

Abstract
Raised serum levels of interferon (IFN)-α have been observed in systemic lupus erythematosus (SLE) patients, and these levels are correlated with both disease activity and severity. The origin of this IFN-α is still unclear, but increasing evidence suggests the critical involvement of activated plasmacytoid predendritic cells (PDCs). In SLE patients, DNA and RNA viruses, as well as immune complexes (ICs), that consist of autoantibodies specific to self-DNA and RNA protein particles can stimulate production of IFN-α. We have developed three series of oligonucleotide (ODN)-based inhibitors of Toll-like receptor (TLR) signaling. These ODNs include inhibitors of TLR9, inhibitors of TLR7 but not TLR9, and sequences that inhibit both TLR7 and TLR9. Specificity of these inhibitors is confirmed by inhibition of IFN-α production by PDCs in response to DNA or RNA viruses. We show that mammalian DNA and RNA, in the form of ICs, are potent self-antigens for TLR9 and TLR7, respectively, and induce IFN-α production by PDCs. This work suggests that TLRs may have a critical role in the promotion of lupus through the induction of IFN-α by PDCs. These inhibitors of TLR signaling thus represent novel therapeutic agents with potential for the treatment of lupus.
After the discovery of the protein Toll as a signaling receptor for immunity in Drosophila melanogaster, several homologous Toll-like receptors (TLRs) have been identified in mammals. TLRs are key receptors of the innate immune system and recognize a diverse range of conserved microbial molecules (1, 2). 4 out of the 10 TLRs identified in humans recognize nucleic acids, demonstrating the fundamental importance of microbial DNA and RNA in triggering innate responses to pathogenic microorganisms (3–6). TLR-mediated activation can initiate rapid and effective control of infection; however, the consequences to the host can be chronic or acute inflammation. Microbial sepsis is the best known example of this, and the roles of TLR2 and TLR4 in sepsis have been clearly demonstrated (7). TLR3 has recently been shown to be required for the central nervous system inflammation leading to the disruption of the blood–brain barrier during West Nile virus infection in mice (8). This demonstrates that the nucleic acid component of a pathogen, in this example an RNA virus, can trigger inflammation destructive to host tissues. In addition, TLR activation by endogenous ligands has been reported in some types of sterile inflammation as well (9, 10). TLR2 and TLR4 are described to respond to endogenous heat-shock proteins, TLR4 to extracellular matrix fragments, fibrinogen and β-defensin (11, 12), and TLR3 to mRNA (13), all of which may be present at elevated levels at sites of tissue injury and inflammation. Similarly, DNA–anti-DNA IgG immune complexes (ICs) have been shown to stimulate autoantibody production in mice by a process involving TLR9 (14). By analogy to the adaptive immune system, the innate immune system appears to require mechanisms for self–nonself discrimination, and failure of these mechanisms may contribute to inflammation and autoimmunity. Discrimination between nucleic acids of mammalian versus microbial origin by TLRs is particularly difficult, and the expression of the DNA- and RNA-specific TLRs in endosomal vesicles, but not on the cell surface (2), may represent one mechanism for restricting the response to nucleic acids from invading microorganisms.
Increased serum levels of IFN-α and elevated expression of a characteristic set of IFN-α–inducible genes in blood cells has been consistently observed in systemic lupus erythematosus (SLE) patients and has been shown to be correlated with disease activity (15–18). These elevated IFN-α levels may have a direct role in the pathology of lupus, because patients with nonautoimmune disorders who are treated with IFN-α can develop antinuclear antibodies, anti–double-stranded (ds) DNA antibodies, and occasionally lupus (19). The source of the elevated IFN-α in SLE may be plasmacytoid pre-DCs (PDCs) (20), the major IFN-α–producing cell type in blood. These cells appear to be chronically activated in SLE patients where a 100-fold decrease in the number of PDCs circulating in the blood has been observed (20, 21). This decrease appears caused by in vivo activation followed by cell migration into peripheral lymphoid tissues and sites of inflammation such as cutaneous lesions (22, 23).
This activation of PDCs could result from signals recognized by TLRs, because the most efficient known inducers of IFN-α by PDCs are synthetic ligands for TLR7 and TLR9 and DNA and RNA viruses, most likely acting through these same TLRs (5, 24). Furthermore, viral infections have been linked to disease exacerbations in SLE patients (18). A second possible signal for IFN-α induction in SLE is triggering by ICs of autoantibodies containing self-DNA or RNA (19, 25, 26). Clearance of apoptotic cells by macrophages is deficient in SLE patients and may promote the formation of these ICs resulting from autoantibodies reactive with autoantigens that contain nucleic acid (RNA and DNA). FcγRII is expressed on PDCs and can internalize ICs, targeting the ICs to endosomal compartments that contain TLR7 and TLR9 (27–29). Ronnblom and Alm have demonstrated the ability of both types of complexes to stimulate IFN-α production from PBMCs in vitro, although the involvement of TLR was not tested (19). The nucleic acids are key components of the anti-dsDNA and antiribonucleoprotein (anti-RNP) ICs because treatment with nucleases completely abolished their ability to induce IFN-α in this system (25), and ICs from serum not containing anti-dsDNA or anti-RNP antibodies do not induce IFN-α (29). Therefore, inhibiting the signaling through TLR7 and TLR9 could control this activation pathway that leads to IFN-α production and should result in strong benefit for lupus patients.
We and others have described several distinct subsets of atypical, nonstimulatory DNA sequences that can inhibit TLR9 stimulation by CpG-containing immunostimulatory sequences (ISS) (30–36). These sequences have been identified from diverse sources, including viral sequences, mutated CpG sequences, or repeats of the TTAGGG motif present in mammalian telomeres (30–38). Most of these studies have been confined to mouse models, but we have recently shown that such TLR9 inhibitors were also active on human cells and, importantly, could inhibit IFN-α production from PDCs in response to CpG-ISS (36). We have termed these inhibitors immunoregulatory DNA sequences (IRS), as they act to regulate immunostimulatory TLR9 ligands (36). No DNA or RNA sequences have yet been reported to inhibit TLR7-mediated activation.
