

Abstract
The molecular immunopathogenesis of West Nile virus (WNV) infection is poorly understood. Here, we characterize a mouse model for WNV using a subcutaneous route of infection and delineate leukocyte subsets and immunoregulatory factors present in the brains of infected mice. Central nervous system (CNS) expression of the chemokine receptor CCR5 and its ligand CCL5 was prominently up-regulated by WNV, and this was associated with CNS infiltration of CD4+ and CD8+ T cells, NK1.1+ cells and macrophages expressing the receptor. The significance of CCR5 in pathogenesis was established by mortality studies in which infection of CCR5−/− mice was rapidly and uniformly fatal. In the brain, WNV-infected CCR5−/− mice had increased viral burden but markedly reduced NK1.1+ cells, macrophages, and CD4+ and CD8+ T cells compared with WNV-infected CCR5+/+ mice. Adoptive transfer of splenocytes from WNV-infected CCR5+/+ mice into infected CCR5−/− mice increased leukocyte accumulation in the CNS compared with transfer of splenocytes from infected CCR5−/− mice into infected CCR5−/− mice, and increased survival to 60%, the same as in infected CCR5+/+ control mice. We conclude that CCR5 is a critical antiviral and survival determinant in WNV infection of mice that acts by regulating trafficking of leukocytes to the infected brain.
West Nile virus (WNV) is an enveloped, neurotropic, single stranded (+) sense RNA flavivirus, first isolated from a woman in Uganda in 1937 (1). Since then it has reemerged multiple times: first in Europe, and most recently during the summer of 1999 in the northeastern United States (2, 3). The virus, which has spread rapidly throughout North America, may cause severe illness especially in the elderly (manifested by meningitis and encephalitis) that may lead to paralysis, coma, and death. During the 1999–2003 outbreaks of WNV in the USA, 13,491 cases were reported, including 533 deaths (∼4%) (3, 4).
A zoonosis, WNV is maintained in a natural cycle between avian hosts and mosquito vectors, although humans, horses, and several other mammals can also serve as hosts (5–8). The level of exposure required for infection and disease is undefined; however, epidemiologic studies indicate that ∼80% of WNV infection remains subclinical whereas ∼20% progresses to a febrile illness. Of these, >30% of cases in the US progress to meningitis and/or encephalitis. Survivors of severe neurological disease typically require prolonged rehabilitation. There is no vaccine or specific antiviral treatment for WNV infection in humans.
Although immunocompromised hosts appear to have increased susceptibility to WNV, specific information on the human innate and adaptive cellular immune response to the virus is limited (2, 9–11). Several mouse models of infection that differ with regard to mouse and viral strain, route of infection, and clinical outcome have been developed to provide insight into immunopathogenesis (12–15). We have used a model in which the highly neurotropic and neurovirulent WNV-NY99 strain is injected s.c., resulting in viral replication and leukocyte infiltration in the brain 4–7 d after infection (13, 16–19). A variant of this model has been used previously to demonstrate that both B and T cells are required for viral clearance, because 100% of both RAG1−/− mice and μMT−/− mice rapidly succumb to viral infection, whereas only ∼35% of mice with intact immune systems die (19, 20). The primary role of humoral immunity is thought to limit viral dissemination to the central nervous system (CNS). CD8+ T cells play a prominent role in viral clearance from the CNS, as infected CD8−/− mice have increased mortality (∼85%) and higher CNS viral burden and viral persistence (13).
Encephalitis caused by other neurotropic/neurovirulent viruses is often characterized by CNS entry of macrophages, CD4+ and CD8+ T cells, NKT cells and dendritic cells. However, little information is available regarding the molecular factors responsible for their recruitment. Here, we provide a profile of the leukocyte subsets and proinflammatory and immunoregulatory molecules present in the CNS of WNV-infected mice. We have used this profile to identify the chemokine receptor CCR5 as a candidate protective factor, and have tested this hypothesis genetically using CCR5 knockout mice.
Results
C57BL/6 mice were infected s.c. with 100–106 focus-forming units (ffu) of strain WNV-NY99, which imitates the natural route of WNV infection in man. All mice infected with 1 or 10 ffu survived. Mortality was first observed in animals injected with 100 ffu and was 30% at that dose (unpublished data). Mortality was relatively insensitive to further increases in WNV dose reaching only 40% at 106 ffu, the highest dose tested. This is consistent with previously published data for WNV-NY99 infection of this strain of mice using a footpad injection (19). Based on these results, we infected mice with 104 ffu WNV s.c. for all subsequent studies. Using this dose, all deaths occurred between 7 and 12 d after infection.
WNV infection induces immunoregulatory mediators in the CNS
To identify molecules that might be important in WNV encephalitis, we screened a total of 45 different immunoregulatory factors for differential expression in the brains of mock- and WNV-infected mice on days 7 and 12 after infection. Of these, 37 (9 cytokines, 8 cytokine receptors, 11 chemokines, and 9 chemokine receptors) were screened at the RNA level by either RPA or RT-PCR of mouse brain (Fig. 1, A and B) and positive samples were quantified using GAPDH or β-actin signals for normalization. Although there was some variability in expression among the mice tested, we accepted as candidates those factors for which expression was increased in at least one mouse. 13 of these factors were expressed in a WNV-dependent manner. 8 out of the 13 (IFNγ, IL-12 receptor β1 and the chemokines CXCL10, MIP-2, CCL2, CCL3, CCL4, and CCL5) were not detected in the brains of mock-infected mice, but were induced specifically by WNV; the other 5 were all chemokine receptors (CCR1, CCR2, CCR5, CXCR3, CX3CR1) and were constitutively expressed in mock-infected mouse brains but were further induced by WNV. 12 out of these 13 factors typically are associated with Th1 immune responses. The major Th2 cytokines IL-4, IL-5, and IL-13 were not detected in the brains from either mock- or virus-infected animals. Thus, WNV appears to generate a Th1 cell–polarized immune response in this model. We identified 10 other factors constitutively expressed at the RNA level that were not further induced by WNV (IL-10 and its receptor; IL-15; IL-11 receptor; gp130; α and β chains of the IFN-γ receptor; the chemokines CXCL4 and CX3CL1; and the chemokine receptor CXCR4).
Figure 1.

West Nile virus induces production of specific immunoregulatory factors in the brains of C57BL/6 mice. (A) RNA analysis by RPA. Leftmost lane in each of the four panels is unprotected probe, and the factors analyzed are named to the left; note that the protected fragments run slightly lower on gel. The names of factors scoring positive in the assay are given to the right of each band. GAPDH is provided as an RNA loading control. Time after infection (days), is indicated at the top of each panel. Mock-infected mice were analyzed on day 7 of the experiment. Each lane on each panel corresponds to RNA from a single mouse brain. The results shown are from two independent experiments with six mice analyzed for each time point, except for mock-infected mice (n = 3). Lanes corresponding in location on each of the four gels represent RNA from the same mouse. (B) RNA analysis by RT-PCR. β-Actin is provided as a loading control. Mock-infected mice were analyzed at day 7. Each lane represents a single mouse. (C) Protein analysis. The factor analyzed is identified at the top left of each graph. Time and virus variables for each factor are specified at the bottom of the panel. Data were pooled from three experiments with three to five in each group, and are presented as mean ± SEM pg/g protein in supernatants from total brain homogenates as a function of time after infection.
