

Abstract
CagA protein is a major virulence factor of Helicobacter pylori, which is delivered into gastric epithelial cells and elicits growth factor–like responses. Once within the cells, CagA is tyrosine phosphorylated by Src family kinases and targets host proteins required to induce the cell responses. We show that the phosphorylated CagA binds Crk adaptor proteins (Crk-II, Crk-I, and Crk-L) and that the interaction is important for the CagA-mediated host responses during H. pylori infection. H. pylori–induced scattering of gastric epithelial cells in culture was blocked by overexpression of dominant-negative Crk and by RNA interference–mediated knockdown of endogenous Crk. H. pylori infection of the gastric epithelium induced disruption of E-cadherin/catenin–containing adherens junctions, which was also dependent on CagA/Crk signaling. Furthermore, inhibition of the SoS1/H-Ras/Raf1, C3G/Rap1/B-Raf, or Dock180/Rac1/Wiskott-Aldrich syndrome protein family verprolin homologous protein pathway, all of which are involved downstream of Crk adaptors, greatly diminished the CagA-associated host responses. Thus, CagA targeting of Crk plays a central role in inducing the pleiotropic cell responses to H. pylori infection that cause several gastric diseases, including gastric cancer.
Helicobacter pylori is present in the stomach of at least half of the world's population, and many studies have indicated that persistent colonization of the stomach by H. pylori causes gastric diseases such as chronic gastritis, peptic ulcer disease, mucosa-associated lymphoid tissue lymphoma, and gastric adenocarcinoma (1–3). CagA protein is secreted into gastric epithelial cells via the type IV secretion system of H. pylori and plays a pivotal role in the etiology of H. pylori–associated gastric diseases (4–8). Recent studies have shown that CagA delivered into the gastric epithelium undergoes phosphorylation on tyrosine residues within EPIYA sequence repeats by Src family kinases (SFKs) (9, 10) and stimulates cell signaling through its interaction with Src homology (SH) 2–containing protein tyrosine phosphatase–2 (SHP-2), Grb2, COOH-terminal Src kinase, hepatocyte growth factor (HGF) receptor/c-Met, and zonula occludens–1 (ZO-1) (11–15). Nonphosphorylated CagA also interacts with host proteins, such as Grb2 and ZO-1, and induces cell responses (13, 14, 16). Although the above studies have shown that CagA has the ability to interact with various host proteins and elicit growth factor–like cell responses, the biological impact of CagA binding to the individual host proteins on H. pylori–associated gastric diseases remains to be elucidated.
Crk-II adaptor protein was originally identified as the mammalian homologue of v-Crk encoded by the v-crk oncogene in avian sarcoma virus CT10 (17), and Crk-I has been identified as an alternative splicing variant of Crk-II mRNA (18). A second homologous protein, Crk-L, was discovered in a subsequent report (19). The Crk proteins possess SH2 and SH3 domains and, as a result, act as adaptor proteins that mediate protein phosphorylation–mediated signaling pathways (20, 21). The SH2 domain of Crk interacts with phosphorylated proteins, such as paxillin, p130Cas, and Gab1, whereas the SH3 domain interacts with guanine nucleotide exchange factors (GEFs), such as C3G (for Rap1), SoS1 (for H-Ras), and Dock180 (for Rac1), via their proline-rich region (PRR) domains (20, 21). Thus, Crk adaptors directly regulate cell signaling downstream of various receptor tyrosine kinases (RTKs) and focal contacts involved in the reorganization of actin cytoskeleton, cell spreading, proliferation, migration, and tumorigenesis (20, 21).
In this context, we investigated whether Crk function is required for the gastric cell responses elicited by H. pylori infection and found that tyrosine-phosphorylated (pY)–CagA interacts with Crk proteins. The binding is biologically essential in promoting H. pylori–elicited cell scattering/hummingbird phenotype and cell–cell dissociation because inhibition of Crk or signaling molecules downstream of Crk considerably abrogated these CagA-dependent cell responses. These findings thus provide the first evidence that interaction between CagA and Crk plays a central role in the diverse activity of CagA that participate in the pleiotropic cell responses to H. pylori infection. We discuss how CagA/Crk signaling contributes to the promotion of H. pylori–associated gastritis and to the generation of a tumor microenvironment.
