

Abstract
Human cytomegalovirus (HCMV) infects endothelial, epithelial, and glial cells in vivo. These cells can express MHC class II proteins, but are unlikely to play important roles in priming host immunity. Instead, it seems that class II presentation of endogenous HCMV antigens in these cells allows recognition of virus infection. We characterized class II presentation of HCMV glycoprotein B (gB), a membrane protein that accumulates extensively in endosomes during virus assembly. Human CD4+ T cells specific for gB were both highly abundant in blood and cytolytic in vivo. gB-specific CD4+ T cell clones recognized gB that was expressed in glial, endothelial, and epithelial cells, but not exogenous gB that was fed to these cells. Glial cells efficiently presented extremely low levels of endogenous gB—expressed by adenovirus vectors or after HCMV infection—and stimulated CD4+ T cells better than DCs that were incubated with exogenous gB. Presentation of endogenous gB required sorting of gB to endosomal compartments and processing by acidic proteases. Although presentation of cellular proteins that traffic into endosomes is well known, our observations demonstrate for the first time that a viral protein sorted to endosomes is presented exceptionally well, and can promote CD4+ T cell recognition and killing of biologically important host cells.
Viruses are contained frequently by cytolytic or cytokine-mediated functions of CD8+ T cells, which recognize peptides that are derived from endogenous viral proteins and are presented on MHC class I molecules. By contrast, CD4+ T cells normally provide “help” to initiate, maintain, or amplify immune responses by surveying for presentation of extracellular proteins by MHC class II molecules. However, it also is well established that class II proteins can present peptides that are derived from endogenous or intracellular proteins. In fact, most peptides that are extracted from class II molecules are derived from endogenous membrane proteins that traffic into exocytic and endocytic pathways (1, 2). Peptides that are derived from nuclear or cytosolic proteins represent a smaller fraction, and have been postulated to reach class II loading compartments after proteasome processing—with or without the involvement of transporter associated with antigen presentation (TAP)—by autophagy or by as yet undefined mechanisms (3–8).
Most studies of class II presentation have focused on professional APCs—DCs, macrophages, or B cells that express copious amounts of class II molecules. Endothelial, epithelial, and glial cells also can express class II proteins, especially after induction by IFN-γ, a cytokine that is elicited commonly during virus infections. These cells act as portals of entry, barriers to movement of viruses between tissues, and “sentinels” that alert the immune system of invasion. Little is known about class II antigen presentation in these cell types and how this functions in control of viruses. It seems unlikely that priming immune responses is the outcome. In contrast to professional APCs, these nonprofessional APCs do not possess well-adapted phagocytic or endocytic machinery, nor do they migrate to primary or secondary lymphoid organs where priming primarily occurs. Instead, it seems more likely that these cells express class II proteins to present endogenous viral antigens and be recognized by CD4+ T cells. This would expand the immune repertoire to recognize and—if these CD4+ T cells were cytolytic or expressed anti-viral cytokines—lead to control of viruses.
Several human viruses apparently are controlled by CD4+ CTLs (9–15). Cytotoxic CD4+ effectors may be especially important with herpesviruses: HSV, varicella-zoster virus, EBV, and HCMV (16–19). These viruses inhibit MHC class I antigen presentation; therefore, class II presentation of viral proteins to CD4+ T cells may be vital to expand the degree to which the immune system can recognize virus-infected cells. In most cases, evidence for CD4+ CTLs has involved T cell clones that could lyse antigen-expressing cells; however, it is possible that cytolytic capacity was acquired during in vitro culture (20, 21). In very few instances have in vivo cytolytic capacity of CD4+ CTLs been demonstrated. Direct ex vivo CD4+ CTLs were described for HIV, although these studies involved the use of superantigens to conjugate target and T cells (14). Recently, mouse CD4+ T cells specific for lymphocytic choriomeningitis virus were shown to be cytotoxic in vivo (22).
HCMV is a ubiquitous herpes virus that promotes the expansion of enormous numbers of CD4+ and CD8+ T cells (23), likely because of periodic reactivation from latency over the course of a lifetime. Although CD8+ T cells clearly play a central role in containing HCMV (24), accumulating evidence (25–27) suggests that CD4+ T cells also can act as effectors directly on virus-infected cells. Patients that generate higher numbers of IFN-γ–producing anti-HCMV CD4+ T cells clear the virus faster and exhibit fewer symptoms (28, 29); CD4+ T cell clones specific to several HCMV antigens are cytolytic (30, 31). In addition, murine cytomegalovirus can be controlled by CD4+ T cells in vivo in the absence of CD8+ T cells (32).
We previously hypothesized that CD4+ T cells can control HCMV infections by recognizing endogenous antigens, viral proteins that are expressed within virus-infected cells (33). This was based on several facets of HCMV biology. First, HCMV infects epithelial cells in the gut, endothelial cells throughout the body, and glial cells in the brain, and causes pathology in each of these tissues (34). Thus, these cells, which are important for HCMV replication and spread in vivo, can express class II proteins, but are unlikely to be involved in priming immunity. Second, HCMV and other herpesviruses assemble virus particles on trans-Golgi network (TGN)/endosomes (35, 36). As part of this process, and as virions bud into cytoplasmic membranes, large quantities of all HCMV structural proteins are delivered into endosomal compartments where they can be processed readily for class II presentation. This is unlike other mammalian viruses (e.g., influenza virus) that bud from the plasma membrane and where antigens are presented by class II pathways involving proteasomes and TAP (7). These observations supported the notion that HCMV proteins might be particularly prone to processing and presentation by class II proteins. Consistent with this premise, two HCMV membrane proteins, glycoprotein B (gB) and glycoprotein H (gH), that are extensively sorted to endosomes, are major CD4+ T cell targets in vivo (23), and most CTL clones that recognize gB and gH are CD4+ (30, 31).
We found that as many as 1% of the total CD4+ T cells in human blood recognized HCMV gB. Most gB-specific CD4+ T cells that are isolated from PBMCs expressed granzyme B, which suggested cytolytic function in vivo. Several gB-specific CD4+ T cell clones also were cytolytic and recognized glial, epithelial, and endothelial cells expressing endogenous gB. However, these cells could not present exogenous (extracellular) gB. Glial cells expressing extremely small quantities of endogenous gB stimulated CD4+ T cell clones. Presentation of endogenous gB required sorting to endosomes and endosomal proteases. Although it is well established that model cellular antigens that are delivered to endosomes can be presented by class II proteins, these observations demonstrate for the first time that an important human pathogen is recognized by a pathway in which viral proteins are sorted specifically to endosomes and are presented efficiently by class II molecules. Moreover, the results support the notion that CD4+ T cells can play an important role in controlling HCMV through cytolytic and cytokine-mediated mechanisms.
RESULTS
Characterization and cloning of HCMV gB-specific CD4+ T cells
Previous studies that involved T cell clones (30, 31) suggested that HCMV glycoprotein gB might be a principal CD4+ T cell antigen. By ELISPOT assays, we found that 1–10% of total CD4+ T cells from several seropositive individuals produced IFN-γ in response to extracts of HCMV-infected cells (unpublished data). In several instances, 10% of the anti-HCMV CD4+ T cells (≤1% of the total CD4+ cells) recognized a soluble form of gB presented by DCs (unpublished data). Given that HCMV expresses >250 polypeptides, gB is a dominant, naturally occurring CD4+ T cell antigen. We tested whether CD4+ T cells, purified directly from blood and without restimulation, were able to express and secrete granzyme B in response to His16 glial cells that expressed gB. His16 cells were derived by stable transfection of the class II transactivator gene into U373-MG cells (37), one of only a very few cells that HCMV infects in vitro and which have been used extensively in studies of HCMV immunity. The frequency of granzyme B–secreting CD4+ T cells was roughly equivalent to that of IFN-γ–secreting T cells (Fig. 1 A). Glial cells expressing a HSV glycoprotein, gI, did not stimulate T cells to secrete either IFN-γ or granzyme B (Fig. 1 A). Secretion of granzyme B requires perforin and is directly correlated with cytolytic activity of T cells (38). Thus, our results are consistent with the notion that anti-gB CD4+ T cells are cytolytic in vivo.