Nine of the factors up-regulated at the RNA level were also tested at the protein level and were positive (Fig. 1 C). Of these, seven were factors induced by WNV (IFN-γ and the chemokines CCL2–5, MIP-2, and CXCL10). CCL5 was particularly strongly and durably induced. We screened an additional seven factors in the brain at the protein level and found that all were highly induced by WNV. Three of the seven—IFN-α, TNF-α and CXCL9—were not constitutively detected. IFN-α and TNF-α expression was transient, with large amounts of cytokine found on day 7 after infection, but not on day 12. Constitutive expression of the other four factors (IL-1β, the adhesion molecule ICAM-1, and the metalloproteinase MMP-9 and its inhibitor TIMP-1) was markedly increased by virus and remained elevated throughout the 12 d period of observation. These molecules may serve to arrest activated leukocytes in cerebral blood vessels and break down the blood brain barrier to facilitate leukocyte penetration into infected brain tissue. With regard to candidate leukocyte chemotactic systems that might guide this process, our data point clearly to the non-ELR CXC chemokines CXCL9 and CXCL10 and their receptor CXCR3; and the CC chemokines CCL2, CCL3, CCL4, and CCL5 and their receptors CCR1, CCR2, and CCR5. The RNA data suggest that the CX3C chemokine CX3CL1 and its receptor CX3CR1 may also play a role. For the remainder of this paper, we focus our analysis on the role of CCR5 in WNV pathogenesis.
CCR5+ leukocytes accumulate in WNV-infected mouse brain
We examined histopathological sections of mock- and WNV-infected mice and visualized intense multifocal cellular infiltrates, primarily localized to hippocampus and external capsule, evident in WNV-infected but not mock-infected mouse brain stained either by hematoxylin and eosin or anti-CD3 (Fig. 2, A and B). Previously published work (21, 22), confirmed by us (unpublished data), has shown that WNV infects neurons.
Figure 2.
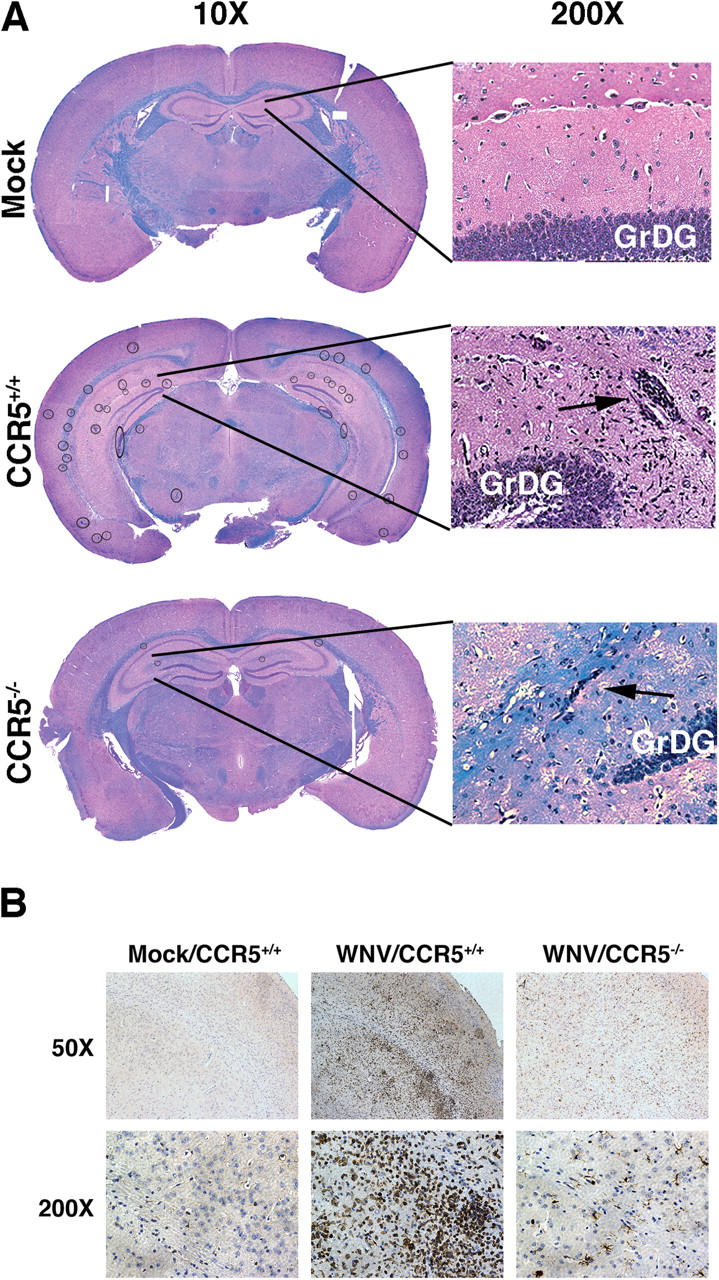
West Nile virus induces multifocal encephalitis in C57BL/6 mice. (A) Histologic analysis. Brains of mock- and WNV-infected CCR5+/+ and CCR5−/− mice harvested at day 12 after infection were fixed in formalin and embedded in paraffin. Coronal sections 6-μm thick were stained with hematoxylin and eosin to visualize cells and luxol fast blue to stain myelin tracts blue. Multiple cellular infiltrates were observed in the infected mice (circled in the 10× image). Typical infiltrates are shown at higher magnification in the hippocampus (arrows). GrDG, granule cell layer of the dentate gyrus. (B) Immunohistochemical analysis. Brain sections, prepared as in A from mock- and WNV-infected mice with the indicated CCR5 genotype, were stained with methyl green and anti–human CD3 mAb. Representative sections of the cortex are shown. CD3+ cells stain brown. Images in A and B are representative of five mock-infected brains, nine WNV-infected CCR5+/+ brains, and six WNV-infected CCR5−/− brains.
Consistent with the histological analysis and immunohistochemistry results, we observed a notable increase in leukocytes isolated from WNV-infected wild-type mouse brains at both 7 and 12 d after infection (Figs. 3 and 4). The total amount of CD4+ T cells rose from 1.2 × 103/brain (0.2% of total leukocytes) in mock-infected brains to 2 × 104/brain (1.8% of total leukocytes) and 9.6 × 104/brain (5.0% of total leukocytes) on days 7 and 12 after infection, respectively. The amount of CD8+ T cells rose from 1.8 × 103/brain (0.3% of total leukocytes) in mock-infected brains to 2.3 × 104/brain (2.0% of total leukocytes) and 1.0 × 105/brain (5.3% of total leukocytes) on days 7 and 12 after infection, respectively. The number of NK1.1+ cells increased from 1.4 × 103/brain (0.3% of total leukocytes) in mock-infected brains to 2.2 × 104/brain (1.9% of total leukocytes) and 4.1 × 104/brain (2.1% of total leukocytes) on days 7 and 12 after infection, respectively. The amount of resident microglia (F4/80+/CD45lo) remained relatively constant throughout the course of disease. However, activated resident microglia (F4/80+/CD45med) rose from a baseline of 0.24 × 105/brain (4.4% of total leukocytes) in mock-infected brains to 1.2 × 105 (11.0% of total leukocytes) and 2.2 × 105 (11.1% of total leukocytes) on days 7 and 12 after infection, respectively. The most impressive increase was seen in infiltrating macrophages (F4/80+/CD45hi). Mock-infected mouse brains had only 7.3 × 103/brain (1.3% of total leukocytes) of these cells, whereas the amount in infected brains increased to 2.7 × 105/brain (23.0% of total leukocytes) and 5.5 × 105/brain (28.3% of total leukocytes) on days 7 and 12 after infection, respectively (Figs. 3 and 4).
Figure 3.

WNV induces accumulation of leukocytes in mouse brain. Representative FACS analysis dot plots of mock- and WNV-infected CCR5+/+ and CCR5−/− mice. Percentages are calculated based on total events; histograms to the right of the corresponding dot plot show CCR5 staining within single positive CD4+ or CD8+ T cells, NK cells, and the three F4/80+/CD45+ subsets. The blue line in the histogram represents isotype control staining, and the red shaded line represents CCR5 staining. Bar shows which portion of the histogram was considered CCR5 positive.
Figure 4.

CCR5 regulates accumulation of T cells, NK cells, and macrophages in the WNV-infected mouse brain. (A) WNV induces accumulation of CCR5+ leukocytes in the mouse brain. CCR5 surface expression was examined on specific leukocyte subsets isolated from mouse brain and the percent positive cells was determined. (B and C) Lack of CCR5 blunts leukocyte accumulation in the mouse brain in response to WNV infection. Data were pooled from three experiments, with three to five mice analyzed at each time point/experiment and are presented as the mean ± SEM. *, P < 0.04 for the indicated comparison by Student's t test.