Results
CagA interacts with Crk adaptors in a phosphorylation-dependent manner
Crk adaptors (Crk-II, Crk-I, and Crk-L) play a pivotal role in signaling pathways downstream of various RTKs (e.g., c-Met, epidermal growth factor receptor [EGFR], and insulin receptor) and integrins (Fig. 1 A) (20, 21). To explore possible involvement of Crk in H. pylori CagA-induced gastric cell responses, we investigated gastric epithelial cell lines AGS, KATOIII, MKN45, and MKN74 for expression of Crk proteins by immunoblotting and found that they express Crk-II, Crk-I, and Crk-L, although the levels of expression varied from line to line (Fig. 1 B). Lysates of AGS cells infected with H. pylori strain NCTC11637, ATCC43579, 26695, or J99 were tested for the presence of pY-CagA by immunoblotting (Fig. 1 C). pY-CagA was detected in all of the lysates, and the amounts of pY-CagA varied with the number of EPYIA motifs in the CagA of the H. pylori strains (e.g., the NCTC11637, ATCC43579, 26695, and J99 strains possess 5, 5, 2, and 1 copies of the motif, respectively). CagA were immunoprecipitated with Crk-II from these H. pylori–infected cells; furthermore, the amount of CagA in the immunoprecipitates increased with the number of EPYIA motifs (Fig. 1 C).
Figure 1.

CagA interacts with Crk adaptor proteins in a phosphorylation-dependent manner. (A) Schematic representations of Crk family adaptors. The numbers are the amino acid positions. (B) Lysates from various gastric epithelial cell lines were subjected to immunoblotting with the indicated antibodies. Note that the Crk antibody recognizes both Crk-II (top band) and Crk-I proteins (bottom band). (C) Lysates from AGS cells infected with various H. pylori strains were immunoprecipitated with anti–Crk-II antibody or normal rabbit IgG (control IgG), and the immunoprecipitates (IP) and lysates were subjected to immunoblotting with the indicated antibodies. Note that antiurease (H. pylori UreA) antibody was used as the bacterial control. (D–F) H. pylori–infected AGS cell lysates prepared in the presence or absence of Na3VO4 were subjected to GST pull-down assay using the indicated GST fusion proteins, and the pulled-down proteins (PD) and lysates (input) were subjected to immunoblotting with the indicated antibodies. (G) Lysate from ΔcagA H. pylori carrying the indicated pHel-FLAG vectors was coincubated for 30 min with AGS cell lysates (with Na3VO4) at 30°C. The lysates were subjected to GST pull-down assay with GST-SH2-Crk, and the PD and lysates were subjected to immunoblotting with the indicated antibodies.
To confirm the binding between CagA and Crk, lysates from infected AGS cells prepared with and without protein tyrosine phosphatase inhibitor Na3VO4 were incubated with glutathione S-transferase (GST)–Crk-II, GST-Crk-I, or GST-Crk-L beads, and a GST pull-down assay was performed. No pY-CagA was detected in the lysates of the cells prepared in the absence of Na3VO4 (Fig. 1 D), and GST-Crk-II, GST-Crk-I, and GST-Crk-L more efficiently pulled down pY-CagA (Fig. 1 E, top) than tyrosine-nonphosphorylated CagA (bottom), suggesting that Crk preferentially interacts with pY-CagA. Because Crk-I is composed of SH2 and SH3 domains (Fig. 1 A), we investigated whether either of these domains interacts with CagA in the GST pull-down assay with GST-Crk-I, GST-SH2-Crk, and GST-SH3-Crk, and the results showed that CagA preferentially interacts with the SH2 domain (Fig. 1 F). To determine whether the conventional function of the SH2 domain is important, an SH2 dominant-negative mutant Crk (R38K-Crk) was tested for binding to CagA, and, as shown in Fig. 1 F, markedly less R38K-Crk than Crk-I or SH2-Crk bound to CagA. To identify the CagA region involved in the binding to Crk, GST-SH2-Crk beads were incubated with lysates from cells infected with ΔcagA H. pylori harboring pHel-FLAG (vector control), pHel-FLAG-FL-CagA (native full-length CagA), pHel-FLAG-F5-CagA (the Tyr residues in the five EPIYA motifs of CagA substituted by Phe [14]), or pHel-FLAG-ΔPY-CagA (lacking the COOH-terminal CagA portion containing the five EPIYA motifs [14]), and the pulled-down proteins were investigated. FL-CagA was pulled down by SH2-Crk, whereas F5-CagA and ΔPY-CagA were not pulled down at all, suggesting that phosphorylated tyrosine residues in the EPIYA motifs of CagA participate in interactions with the Crk SH2 domain (Fig. 1 G).