Figure 1.

Anti-gB CD4+ T cells are cytotoxic. (A) His16 cells were infected with 30 PFUs/cell of Ad viruses expressing HCMV gB (AdtetgB) or HSV gI (AdtetgI) for 24 h, incubated with CD4+ T cells purified from PBMCs, and ELISPOT assays were performed for IFN-γ or granzyme B (GrB). Error bars denote standard deviations, and data points without error bars represent differences that are too small. His16 cells were infected with 10 PFUs/cell of AdtetgB (B) or 100 PFUs/cell of AdTbH9 (C) for 24 h, labeled with Na2 51CrO4, and then incubated with anti-gB CD4+ T cell clones (gB10/G3, gB10/H5, or gB100/2) or with mtb39-specific CD4+ T cell clone, TbH9-9, at various effector/target ratios for 24 h, and the specific release of 51Cr was determined.
24 HLA-DR17–restricted gB-specific CD4+ T cell clones were isolated by limiting dilution using DCs that were incubated with soluble gB. Three CD4+ T cell clones were tested in CTL assays. His16 glial cells expressing gB after infection with a nonreplicating adenovirus (Ad) vector (AdtetgB) (39) were lysed by the gB-specific clones, but cells that were infected with a control Ad vector were not (Fig. 1 B). These results support the notion that a substantial fraction of HCMV-specific CD4+ T cells can recognize gB. Given that all three clones were cytotoxic, and that most gB-specific CD4+ T cells—as judged by IFN-γ secretion—expressed granzyme, it is likely that a substantial fraction of gB-specific CD4+ T cells is cytotoxic.
Presentation of exogenous and endogenous HCMV gB to CD4+ T cells by glial, epithelial, and endothelial cells
His16 cells infected with AdtetgB, that expresses full-length, membrane-anchored gB, stimulated the gB-specific clone gB3/F11 to produce IFN-γ at levels similar to DCs that present soluble gB (Fig. 2 A). His16 cells did not present soluble gB, even at 10 μg/ml, a dose that is 100-fold greater than that required for substantial stimulation of gB3/F11 by DCs that are incubated with soluble gB (Fig. 2 A and not depicted). When supernatants from AdtetgB-infected His16 cells were transferred to other His16 cells, they failed to stimulate CD4+ T cells (unpublished data). It was impossible to express endogenous gB in DCs because of poor transduction of DCs with Ad vectors, as observed previously (40). Several other clones similarly recognized gB that was expressed endogenously in His16 cells (four shown in Fig. 2 B), but most did not recognize exogenous gB or were stimulated only weakly with high concentrations (50–100 μg/ml) of soluble gB that was incubated for extended periods (not depicted).
Figure 2.
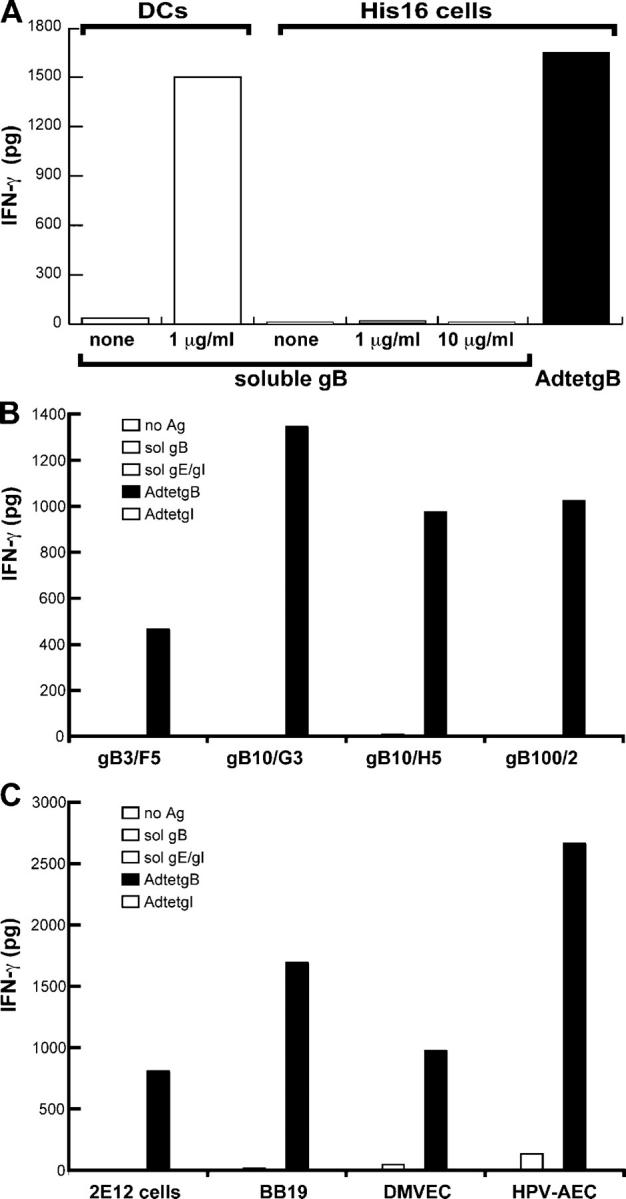
Presentation of endogenous and exogenous gB to CD4+ T cell clones. (A) Autologous DCs (104) were incubated with 1 μg of soluble gB and 4 × 104 anti-gB CD4+ T cell clone gB3/F11. His16 glial cells were infected with AdtetgB (200 PFUs/cell) for 24 h, then incubated with 4 × 104 clone gB3/F11 or incubated with 1 or 10 μg of soluble gB and simultaneously with 4 × 104 clone gB3/F11. (B) His16 cells were infected with AdtetgB or AdtetgI (30 PFUs/cell) for 24 h before addition of T cells, or incubated with 10 μg/ml of soluble gB or soluble gE/gI and with gB-specific clones gB3/F5, gB10/G3, gB10/H5, or gB100/2. (C) Class II–expressing gastric epithelial cells (2E12) or IFN-γ–stimulated endothelial cells (BB19, brain; DMVEC, dermal vascular; HPV-AEC, aortic) were infected with AdtetgB or AdtetgI for 24 h or incubated with 10 μg of soluble gB or gE/gI and with CD4+ T cell clone gB10/H5. IFN-γ produced by the CD4+ T cells was measured by ELISA after 24 h.
These observations were extended to epithelial and endothelial cells. Class II transactivator–transfected 2E12 gastric epithelial cells, which constitutively express class II proteins and three different endothelial cell lines that were induced to express class II proteins with IFN-γ, also presented endogenous gB, but not soluble gB (Fig. 2 C). Similar results were obtained with other endothelial cells (unpublished data). These cells are immortal lines but mimic properties of untransformed epithelial and endothelial cells; 2E12 cells can be infected by EBV (41) and the endothelial lines can be infected by HCMV (unpublished data). We concluded that glial, epithelial, and endothelial cells proficiently presented endogenous, but not exogenous, gB—by class II proteins—to CD4+ T cells.
Glial cells can internalize soluble gB and present gB peptides and other exogenous antigens
The failure of glial, endothelial, and epithelial cells to present exogenous gB might relate to an inability to internalize gB. HCMV gB binds to cell surface heparan sulfate proteoglycans (42), which are internalized slowly (43). We used iodinated gB to evaluate uptake by His16 glial cells, and found that these cells internalized soluble 125I-labeled gB, albeit slowly; ∼40% of the bound gB was internalized after 90 min (Fig. 3 A).
Figure 3.