We describe novel classes of oligonucleotide (ODN)-based inhibitors of TLRs, including inhibitors of both TLR7 and TLR9 signaling. We show that these IRS can inhibit IFN-α production from human PDCs in response to both DNA and RNA viruses as well as to stimulation by ICs isolated form lupus patients.
Results
Identification of inhibitors specific for mouse TLR7 and TLR9 signaling
Our prototype for TLR9 inhibitors is IRS 869 (36). This sequence efficiently inhibits TLR9 activation by ISS but has no effect when splenocytes are activated with the TLR7 ligand R848 (Fig. 1, A and B). We now describe two new classes of IRS: inhibitors of TLR7 but not TLR9 and inhibitors of stimulation through these two receptors (Fig. 1 and Table I). One type of inhibitor, represented by IRS 661, specifically reduced responses of mouse splenocytes to the TLR7 agonist R848, but not to a CpG-ISS that signal through TLR9 ligands (Fig. 1, A and B). The second new type of inhibitors, exemplified by IRS 954, was derived by combining sequence elements from both IRS 661 and IRS 869 (Table I) and provides considerable inhibition of splenocyte activation by both TLR7 and TLR9 ligands (Fig. 1). As shown in Table I, IRS 967 comprises the TLR7 inhibitor IRS 661 and the TLR9 inhibitor IRS 869 and inhibits signaling through either TLR. IRS 957 and IRS 954 show that the ODN can be shortened such that only the three 5′ residues from IRS 661 are needed to confer complete TLR7 inhibitory activity on an ODN with TLR9 inhibitory activity only. Further reduction or alteration of this motif results in progressive loss of the ability to inhibit TLR7 (IRS 986, 987, and 988). Given that many potent agonists for TLR7 are molecules the size of a single nucleotide, such as R848, and many are adenosine or guanosine analogues, perhaps it is not surprising that a motif of a few nucleotides might confer an antagonist activity. Substantially more IRS was necessary to block the response to R848 versus 1018 ISS, which may reflect the relative affinities of agonist and antagonist for these two receptors. However, cellular uptake of R848, a small molecule, and larger ODNs may be quite different, and external concentrations of each may not reflect the relative concentrations in the endosome. Interestingly, the inhibition by IRS 954 did not extend to other TLR ligands because there was little to no inhibition of CD11c+ splenocytes activated by Pam3Cys, Poly I:C, LPS, or Flagellin, which use TLR2, -3, -4, and -5, respectively (Fig. 2). We have shown that IRS could inhibit TLR9 activation in vivo (36). We then asked whether IRS 954 could also block TLR7 activation. Mice were injected with the TLR7 ligand R848, and IL-12 levels in the serum were measured 2 h after the injection. When mice were preinjected with IRS 954 with varying amounts, a substantial reduction in IL-12 response was observed, whereas the inactive control ODN did not inhibit (Fig. 3).
Figure 1.
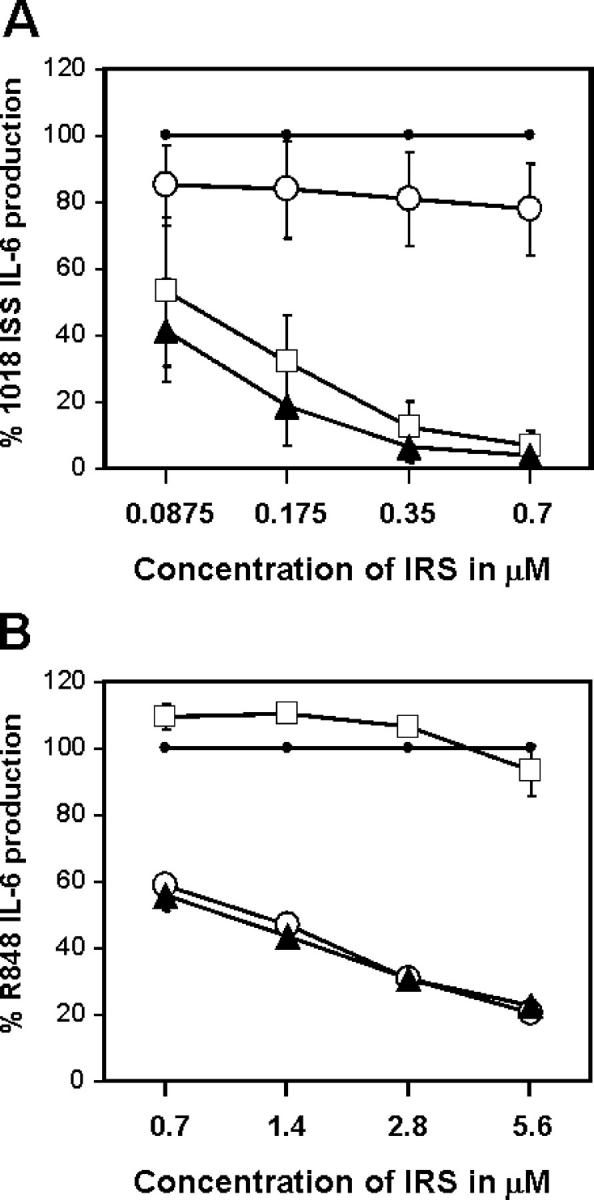
Identification of inhibitors specific for mouse TLR7 and TLR9 signaling. Splenocytes from BALB/c mice were stimulated for 48 h with (A) 0.7 μM 1018 ISS or (B) 1 μM R848 alone or in the presence of IRS 869 (open squares), IRS 661 (open circles), or IRS 954 (closed triangles) at different concentrations. IL-6 production was measured by ELISA and plotted as a percentage of 1018 ISS (A) or R848 alone (B). Maximum levels of IL-6 varied between 1,663 and 4,266 pg/ml for 1018 ISS and between 924 and 2,721 pg/ml for R848. Values represent the means ± SEM of four independent experiments.
