:32783–32792. [DOI] [PubMed] [Google Scholar]
- 24.Harizi, H., M. Juzan, V. Pitard, J.F. Moreau, and N. Gualde. 2002. Cyclooxygenase-2-issued prostaglandin e(2) enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J. Immunol. 168:2255–2263. [DOI] [PubMed] [Google Scholar]
- 25.Stolina, M., S. Sharma, Y. Lin, M. Dohadwala, B. Gardner, J. Luo, L. Zhu, M. Kronenberg, P.W. Miller, J. Portanova, et al. 2000. Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J. Immunol. 164:361–370. [DOI] [PubMed] [Google Scholar]
- 26.Huang, M., M. Stolina, S. Sharma, J.T. Mao, L. Zhu, P.W. Miller, J. Wollman, H. Herschman, and S.M. Dubinett. 1998. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res. 58:1208–1216. [PubMed] [Google Scholar]
- 27.Chouaib, S., K. Welte, R. Mertelsmann, and B. Dupont. 1985. Prostaglandin E2 acts at two distinct pathways of T lymphocyte activation: inhibition of interleukin 2 production and down-regulation of transferrin receptor expression. J. Immunol. 135:1172–1179. [PubMed] [Google Scholar]
- 28.Kaur, K., S.G. Harris, J. Padilla, B.A. Graf, and R.P. Phipps. 1999. Prostaglandin E2 as a modulator of lymphocyte mediated inflammatory and humoral responses. Adv. Exp. Med. Biol. 469:409–412. [DOI] [PubMed] [Google Scholar]
- 29.Koga, Y., K. Taniguchi, C. Kubo, and K. Nomoto. 1982. Peritoneal adherent cell inhibit the generation of cytotoxic T lymphocytes with prostaglandin-mediated system. Cell. Immunol. 66:195–201. [DOI] [PubMed] [Google Scholar]
- 30.Ting, C.C., and M.E. Hargrove. 1982. Tumor cell-triggered macrophage-mediated suppression of the T-cell cytotoxic response to tumor-associated antigens. II. Mechanisms for induction of suppression. J. Natl. Cancer Inst. 69:873–878. [PubMed] [Google Scholar]
- 31.Coleman, R.A., W.L. Smith, and S. Narumiya. 1994. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 46:205–229. [PubMed] [Google Scholar]
- 32.Fujino, H., W. Xu, and J.W. Regan. 2003. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem. 278:12151–12156. [DOI] [PubMed] [Google Scholar]
- 33.Dohadwala, M., J. Luo, L. Zhu, Y. Lin, G.J. Dougherty, S. Sharma, M. Huang, M. Pold, R.K. Batra, and S.M. Dubinett. 2001. Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J. Biol. Chem. 276:20809–20812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mutoh, M., K. Watanabe, T. Kitamura, Y. Shoji, M. Takahashi, T. Kawamori, K. Tani, M. Kobayashi, T. Maruyama, K. Kobayashi, et al. 2002. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 62:28–32. [PubMed] [Google Scholar]
- 35.Sinha, P., V.K. Clements, and S. Ostrand-Rosenberg. 2005. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J. Immunol. 174:636–645. [DOI] [PubMed] [Google Scholar]
- 36.Zea, A.H., P.C. Rodriguez, M.B. Atkins, C. Hernandez, S. Signoretti, J. Zabaleta, D. McDermott, D. Quiceno, A. Youmans, A. O'Neill, et al. 2005. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 65:3044–3048. [DOI] [PubMed] [Google Scholar]
- 37.Hida, T., Y. Yatabe, H. Achiwa, H. Muramatsu, K. Kozaki, S. Nakamura, M. Ogawa, T. Mitsudomi, T. Sugiura, and T. Takahashi. 1998. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res. 58:3761–3764. [PubMed] [Google Scholar]
- 38.Riedl, K., K. Krysan, M. Pold, H. Dalwadi, N. Heuze-Vourc'h, M. Dohadwala, M. Liu, X. Cui, R. Figlin, J. T. Mao, et al. 2004. Multifaceted roles of cyclooxygenase-2 in lung cancer. Drug Resist. Updat. 7:169–184. [DOI] [PubMed] [Google Scholar]
- 39.Dannenberg, A.J., and K. Subbaramaiah. 2003. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 4:431–436. [DOI] [PubMed] [Google Scholar]