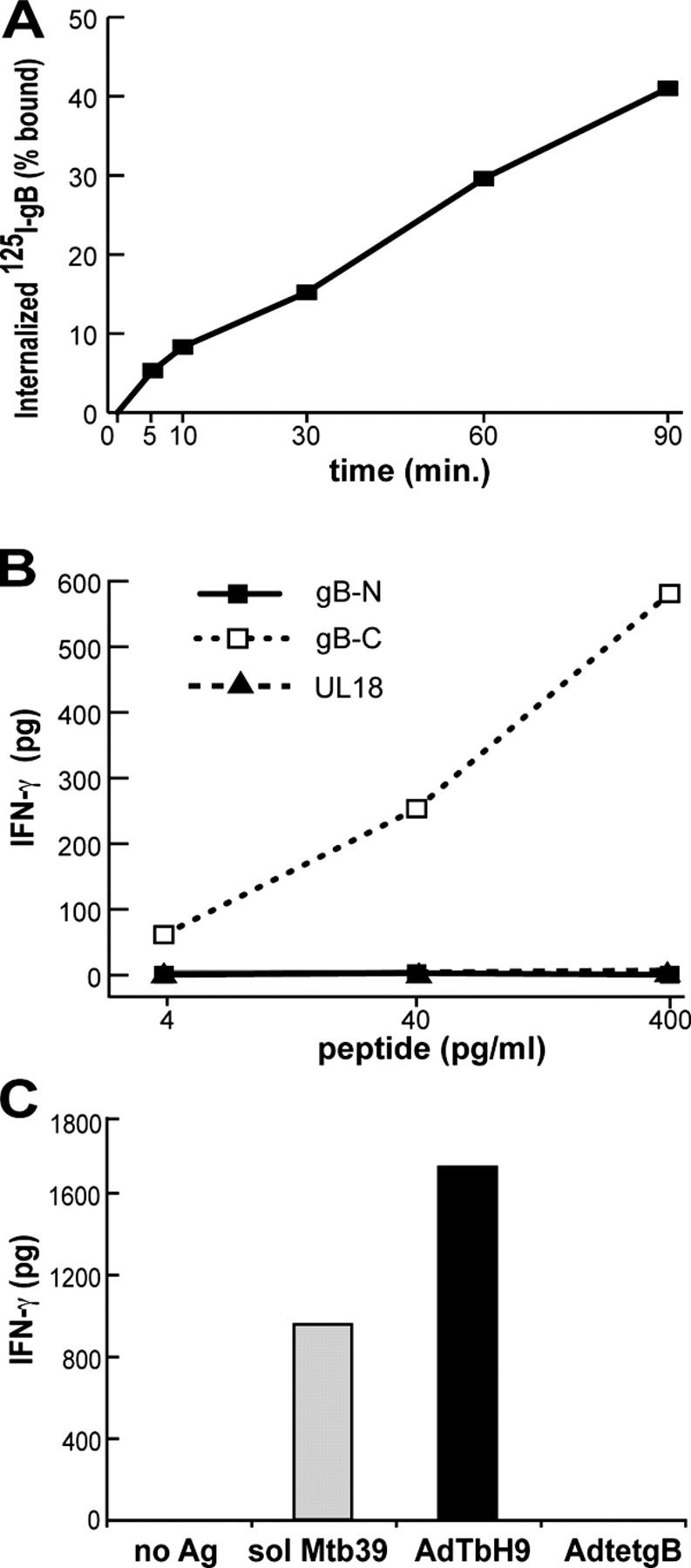
Glial cells internalize soluble gB, and present gB peptides and a soluble TB antigen. (A) His16 cells were incubated with 125I-gB at 4°C, washed, warmed to 37°C, and the cell surface gB was removed with citrate buffer before counting cell-associated (internalized) 125I. Background (cells not warmed to 37°C) was subtracted from each value. (B) His16 cells were incubated with pools of peptides (15 mers overlapping by 10 residues) making up the NH2-terminal (gB-N, residues 1–440) or the COOH-terminal (gB-C, residues 430–907) half of gB or all of UL18, for 6 h before incubation with gB10/G3 CD4+ T cells for 24 h. (C) His16 cells were incubated with medium alone (no Ag) or with 1 μg/ml of soluble mtb39, and TbH9-9 (mtb39-specific) T cells, or were infected with 100 PFUs/cell of AdTbH9 or AdtetgB for 24 h before addition of TbH9-9 T cells. IFN-γ was measured in B and C.
To determine whether His16 cells could present gB peptides, cells were incubated with pools of 15-mers, overlapping by 10 residues, encompassing residues 1–440 (gB-N) or 431–907 (gB-C). A pool of peptides, including all of HCMV UL18, served as a negative control. Three different clones recognized gB-C, but not gB-N or UL18 peptide pools (Fig. 3 B; only one clone shown). The level of stimulation of all three clones at 400 pg/ml of peptides was similar to that obtained with His16 cells that were infected with 3–5 PFUs/cell of AdtetgB (unpublished data). Because our clones recognize soluble gB (residues 25–692) and a peptide mixture encompassing residues 431–907, one or more epitope(s) is located within residues 431–692.
To ascertain whether His16 cells could present another exogenous antigen, the cells were incubated with a 40-kD soluble, Escherichia coli-produced, recombinant tuberculosis (TB) protein, mtb39—a member of a family of related TB proteins (44). Presentation to mtb39-specific CD4+ T cell clone TbH9-9 (which recognizes residues 133–147) was assessed (37, 45). His16 cells that were incubated with 1 μg/ml of mtb39 stimulated TbH9-9 cells well compared with cells that expressed endogenous, Ad-delivered mtb39 (Fig. 3 C). Note that cells were infected with relatively high doses of AdTbH9, and in this case, mtb39 is linked to a signal sequence and is secreted, but also likely fills the exocytic pathway (46). Together, these results show that His16 cells are capable of internalizing and presenting another soluble protein to CD4+ T cells, but do not present extracellular gB well.
Relative efficiency of presentation of endogenous gB
Nonprofessional APCs are not as capable as professional APCs at antigen capture, processing, and presentation. Nevertheless, several gB-specific CD4+ T cell clones were stimulated to similar or higher levels by AdtetgB-infected His16 cells, compared with DCs that presented soluble gB (Fig. 2 A, and not depicted). When APCs were limiting, His16 cells expressing gB stimulated gB100/2 T cells better than did DCs that were incubated with exogenous gB (Fig. 4 A). Additionally, His16 cells expressing endogenous mtb39, expressed using an Ad vector, also stimulated TbH9-9 T cells approximately as well as syngeneic lymphoblastoid cell lines (LCLs) that were incubated with soluble mtb39 (unpublished data). As a second measure of the efficiency of presentation of endogenous gB, His16 cells were infected with low doses of AdtetgB. Significant stimulation of two different gB-specific clones was observed with His16 cells that were infected using as little as 0.78–1.5 PFUs/cell of AdtetgB; half-maximal presentation occurred at ∼5 PFUs/cell (Fig. 4 B). These Ad vectors do not replicate in His16 cells and at these low virus doses, gB expression could not be detected by radiolabeling or Western blots (unpublished data). Thus, presentation of gB by His16 cells—as measured by stimulation of T cell clones—is dependent on gB concentrations in the APCs, and extremely low quantities of gB are sufficient for T cell stimulation. Although the APCs and the source of antigen were different in Fig. 4 A, when APCs were limiting, T cell stimulation was reduced or extinguished. Conclusions about antigen presentation must be tempered by the fact that antigen presentation was measured indirectly and by using T cell clones that were selected for gB recognition. However, coupled with the results in Fig. 7, our observations are consistent with the conclusion that His16 cells can present endogenous gB very efficiently as compared with professional APCs.
Figure 4.

Efficiency of presentation of exogenous gB by DCs versus endogenous gB by glial cells. (A) Various numbers of His16 cells or autologous DCs were incubated with 1 μg/ml of soluble gB (sol gB) at the time of adding T cells or were infected with 10 PFUs/cell of AdtetgB for 24 h before adding T cells. 4 × 104 gB100/2 anti-gB CD4+ T cells were used in all cases. (B) His16 cells were infected with various doses of AdtetgB for 24 h before incubation with gB10/G3 or gB10/H5 T cell clones for 24 h. In both cases, IFN-γ was measured.
Figure 7.

Presentation of gB in HCMV-infected cells. His16 cells were infected with WT HCMV strain TR or TR-BAC lacking the US2 and US3 genes using 0.5 PFU/cell (A) or 0.05 PFU/cell (B) for 72 h and incubated with anti-gB T cells gB10/G3 or gB10/H5 for 24 h, and IFN-γ was measured.