Table I.
TLR7 and TLR9 inhibition is associated with specific sequence motifs
| 0.7 μM 1018 ISS
|
1 μM R848
|
|||||
|---|---|---|---|---|---|---|
| ODN concentration | ODN concentration | |||||
| 1.4 μM | 0.35 μM | 0.08 μM | 5.6 μM | 1.4 μM | 0.35 μM | |
| Alone | 100 | 100 | 100 | 100 | 100 | 100 |
| IRS 869: 5′-TCCTGGAGGGGTTGT-3′ | 7 | 12 | 32 | 93 | 110 | 104 |
| IRS 661: 5′-TGCTTGCAAGCTTGCAAGCA-3′ | 78 | 81 | 84 | 21 | 47 | 77 |
| IRS 967: 5′-TGCTTGACAGCTTGACAGCATCCTGGAGGGGTTGT-3′ | 1 | 2 | 8 | 16 | 49 | 80 |
| IRS 957: 5′-TGCTTGACATCCTGGAGGGGTTGT-3′ | 2 | 4 | 10 | 17 | 39 | 78 |
| IRS 954: 5′-TGCTCCTGGAGGGGTTGT-3′ | 4 | 7 | 19 | 23 | 44 | 72 |
| IRS 986: 5′-GCTCCTGGAGGGGTTGT-3′ | 1 | 2 | 33 | 38 | 85 | 128 |
| IRS 987: 5′-CTCCTGGAGGGGTTGT-3′ | 1 | 1 | 11 | 107 | 128 | 142 |
| IRS 988: 5′-AAATCCTGGAGGGGTTGT-3′ | 3 | 32 | 84 | 66 | 95 | 117 |
| Ctrl-ODN: 5′-TCCTGCAGGTTAAGT-3′ | 104 | 102 | 97 | 108 | 108 | 105 |
Splenocytes from BALB/c mice were stimulated for 48 h with 0.7 μM 1018 ISS or 1 μM R848 alone or in the presence of different IRS sequences at variable concentrations as indicated. Underlined motifs are responsible for TLR7 inhibition. IL-6 production was measured by ELISA and plotted as a percentage of 1018 ISS or R848 alone. Means of four independent experiments are shown.
Figure 2.

IRS do not affect signaling through other TLRs. Purified CD11c-positive splenocytes from BALB/c mice were stimulated for 48 h with either 5 μg/ml LPS, 0.1 μg/ml Pam3Cys, 1 μg/ml Flagellin, or 50 μg/ml Poly I:C alone or in the presence of IRS 954 at different concentrations. IL-6 production was measured by ELISA and plotted as a percentage of the stimuli alone. Means of maximum levels were 1,620 pg/ml (LPS), 2,463 pg/ml (Pam3Cys), 880 pg/ml (Flagellin), and 678 pg/ml (Poly I:C). Values represent means ± SEM of five independent experiments.
Figure 3.

IRS are active in vivo to block TLR7 stimulation. BALB/c mice were injected i.p. with inactive control ODN or various amounts of IRS 954 as indicated. 2 h later, mice were injected i.p. route R848. 2 h after the R848 injection, the serum was collected, and IL-12 was measured by immunoassay. Values represent means ± SEM of a group of 10 mice. ***, P < 0.001.
IRS retain function and specificity in human cells
We next investigated whether these IRS had similar activity and specificity in humans by examining their ability to block human B cell activation. As observed in the mouse system, IRS 869 inhibits TLR9 but not TLR7, IRS 661 inhibits TLR7 but not TLR9, and IRS 954 inhibits responses to ligands for either receptor (Fig. 4). TLR8 is also a functional receptor for R848 and RNA in humans, but not in mice (2). Although PDCs and B cells express little TLR8, other cell types in the blood do express TLR8 and can be activated by SLE serum. In particular, human monocytes, have little TLR7 but express high levels of TLR8 (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20050914/DC1). The TLR7 inhibitor IRS 661 has little to no effect on activation of highly purified monocytes by R848 (Fig. S1 B), suggesting that TLR8 responses are not inhibited by such IRS. We cannot rule out, however, that IRS are inactive in monocytes for reasons unrelated to receptor specificity.
Figure 4.

IRS retain similar specificity in human cells. Purified human B cells were stimulated with 0.7 μM 1018 ISS (left) or 1 μM R848 (right) alone or in the presence of IRS 869, IRS 661, or IRS 954 at 0.7 μM and 2.8 μM, respectively. IL-6 production was measured by ELISA and plotted as a percentage of 1018 ISS or R848 alone. Means of maximum levels of IL-6 were 2,904 pg/ml (R848) and 851 pg/ml (1018 ISS). Values represent means ± SEM of 10 (1018 ISS) and 6 (R848) independent donors. ***, P < 0.001.
IRS specific for TLR7 act independently of TLR9
The ability of short DNA sequences to inhibit signaling through TLR7, a receptor specific for single-stranded RNA, was unexpected. One possible explanation is that IRS is recognized by TLR9, and this leads to blockade of signaling through components shared by TLR7 and TLR9, such as MyD88 or IFN regulatory factor–7 (2, 39). We tested whether TLR9 was involved in the inhibition by IRS of TLR7 stimulation by testing IRS activity on cells from TLR9-deficient mice (3). Splenocytes from WT or TLR9-deficient mice were stimulated with R848 alone or in the presence of the TLR7 inhibitors IRS 661 or IRS 954. Both types of IRS decreased R848-induced IL-6 equally well in WT and TLR9-deficient mice (Fig. 5 A), excluding a role for TLR9 in the inhibition of TLR7-mediated responses. The response to 1018 ISS was inhibited by IRS 954, but not IRS 661, in WT mice and was absent in TLR9 deficient mice (Fig. 5 B).
Figure 5.