CCR5 has been reported to be expressed mainly on subsets of monocytes, macrophages, NK cells, and T lymphocytes. Consistent with this, on day 7 after infection, 12% of CD8+ T cells, and 25% each of CD4+ T cells, NK1.1+ NK cells, and F4/80+ CD45hi activated infiltrating macrophages, which were induced to accumulate in the brain by WNV, expressed CCR5 (Figs. 3 and 4 A). On day 12 after infection, expression of CCR5 on NK cells and activated macrophages in WNV-infected CNS had decreased to 16% and 15%, respectively, whereas expression on CD4+ and CD8+ T cells was not statistically significantly different from expression on day 7. In mock-infected mouse brain, of the few leukocytes present, 4% of resident microglial cells (F4/80+/CD45lo) expressed CCR5 (Fig. 3 and 4 A). Together, these data support the hypothesis that CCR5 may play a role in the migration of a portion of CD4+ and CD8+ T cells, NK cells and activated tissue-infiltrating macrophages to brains infected with WNV.
Loss of CCR5 results in impaired leukocyte trafficking to the CNS
We tested this hypothesis by comparing the outcomes of WNV infection between CCR5+/+ and CCR5−/− mice. On day 12 after infection, CCR5−/− mice had markedly reduced cellular infiltrates (<1 lesion per 200× hpf in coronal sections of the hippocampus and external capsule compared with 5–8 lesions per 200× hpf for WNV-infected wild type mice (Fig. 2 A). This was confirmed by direct immunohistochemical analysis of the T cell antigen CD3, which revealed markedly decreased staining of infected CCR5−/− mouse brains compared with the brains of infected CCR5+/+ mice (Fig. 2 B). FACS analysis allowed for direct quantitation of this decrease (Figs. 3 and 4). Specifically, on day 7 in CCR5−/− mice, we found a >50% decrease in the number of CD4+ T cells, NK1.1+ cells, F4/80+/CD45med activated microglia and F4/80+/CD45hi infiltrating macrophages compared with values for WNV-infected CCR5+/+ controls. The number of CD8+ T cells was not significantly decreased at this time point. By day 12 after infection, the reduction in the cell infiltrate to the WNV-infected CCR5−/− mouse brain was even more evident: 60% reduction in CD4+ T cells, 46% reduction in CD8+ T cells, 84% reduction in NK1.1+ cells, 63% reduction in activated microglia, and a 76% reduction in infiltrating macrophages compared with infected CCR5+/+ control mice (Figs. 3 and 4).
To determine whether reduced leukocyte accumulation in the brain could be attributed indirectly to the loss of inflammatory mediator expression in CCR5−/− mice, we measured the effect of CCR5 gene inactivation on the expression of these molecules in the WNV-infected brain. No significant differences were found for CCL3, CCL4, CCL5, MIP-2, CXCL9 or CXCL10 (Tables I and II). On day 12, the protein levels of the CCR2 ligand CCL2 were decreased by ∼50% in brains of CCR5−/− mice infected with WNV as compared with infected CCR5+/+ mouse brain; however, CCL2 was still expressed at high levels, ∼2.5 ng/g brain tissue. We also did not detect CCR5-dependent differences in the expression of the cytokines IFN-γ, IFN-α, TNF-α, or IL-1β or the adhesion molecule ICAM-1 in WNV-infected mouse brain. There was a notably lower expression of the metalloproteinase inhibitor TIMP-1 (but not MMP-9) on day 12 after infection in WNV-infected CCR5−/− mouse brains compared with infected CCR5+/+ controls. However, the blood brain barrier was clearly already compromised by day 12, as evidenced by viral burden and leukocyte accumulation in the CNS by day 7 after infection. Furthermore, the serum levels of TNF-α, IFN-γ, IL-4, IL-5, IL-2, CCL5 and CXCL10 were not significantly different between WNV-infected CCR5+/+ and CCR5−/− mice throughout the course of the experiment (Fig. 5 A, P < 0.02).
Table I.
Effect of CCR5 inactivation on induction of chemokines in the brain by WNV
| DAI | Infection | Strain | CCL2 | CCL3 | CCL4 | CCL5 | CCL11 | CXCL9 | CXCL10 |
|---|---|---|---|---|---|---|---|---|---|
| 7 | mock | CCR5+/+ | BLD | BLD | BLD | BLD | BLD | BLD | BLD |
| 7 | mock | CCR5−/− | BLD | BLD | BLD | BLD | BLD | BLD | BLD |
| 7 | WNV | CCR5+/+ | 4.1 ± 1.1 | 3.2 ± 0.6 | 2.9 ± 0.5 | 4.3 ± 0.5 | 0.3 ± 0.05 | 12.6 ± 1.8 | 24.9 ± 0.5 |
| 7 | WNV | CCR5−/− | 2.9 ± 0.9 | 3.6 ± 0.9 | 3.2 ± 0.7 | 4.9 ± 0.8 | 0.3 ± 0.09 | 11.2 ± 3.2 | 22.6 ± 1.8 |
| 12 | WNV | CCR5+/+ | 4.8 ± 0.6 | 0.3 ± 0.06 | 0.4 ± 0.1 | 3.6 ± 0.4 | 0.2 ± 0.05 | 8.2 ± 0.9 | 22.5 ± 0.7 |
| 12 | WNV | CCR5−/− | 2.5 ± 0.7a | 0.6 ± 0.2 | 0.9 ± 0.5 | 3.3 ± 1.5 | 0.2 ± 0.1 | 5.8 ± 3.3 | 15.6 ± 5.5 |
Data are pooled from two to three individual experiments with two to three mice in each group and are presented as the mean ± SEM. Units are expressed as nanograms per gram of brain tissue.
P < 0.02 when compared to CCR5+/+ mice at the same day after infection.
BLD; below the level of detection; DAI, days after infection.
Table II.
Effect of CCR5 inactivation on induction of immunoregulatory factors in the brain by WNV
| DAI | Infection | Strain | IL-1β | IL-4 | IL-10 | IFN-α | IFN-γ | TNF-α | ICAM-1 | MMP-9 | TIMP-1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | mock | CCR5+/+ | 0.04 ± 0.02 | BLD | BLD | BLD | BLD | BLD | 14.6 ± 0.5 | 4.8 ± 0.6 | 0.4 ± 0.03 |
| 7 | mock | CCR5−/− | 0.2 ± 0.02 | BLD | BLD | BLD | 0.07 ± 0.01 | BLD | 10.6 ± 0.3 | 4.1 ± 0.4 | 0.4 ± 0.04 |
| 7 | WNV | CCR5+/+ | 0.3 ± 0.01 | BLD | BLD | 9.0 ± 2.0 | 0.08 ± 0.05 | 0.7 ± 0.3 | 47.5 ± 7.6 | 53.9 ± 10.1 | 11.8 ± 3.0 |
| 7 | WNV | CCR5−/− | 0.2 ± 0.07 | BLD | BLD | 6.0 ± 2.1 | 0.14 ± 0.08 | 0.3 ± 0.2 | 41.1 ± 7.3 | 28.9 ± 7.6 | 10.9 ± 3.2 |
| 12 | WNV | CCR5+/+ | 0.3 ± 0.08 | BLD | BLD | 0.2 ± 0.1 | 0.09 ± 0.07 | BLD | 41.2 ± 3.7 | 25.1 ± 12.8 | 9.1 ± 1.7 |
| 12 | WNV | CCR5−/− | 0.2 ± 0.07 | BLD | BLD | 0.1 ± 0.07 | 0.06 ± 0.05 | BLD | 29.5 ± 8.5 | 12.5 ± 4.2 | 1.3 ± 0.01a |
Data are pooled from two to three individual experiments with two to three mice in each group and are presented as the mean ± SEM. Units are expressed as nanograms per gram of brain tissue.