Endogenous presentation requires endosomal proteases and not proteasomes
Endogenous, cytoplasmic proteins can be degraded by proteasomes and reach endosomes/lysosomes through TAP or by undefined mechanisms (3, 7, 8). Processing and presentation of endogenous gB was not affected by the proteasome inhibitors, lactacystin (Fig. 5 A) or MG132 (not depicted). Similarly, HSV ICP47, a specific inhibitor of TAP (47), had no effect (unpublished data). By contrast, leupeptin, which inhibits cysteine and serine proteases, completely blocked gB presentation (Fig. 5 A). Cathepsins contribute to progressive fragmentation of the protein invariant chain (Ii); therefore, it was reasonable that 50 μM leupeptin inhibited processing of Ii and reduced loading of class II complexes. However, presentation of exogenous mtb39 was unaffected at lower leupeptin concentrations (2.5–5 μM), yet presentation of endogenous gB was reduced substantially (Fig. 5 B). A second inhibitor, pepstatin, which blocks aspartyl proteases, did not inhibit gB presentation (Fig. 5 A). Brefeldin A, an inhibitor of ER to Golgi transport, and bafilomycin A, an inhibitor of vacuolar acidification, also blocked presentation of gB but both potentially can inhibit transport or maturation of class II proteins. We concluded that endogenous gB is processed by endosomal proteases and not by proteasomes.
Figure 5.

Presentation of endogenous gB occurs in endosomes. (A) His16 cells were treated with 50 μM leupeptin or pepstatin, or 10 μM brefeldin A, bafilomycin, or lactacystin for 30 min, infected with AdtetgB (3 PFUs/cell) for 24 h in the presence of the inhibitors, washed, fixed with 0.1% p-formaldehyde, and then incubated for 24 h with gB100/2 T cells. (B) His16 cells were treated with the indicated concentrations of leupeptin, infected with AdtetgB, and used in T cell assays similar to those in (A), using gB10/H5 and gB100/2 T cell clones. Other His16 cells were incubated with 2 μg/ml soluble mtb39 (sol mtb39) and TbH9-9 (mtb39-specific) T cells. IFN-γ was measured in both cases.
Presentation of gB requires cytoplasmic sorting sequences
The cytoplasmic tail (CT) domain of HCMV gB contains several sorting sequences that are responsible for extensive accumulation of gB in the TGN/endosomes (48), a prelude to virion assembly (35, 36). We reasoned that the targeting of gB to TGN/endosomes would result in efficient class II–mediated presentation of gB. To examine this, Ad vectors expressing mutant forms of gB were constructed: (a) gBΔCT lacks all of the CT domain, except for three juxtamembrane residues; (b) gB-KKSL contains four additional residues at the extreme COOH terminus that recycle the protein back to the ER (49); and (c) gB-KKSLAL is a control protein that contains two additional residues that revert the KKSL phenotype to WT (Fig. 6 A). FACS analyses indicated extensive accumulation of gBΔCT on the cell surface, but WT gB, gBKKSL, and gBKKSLAL were internal (Fig. 6 B). Confocal analyses indicated that WT gB was in perinuclear vesicles, some of which stained with anti–HLA-DM antibodies (Fig. 6 C), as well as with several TGN and endosome markers (not depicted). gB-KKSL colocalized with the ER marker protein disulfide isomerase (Fig. 6 C), and unlike WT gB and gB-KKSLAL, gB-KKSL was not proteolytically cleaved by furin, a TGN-localized protease (not depicted).
Figure 6.
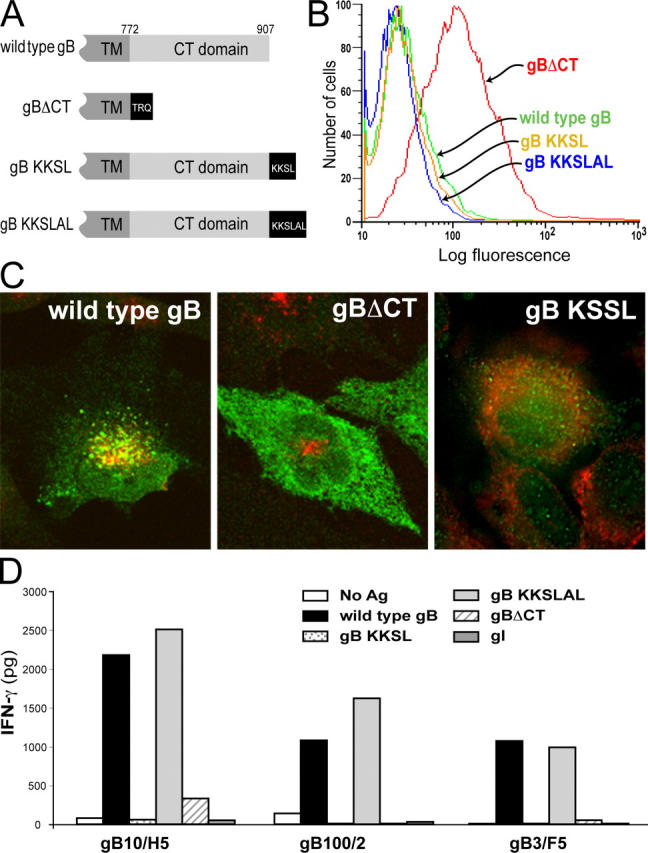
Endosomal targeting of endogenous gB is required for presentation. (A) WT gB contains a 135-residue cytosolic domain. gBΔCT is truncated three residues after the transmembrane (TM) domain. gB-KKSL contains the ER retention motif KKSL at the COOH terminus. gB-KKSLAL contains two additional residues that reverse the effects of KKSL. (B) His16 cells were infected with Ad vectors expressing WT gB, gB-KKSL, gB-KKSLAL, or gBΔCT (50 PFUs/cell), and cell surface expression of gB was assessed by FACS. (C) His16 cells were infected with Ad viruses (50 PFUs/cell) for 24 h, fixed, permeabilized, and stained for gB (green) and HLA-DM (red) in the case of WT gB and gBΔCT, or protein disulfide isomerase (red) in the case of gB-KKSL. (D) His16 cells were infected with Ad viruses (1 PFU/cell) expressing WT gB, gBΔCT, gB-KKSL, gB-KKSLAL, or HSV gI for 24 h then incubated with anti-gB CD4+ T cell clones. IFN-γ was measured after 24 h.
Three CD4+ T cell clones recognized WT gB and gB-KKSLAL, but not gB-KKSL or gBΔCT (Fig. 6 D). We concluded that gB must be sorted to the TGN/endosomes in order to be presented, and gB that was transported to the cell surface or retained in the ER was not presented.
Class II presentation of gB in HCMV-infected cells and effects of US2 and US3
Laboratory strains of HCMV replicate efficiently only in cultured human fibroblasts (34). Recently, HCMV clinical isolates that infect other cell types were described (50). The clinical strain, HCMV TR efficiently infected His16 cells (not depicted) and stimulated gB-specific CD4+ T cell clones, even at relatively low input virus doses (Fig. 7 A). The levels of IFN-γ (500–1000 pg) that were produced by CD4+ T cells that were in contact with His16 cells that were infected with only 0.5 PFU/cell of HCMV TR (Fig. 7 A) were similar to those produced frequently with His16 cells that were infected with AdtetgB or with DCs and exogenous gB (Fig. 4). Moreover, there was substantial stimulation of CD4+ T cells when His16 cells were infected with extremely low doses (0.05 PFU/cell) of HCMV TR (Fig. 7 B). Even at these low virus doses, it is likely that gB expression is attained in a reasonable fraction of cells, because the HCMV particle/PFU ratio is 150–1,000. It is possible that gB that was part of the input virus was presented here, although this might be unlikely given the very low input doses. In other experiments, cells that were infected with UV-inactivated HCMV TR were recognized poorly by T cells (unpublished data). However, these UV-inactivated viruses were not characterized in terms of gene expression and for the integrity of virion proteins, and it is difficult to reach solid conclusions. More importantly, these results testify to the conclusion that these glial cells can present gB and stimulate CD4+ T cells when extremely low concentrations of gB are present. Given previous observations that high protein concentrations were required for cytoplasmic antigens to be presented (51, 52), this supports efficient presentation.