IRS specific for TLR7 act independently of TLR9. Splenocytes from C57BL/6 (closed bars) and TLR9-deficient (hatched bars) mice were stimulated for 48 h with R848 (A) or 1018 ISS (B) alone or in the presence of IRS 661 or IRS 954 at different concentrations. IL-6 production was measured by ELISA. Values represent means ± SEM of three independent experiments. **, P < 0.01; ***, P < 0.001.
IRS inhibit IFN-α from human PDCs after stimulation with DNA or RNA viruses
We have shown recently that IRS can inhibit IFN-α production from human PDCs in response to CpG-containing ISS (36). This latter activity is of particular importance to lupus, as IFN-α produced by PDCs in response to virus or ICs may play an important role in the pathogenesis of this disease. To test the ability of IRS to inhibit IFN-α induction by more complex but more physiologically relevant stimuli, purified PDCs were stimulated with either UV-irradiated HSV (a DNA virus) or inactivated influenza virus (a single-stranded RNA virus, also known as flu), both of which are strong IFN-α inducers in PDCs. Levels of IFN-α production ranged between 5 and 95 ng/ml (mean = 25.6 ng/ml) in response to HSV and between 3.5 and 63 ng/ml (mean = 30.8 ng/ml) in response to flu. IRS 954, but not IRS 661, inhibited IFN-α production in a dose-dependent manner from PDCs stimulated with HSV (Fig. 6, A and B), whereas both IRS 661 and IRS 954 could significantly inhibit IFN-α production in response to flu (P < 0.001; Fig. 6, C and D). PDCs represent only a small proportion of the total blood cells; however, similar findings were obtained using total PBMCs (unpublished data). These results demonstrate that DNA and RNA viruses can induce IFN-α by human PDCs through the stimulation of TLR9 and TLR7, respectively (2). This is consistent with studies in TLR-deficient mice showing important roles for TLR7 and TLR9 in response to flu and HSV, respectively (5, 24). In addition, we could not detect any biological response to either virus using DCs from MyD88-deficient mice (unpublished data). These results also demonstrate the ability of IRS 954 to inhibit IFN-α production by viruses, an important finding given the association of virus infections and exacerbations of lupus (18).
Figure 6.

IRS inhibit IFN-α from human PDCs after stimulation with DNA or RNA viruses. Purified human PDCs were stimulated with (A and B) UV-irradiated HSV (MOI = 5) or (C and D) heat-inactivated influenza virus (MOI = 2) alone or in the presence of different concentrations of IRS 954, IRS 661, or a control sequence. After 48 h of culture, supernatants were harvested and IFN-α was measured by ELISA and plotted as a percentage of virus stimulation alone. Means of maximum levels of IFN-α were 34,308 pg/ml (HSV) and 40,599 pg/ml (Influenza). Values represent means ± SEM of 13 (A), 12 (B), 8 (C), and 13 (D) donors. *, P < 0.1; ***, P < 0.001.
IRS inhibit IFN-α from human PDCs after stimulation with either anti-dsDNA or anti-RNP ICs
Host-derived nucleic acids, in the form of ICs, potentially represent a second type of TLR-mediated signal for the production of IFN-α by PDCs. ICs isolated from SLE patients that are composed of either anti-DNA antibodies and DNA or anti-RNP antibodies and RNP derived from apoptotic cells have been shown to be potent inducers of IFN-α by PBMCs from normal individuals (19). The roles of DNA- and RNA-specific TLRs in this induction were not evaluated; however, stimulation of proliferation and antibody production of mouse B cells by DNA-containing ICs have been shown to involve TLR9 and recognition of CpG motifs (14). This stimulation by self–nucleic acids could explain the chronic elevation of IFN-α and IFN-α–induced genes and the enhanced activation of PDCs frequently associated with lupus. Isolated PDCs from healthy donors were cultured in the presence of ICs (either anti-dsDNA or RNA-containing anti-RNP antibodies) and UV-treated apoptotic U937 cells as a source of nucleic acids and associated proteins. Sera from anti-dsDNA–positive SLE patients (Fig. 7 A) and purified IgG from anti-RNP–positive SLE patients (Fig. 7 C) induced substantial levels of IFN-α from PDCs, which demonstrates that IFN-α production induced by ICs in PBMCs originates from the PDCs. As previously described, in contrast to anti-DNA–positive serum, ICs from anti-RNP–positive patients need to be purified in order to effectively stimulate IFN-α (19). Strikingly, when added to the cultures, the TLR7 inhibitor IRS 661 efficiently blocked anti-RNP– but not anti-DNA–induced IFN-α, whereas IRS 954, which inhibits TLR7 and TLR9, blocked stimulation by both types of ICs (Fig. 7, B and D). The addition of nucleases has been shown to dramatically reduce the IFN-α response to ICs (25). This demonstrates that nucleic acids from mammalian origin together with autoantibodies can activate TLRs and induce the production of IFN-α by PDCs. DNA–anti-dsDNA ICs induced IFN-α through TLR9 because IRS 954 but not IRS 661 could block the response (Fig. 7 B), whereas RNA protein particles–anti-RNP ICs induced IFN-α through the stimulation of TLR7, as both IRS 661 and IRS 954 could inhibit the IFN-α response (Fig, 7 D).
Figure 7.

IRS inhibit IFN-α from human PDCs after stimulation with either anti-dsDNA or anti-RNP ICs. Purified human PDCs were cultured for 48 h with UV-irradiated U937 cells and (A) serum (10% final concentration) from six different anti-dsDNA–positive SLE patients, (B) serum (10% final concentration) from an anti-dsDNA–positive SLE patient (260 IU/ml of anti-dsDNA antibodies and negative for anti-RNP autoantibodies), (C) purified IgG (0.5 mg/ml final concentration) from three different anti-RNP–positive SLE patients, or (D) purified IgG (0.5 mg/ml final concentration) from an anti-RNP–positive SLE patient (142 U/ml of antiautoantibodies and negative for anti-dsDNA antibodies). (B and D) Cells were stimulated with ICs alone or in the presence of control ODN, IRS 661, or IRS 954. Patient D (B) and patient A (D) were used as sources of ICs. IFN-α was measured by ELISA. Values represent means ± SEM of eight (B) and six (D) PDC donors. *, P < 0.1; **, P < 0.01; ***, P < 0.001.