P < 0.02 when compared to CCR5+/+ mice at the same day after infection.
BLD; below the level of detection; DAI, days after infection.
Figure 5.

CCR5 deficiency affects immune responses to WNV selectively in the brain. (A) Serum cytokine analysis. The factor analyzed is identified at the bottom of the graph. Data were pooled from two experiments with five to seven mice in each group, and are presented as mean ± SEM pg/ml serum as a function of time after infection. (B and C) Intracellular cytokine staining for WNV-induced IFN-γ. Splenocytes were isolated from mice at the indicated days after WNV infection and assayed without exogenous stimulation (B) or after 4 h of stimulation with ionomycin and PMA (C). Data presented are from one experiment with five mice at each time point. Values at the top of each bar are the percentage of total cells represented by the indicated cell type. (D) Adoptive transfer of CCR5+/+ and CCR5−/− splenocytes from WNV-infected mice into WNV-infected CCR5−/− mice. Infected CCR5−/− recipient mice, designated (WT → KO) and (KO → KO) received splenocytes from infected CCR5+/+ or infected CCR5−/− donor mice, respectively, 4 d after s.c. infection. FACS analysis was performed 4 d after transfer (8 d after infection). Data are presented as the mean ± SEM and are from one experiment with five mice in each group. (B and C) Percents shown are the percentage of total cells. *, P < 0.02 for the indicated comparison (of total cell number) by Student's t test.
To determine whether the diminished cell accumulation seen in the CNS of WNV-infected CCR5−/− mice could be the result of an altered splenic response to the virus, we directly compared the number of IFN-γ–producing CD4+ and CD8+ T cells from spleens of CCR5+/+ and CCR5−/− mice ex vivo both with and without exogenous stimulation at several time points after infection. The numbers and percentage of CD4+/IFN-γ+ as well as CD8+/IFN-γ+ cells were nearly identical between the two groups at days 3, 5, and 7 after infection when no exogenous stimulation was applied; this result suggests that both groups of mice have the same splenic immune response to WNV infection (Fig. 5 B). When splenocytes were stimulated ex vivo with PMA and ionomycin, the IFN-γ response was similar for CCR5−/− and CCR5+/+ mice infected with WNV (Fig. 5 C).
To more directly determine whether defective trafficking of the cells to the CNS is the cause of the cell deficit seen in WNV-infected CCR5−/− mouse brains, we adoptively transferred splenocytes from WNV-infected CCR5+/+ donor mice intravenously into WNV-infected CCR5−/− recipients and compared cell accumulation in the CNS to CCR5−/− recipients of the same number of splenocytes from WNV-infected CCR5−/− donors. WNV-infected CCR5−/− mice that received CCR5+/+ splenocytes had a significantly greater amount of CD4+ and CD8+ T cells, NK1.1+ cells, and infiltrating macrophages (increased 2.3-, 5.3-, 3.1-, and 7.9-fold, respectively) compared with WNV-infected CCR5−/− mice that received CCR5−/− splenocytes (Fig. 5 D, P < 0.02). In the CCR5−/− mice that received CCR5+/+ splenocytes, we were able to determine by FACS that 4.7% of the CD4+ T cells, 9.6% of CD8+ T cells, and 0.9% of NK1.1+ cells in the brain expressed CCR5, demonstrating that the transferred cells were able to migrate to the brain.
CCR5 is critical to viral control and survival
We next tested the importance of CCR5 on viral clearance and survival. Viral load in the spleens of CCR5+/+ and CCR5−/− mice was not statistically different in onset, magnitude, duration, or clearance as both groups had a maximal viral load on day 3 after infection and surviving mice had cleared virus from the spleen by day 9 after infection (Fig. 6 A), further indicating that the immune response to WNV in CCR5−/− mice is not altered. WNV-infected CCR5−/− mice had nearly identical CNS viral loads as CCR5+/+ controls on day 7 after infection (Fig. 6 B). However, whereas surviving WNV-infected CCR5+/+ mice began to clear virus by day 12, the few surviving CCR5−/− mice available for analysis showed no decrease in CNS viral load on day 12, but rather a 32-fold increase relative to controls. This was evident even when CCR5+/+ mice were selected to match the poor clinical status of CCR5−/− mice at this time point (Fig. 6 B). Consistent with this result, infection with WNV was uniformly fatal in CCR5−/− mice (n = 38), whereas in CCR5+/+ mice mortality was 35% (Fig. 6 C, n = 50). All infected CCR5−/− mice were dead by day 13. In contrast, WNV-infection in mice lacking either CCR1 or CX3CR1, which are two chemokine receptors related to CCR5 and shown by RNA analysis (Fig. 1) to be up-regulated by WNV infection, resulted in mortality that was only slightly increased relative to that observed in wild-type control mice (Fig. 6 C). Finally, adoptive transfer of splenocytes from WNV-infected CCR5+/+ donor mice to WNV-infected CCR5−/− recipients (WT → KO) reduced mortality to the level observed for wild-type C57BL/6 mice. In contrast, transfer of the same number of splenocytes from WNV-infected CCR5−/− mice to WNV-infected CCR5−/− recipients (KO → KO) had only a minor protective effect (Fig. 6 D).
Figure 6.
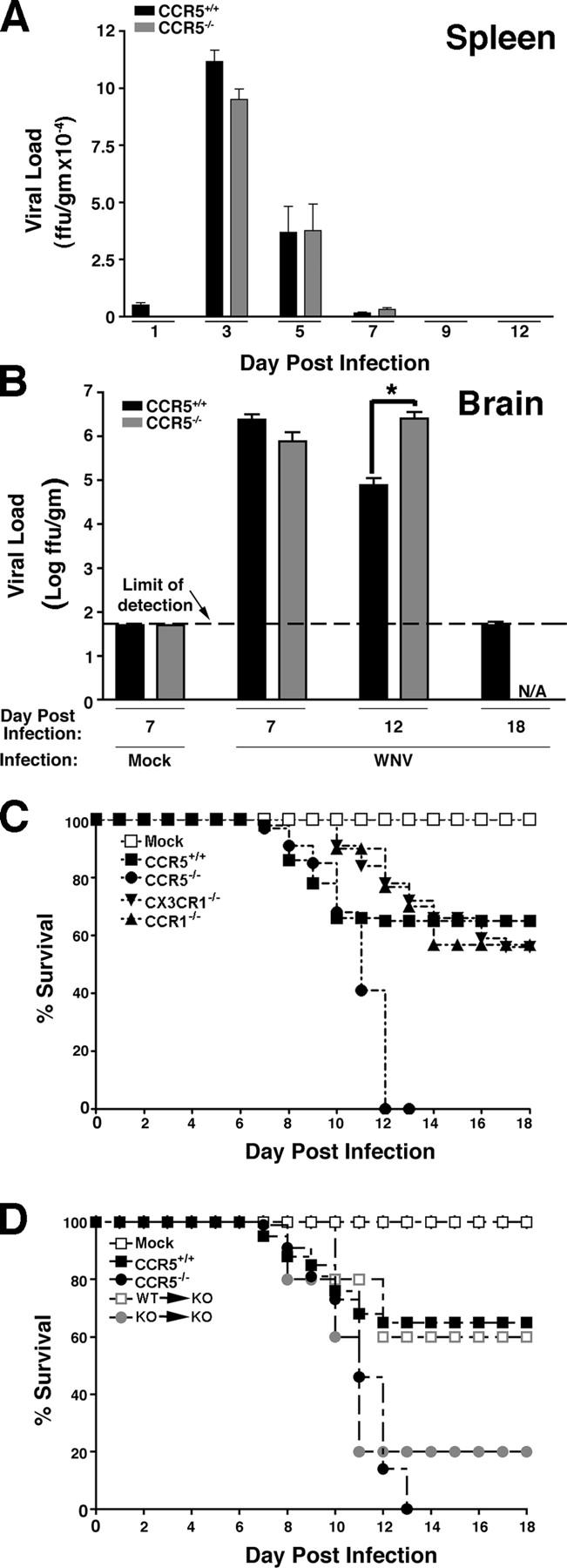
West Nile virus infection is uniformly fatal in CCR5 knockout mice. (A) Time course of viral clearance from spleen. Virus was quantified in the spleens of the indicated strains of surviving mice at the indicated times by focus formation assay. Mock-infected mice were analyzed on day 7 of the experiment. (B) Time course of viral clearance from brain. Virus was quantified in the brain of the indicated strains of mice at the indicated times by focus formation assay. Data were pooled from three to four experiments with three to five mice at each time point indicated and are presented as the mean ± SEM. *, P < 0.02 for the indicated comparison by Student's t test. N/A, not applicable, because all infected CCR5−/− die by day 13. (C) Kaplan-Meier analysis of survival of WNV-infected mice. Results shown are pooled data from three independent experiments for CCR5+/+ mice (n = 50 total mice) and CCR5−/− mice (n = 34), and two independent experiments for CCR1−/− mice (n = 30) and CX3CR1−/− mice (n = 32). Similar results were obtained for both wild-type C57BL6/J and C57BL/6NTac control mice; however, only the C57BL6/J results are shown (CCR5+/+). (D) Kaplan-Meier analysis of survival of adoptive transfer mice. WT → KO, transfer of splenocytes from WNV-infected CCR5+/+ mice into WNV-infected CCR5−/− mice; KO → KO, transfer of splenocytes from WNV-infected CCR5−/− mice to WNV-infected CCR5−/− mice. The following control groups were included that did not undergo adoptive transfer: Mock, mock-infected CCR5+/+mice; CCR5+/+, WNV-infected CCR5+/+ mice; and CCR5−/−, WNV-infected CCR5−/− mice. Data represent a single experiment with 10 mice in each group.