HCMV TR was cloned recently by inserting a bacterial artificial chromosome (BAC) into the HCMV US2-US6 genes to derive a virus denoted TR-BAC, which does not express the unique short (US)2 and US3 proteins (50). We showed previously, by using Ad vectors, that US2 and US3 can inhibit the class II pathway and block presentation of exogenous mtb39 to CD4+ T cells (37, 45). However, effects of US2 and US3 have been difficult to study in the context of HCMV because of inefficient infection of any class II–expressing cells. Here, we show that low doses of TR-BAC (a US2−/US3− mutant) reproducibly stimulated two different CD4+ T cell clones 2.5-fold better than did WT TR that expresses US2 and US3 (Fig. 7 B). This effect was not observed at higher doses of virus (Fig. 7 A), probably because higher levels of gB (perhaps in the input virus) overwhelmed the effects of US2 and US3. This is the first demonstration of the effects of US2 and US3 on class II presentation, in the context of HCMV infection. Together, these data are consistent with the notion that gB can be presented efficiently to CD4+ T cells when cells are infected with very low HCMV doses, and that presentation in the context of HCMV infected cells can be reduced by US2 and US3.
DISCUSSION
HCMV elicits a robust and long-lasting cellular immune response that includes enormous numbers of CD4+ and CD8+ T cells (23). These T cells are essential for the control of virus in vivo. Clearly, anti-HCMV CD4+ T cells play an important role in expanding CD8+ T cell populations, as evidenced by the failure of anti-HCMV CD8+ T cell therapy in immunosuppressed patients who lack CD4+ T cell “help” (53). However, there also is evidence that HCMV-specific CD4+ T cells can play a key role as effectors to contain or eliminate the virus. CD4+ T cell clones that are specific for several HCMV proteins were described to possess cytolytic activity (17, 29–31), although it might be argued that this was acquired in vitro. Observations that HCMV assembly and class II antigen presentation pathways intersect in endosomal compartments predict that there is extensive presentation of endogenous viral proteins by class II molecules. It is highly unlikely that in important host cells—epithelial, endothelial, and glial cells—this presentation is associated with priming of host immunity. Instead, we hypothesized that class II presentation in these cells relates to recognition of HCMV infection by CD4+ effector T cells, and provides a mechanism for control of virus in addition to CD8+ T cells.
Based on these considerations, we initially asked whether HCMV-specific CD4+ T cells were cytolytic in vivo. A substantial fraction of freshly isolated CD4+ T cells responded to glial cells presenting endogenous gB by expressing granzyme B. Although, it also is possible that gB-specific CD4+ T cells use the Fas-Fas ligand–mediated lytic mechanism, the observation that the frequency of granzyme B–secreting T cells was similar to those producing IFN-γ supported the notion that granule exocytosis is a major pathway for cytotoxicity of anti-gB CD4+ CTLs. Granzyme release is an event downstream of polyperforin pore formation on target cell membranes, and these concerted events directly correlate with the lytic activity of CTLs (38, 54). The observation that endogenous gB presented by glial cells is recognized by circulating CD4+ T cell effectors supports a direct role for these immune cells in controlling the virus. In addition, our study connects HCMV replication in these biologically relevant cells to potentially abundant and functionally important CD4+ CTLs. This is one of a very few examples in which evidence supports the existence of anti-viral CD4+ CTLs in vivo, and suggests that these cells are more widespread than believed previously.
Endogenous HCMV gB was presented efficiently by endothelial, glial, and epithelial cells. Glial cells expressing extremely low levels of gB—delivered by using an Ad vector or after HCMV infection—stimulated CD4+ T cells as well as, or better than, DCs that were incubated with exogenous gB. This presentation required sorting to endosomes and endosomal proteases. gB targeted to the plasma membrane or retained in the ER was not presented well. Given that HCMV assembles in endosomes, it is highly probable that this pathway extends to most other HCMV structural proteins. Consistent with this, a second HCMV glycoprotein, gH, is an important target of anti-HCMV CD4+ T cells (31). Other herpesviruses (HSV, EBV, varicella-zoster virus, and Kaposi's sarcoma–associated herpesvirus) also infect endothelial, glial, or epithelial cells and similarly assemble in TGN/endosomes. Thus, we believe that class II presentation of endosome-sorted herpesvirus proteins probably is a prevalent process.
Endothelial, epithelial, and glial cells failed to present exogenous, soluble gB, yet His16 glial cells could present a soluble TB protein. Uptake of gB was slow, and it is reasonable that delivery to loading compartments also was slow or inefficient. Alternatively, exogenous gB may be delivered into endosomal subcompartments that contain a different set of proteases compared with those that endogenous gB encounters. Endogenous, membrane-anchored gB traffics internally in recycling loops between the TGN and endosomes (39). Previous studies observed that different subcellular compartments produce distinct epitopes from the same protein, or may destroy other epitopes (7, 55, 56). Mapping of gB epitopes, combined with cell fractionation studies, might produce a better picture of why soluble gB is not presented. However, this is beyond the scope of present studies and is not directly relevant because endogenous gB, and not soluble gB, is produced during HCMV infection.
Several studies have described class II presentation of endogenous viral antigens by mechanisms that differ from that described here. Measles virus proteins were presented by TAP-dependent or -independent pathways, although the cytoplasmic matrix protein required high and sustained levels of expression compared with class I presentation (51, 52). Certain epitopes derived from influenza virus membrane proteins were presented in a proteasome- and TAP-dependent class II pathway, whereas other epitopes were presented only as exogenous antigens (7, 10). Unlike HCMV and other herpesviruses, influenza and measles viruses assemble at the plasma membrane without the requirement for sorting to, or accumulation in, endosomes. EBV expresses a limited set of viral proteins in latently infected B cells, and a nuclear antigen, EBV nuclear antigen-1, is delivered to endosomes by autophagy and then is presented by class II molecules (6). These observations, together with our results, provide an appreciation that CD4+ T cells recognize endogenous viral antigens by a variety of mechanisms, and that this recognition has important implications for the control of viral infections.
Class II presentation of endogenous HCMV antigens provides an explanation for previous observations that this virus also inhibits the MHC class II pathway (33, 37, 45). At the outset, these findings might seem to be a contradiction. However, it is well established that HCMV expresses inhibitors of the class I pathway and NK cell recognition, and yet, the virus is controlled amply by CD8+ and NK cells (33, 57, 58). HCMV immune evasion proteins do not affect immune priming as reflected by robust CD8+ and CD4+ T responses, and inhibition of antigen presentation is inefficient in professional APCs (40, 59, 60). Instead, this avoidance reduces the recognition of virus-infected cells, likely within a narrow window of time (e.g., following reactivation) or involving specific cell types (e.g., nonprofessional APCs, such as epithelial, endothelial, or glial cells) (57). Thus, class II presentation of endogenous viral proteins to CD4+ T cells increases the host's capacity to recognize the virus in the face of inhibition of CD8+ T and NK cell recognition, and is, itself, the subject of immune evasion. Here, by using a clinical strain of HCMV, we showed that US2 and US3 reduced class II presentation of gB in HCMV-infected glial cells. The effects of US2 and US3 were most prominent at low doses of HCMV, conditions that may reflect infection in vivo where modest differences may provide a substantial selective advantage, especially in cells that express lower levels of class II proteins. The fact that HCMV can inhibit the class II pathway, under certain circumstances, attests to the notion that presentation of endogenous proteins in nonprofessional APCs is an important mechanism by which HCMV is recognized by the immune system, and is something that must be thwarted.
MATERIALS AND METHODS
Cells and media.
His16 and 2E12 cells were derived by stable transfection of U373-MG or AGS cells, respectively, with the human CIITA gene (37, 40, 41). BB19 (61), DMVEC (62), and HPV-AEC endothelial cell lines were obtained from A. Moses (Oregon Health & Science University, Portland, Oregon), and were grown in endothelial cell basal medium-2 and supplements. 293 cells used to propagate adenovirus (Ad) vectors were maintained in Eagle's minimum essential medium plus 10% FBS, and fibroblasts were grown in DMEM plus 10% FBS. DCs were prepared and maintained as described (46). Tn5 insect cells were grown in Grace's insect cell media.