Discussion
TLRs are among the most widely expressed recognition receptors of the innate immune system and recognize a very diverse set of molecules derived from pathogenic microorganisms known as pathogen-associated molecule patterns. 10 different TLRs have been identified in humans that recognize conserved microbial components, initiate specific biological responses, and are thus essential components of the innate response to infection. Interestingly, 4 of these 10 TLRs have been implicated in the binding of nucleic acids. TLR3 recognizes dsRNA from viruses and can also be stimulated by Poly I:C; TLR7/8 recognize ssRNA; and TLR9 recognizes bacterial and viral DNA and synthetic oligonucleotides containing unmethylated CG dinucleotides (1, 2). Activation of TLRs without appropriate control, however, can lead to substantial inflammation resulting in tissue damage and autoimmunity (9, 10). Recently, Wang et al. reported that the production of proinflammatory cytokines in response to West Nile virus, a dsRNA virus, was reduced in TLR3-deficient mice. This reduction did not affect viral load in the mice but rather protected them from the TLR3-dependent inflammation that promoted destruction of the blood–brain barrier (8). Examples of TLR4 involvement in the induction of colitis or microbial sepsis are well defined (7). Other examples link TLR stimulation and atherosclerosis (10). Various mechanisms of control have been established to avoid unwanted TLR activation. Their expression is restricted to certain subsets of cells, and their level of expression is usually down-regulated after activation through a negative feedback loop. Furthermore, TLR and regulatory T cells cross-regulate each other in order to balance TLR-induced inflammation and suppression by regulatory T cells. As shown by Pasare et al., APCs, when stimulated through their TLRs, induce signals that will block the suppressive action of regulatory T cells (40). Conversely, regulatory T cells can secrete cytokines such as IL-10 that are known to inhibit TLR stimulation (41, 42).
In addition, there is increasing evidence that TLRs can also be activated by endogenous ligands (9), and it is essential that these receptors can discriminate self from nonself to prevent undesirable inflammation. For TLRs recognizing nucleic acids, the distinction between nucleic acids from mammalian versus microbial origin might be very difficult. The major difference between microbial and mammalian DNA is the level of methylation, whereas there are no clear sequential or structural differences for RNA. All four receptors that recognize RNA and DNA products are localized in intracellular compartments that could be one of the mechanisms to prevent undesirable TLR activation by endogenous RNA and DNA. Collectively, this suggests that TLR stimulation can be an important factor in the development of autoimmunity, and the ability to control TLR activation may have great therapeutic potential.
We and others have reported oligonucleotide sequences that can inhibit TLR9 activation mediated by CpG-containing oligonucleotides (30–36). These sequences are active on both mouse and human cells in vitro and in mice in vivo. We recently extended this to show that IRS could prevent IFN-α production from PDCs after TLR9 activation (36). Furthermore, coinjection of IRS with ISS in mice treated with D-galactosamine prevented death, demonstrating that IRS are potent enough in vivo to prevent massive systemic inflammation caused by TLR9 activation (36). Some IRS have been shown to have efficacy in vivo in severe autoimmune models (34, 37, 38, 43). However, the use of complete Freund's adjuvant in some of these models, which includes mycobacterial DNA, renders these experiments difficult to interpret, because the observed effect of IRS may be by inhibiting the adjuvant and not the resulting pathology. Further experiments using complete Freund's adjuvant–free models will be necessary to address the potential of IRS to prevent autoimmunity. We have now developed three series of IRS, including inhibitors of TLR9, inhibitors of TLR7 but not TLR9, and, most interestingly, sequences that inhibit signals through these two receptors. The mechanism of action of IRS is not known; however, evidence to date and the pattern of specificity of these inhibitors suggest that the effect is targeted to the receptor either through competitive antagonism or another mechanism. We have yet to find any activity of IRS when used alone, and broader assays such as microarray analysis may be necessary to conclude whether IRS have direct activities.
SLE affects >1,000,000 people in the United States alone, primarily young and middle-aged women. The etiology of the disease is unknown, but a strong genetic component appears to be involved. SLE is a relapsing, remitting disease with devastating consequences and is poorly treated or prevented with existing therapies. Patients suffer from kidney dysfunction leading to renal failure and a wide and variable range of symptoms, including arthritis, fever, skin rashes, and brain inflammation. ICs of autoantibodies to chromatin and RNA protein particles (snRNP) are diagnostic for SLE and are thought to play an important role in the pathogenesis of the disease. Increased serum levels of IFN-α have been observed in many SLE patients and correlate with both disease activity and key disease markers, such as anti-DNA antibodies (15–17, 19). Furthermore, a set of characteristic IFN-α–inducible genes are constitutively up-regulated in blood cells of SLE patients (20, 44–46). These elevated IFN-α levels may have a direct role in the pathology of lupus because patients with nonautoimmune disorders who are treated with IFN-α can develop antinuclear antibodies, anti-dsDNA antibodies, and occasionally SLE as well. Viral infections, UV skin injury, or other events leading to IFN-α induction are known to be activators of flares of SLE. In addition, NZB mice, which spontaneously develop a lupus-like disease, have less severe disease with delayed onset when made deficient for the IFN-α receptor (47).