Discussion
This study provides a detailed analysis of the molecular and cellular immune response in the brain during WNV infection and demonstrates that expression of the chemokine receptor CCR5 is crucial for viral clearance and survival in a mouse model of disease. WNV induced production of all three CCR5 ligands in the brain, with particularly high and durable induction of CCL5, which is consistent with local accumulation of CCR5-expressing NK cells, macrophages, and CD4+ and CD8+ T lymphocytes that we observed in the model. Several lines of evidence lead us to believe that trafficking of leukocytes expressing CCR5 to the WNV-infected mouse brain is critical for survival. First, genetic disruption of CCR5 markedly reduced accumulation of these cell types in brains of WNV-infected mice, without significantly affecting CNS expression of other immunoregulatory factors, including the CCR5 ligands CCL3, CCL4, and CCL5. Second, there was no defect in clearance of the virus from the spleen in CCR5−/− mice. Third, WNV-induced production of IFN-γ by CD4+ and CD8+ T cells in the spleen was identical for CCR5+/+ and CCR5−/− mice. Fourth, when splenocytes from WNV-infected CCR5+/+ or CCR5−/− mice were adoptively transferred into WNV-infected CCR5−/− mice, we found much greater leukocyte accumulation in the brain after CCR5+/+ than after CCR5−/− cell transfer and detected CCR5+ leukocytes in the brains of CCR5−/− recipients. This experiment, which involves direct injection of WNV-activated CCR5+/+ or CCR5−/− splenocytes into the blood, bypasses the splenic egress step, and suggests strongly, when coupled with unaltered splenic T cell or viral clearance responses in CCR5−/− mice, that CCR5 functions by promoting trafficking of leukocytes from the blood to the brain in response to WNV infection, for the purpose of containing and clearing the virus. This is consistent with previously published work on WNV which demonstrated that RAG−/− mice have dramatically increased CNS viral burden and succumb to infection within 12 d, the same time frame that we found for CCR5−/− mice (19). Furthermore, mice lacking CD8+ T cells also have increased viral burden in the CNS and mortality when infected with WNV (13, 18, 23–25). Shirato et al. have recently reported an increased expression of mRNA for CCR5 ligands when mice were infected i.p. with a lethal versus nonlethal strain of WNV (14). Our data confirm and extend these results in a distinct model by providing mRNA and protein analysis for these and numerous other immunoregulatory molecules and by directly testing the importance and role of CCR5.
The critical role of CCR5 in WNV pathogenesis appears to be unique compared with other neurotropic viruses that have been tested to date. CNS infection with lymphocytic choriomeningitis virus results in equivalent viral burden and mortality in CCR5−/− and CCR5+/+ mice suggesting that this receptor does not play a role in pathogenesis in this model (26). Likewise, infection with the neurovirulent retrovirus FR98 results in identical mortality rates and CNS viral burden in CCR5−/− and CCR5+/+ mice (27). Infection with a neurovirulent strain of mouse hepatitis virus (MHV) results in similar viral titers in the CNS of CCR5−/− and CCR5+/+ mice, but knockouts have reduced demyelination, the major manifestation of disease in this model. The mechanism involves CCR5-dependent recruitment of macrophages and CD4+ and CD8+ T cells to the CNS (28). Thus, CCR5 promotes demyelinating disease in MHV infection, not antiviral host defense as it does in WNV infection. CCR5 has also been demonstrated to regulate leukocyte trafficking to the brain, but not the lung, after Cryptococcus neoformans infection (29).
The importance of CCR5 also varies among noninfectious, immunologically mediated, CNS disease. CCR5 deficiency has no effect on experimental autoimmune encephalitis (EAE), despite the fact that CCR5 ligands are highly expressed in the CNS of mice with EAE (30). In addition, wild-type and CCR5-deficient mice have similar outcomes in experimental autoimmune neuritis, including similar levels of leukocytes recruited to the cauda equina (31). These examples show the potential for chemokine receptors to play specific and important roles or more redundant roles depending on the disease context, and the spatial, temporal, and quantitative details of cognate ligand expression.
Our data raise new questions regarding whether CCR5 may play a more general role in the immune response to other flaviviruses. In this regard, in a recent study of 10 cytokines in patients infected with Japanese Encephalitis virus (the prototype flavivirus), increased serum CCL5 alone was associated with a fatal outcome (32). The mechanism was not established, but could conceivably involve desensitization of leukocyte CCR5 in the bloodstream, thereby preventing trafficking to the infected CNS.
We found that CCR1, CCR2, CXCR3, and CX3CR1 and their ligands were also up-regulated in mouse brains by WNV. These systems could work cooperatively with CCR5 and could explain why leukocytes accumulate in small numbers in the CNS of WNV-infected CCR5−/− mice and why many of the leukocytes in the WNV-infected brain are CCR5 negative. Induction of CXCR3 was particularly strong, and is consistent with induction of a Th1-polarized cytokine response in the brain (18, 23, 24). However, our results using knockout mice do not support an important, nonredundant role for CCR1 or CX3CR1 in this model. Our gene and protein expression data also suggest new and testable hypotheses regarding the role in control of WNV infection of TNF-α, the IL-12/IFN-γ axis, IFN-α, the leukocyte adhesion molecule ICAM-1, the matrix metalloproteinase MMP-9, and the metalloproteinase inhibitor TIMP-1 (33). WNV has been reported to be very sensitive to both exogenous type I and II interferons in vitro (34), but they are only able to prevent infection in vivo in mice when given before challenge (35). IFN-α may be even less effective in humans because infection with WNV has been reported in patients actively being treated with IFN-α and ribavirin for hepatitis C (36). Matrix metalloproteinases may be important in disrupting the blood brain barrier to allow hematogenous spread of WNV to the CNS, as has been previously suggested (19). Although the role of TNF-α in the brain is unclear, recent work has clarified its importance in the periphery where it is produced in a TLR3-dependent manner and mediates permeabilization of the blood brain barrier to facilitate WNV entry (12). The specific roles in pathogenesis of these and other factors induced in the brain by WNV are currently under investigation.