Crk/CagA binding induces cell scattering/hummingbird phenotype during H. pylori infection
To confirm the role of Crk function in the CagA signaling pathway, AGS cells expressing the dominant-negative Crk-I mutants R38K-Crk (a mutation within SH2), W170K-Crk (a mutation within SH3), and R38K/W170K-Crk (mutations in SH2 and SH3) were investigated for their effect on H. pylori–induced cell scattering/hummingbird phenotype, which is a hallmark of the CagA-dependent cell response (7, 12, 14, 15, 22). Scattering was found to be diminished in cells expressing R38K-Crk or W170K-Crk, but not in cells expressing R38K/W170K-Crk (Fig. 2 A). Indeed, H. pylori infection of AGS cells transfected with the mock control–induced scattering of ∼80% of the cells, whereas the cells expressing R38K-Crk or W170K-Crk scattered <20% of the cells (Fig. 2 B). Under these conditions, the cells expressing nondominant-negative Crk (R38K/W170-Crk) scattered ∼60% of the cells (Fig. 2 B). Pseudomonas aeruginosa ExoT has been found to specifically inhibit Crk through its ADP-ribosyl transferase (ADPRT) activity (23). ExoT is composed of an NH2-terminal Rho–GTPase-activating protein (GAP) and COOH-terminal ADPRT domain. We therefore created GFP-ExoT together with GFP-ADPRT-ExoT (inactivated RhoGAP but intact ADPRT activity) and GFP-EEDD-ExoT (the nontoxic mutant as the negative control; Fig. 2 C) and tested them for an effect on H. pylori–induced cell scattering/hummingbird phenotype. Cell scattering was almost completely abolished when GFP-ADPRT-ExoT was expressed, whereas expression of GFP-EEDD-ExoT had only a slight inhibitory effect on the cell response (Fig. 2 D and Fig. S1 A, a–c, available at http://www.jem.org/cgi/content/full/jem.20051027/DC1). Thus, the results of the series of experiments suggested that Crk plays a crucial role in CagA-dependent cell scattering/hummingbird phenotype.
Figure 2.

Crk signaling is critical for CagA activity. (A) AGS cells transiently transfected with Myc-tagged Crk-I (a), R38K-Crk (b), W170K-Crk (c), or R38K/W170K-Crk (d) expression plasmids were infected with H. pylori at an MOI of 100. The cells were fixed 5 h after infection and stained with anti-Myc tag antibody (green), phalloidin (red), and anti–H. pylori antibody (blue). Arrowheads point to long protruding structures of scattered cells. Bar, 20 μm. (B) Percentages of scattered cells in A. The black bar indicates the control. (C) Schematic representations of ExoT mutants. ADPRT-ExoT has a mutation in the GAP domain alone (R149K), whereas EEDD-ExoT has a mutation in both the GAP domain and the ADPRT domain (E383D and E385D). The numbers are the amino acid positions. (D) AGS cells transiently transfected with the indicated GFP plasmids were infected with H. pylori at an MOI of 100. The percentages of scattered GFP-positive cells were calculated 5 h after infection. At least 100 cells were counted in each experiment. The black bar indicates the control. The data shown represent means and SDs of triplicate experiments.
Endogenous Crk is required for H. pylori–induced cell scattering/hummingbird phenotype
Next, we used a small interfering RNA (siRNA)–mediated RNA interference (RNAi) approach to achieve knockdown of endogenous Crk level in AGS cells. In the RNAi experiment, transfection of Luc-siRNA (as the negative control) had no effect on Crk-II/Crk-L expression, but transfection of Crk-siRNA (specific for the crk-II and crk-I genes) or of Crk-L-siRNA (specific for the crk-L gene) greatly decreased their expression to ∼30% of the control level (Fig. 3 A). Furthermore, AGS cells cotransfected with both Crk-siRNA and Crk-L-siRNA expressed hardly any Crk at all (Fig. 3 A). H. pylori–induced cell scattering/hummingbird phenotype of Crk/Crk-L knockdown cells was substantially decreased to 37% of the control level (Fig. 3 B). To ensure the specificity of the Crk-siRNA inhibition, we created RNAi-resistant Crk (CrkR; Fig. 3 C) and introduced GFP-CrkR into Crk-RNAi–treated AGS cells. Although endogenous Crk-II was expressed at a low level by Crk-RNAi, the level of ectopic GFP-CrkR expression in the Crk-RNAi–treated cells was similar to the level of expression in the control RNAi-treated cells (Fig. 3 D). Under these conditions AGS cells expressing CrkR restored the ability to scatter in response to H. pylori infection even when treated with Crk-RNAi (Fig. 3 E). However, the inhibition of cell scattering by Crk-RNAi was not reversed when W170K-CrkR (RNAi-resistant dominant-negative Crk) was expressed (Fig. 3 E). Time-lapse microscopy revealed that AGS cell colonies treated with control RNAi lost cell–cell adhesion within 3 h after infection, and cell scattering/hummingbird phenotype occurred after 5–6 h (Fig. 3 F, a–d and Video S1, available at http://www.jem.org/cgi/content/full/jem.20051027). The cells treated with Crk/Crk-L-RNAi, on the other hand, exhibited less cell–cell dissociation and subsequent dispersal than the control cells (Fig. 3 F, e–h, red asterisks and Video S2). However, the decrease in the response to H. pylori infection of AGS cells caused by Crk/Crk-L-RNAi was restored to the control level when CrkR was expressed (Fig. 3, F and G, green numbers).