There is considerable evidence that chronically activated PDCs and the IFN-α that they produce in response to TLR stimulation are involved in the pathogenesis of SLE. Patients with SLE have a 50–100-fold decrease in the number of PDCs circulating in the blood (20, 21). This decrease appears caused by in vivo activation followed by cell migration into peripheral lymphoid tissues and sites of inflammation. Indeed, activated PDCs have been observed to accumulate in cutaneous lupus erythematosus lesions (22, 23). These cells, when activated with viruses, can produce large amounts of IFN-α. In addition, it was recently shown that ICs present in serum samples from SLE patients can cause the production of IFN-α by PBMCs in vitro. A growing body of evidence supports the idea that TLR activation plays a central role in the maintenance and progression of the disease by promoting elevated IFN-α levels. TLR7 and TLR9 are particularly relevant to SLE, as they are expressed by human PDCs, and stimulation through these receptors leads to very high levels of IFN-α production by PDCs. Exogenous viruses acting through these TLRs would be expected to induce IFN-α and thus exacerbate the disease, and this is consistent with the observed association of lupus flares with viral infections. We show that ICs associated with self-DNA and RNA can directly activate PDCs to make IFN-α. The recognition by TLRs is likely facilitated by the expression of FcγRII on PDCs, allowing efficient uptake of the self–nucleic acid into the TLR-containing endosomal compartments (29, 48). These ICs are thus endogenous IFN-α inducers, and the resulting IFN-α production could perpetuate the autoimmune process. In addition, the role of ICs in the pathogenesis of lupus is complex, and a similar mode of activation could occur for B cells, as previously shown in mice (14).
We show that immunoregulatory sequences such as IRS 954 can inhibit signaling through TLR7 and TLR9 and decrease IFN-α produced by PDCs either in response to DNA and RNA viruses or to both types of circulating ICs isolated from lupus patients. IRS 954 also inhibits the activation of B cells through TLR7/8 and TLR9, a process that, in concert with a signal through the B cell receptor, can lead to production of autoantibodies specific for both the nucleic acid and immunoglobulin components of ICs (14). Our work demonstrates that mammalian RNA and DNA, when complexed with autoantibodies, can represent potent self-antigens for TLR7 or TLR9, respectively, and that this inappropriate self-recognition by the innate immune system may play a substantial role in autoimmune diseases such as lupus. Treatment with an IRS has the potential to modulate the major source of excessive IFN-α in SLE, without completely preventing the acute IFN-α responses to viral infection mediated by other recognition mechanisms, such as TLR3 and protein kinase R. This approach could thus be less immunosuppressive than therapies aimed at blocking IFN-α interaction with its receptor; however, this will have to be evaluated in further studies. Using inhibitors of TLR represents a new approach to treatment of SLE with the potential to reduce symptoms and prevent relapses through inhibition of a key step in disease pathogenesis.
Materials and methods
ODN synthesis
Phosphorothioate ODNs were prepared as previously described (42). The prototypes for the IRS classes used were IRS 869 (5′-TCCTGGAGGGGTTGT-3′), IRS 661 (5′-TGCTTGCAAGCTTGCAAGCA-3′), and IRS 954, 5′-TGCTCCTGGAGGGGTTGT-3′. Control oligonucleotides were 5′-TCCTGCAGGTTAAGT-3′and 5′-TCCTGGCGGAAAAGT-3′. CpG-containing ISS was 1018 (5′-TGACTGTGAACGTTCGAGATGA-3′). All ODNs had <5 endotoxin U/mg ODN, determined by Limulus amebocyte lysate assay (BioWhittaker).
Purification of ICs of SLE patients
Serum samples from SLE patients were obtained from Golden West Biologicals, the Binding Site, and SeraCare. All samples were filtered through a 0.45-μm polyvinylidene fluoride syringe before purification. Anti-dsDNA and anti-RNP titer levels were measured using commercially available ELISA kits (Corgenix). Samples were compared with a positive control provided by the manufacturer. Positive anti-dsDNA samples had an activity greater than 122 IU/ml. Positive anti-RNP samples had an activity greater than 22 U/ml. IgG from anti-RNP–positive patients was purified using HiTrap Protein G HP column (GE Healthcare). Once purified, IgG were desalted and then quantified.
In vitro stimulation of mouse cell subsets
Spleens from 6- to 12-wk-old BALB/c mice were harvested, and the splenocytes were prepared using standard techniques. Cells were stimulated with 0.7 μM 1018 ISS, 1 μM R848, 5 μg/ml LPS, 0.1 μg/ml Pam3Cys, 1 μg/ml Flagellin, or 50 μg/ml Poly I:C (Invivogen) in the presence of the amount of IRS indicated in the figures. Experiments with mice were approved by the Institutional Animal Care and Use Committee of Northview Pacific Laboratories.
In vivo experiments
6–12-wk-old BALB/c mice were used for all in vivo experiments. Mice were first injected i.p. with variable quantities of IRS as mentioned in the figure legends, followed 2 h later by 2.5 μg R848. All injections used ODN in saline. 2 h after injections, blood was harvested, and serum was prepared using standard procedures.
Isolation and in vitro stimulation of purified human PDCs
Buffy coats were obtained from the Stanford Blood Center. All cells were used under protocols approved by the Institutional Review Board. PDCs were isolated using BDCA-4 enrichment as previously described (42). Purity was routinely >97%. PDC experiments were conducted with 3–5 × 104 PDC/well cultured in 96-well roundbottom plates. HSV-1 (gift from R. Pyles, University of Texas Medical Branch, Galveston, TX) was UV inactivated and used at a multiplicity of infection (MOI) of 5. Influenza virus (H1N1, strain A/PR/8/34) from American Type Culture Collection was inactivated for 30 min at 56°C and used at an MOI of 2. Alternatively, 30,000 PDCs were cultured with 50,000 UV-irradiated (60 mJ) U937 cells in the presence of 10% serum from anti-dsDNA–positive SLE patients or 0.5 mg/ml of purified IgG from anti-RNP–positive SLE patients. Cells were cultured for 48 h and supernatants harvested for ELISA.
Isolation and in vitro stimulation of purified human B cells and monocytes
B cells and monocytes were isolated using CD19 and CD14 enrichment, respectively, as previously described (36). Purity was routinely 99%. Experiments were conducted with 2–4 × 105 B cells or 2 × 105 monocytes per well cultured in 96-well flatbottom plates.