An important question raised by our findings is whether CCR5 is also protective in WNV infection of humans. In limited studies, T cells and macrophages have been reported to accumulate in WNV-infected human brains as they do in our mouse model (25, 37). Moreover, like humans, the mouse is easily infected with WNV, and the major clinical manifestations of disease are similar in both species: encephalitis and mortality in a subset of infected individuals. If CCR5 is protective in humans, individuals homozygous for the CCR5Δ32 mutation, who lack functional CCR5 and include 1% of North American Caucasians (38), may be at greater risk for fatal encephalitis from WNV infection. A related issue is the safety of CCR5 blocking agents under development for patients with HIV/AIDS who become infected with WNV.
In conclusion, our data identify CCR5 as a critical protective factor in fatal encephalitis caused by WNV in a mouse model. The effect is highly specific relative to other neurotropic viruses and at least two other relevant chemokine receptors. Loss of CCR5 results in decreased ability to recruit/maintain leukocytes into the infected CNS where they may function to clear the virus. The data also raise important new questions about the potential roles of other immunoregulatory systems that are induced by WNV in this model and in man.
Materials and Methods
Mouse strains
Mouse studies were performed in an animal biosafety level three facility under a protocol approved by the National Institute of Allergy and Infectious Diseases/National Institutes of Health (NIH) Animal Care and Use Committee. CCR5−/− mice (B6;129P2-Ccr5 tm1Kuz), its approximate genetic match mouse strain B6129PF2 and C57BL6/J mice were all purchased from the Jackson Laboratory. Infection of B6129PF2 and C57BL6/J mice with 104 ffu WNV induced similar changes and time courses with regard to all parameters that were compared (mortality, CNS viral load, CNS accumulation of leukocyte subsets [total and CCR5+ T cells, NK cells, and macrophages], and CNS chemokine protein [CCL3, CCL4, CCL5, and CXCL10]; unpublished data). In all experiments shown, data for CCR5+/+ mice were generated using the C57BL6/J mouse. CX3CR1−/− mice (F10 backcross on the C57BL/6NTac background), CCR1−/− mice (F10 backcross on the C57BL/6NTac background), and control C57BL/6NTac mice were obtained from Taconic. All experiments were initiated using female mice 8–12 wk old. Mice were selected randomly for phenotypic analysis at day 7 after infection with WNV. On day 12 after WNV infection, all surviving CCR5−/− mice were severely ill (limp tail, hunched back, ruffled fur, minimal activity), and therefore infected C57BL6/J mice with similar clinical signs were selected for phenotypic analysis at this time point.
Viral infections
WNV strain NY99-35262 was provided by R. Lanciotti (Centers for Disease Control and Prevention, Fort Collins, CO). Originally isolated from a Chilean flamingo at the Bronx Zoo in 1999 (3), this isolate had been passaged once in Vero cell culture that contained 10% FBS. It was then reamplified by two passages in Vero cells (WHO seed passage 143) that did not contain FBS. A virus suspension was prepared that had a titer of 2 × 107 ffu/ml, as determined using an immunostaining focus-forming assay and WNV-specific mouse antibodies (39). Mice were injected s.c. in the back of the neck with either 104 ffu WNV-NY99 suspended in 50 μl HBSS (Invitrogen) or with HBSS alone (mock infected). Mice were monitored visually and weighed daily. Brains of killed mice were aseptically removed, weighed and placed directly in either 2 ml HBSS (for ELISA and viral titer assay) or 5 ml FACS buffer (HBSS + 1% BSA + 0.1% NaN3; Sigma-Aldrich).
ELISA
Brains were homogenized in sterile HBSS and frozen at −80°C. Samples were thawed and centrifuged at 1,500 g for 25 min. The aqueous fraction of the homogenate was serially diluted, and then analyzed for the presence of specific cytokine and chemokine proteins using murine Quantikine Immunoassay kits according to the manufacturer's recommendations (R&D systems). Samples were used only once.
RT-PCR
Total RNA was extracted from homogenized brain tissue using TRIzol (Invitrogen). 2 ng of total RNA was converted to cDNA using a Superscript III RT kit (Invitrogen), with random primers. PCR was performed using 2 μl cDNA in a reaction mix with Taq polymerase (Invitrogen). PCR reactions were optimized to determine the number of cycles where the visual signal was in the linear range. These experiments determined that at ∼35 cycles, the signal plateaued for all targets tested. Therefore, all PCR reactions were run for 30 cycles using the following primer pairs: β-actin forward (5′-AGCCATGTACGTAGCCATCC-3′), β-actin reverse (5′-TCTCAGCTGTGGTGGTGAAG-3′); CXCR3 forward (5′-TGCTAGATGCCTCGGACTTT-3′), CXCR3 reverse (5′-CGCTGACTCAGTAGCACAGC-3′); CXCL4 forward (5′-AGTCCTGAGCTGCTGCTTCT-3′), CXCL4 reverse (5′-GGCAAATTTTCCTCCCATTC-3′); CXCR4 forward (5′-TCAGTGGCTGACCTCCTCTT-3′), CXCR4 reverse (5′-TTTCAGCCAGCAGTTTCCTT-3′); CX3CL1 forward (5′-AGCCCTGTGACATTTTCTGG-3′), CX3CL1 reverse (5′-CCTCACTCTCAGGAGCCAAC-3′); and CX3CR1 forward (5′-GGAGACTGGAGCCAACAGAG-3′), CX3CR1 reverse (5′-TCTTGTCTGGCTGTGTCCTG-3′). PCR products were separated and visualized on ethidium bromide-stained 2% agarose/TBE gels.
Ribonuclease protection assay
Cytokine, chemokine, cytokine receptor, and chemokine receptor mRNA transcripts were analyzed from total brain RNA using mCK-1, mCK-5, mCR-1, and mCR-5 multi-template probe sets, respectively (BD Biosciences). RPA analysis was performed using 10 μg of total RNA using a previously described protocol (28). For quantitation of signal intensity, autoradiographs were scanned and individual chemokine, cytokine, or chemokine/cytokine receptor transcript bands were normalized as the ratio of band intensity to the GAPDH control. Analysis was performed using NIH Image J1.32i software.
Cell isolation and FACS analysis
Mice were killed by cervical dislocation. Organs were aseptically removed but not perfused before analysis. Single cell suspensions of leukocytes were obtained from brain using a previously described method (40). In brief, brains were collected in FACS buffer (HBSS + 1% BSA + 0.1% NaN3) and homogenized using the plunger portion of a 6-cc syringe on ice in a petri dish. The suspension was brought to 7 ml with FACS buffer and 3 ml of 90% percoll in PBS was added. After 5 min at 22°C, the suspension was underlaid with 1 ml of 70% Percoll (in RPMI), and was then centrifuged at 2,470 rpm for 20 min at 22°C at room temperature. The leukocytes at the interphase were isolated and washed three times in FACS buffer. Cells were fixed in 100 μl BD Cytofix (BD Biosciences) at 4°C for 20 min, counted using a hemacytometer, and then filtered though a 70-μm mesh screen. Cells were washed three times with FACS buffer and incubated with Fc block (Becton Dickinson) in 100 μl CD16/32 Ab (BD Biosciences) diluted 1:200 in FACS buffer. Cells were then incubated with specific antibodies for 45 min at 4°C, washed three times and suspended in 400 μl FACS buffer. Antibodies used for flow cytometry were FITC-conjugated anti–mouse CD4 (GK1.5), CD19 (1D3), CD45 (LCA, Ly-5; BD Biosciences) and F4/80 (Serotec); PE-conjugated anti–mouse CCR5, NK1.1 (PK136), and CD45 (LCA, Ly-5); APC-conjugated anti-mouse CD8 (Ly-2) and NK1.1 (PK136; BD Biosciences); and PerCP-conjugated anti-mouse CD4 (L3T4) and CD45 (LCA, Ly-5). In all cases, an isotype-matched FITC-, PE-, APC-, or PerCP-conjugated antibody was used as the control. Cells were analyzed on a FACSCalibur (Becton Dickinson) where leukocytes were further refined by gating on cells with the appropriate forward and side scatter profile. 100,000 events were captured for analysis for each brain and each staining set. Data were analyzed using FlowJo software and are presented as the percentage of positive cells within the gated population.