Cloning and expansion of T cells.
Human blood and sera were obtained according to human subjects protocol approved by Institutional Review Boards at Oregon Health & Science University and Portland Veterans Administration Medical Center. CD4+ T cells, from HLA-DRB17+ individuals to match the DR type of His16 cells, were purified from PBMCs using CD8 midi-MACS columns (Dynal). T cell frequencies were assessed by ELISPOT, using PBMCs. T cells were expanded, using autologous DCs and macrophage, cloned by limiting dilution using allo-PBMCs and allo-lymphoblastoid cell lines (LCLs) (46, 63), and tested by ELISPOT for reactivity with soluble gB using DCs. Clones were expanded using γ-irradiated allo-LCLs and allo-PBMCs, anti-CD3 mAb (Chiron), and IL-2 (46, 63).
Viruses.
WT and mutant human CMV (HCMV) strain TR (50) were propagated and titered on fibroblasts. The virus was purified using sorbitol cushion and sucrose gradients. Ad vectors expressing mutant forms of HCMV gB were constructed similar to those of HCMV US2 and US3 (37, 45), HSV gI (64), and WT gB (48). The promoter for all Ad vectors is induced when cells are co-infected with a second vector, Adtet-trans. An Ad vector expressing mtb39 fused to IL-1 signal sequence (AdTbH9) (46), as well as a baculovirus encoding His-tagged HCMV gB extracellular domain encompassing residues 1–692 (42), have been described.
Protein and peptide antigens.
HCMV-infected cell extracts were prepared by freeze-thawing infected fibroblasts. Soluble recombinant HCMV gB and HSV gE/gI dimer were produced and purified as described (42, 65). Soluble mtb39 was obtained from Corixa. HCMV gB and UL18 peptide pools consisting of 15-mers overlapping by 10 residues, synthesized at Mimotopes Inc., were obtained from L. Picker (Oregon Health & Science University, Portland, Oregon).
Antibodies, confocal microscopy, and flow cytometry.
mAb 27–78 (66) and rabbit antisera to gB were obtained from W. Britt (University of Alabama, Birmingham, AL). Antibodies to protein disulfide isomerase (StressGen Biotechnologies), TGN46 (Serotec), and HLA-DMβ (BD Biosciences), as well as Alexa 488-conjugated donkey anti–sheep and goat anti–mouse IgG and Alexa 594-conjugated goat anti–rabbit IgG (Invitrogen) were purchased commercially. Immunofluorescence and flow cytometry were performed as described (45).
Endocytosis assay.
Soluble gB was labeled with Na125I using Iodobeads (Pierce Chemical Co.). Cells were incubated with 125I-gB at 4°C for 15 min, warmed to 37°C to allow internalization and, at various times, cell surface gB was stripped from cells by a 10-min incubation with citrate buffer (40 mM citric acid, 10 mM KCl, 135 mM NaCl, pH 3.0) at 4°C. Internalized gB was determined by counting cell-associated radioactivity.
CD4+ T cell assays.
Antigen presentation assays were performed in 96-well plates (37, 45). In brief, 104 APCs were infected with Ad vectors for 18 h or were left uninfected, and then were incubated for 18–24 h with 4 × 104 CD4+ T cells. Endothelial cells were treated with IFN-γ (50 U/ml, Immunex) for 24 h before infection. Soluble antigens were added 0–4 h before addition of T cells. For drug inhibition studies, cells were incubated with the compounds for 30 min before and during Ad infection or soluble protein addition, fixed with 0.1% p-formaldehyde for 5 min, and washed and incubated with T cells for 18–24 h. For studies with HCMV, cells were infected for 72 h before T cells were added. Secretion of IFN-γ by the T cells was detected by a sandwich ELISA (45). For granzyme B ELISPOT assays (67), MACS-purified CD4+ T cells were incubated with His16 cells that were infected with AdtetgB or AdtetgI.
CTL assays.
51Cr release assays were used to measure cytotoxicity (68). In brief, His16 cells were infected with Ad vectors for 18 h, labeled with Na2 51CrO4 (100 μCi/106 cells) for 1–2 h, and plated at 104/well in triplicate with various numbers of CD4+ T cells for 24 h. Percentage specific lysis was calculated based on maximal release measured by using 2% Nonidet P-40. Spontaneous release was never >30%.
Acknowledgments
We are grateful to L. Hutt-Fletcher, A. Moses, and T. Shenk for cells; T. Compton for the baculovirus expressing soluble gB; W. Britt for antibodies; L. Picker for peptides and advice; and K. Frueh for valuable comments. We are thankful to R. Lines and G. Swarbrick for support with T cell cloning and ELISPOT assays, T. Wisner and A. Snyder for help with confocal microscopy, and A. Pinto for assistance with CTL assays.
This research was supported by grants from the National Institutes of Health nos. EY11245 and AI055051 (to D.C. Johnson), AI21640 (to J.A. Nelson), and AI01644 and AI48090 (to D.M. Lewinsohn).
The authors have no conflicting financial interests.
Abbreviations used: Ad, adenovirus; BAC, bacterial artificial chromosome; CT, cytoplasmic tail; gB, glycoprotein B; gH, glycoprotein H; HCMV, human CMV; LCL, lymphoblastoid cell line; TAP, transporter associated with antigen presentation; TB, tuberculosis; TGN, trans-Golgi network; US, unique short.
N.R. Hegde and C. Dunn contributed equally to this work.
References
- 1.Rudensky, A., P. Preston-Hurlburt, S.C. Hong, A. Barlow, and C.A. Janeway Jr. 1991. Sequence analysis of peptides bound to MHC class II molecules. Nature. 353:622–627. [DOI] [PubMed] [Google Scholar]
- 2.Chicz, R.M., R.G. Urban, J.C. Gorga, D.A. Vignali, W.S. Lane, and J.L. Strominger. 1993. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J. Exp. Med. 178:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lich, J.D., J.F. Elliott, and J.S. Blum. 2000. Cytoplasmic processing is a prerequisite for presentation of an endogenous antigen by major histocompatibility complex class II proteins. J. Exp. Med. 191:1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nimmerjahn, F., S. Milosevic, U. Behrends, E.M. Jaffee, D.M. Pardoll, G.W. Bornkamm, and J. Mautner. 2003. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33:1250–1259. [DOI] [PubMed] [Google Scholar]
- 5.Dani, A., A. Chaudhry, P. Mukherjee, D. Rajagopal, S. Bhatia, A. George, V. Bal, S. Rath, and S. Mayor. 2004. The pathway for MHCII-mediated presentation of endogenous proteins involves peptide transport to the endo-lysosomal compartment. J. Cell Sci. 117:4219–4230. [DOI] [PubMed] [Google Scholar]
- 6.Paludan, C., D. Schmid, M. Landthaler, M. Vockerodt, D. Kube, T. Tuschl, and C. Munz. 2004. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 307:593–596. [DOI] [PubMed] [Google Scholar]
- 7.Tewari, M.K., G. Sinnathamby, D. Rajagopal, and L.C. Eisenlohr. 2005. A cytosolic pathway for MHC class II-restricted antigen processing that is proteasome and TAP dependent. Nat. Immunol. 6:287–294. [DOI] [PubMed] [Google Scholar]