Real-time quantitative PCR (TaqMan) analysis
RNA from purified human monocytes was isolated and converted to cDNA, and PCR reactions were performed as described previously (42). The human sequences for synthesized primers and the calculation method were as previously described (42). In brief, threshold cycle (CT) values for each gene were normalized to the housekeeping gene HPRT using the formula 1.8 × (HKG−GENE) × 1,000, where HKG is the mean CT of triplicate HPRT runs, GENE is the mean CT of duplicate runs of the gene of interest, and 1,000 is arbitrarily chosen as a factor to bring all values above 0.
Online supplemental material.
Fig. S1 shows the effect of TLR7 inhibitors on purified human monocytes activated through TLR8. Expression levels of TLR7, TLR8, and TLR9 were evaluated using quantitative PCR. IL-6 and TNF-α production were measured by ELISA. Online supplemental material is available at http://www.jem.org/cgi.content/full/jem.20050914/DC1.
Acknowledgments
We would like to thank our colleagues at Dynavax Technologies for their critical reading of the manuscript.
This work was supported by the Alliance for Lupus Research.
The authors have no conflicting financial interests.
Abbreviations used: CT, threshold cycle; ds, double-stranded; IC, immune complex; IRS, immunoregulatory DNA sequences; ISS, immunostimulatory sequences; MOI, multiplicity of infection; ODN, oligonucleotide; PDC, plasmacytoid pre-DC; RNP, ribonucleoprotein; SLE, systemic lupus erythematosus; TLR, Toll-like receptor.
References
- 1.Janeway, C.A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. [DOI] [PubMed] [Google Scholar]
- 2.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511. [DOI] [PubMed] [Google Scholar]
- 3.Hemmi, H., O. Takeuchi, T. Kawai, T. Kaisho, S. Sato, H. Sanjo, M. Matsumoto, K. Hoshino, H. Wagner, K. Takeda, and S. Akira. 2000. A Toll-like receptor recognizes bacterial DNA. Nature. 408:740–745. [DOI] [PubMed] [Google Scholar]
- 4.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 5.Diebold, S.S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 303:1529–1531. [DOI] [PubMed] [Google Scholar]
- 6.Heil, F., H. Hemmi, H. Hochrein, F. Ampenberger, C. Kirschning, S. Akira, G. Lipford, H. Wagner, and S. Bauer. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 303:1526–1529. [DOI] [PubMed] [Google Scholar]
- 7.Beutler, B. 2004. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 430:257–263. [DOI] [PubMed] [Google Scholar]
- 8.Wang, T., T. Town, L. Alexopoulou, J.F. Anderson, E. Fikrig, and R.A. Flavell. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10:1366–1373. [DOI] [PubMed] [Google Scholar]
- 9.Andreakos, E., B. Foxwell, and M. Feldmann. 2004. Is targeting Toll-like receptors and their signaling pathway a useful therapeutic approach to modulating cytokine-driven inflammation? Immunol. Rev. 202:250–265. [DOI] [PubMed] [Google Scholar]
- 10.Rifkin, I.R., E.A. Leadbetter, L. Busconi, G. Viglianti, and A. Marshak-Rothstein. 2005. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 204:27–42. [DOI] [PubMed] [Google Scholar]
- 11.Smiley, S.T., J.A. King, and W.W. Hancock. 2001. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 167:2887–2894. [DOI] [PubMed] [Google Scholar]
- 12.Biragyn, A., P.A. Ruffini, C.A. Leifer, E. Klyushnenkova, A. Shakhov, O. Chertov, A.K. Shirakawa, J.M. Farber, D.M. Segal, J.J. Oppenheim, and L.W. Kwak. 2002. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 298:1025–1029. [DOI] [PubMed] [Google Scholar]
- 13.Kariko, K., H. Ni, J. Capodici, M. Lamphier, and D. Weissman. 2004. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 279:12542–12550. [DOI] [PubMed] [Google Scholar]
- 14.Leadbetter, E.A., I.R. Rifkin, A.M. Hohlbaum, B.C. Beaudette, M.J. Shlomchik, and A. Marshak-Rothstein. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 416:603–607. [DOI] [PubMed] [Google Scholar]
- 15.Hooks, J.J., H.M. Moutsopoulos, S.A. Geis, N.I. Stahl, J.L. Decker, and A.L. Notkins. 1979. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 301:5–8. [DOI] [PubMed] [Google Scholar]
- 16.Ytterberg, S.R., and T.J. Schnitzer. 1982. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 25:401–406. [DOI] [PubMed] [Google Scholar]
- 17.Bengtsson, A.A., G. Sturfelt, L. Truedsson, J. Blomberg, G. Alm, H. Vallin, and L. Ronnblom. 2000. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 9:664–671. [DOI] [PubMed] [Google Scholar]
- 18.Banchereau, J., V. Pascual, and A.K. Palucka. 2004. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 20:539–550. [DOI] [PubMed] [Google Scholar]
- 19.Ronnblom, L., and G.V. Alm. 2003. Systemic lupus erythematosus and the type I interferon system. Arthritis Res. Ther. 5:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanco, P., A.K. Palucka, M. Gill, V. Pascual, and J. Banchereau. 2001. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 294:1540–1543. [DOI] [PubMed] [Google Scholar]
- 21.Cederblad, B., S. Blomberg, H. Vallin, A. Perers, G.V. Alm, and L. Ronnblom. 1998. Patients with systemic lupus erythematosus have reduced numbers of circulating natural interferon-alpha-producing cells. J. Autoimmun. 11:465–470. [DOI] [PubMed] [Google Scholar]
- 22.Farkas, L., K. Beiske, F. Lund-Johansen, P. Brandtzaeg, and F.L. Jahnsen. 2001. Plasmacytoid dendritic cells (natural interferon-alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am. J. Pathol. 159:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blomberg, S., M.L. Eloranta, B. Cederblad, K. Nordlin, G.V. Alm, and L. Ronnblom. 2001. Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus. 10:484–490. [DOI] [PubMed] [Google Scholar]