- 40.Duperron, C., and A. Castonguay. 1997. Chemopreventive efficacies of aspirin and sulindac against lung tumorigenesis in A/J mice. Carcinogenesis. 18:1001–1006. [DOI] [PubMed] [Google Scholar]
- 41.Levin, G., N. Kariv, E. Khomiak, and A. Raz. 2000. Indomethacin inhibits the accumulation of tumor cells in mouse lungs and subsequent growth of lung metastases. Chemotherapy. 46:429–437. [DOI] [PubMed] [Google Scholar]
- 42.Leahy, K.M., R.L. Ornberg, Y. Wang, B.S. Zweifel, A.T. Koki, and J.L. Masferrer. 2002. Cyclooxygenase-2 inhibition by celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res. 62:625–631. [PubMed] [Google Scholar]
- 43.Tsubouchi, Y., S. Mukai, Y. Kawahito, R. Yamada, M. Kohno, K. Inoue, and H. Sano. 2000. Meloxicam inhibits the growth of non-small cell lung cancer. Anticancer Res. 20:2867–2872. [PubMed] [Google Scholar]
- 44.Suer Gokmen, S., Y. Yoruk, E. Cakir, F. Yorulmaz, and S. Gulen. 1999. Arginase and ornithine, as markers in human non-small cell lung carcinoma. Cancer Biochem. Biophys. 17:125–131. [PubMed] [Google Scholar]
- 45.Singh, R., S. Pervin, A. Karimi, S. Cederbaum, and G. Chaudhuri. 2000. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 60:3305–3312. [PubMed] [Google Scholar]
- 46.Kusmartsev, S., and D.I. Gabrilovich. 2005. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J. Immunol. 174:4880–4891. [DOI] [PubMed] [Google Scholar]
- 47.Caso, G., M.A. McNurlan, N.D. McMillan, O. Eremin, and P.J. Garlick. 2004. Tumour cell growth in culture: dependence on arginine. Clin. Sci. (Lond.) 107:371–379. [DOI] [PubMed] [Google Scholar]
- 48.Terayama, H., T. Koji, M. Kontani, and T. Okumoto. 1982. Arginase as an inhibitory principle in liver plasma membranes arresting the growth of various mammalian cells in vitro. Biochim. Biophys. Acta. 720:188–192. [DOI] [PubMed] [Google Scholar]
- 49.De Santo, C., P. Serafini, I. Marigo, L. Dolcetti, M. Bolla, S.P. Del, C. Melani, C. Guiducci, M.P. Colombo, M. Iezzi, et al. 2005. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc. Natl. Acad. Sci. USA. 102:4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bronte, V., T. Kasic, G. Gri, K. Gallana, G. Borsellino, I. Marigo, L. Battistini, M. Iafrate, T. Prayer-Galetti, F. Pagano, et al. 2005. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med. 201:1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barbul, A. 1990. Arginine and immune function. Nutrition. 6:53–58. [PubMed] [Google Scholar]
- 52.Gobert, A.P., D.J. McGee, M. Akhtar, G.L. Mendz, J.C. Newton, Y. Cheng, H.L. Mobley, and K.T. Wilson. 2001. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc. Natl. Acad. Sci. USA. 98:13844–13849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iniesta, V., L.C. Gomez-Nieto, and I. Corraliza. 2001. The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J. Exp. Med. 193:777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iniesta, V., L. Carlos Gomez-Nieto, I. Molano, A. Mohedano, J. Carcelen, C. Miron, C. Alonso, and I. Corraliza. 2002. Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol. 24:113–118. [DOI] [PubMed] [Google Scholar]
- 55.Munder, M., F. Mollinedo, J. Calafat, J. Canchado, C. Gil-Lamaignere, J.M. Fuentes, C. Luckner, G. Doschko, G. Soler, K. Eichmann, et al. 2005. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 105:2549–2556. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez, P.C., A.H. Zea, J. DeSalvo, K.S. Culotta, J. Zabaleta, D.G. Quiceno, J.B. Ochoa, and A.C. Ochoa. 2003. L-arginine consumption by macrophages modulates the expression of CD3zeta chain in T lymphocytes. J. Immunol. 171:1232–1239. [DOI] [PubMed] [Google Scholar]