- 8.Zhou, D., P. Li, Y. Lin, J.M. Lott, A.D. Hislop, D.H. Canaday, R.R. Brutkiewicz, and J.S. Blum. 2005. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 22:571–581. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson, S., J.R. Richert, W.E. Biddison, A. Satinsky, R.J. Hartzman, and H.F. McFarland. 1984. Measles virus-specific T4+ human cytotoxic T cell clones are restricted by class II HLA antigens. J. Immunol. 133:754–757. [PubMed] [Google Scholar]
- 10.Morrison, L.A., A.E. Lukacher, V.L. Braciale, D.P. Fan, and T.J. Braciale. 1986. Differences in antigen presentation to MHC class I- and class II-restricted influenza virus-specific cytolytic T lymphocyte clones. J. Exp. Med. 163:903–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Littaua, R.A., A. Takeda, J. Cruz, and F.A. Ennis. 1992. Vaccinia virus-specific human CD4+ cytotoxic T-lymphocyte clones. J. Virol. 66:2274–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penna, A., P. Fowler, A. Bertoletti, S. Guilhot, B. Moss, R.F. Margolskee, A. Cavalli, A. Valli, F. Fiaccadori, F.V. Chisari, et al. 1992. Hepatitis B virus (HBV)-specific cytotoxic T-cell (CTL) response in humans: characterization of HLA class II-restricted CTLs that recognize endogenously synthesized HBV envelope antigens. J. Virol. 66:1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green, S., I. Kurane, S. Pincus, E. Paoletti, and F.A. Ennis. 1997. Recognition of dengue virus NS1-NS2a proteins by human CD4+ cytotoxic T lymphocyte clones. Virology. 234:383–386. [DOI] [PubMed] [Google Scholar]
- 14.Appay, V., J.J. Zaunders, L. Papagno, J. Sutton, A. Jaramillo, A. Waters, P. Easterbrook, P. Grey, D. Smith, A.J. McMichael, et al. 2002. Characterization of CD4(+) CTLs ex vivo. J. Immunol. 168:5954–5958. [DOI] [PubMed] [Google Scholar]
- 15.Wahid, R., M.J. Cannon, and M. Chow. 2005. Virus-specific CD4+ and CD8+ cytotoxic T-cell responses and long-term t-cell memory in individuals vaccinated against polio. J. Virol. 79:5988–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arvin, A.M., M. Sharp, S. Smith, C.M. Koropchak, P.S. Diaz, P. Kinchington, W. Ruyechan, and J. Hay. 1991. Equivalent recognition of a varicella-zoster virus immediate early protein (IE62) and glycoprotein I by cytotoxic T lymphocytes of either CD4+ or CD8+ phenotype. J. Immunol. 146:257–264. [PubMed] [Google Scholar]
- 17.Gyulai, Z., V. Endresz, K. Burian, S. Pincus, J. Toldy, W.I. Cox, C. Meric, S. Plotkin, E. Gonczol, and K. Berencsi. 2000. Cytotoxic T lymphocyte (CTL) responses to human cytomegalovirus pp65, IE1-Exon4, gB, pp150, and pp28 in healthy individuals: reevaluation of prevalence of IE1-specific CTLs. J. Infect. Dis. 181:1537–1546. [DOI] [PubMed] [Google Scholar]
- 18.Munz, C., K.L. Bickham, M. Subklewe, M.L. Tsang, A. Chahroudi, M.G. Kurilla, D. Zhang, M. O'Donnell, and R.M. Steinman. 2000. Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 191:1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmid, D.S. 1988. The human MHC-restricted cellular response to herpes simplex virus type 1 is mediated by CD4+, CD8− T cells and is restricted to the DR region of the MHC complex. J. Immunol. 140:3610–3616. [PubMed] [Google Scholar]
- 20.Bourgault, I., A. Gomez, E. Gomard, F. Picard, and J.P. Levy. 1989. A virus-specific CD4+ cell-mediated cytolytic activity revealed by CD8+ cell elimination regularly develops in uncloned human antiviral cell lines. J. Immunol. 142:252–256. [PubMed] [Google Scholar]
- 21.Sun, Q., R.L. Burton, K.E. Pollok, D.J. Emanuel, and K.G. Lucas. 1999. CD4(+) Epstein-Barr virus-specific cytotoxic T-lymphocytes from human umbilical cord blood. Cell. Immunol. 195:81–88. [DOI] [PubMed] [Google Scholar]
- 22.Jellison, E.R., S.K. Kim, and R.M. Welsh. 2005. Cutting edge: MHC class II-restricted killing in vivo during viral infection. J. Immunol. 174:614–618. [DOI] [PubMed] [Google Scholar]
- 23.Sylwester, A.W., B.L. Mitchell, J.B. Edgar, C. Taormina, C. Pelte, F. Ruchti, P.R. Sleath, K.H. Grabstein, N.A. Hosken, F. Kern, et al. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 202:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riddell, S.R., K.S. Watanabe, J.M. Goodrich, C.R. Li, M.E. Agha, and P.D. Greenberg. 1992. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 257:238–241. [DOI] [PubMed] [Google Scholar]
- 25.Rentenaar, R.J., L.E. Gamadia, N. van DerHoek, F.N. van Diepen, R. Boom, J.F. Weel, P.M. Wertheim-van Dillen, R.A. van Lier, and I.J. ten Berge. 2000. Development of virus-specific CD4(+) T cells during primary cytomegalovirus infection. J. Clin. Invest. 105:541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Leeuwen, E.M., E.B. Remmerswaal, M.T. Vossen, A.T. Rowshani, P.M. Wertheim-van Dillen, R.A. van Lier, and I.J. ten Berge. 2004. Emergence of a CD4+CD28− granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J. Immunol. 173:1834–1841. [DOI] [PubMed] [Google Scholar]
- 27.Gamadia, L.E., R.J. Rentenaar, R.A. van Lier, and I.J. ten Berge. 2004. Properties of CD4(+) T cells in human cytomegalovirus infection. Hum. Immunol. 65:486–492. [DOI] [PubMed] [Google Scholar]
- 28.Sester, M., U. Sester, B. Gartner, G. Heine, M. Girndt, N. Mueller-Lantzsch, A. Meyerhans, and H. Kohler. 2001. Levels of virus-specific CD4 T cells correlate with cytomegalovirus control and predict virus-induced disease after renal transplantation. Transplantation. 71:1287–1294. [DOI] [PubMed] [Google Scholar]
- 29.Gamadia, L.E., E.B. Remmerswaal, J.F. Weel, F. Bemelman, R.A. van Lier, and I.J. Ten Berge. 2003. Primary immune responses to human CMV: a critical role for IFN-gamma-producing CD4+ T cells in protection against CMV disease. Blood. 101:2686–2692. [DOI] [PubMed] [Google Scholar]
- 30.Hopkins, J.I., A.N. Fiander, A.S. Evans, M. Delchambre, D. Gheysen, and L.K. Borysiewicz. 1996. Cytotoxic T cell immunity to human cytomegalovirus glycoprotein B. J. Med. Virol. 49:124–131. [DOI] [PubMed] [Google Scholar]
- 31.Elkington, R., N.H. Shoukry, S. Walker, T. Crough, C. Fazou, A. Kaur, C.M. Walker, and R. Khanna. 2004. Cross-reactive recognition of human and primate cytomegalovirus sequences by human CD4 cytotoxic T lymphocytes specific for glycoprotein B and H. Eur. J. Immunol. 34:3216–3226. [DOI] [PubMed] [Google Scholar]
- 32.Jonjic, S., I. Pavic, P. Lucin, D. Rukavina, and U.H. Koszinowski. 1990. Efficacious control of cytomegalovirus infection after long-term depletion of CD8+ T lymphocytes. J. Virol. 64:5457–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hegde, N.R., M.S. Chevalier, and D.C. Johnson. 2003. Viral inhibition of MHC class II antigen presentation. Trends Immunol. 24:278–285. [DOI] [PubMed] [Google Scholar]
- 34.Britt, W.J., and C.A. Alford. 1996. Cytomegalovirus. Fields Virology. B.N. Fields, D.M. Knipe, P.M. Howley, R.M. Chanock, J.L. Melnick, T.P. Monath, B. Roizman, and S.E. Strauss, editors. Lippincott-Raven, Philadelphia. 2493–2523.