Intracellular cytokine staining
Mouse splenocytes (3 × 106 cells/well) were incubated without exogenous stimulation for 4 h or with 50 ng/ml PMA (Sigma-Aldrich) plus 500 ng/ml ionomycin (Sigma-Aldrich) for 4 h at 37°C. Cells were then washed and blocked with Fc block for 20 min at 4°C. Cells were washed twice with FACS buffer, and surface markers were stained with CD19-FITC, CD8-PE, and CD4-PerCP for 30 min at 4°C. Cells were washed twice with FACS buffer and resuspended in 100 μl Cytofix/Cytoperm (Becton Dickinson) for 20 min at 4°C to permeabilize the plasma membrane and facilitate antibody staining of IFN-γ. We washed the cells twice with Perm/Wash solution and stained them with an APC-conjugated mAb to mouse IFN-γ (Becton Dickinson) for 30 min at 4°C. Finally, cells were washed twice and resuspended in FACS buffer and analyzed on a FACSCalibur (Becton Dickinson). In all samples 100,000 events were captured. Data were analyzed using FloJo software.
Adoptive transfer
CCR5−/− and CCR5+/+ donor mice were infected with 104 ffu WNV s.c. 3 d later, CCR5−/− recipient mice were infected with 104 ffu WNV s.c. After recipients had been infected for 4 d, a duration sufficient for virus to enter the brain, 107 splenocytes from donors (infected for 7 d at this time point) were transferred IV to recipients. Splenocytes were isolated using lympholyte M (Cedarlane Labs). CCR5−/− recipients of donor CCR5−/− splenocytes are referred to as KO → KO. CCR5−/− recipients of donor CCR5+/+ splenocytes are referred to as WT → KO.
Viral titers
Vero cells were maintained in OptiPro SFM (Invitrogen) until passaged for the viral titer assay at which time they were grown in OptiPro SFM + 2% FBS + 50 μg/ml gentamicin. The virus was quantitated by growth on confluent Vero cell monolayers in 12 well plates. 200 μl of brain homogenate supernatant was diluted in culture media and placed on the cells in duplicate. Plates were incubated for 1 h at 37°C to allow virus attachment, and then cells were overlaid with 2 ml of sterile Opti-MEM (Invitrogen) + methyl cellulose 8 g/liter (EM Science) + 2% FBS + 50 μg/ml gentamicin. Plates were incubated for 2 d at 37°C, and then washed three times with PBS (Biosource International) and incubated for 1 h at 37°C with 500 μl of diluted anti-WNV anti-sera/well (HMAF; American Type Culture Collection VR-82). After washing, samples were incubated at 37°C with 500 μl of diluted anti–mouse/anti–rabbit (1:10)-labeled polymer (DAKO Cytomation) for 1 h. After washing, 1 ml DAB mix (4.5 mg DAB [Sigma-Aldrich] /10 ml PBS + 4.5 μl 30% H202 [Fisher Scientific]/10 ml) was added at 22°C for 10 min to 1 h to visualize WNV foci. The reaction was stopped by the addition of water. The average of duplicate assays was used to determine viral titers, which are expressed as ffu per gram of brain tissue.
Histology
Brains were aseptically removed from mice after cervical dislocation and placed directly into 10% normal buffered formalin (Fisher Chemicals) for 24 h. Samples were paraffin-embedded and cut into 6-μm sections (Histoserve). RNase protection (gloves, RNase-free water and sterilized instruments) was used at all stages and slides were baked at 60°C for 1 h after sectioning. Sections were stained with hematoxylin and eosin (for visualization of cells) as well as luxol fast blue (for visualization of myelin). Separate sections were stained with an anti–human CD3 antibody (DAKO Cytomation) or with hyperimmune mouse ascites fluid collected after inoculation with WNV (HMAF). In brief, slides were deparaffinized with Citrosolve (National Diagnostics) and graded ethanol followed by a water wash. Slides were heated to 90°C and washed with water twice, blocked with hydrogen peroxide for 10 min, and washed in water. Slides were blocked with BSA for 20 min, and then incubated with primary antibody overnight at a 1:100 dilution. Next, we washed the slides, incubated them with biotinylated secondary antibody for 30 min at 1:500 dilution, and then rewashed. Slides were incubated with streptavidin for 30 min and washed and developed with DAB (DAKO Cytomation). Finally, slides were counterstained with hematoxylin and dehydrated with graded ethanol and Citrosolve.
Statistical analysis
Statistically significant differences between groups of mice were determined by Student's t test using Microsoft Excel MAC v.X software, and p-values <0.05 were considered significant.
Acknowledgments
We thank the animal care staff at the NIH for technical assistance. We also thank J. Ward and L. Brinster for expert analysis of neuropathology.
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH.
The authors have no conflicting financial interests.
Abbreviations used: CNS, central nervous system; ffu, focus-forming unit; WNV, West Nile virus.
W.G. Glass and J.K. Lim contributed equally to this work.
W.G. Glass's present address is Centocor Global R&D, Infectious Diseases, Radnor, PA 19087.
References
- 1.Smithburn, K.C., T.P. Hughes, A.W. Burke, and J.H. Paul. 1940. Neurotropic virus isolated from the blood of a native of Uganda. Am. J. Trop. Med. 20:471–472. [Google Scholar]
- 2.Hubalek, Z., and J. Halouzka. 1999. West Nile fever–a reemerging mosquito-borne viral disease in Europe. Emerg. Infect. Dis. 5:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lanciotti, R.S., J.T. Roehrig, V. Deubel, J. Smith, M. Parker, K. Steele, B. Crise, K.E. Volpe, M.B. Crabtree, J.H. Scherret, et al. 1999. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 286:2333–2337. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2004. West Nile virus activity–United States, October 6-12, 2004. MMWR Morb. Mortal. Wkly. Rep. 53:950–951. [PubMed]
- 5.van der Poel, W.H. 1999. West Nile-like virus is the cause of encephalitis in humans and horses and the death of hundreds of birds in New York. [In Dutch.] Tijdschr. Diergeneeskd. 124:704–705 [PubMed] [Google Scholar]
- 6.Steele, K.E., M.J. Linn, R.J. Schoepp, N. Komar, T.W. Geisbert, R.M. Manduca, P.P. Calle, B.L. Raphael, T.L. Clippinger, T. Larsen, et al. 2000. Pathology of fatal West Nile virus infections in native and exotic birds during the 1999 outbreak in New York City, New York. Vet. Pathol. 37:208–224. [DOI] [PubMed] [Google Scholar]
- 7.Ostlund, E.N., J.E. Andresen, and M. Andresen. 2000. West Nile encephalitis. Vet. Clin. North Am. Equine Pract. 16:427–441. [DOI] [PubMed] [Google Scholar]
- 8.Briese, T., W.G. Glass, and W.I. Lipkin. 2000. Detection of West Nile virus sequences in cerebrospinal fluid. Lancet. 355:1614–1615. [DOI] [PubMed] [Google Scholar]
- 9.Asnis, D.S., R. Conetta, G. Waldman, and A.A. Teixeira. 2001. The West Nile virus encephalitis outbreak in the United States (1999-2000): from Flushing, New York, to beyond its borders. Ann. NY Acad. Sci. 951:161–171. [DOI] [PubMed] [Google Scholar]
- 10.Asnis, D.S., R. Conetta, A.A. Teixeira, G. Waldman, and B.A. Sampson. 2000. The West Nile Virus outbreak of 1999 in New York: the Flushing Hospital experience. Clin. Infect. Dis. 30:413–418. [DOI] [PubMed] [Google Scholar]