- 24.Lund, J., A. Sato, S. Akira, R. Medzhitov, and A. Iwasaki. 2003. Toll-like receptor 9–mediated recognition of herpes simplex virus–2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallin, H., A. Perers, G.V. Alm, and L. Ronnblom. 1999. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J. Immunol. 163:6306–6313. [PubMed] [Google Scholar]
- 26.Bave, U., G.V. Alm, and L. Ronnblom. 2000. The combination of apoptotic U937 cells and lupus IgG is a potent IFN-alpha inducer. J. Immunol. 165:3519–3526. [DOI] [PubMed] [Google Scholar]
- 27.Regnault, A., D. Lankar, V. Lacabanne, A. Rodriguez, C. Thery, M. Rescigno, T. Saito, S. Verbeek, C. Bonnerot, P. Ricciardi-Castagnoli, and S. Amigorena. 1999. Fcγ receptor–mediated induction of dendritic cell maturation and major histocompatibility complex class I–restricted antigen presentation after immune complex internalization. J. Exp. Med. 189:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalergis, A.M., and J.V. Ravetch. 2002. Inducing tumor immunity through the selective engagement of activating Fcγ receptors on dendritic cells. J. Exp. Med. 195:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Means, T.K., E. Latz, F. Hayashi, M.R. Murali, D.T. Golenbock, and A.D. Luster. 2005. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieg, A.M., T. Wu, R. Weeratna, S.M. Efler, L. Love-Homan, L. Yang, A.K. Yi, D. Short, and H.L. Davis. 1998. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc. Natl. Acad. Sci. USA. 95:12631–12636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada, H., I. Gursel, F. Takeshita, J. Conover, K.J. Ishii, M. Gursel, S. Takeshita, and D.M. Klinman. 2002. Effect of suppressive DNA on CpG-induced immune activation. J. Immunol. 169:5590–5594. [DOI] [PubMed] [Google Scholar]
- 32.Zhu, F.G., C.F. Reich, and D.S. Pisetsky. 2002. Inhibition of murine dendritic cell activation by synthetic phosphorothioate oligodeoxynucleotides. J. Leukoc. Biol. 72:1154–1163. [PubMed] [Google Scholar]
- 33.Stunz, L.L., P. Lenert, D. Peckham, A.K. Yi, S. Haxhinasto, M. Chang, A.M. Krieg, and R.F. Ashman. 2002. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur. J. Immunol. 32:1212–1222. [DOI] [PubMed] [Google Scholar]
- 34.Ho, P.P., P. Fontoura, P.J. Ruiz, L. Steinman, and H. Garren. 2003. An immunomodulatory GpG oligonucleotide for the treatment of autoimmunity via the innate and adaptive immune systems. J. Immunol. 171:4920–4926. [DOI] [PubMed] [Google Scholar]
- 35.Gursel, I., M. Gursel, H. Yamada, K.J. Ishii, F. Takeshita, and D.M. Klinman. 2003. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J. Immunol. 171:1393–1400. [DOI] [PubMed] [Google Scholar]
- 36.Duramad, O., K.L. Fearon, B. Chang, J.H. Chan, J. Gregorio, R.L. Coffman, and F.J. Barrat. 2005. Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflamation. J. Immunol. 174:5193–5200. [DOI] [PubMed] [Google Scholar]
- 37.Zeuner, R.A., K.J. Ishii, M.J. Lizak, I. Gursel, H. Yamada, D.M. Klinman, and D. Verthelyi. 2002. Reduction of CpG-induced arthritis by suppressive oligodeoxynucleotides. Arthritis Rheum. 46:2219–2224. [DOI] [PubMed] [Google Scholar]
- 38.Dong, L., S. Ito, K.J. Ishii, and D.M. Klinman. 2004. Suppressive oligonucleotides protect against collagen-induced arthritis in mice. Arthritis Rheum. 50:1686–1689. [DOI] [PubMed] [Google Scholar]
- 39.Honda, K., H. Yanai, H. Negishi, M. Asagiri, M. Sato, T. Mizutani, N. Shimada, Y. Ohba, A. Takaoka, N. Yoshida, and T. Taniguchi. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 434:772–777. [DOI] [PubMed] [Google Scholar]
- 40.Pasare, C., and R. Medzhitov. 2003. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 299:1033–1036. [DOI] [PubMed] [Google Scholar]
- 41.Moore, K.W., R. de Waal Malefyt, R.L. Coffman, and A. O'Garra. 2001. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19:683–765. [DOI] [PubMed] [Google Scholar]
- 42.Duramad, O., K.L. Fearon, J.H. Chan, H. Kanzler, J.D. Marshall, R.L. Coffman, and F.J. Barrat. 2003. IL-10 regulates plasmacytoid dendritic cell response to CpG-containing immunostimulatory sequences. Blood. 102:4487–4492. [DOI] [PubMed] [Google Scholar]
- 43.Dong, L., S. Ito, K.J. Ishii, and D.M. Klinman. 2005. Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus-prone NZB x NZW mice. Arthritis Rheum. 52:651–658. [DOI] [PubMed] [Google Scholar]
- 44.von Wussow, P., D. Jakschies, H. Hochkeppel, M. Horisberger, K. Hartung, and H. Deicher. 1989. MX homologous protein in mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 32:914–918. [PubMed] [Google Scholar]
- 45.Bennett, L., A.K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, and V. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 197:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baechler, E.C., F.M. Batliwalla, G. Karypis, P.M. Gaffney, W.A. Ortmann, K.J. Espe, K.B. Shark, W.J. Grande, K.M. Hughes, V. Kapur, et al. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA. 100:2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santiago-Raber, M.L., R. Baccala, K.M. Haraldsson, D. Choubey, T.A. Stewart, D.H. Kono, and A.N. Theofilopoulos. 2003. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J. Exp. Med. 197:777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bave, U., M. Magnusson, M.L. Eloranta, A. Perers, G.V. Alm, and L. Ronnblom. 2003. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J. Immunol. 171:3296–3302. [DOI] [PubMed] [Google Scholar]