- 35.Sanchez, V., K.D. Greis, E. Sztul, and W.J. Britt. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Homman-Loudiyi, M., K. Hultenby, W. Britt, and C. Soderberg-Naucler. 2003. Envelopment of human cytomegalovirus occurs by budding into Golgi-derived vacuole compartments positive for gB, Rab 3, trans-golgi network 46, and mannosidase II. J. Virol. 77:3191–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomazin, R., J. Boname, N.R. Hegde, D.M. Lewinsohn, Y. Altschuler, T.R. Jones, P. Cresswell, J.A. Nelson, S.R. Riddell, and D.C. Johnson. 1999. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat. Med. 5:1039–1043. [DOI] [PubMed] [Google Scholar]
- 38.Catalfamo, M., and P.A. Henkart. 2003. Perforin and the granule exocytosis cytotoxicity pathway. Curr. Opin. Immunol. 15:522–527. [DOI] [PubMed] [Google Scholar]
- 39.Jarvis, M.A., K.N. Fish, C. Soderberg-Naucler, D.N. Streblow, H.L. Meyers, G. Thomas, and J.A. Nelson. 2002. Retrieval of human cytomegalovirus glycoprotein B from cell surface is not required for virus envelopment in astrocytoma cells. J. Virol. 76:5147–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hegde, N.R., and D.C. Johnson. 2003. Human cytomegalovirus US2 causes similar effects on both major histocompatibility complex class I and II proteins in epithelial and glial cells. J. Virol. 77:9287–9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borza, C.M., and L.M. Hutt-Fletcher. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594–599. [DOI] [PubMed] [Google Scholar]
- 42.Carlson, C., W.J. Britt, and T. Compton. 1997. Expression, purification, and characterization of a soluble form of human cytomegalovirus glycoprotein B. Virology. 239:198–205. [DOI] [PubMed] [Google Scholar]
- 43.Saxena, U., M.G. Klein, and I.J. Goldberg. 1990. Metabolism of endothelial cell-bound lipoprotein lipase. Evidence for heparan sulfate proteoglycan-mediated internalization and recycling. J. Biol. Chem. 265:12880–12886. [PubMed] [Google Scholar]
- 44.Dillon, D.C., M.R. Alderson, C.H. Day, D.M. Lewinsohn, R. Coler, T. Bement, A. Campos-Neto, Y.A. Skeiky, I.M. Orme, A. Roberts, et al. 1999. Molecular characterization and human T-cell responses to a member of a novel Mycobacterium tuberculosis mtb39 gene family. Infect. Immun. 67:2941–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hegde, N.R., R.A. Tomazin, T.W. Wisner, C. Dunn, J.M. Boname, D.M. Lewinsohn, and D.C. Johnson. 2002. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J. Virol. 76:10929–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewinsohn, D.A., R.A. Lines, and D.M. Lewinsohn. 2002. Human dendritic cells presenting adenovirally expressed antigen elicit Mycobacterium tuberculosis–specific CD8+ T cells. Am. J. Respir. Crit. Care Med. 166:843–848. [DOI] [PubMed] [Google Scholar]
- 47.Hill, A., P. Jugovic, I. York, G. Russ, J. Bennink, J. Yewdell, H. Ploegh, and D. Johnson. 1995. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 375:411–415. [DOI] [PubMed] [Google Scholar]
- 48.Jarvis, M.A., T.R. Jones, D.D. Drummond, P.P. Smith, W.J. Britt, J.A. Nelson, and C.J. Baldick. 2004. Phosphorylation of human cytomegalovirus glycoprotein B (gB) at the acidic cluster casein kinase 2 site (Ser900) is required for localization of gB to the trans-Golgi network and efficient virus replication. J. Virol. 78:285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Browne, H., S. Bell, T. Minson, and D.W. Wilson. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J. Virol. 70:4311–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy, E., D. Yu, J. Grimwood, J. Schmutz, M. Dickson, M.A. Jarvis, G. Hahn, J.A. Nelson, R.M. Myers, and T.E. Shenk. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA. 100:14976–14981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaraquemada, D., M. Marti, and E.O. Long. 1990. An endogenous processing pathway in vaccinia virus-infected cells for presentation of cytoplasmic antigens to class II-restricted T cells. J. Exp. Med. 172:947–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malnati, M.S., S. Ceman, M. Weston, R. DeMars, and E.O. Long. 1993. Presentation of cytosolic antigen by HLA-DR requires a function encoded in the class II region of the MHC. J. Immunol. 151:6751–6756. [PubMed] [Google Scholar]
- 53.Walter, E.A., P.D. Greenberg, M.J. Gilbert, R.J. Finch, K.S. Watanabe, E.D. Thomas, and S.R. Riddell. 1995. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 333:1038–1044. [DOI] [PubMed] [Google Scholar]
- 54.Lord, S.J., R.V. Rajotte, G.S. Korbutt, and R.C. Bleackley. 2003. Granzyme B: a natural born killer. Immunol. Rev. 193:31–38. [DOI] [PubMed] [Google Scholar]
- 55.Griffin, J.P., R. Chu, and C.V. Harding. 1997. Early endosomes and a late endocytic compartment generate different peptide-class II MHC complexes via distinct processing mechanisms. J. Immunol. 158:1523–1532. [PubMed] [Google Scholar]
- 56.Bonifaz, L.C., S. Arzate, and J. Moreno. 1999. Endogenous and exogenous forms of the same antigen are processed from different pools to bind MHC class II molecules in endocytic compartments. Eur. J. Immunol. 29:119–131. [DOI] [PubMed] [Google Scholar]
- 57.Johnson, D.C., and G. McFadden. 2002. Viral immune evasion. Immunology of Infectious Diseases. S.H.E. Kaufmann, A. Sher, and R. Ahmed, editors. American Society for Microbiology, Washington, D.C. 357–377.
- 58.Yewdell, J.W., and A.B. Hill. 2002. Viral interference with antigen presentation. Nat. Immunol. 3:1019–1025. [DOI] [PubMed] [Google Scholar]
- 59.Hengel, H., U. Reusch, G. Geginat, R. Holtappels, T. Ruppert, E. Hellebrand, and U.H. Koszinowski. 2000. Macrophages escape inhibition of major histocompatibility complex class I-dependent antigen presentation by cytomegalovirus. J. Virol. 74:7861–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manley, T.J., L. Luy, T. Jones, M. Boeckh, H. Mutimer, and S.R. Riddell. 2004. Immune evasion proteins of human cytomegalovirus do not prevent a diverse CD8+ cytotoxic t cell response in natural infection. Blood. 104:1075–1082. [DOI] [PubMed] [Google Scholar]
- 61.Prudhomme, J.G., I.W. Sherman, K.M. Land, A.V. Moses, S. Stenglein, and J.A. Nelson. 1996. Studies of Plasmodium falciparum cytoadherence using immortalized human brain capillary endothelial cells. Int. J. Parasitol. 26:647–655. [DOI] [PubMed] [Google Scholar]
- 62.Moses, A.V., K.N. Fish, R. Ruhl, P.P. Smith, J.G. Strussenberg, L. Zhu, B. Chandran, and J.A. Nelson. 1999. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J. Virol. 73:6892–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riddell, S.R., and P.D. Greenberg. 1990. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J. Immunol. Methods. 128:189–201. [DOI] [PubMed] [Google Scholar]
- 64.Collins, W.J., and D.C. Johnson. 2003. Herpes simplex virus gE/gI expressed in epithelial cells interferes with cell-to-cell spread. J. Virol. 77:2686–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chapman, T.L., I. You, I.M. Joseph, P.J. Bjorkman, S.L. Morrison, and M. Raghavan. 1999. Characterization of the interaction between the herpes simplex virus type I Fc receptor and immunoglobulin G. J. Biol. Chem. 274:6911–6919. [DOI] [PubMed] [Google Scholar]
- 66.Britt, W.J. 1984. Neutralizing antibodies detect a disulfide-linked glycoprotein complex within the envelope of human cytomegalovirus. Virology. 135:369–378. [DOI] [PubMed] [Google Scholar]
- 67.Salerno-Goncalves, R., M. Fernandez-Vina, D.M. Lewinsohn, and M.B. Sztein. 2004. Identification of a human HLA-E-restricted CD8+ T cell subset in volunteers immunized with Salmonella enterica serovar Typhi strain Ty21a typhoid vaccine. J. Immunol. 173:5852–5862. [DOI] [PubMed] [Google Scholar]
- 68.York, I.A., C. Roop, D.W. Andrews, S.R. Riddell, F.L. Graham, and D.C. Johnson. 1994. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 77:525–535. [DOI] [PubMed] [Google Scholar]