- 11.Tsai, T.F., F. Popovici, C. Cernescu, G.L. Campbell, and N.I. Nedelcu. 1998. West Nile encephalitis epidemic in southeastern Romania. Lancet. 352:767–771. [DOI] [PubMed] [Google Scholar]
- 12.Wang, T., T. Town, L. Alexopoulou, J.F. Anderson, E. Fikrig, and R.A. Flavell. 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 10:1366–1373; 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 13.Shrestha, B., and M.S. Diamond. 2004. Role of CD8+ T cells in control of West Nile virus infection. J. Virol. 78:8312–8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shirato, K., T. Kimura, T. Mizutani, H. Kariwa, and I. Takashima. 2004. Different chemokine expression in lethal and non-lethal murine West Nile virus infection. J. Med. Virol. 74:507–513. [DOI] [PubMed] [Google Scholar]
- 15.Mashimo, T., M. Lucas, D. Simon-Chazottes, M.P. Frenkiel, X. Montagutelli, P.E. Ceccaldi, V. Deubel, J.L. Guenet, and P. Despres. 2002. A nonsense mutation in the gene encoding 2′-5′-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc. Natl. Acad. Sci. USA. 99:11311–11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Licon Luna, R.M., E. Lee, A. Mullbacher, R.V. Blanden, R. Langman, and M. Lobigs. 2002. Lack of both Fas ligand and perforin protects from flavivirus-mediated encephalitis in mice. J. Virol. 76:3202–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamond, M.S., E.M. Sitati, L.D. Friend, S. Higgs, B. Shrestha, and M. Engle. 2003. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 198:1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diamond, M.S., B. Shrestha, E. Mehlhop, E. Sitati, and M. Engle. 2003. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 16:259–278. [DOI] [PubMed] [Google Scholar]
- 19.Diamond, M.S., B. Shrestha, A. Marri, D. Mahan, and M. Engle. 2003. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 77:2578–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ceccaldi, P.E., M. Lucas, and P. Despres. 2004. New insights on the neuropathology of West Nile virus. FEMS Microbiol. Lett. 233:1–6. [DOI] [PubMed] [Google Scholar]
- 21.Shrestha, B., D. Gottlieb, and M.S. Diamond. 2003. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 77:13203–13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lucas, M., T. Mashimo, M.P. Frenkiel, D. Simon-Chazottes, X. Montagutelli, P.E. Ceccaldi, J.L. Guenet, and P. Despres. 2003. Infection of mouse neurones by West Nile virus is modulated by the interferon-inducible 2′-5′ oligoadenylate synthetase 1b protein. Immunol. Cell Biol. 81:230–236. [DOI] [PubMed] [Google Scholar]
- 23.Wang, Y., M. Lobigs, E. Lee, and A. Mullbacher. 2003. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 77:13323–13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ben-Nathan, D., I. Huitinga, S. Lustig, N. van Rooijen, and D. Kobiler. 1996. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch. Virol. 141:459–469. [DOI] [PubMed] [Google Scholar]
- 25.Omalu, B.I., A.A. Shakir, G. Wang, W.I. Lipkin, and C.A. Wiley. 2003. Fatal fulminant pan-meningo-polioencephalitis due to West Nile virus. Brain Pathol. 13:465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nansen, A., J.P. Christensen, S.O. Andreasen, C. Bartholdy, J.E. Christensen, and A.R. Thomsen. 2002. The role of CC chemokine receptor 5 in antiviral immunity. Blood. 99:1237–1245. [DOI] [PubMed] [Google Scholar]
- 27.Peterson, K.E., J.S. Errett, T. Wei, D.E. Dimcheff, R. Ransohoff, W.A. Kuziel, L. Evans, and B. Chesebro. 2004. MCP-1 and CCR2 contribute to non-lymphocyte-mediated brain disease induced by Fr98 polytropic retrovirus infection in mice: role for astrocytes in retroviral neuropathogenesis. J. Virol. 78:6449–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glass, W.G., M.T. Liu, W.A. Kuziel, and T.E. Lane. 2001. Reduced macrophage infiltration and demyelination in mice lacking the chemokine receptor CCR5 following infection with a neurotropic coronavirus. Virology. 288:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huffnagle, G.B., L.K. McNeil, R.A. McDonald, J.W. Murphy, G.B. Toews, N. Maeda, and W.A. Kuziel. 1999. Cutting edge: role of C-C chemokine receptor 5 in organ-specific and innate immunity to Cryptococcus neoformans. J. Immunol. 163:4642–4646. [PubMed] [Google Scholar]
- 30.Tran, E.H., W.A. Kuziel, and T. Owens. 2000. Induction of experimental autoimmune encephalomyelitis in C57BL/6 mice deficient in either the chemokine macrophage inflammatory protein-1alpha or its CCR5 receptor. Eur. J. Immunol. 30:1410–1415. [DOI] [PubMed] [Google Scholar]
- 31.Duan, R.S., Z. Chen, L. Bao, H.C. Quezada, I. Nennesmo, B. Winblad, and J. Zhu. 2004. CCR5 deficiency does not prevent P0 peptide 180-199 immunized mice from experimental autoimmune neuritis. Neurobiol. Dis. 16:630–637. [DOI] [PubMed] [Google Scholar]
- 32.Winter, P.M., N.M. Dung, H.T. Loan, R. Kneen, B. Wills, T. Thu le, D. House, N.J. White, J.J. Farrar, C.A. Hart, and T. Solomon. 2004. Proinflammatory cytokines and chemokines in humans with Japanese encephalitis. J. Infect. Dis. 190:1618-1626. [DOI] [PubMed] [Google Scholar]
- 33.Rosenzweig, S.D., and S.M. Holland. 2004. Congenital defects in the interferon-gamma/interleukin-12 pathway. Curr. Opin. Pediatr. 16:3–8. [DOI] [PubMed] [Google Scholar]
- 34.Anderson, J.F., and J.J. Rahal. 2002. Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg. Infect. Dis. 8:107–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrey, J.D., C.W. Day, J.G. Julander, L.M. Blatt, D.F. Smee, and R.W. Sidwell. 2004. Effect of interferon-alpha and interferon-inducers on West Nile virus in mouse and hamster animal models. Antivir. Chem. Chemother. 15:101–109. [DOI] [PubMed] [Google Scholar]
- 36.Hrnicek, M.J., and M.E. Mailliard. 2004. Acute west nile virus in two patients receiving interferon and ribavirin for chronic hepatitis C. Am. J. Gastroenterol. 99:957. [DOI] [PubMed] [Google Scholar]
- 37.Yang, J.S., J.J. Kim, D. Hwang, A.Y. Choo, K. Dang, H. Maguire, S. Kudchodkar, M.P. Ramanathan, and D.B. Weiner. 2001. Induction of potent Th1-type immune responses from a novel DNA vaccine for West Nile virus New York isolate (WNV-NY1999). J. Infect. Dis. 184:809–816. [DOI] [PubMed] [Google Scholar]
- 38.Lu, Y., V.R. Nerurkar, W.M. Dashwood, C.L. Woodward, S. Ablan, C.M. Shikuma, A. Grandinetti, H. Chang, H.T. Nguyen, Z. Wu, et al. 1999. Genotype and allele frequency of a 32-base pair deletion mutation in the CCR5 gene in various ethnic groups: absence of mutation among Asians and Pacific Islanders. Int. J. Infect. Dis. 3:186–191. [DOI] [PubMed] [Google Scholar]
- 39.Pletnev, A.G., R. Putnak, J. Speicher, E.J. Wagar, and D.W. Vaughn. 2002. West Nile virus/dengue type 4 virus chimeras that are reduced in neurovirulence and peripheral virulence without loss of immunogenicity or protective efficacy. Proc. Natl. Acad. Sci. USA. 99:3036–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lane, T.E., M.T. Liu, B.P. Chen, V.C. Asensio, R.M. Samawi, A.D. Paoletti, I.L. Campbell, S.L. Kunkel, H.S. Fox, and M.J. Buchmeier. 2000. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 74:1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]