Figure 3.

H. pylori–induced cell scattering/hummingbird phenotype is blocked by Crk-RNAi. (A) Lysates from AGS cells transfected for 72 h with the siRNAs as indicated were subjected to immunoblotting with the indicated antibodies. (B) AGS cells in A were infected with H. pylori at an MOI of 100, and the percentages of scattered cells were calculated 5 h after infection. The black bar indicates the control. The data shown represent means and SDs of triplicate experiments with at least 100 cells each. (C) Comparison between the nucleotide sequences of crk coding sequences (cds) and crk R cds. To generate crk R, two adenine nucleotides were replaced by thymine nucleotides (red letters) in the Crk-siRNA–targeted sequence of the crk gene. (D) Lysates from AGS cells transfected with Luc-siRNA or Crk-siRNA at 200 nM for 72 h and with GFP or GFP-CrkR plasmid for 20 h were subjected to immunoblotting with the indicated antibodies. (E) AGS cells transfected with the indicated siRNAs for 72 h and with the indicated GFP plasmids for 20 h were infected with H. pylori at an MOI of 100. The percentages of scattered GFP-positive cells were counted 5 h after infection. The black bar indicates the control. The data shown represent means and SDs of triplicate experiments with at least 100 cells each. (F) AGS cells transfected with Luc-siRNA at 400 nM for 72 h (a–d) or Crk/Crk-L-siRNA at 400 nM for 72 h (e–h) and with GFP-CrkR plasmid for 20 h (a–h) were infected with H. pylori at an MOI of 100. Phase-contrast images of each type of infected cell were monitored by time-lapse microscopy 90 (a and e), 180 (b and f), 270 (c and g), and 360 (d and h) min after infection (the entire sequence is shown in Videos S1 and S2). Green numbers point to the same GFP-CrkR–expressing cells, and red asterisks point to the same RNAi-treated cells. Bar, 20 μm. (G, a and b) GFP fluorescence of GFP-CrkR in F (a and e) were observed, respectively.
CagA-induced cell–cell dissociation depends on Crk activity
When a Madin-Darby canine kidney (MDCK) cell monolayer was infected with H. pylori, CagA colocalized to sites of bacterial attachment with the tight junction (TJ) scaffold protein ZO-1 in a CagA phosphorylation–independent manner (13). A more recent study indicated that H. pylori targets cell–cell junctions and promotes destruction of adherens junctions (AJs); however, the signaling pathway involved in the destruction is unknown (24, 25). Although AGS cells do not express cadherins, they adhere to each other through ZO-1/occludin-containing TJs (Fig. S2, A and B, available at http://www.jem.org/cgi/content/full/jem.20051027). We therefore used MKN74 cells, a gastric epithelial cell line, to investigate the effect of CagA on AJs, because MKN74 cells develop functional AJs with E-cadherin and β-catenin (Fig. 4 A and Fig. S2 A). MKN74 cells were infected with wild-type H. pylori or the ΔcagA strain, and the intracellular distribution of β-catenin was investigated. As shown Fig. 4 A, the cortical actin and β-catenin had accumulated along to cell–cell junctions in the cells infected with the mock or ΔcagA strain (a–c and g–i), whereas β-catenin had diffused within the cytoplasm of the cells infected with wild-type H. pylori (d–f). Under the same condition, CagA secreted by H. pylori was enriched in cell–cell adhesion sites and sustained the tyrosine phosphorylation in the infected cells (Fig. 4 B). Similarly, diffuse cytoplasmic localization of E-cadherin was also seen in MKN74 cells infected with wild-type H. pylori but not in the cells infected with the ΔcagA strain (Fig. S2 D, a and b), and the same was true of T47D cells (a breast cancer cell line) and NCI-N87 cells (another gastric epithelial cell line) infected with H. pylori (Fig. S3, A and B). To further pursue the results, we used E-cadherin–expressing L (EL) cells to generate a cell line that stably expresses CagA (EL/pMX-CagA; Fig. 4, C and D). In contrast to the control EL/pMX cells, the EL/pMX-CagA cells lost E-cadherin–mediated cell–cell adhesion and grew separately (Fig. 4 D), suggesting that CagA protein regulates the dissociation of AJs. In addition, when GFP-CagA, but not GFP-ΔPY-CagA, was expressed in MDCK cells, β-catenin was diffusely localized within the cytoplasm (Fig. 4 E). As shown in Fig. 4 F, there was less H. pylori–induced destruction of AJs in MKN74 cells expressing DsRed2-W170K-Crk-I (c, arrowheads) than in the control cells expressing DsRed2 alone (a, arrowheads). These results strongly indicated that CagA/Crk-stimulated signaling contributes to the dissociation of AJs during H. pylori infection.
Figure 4.
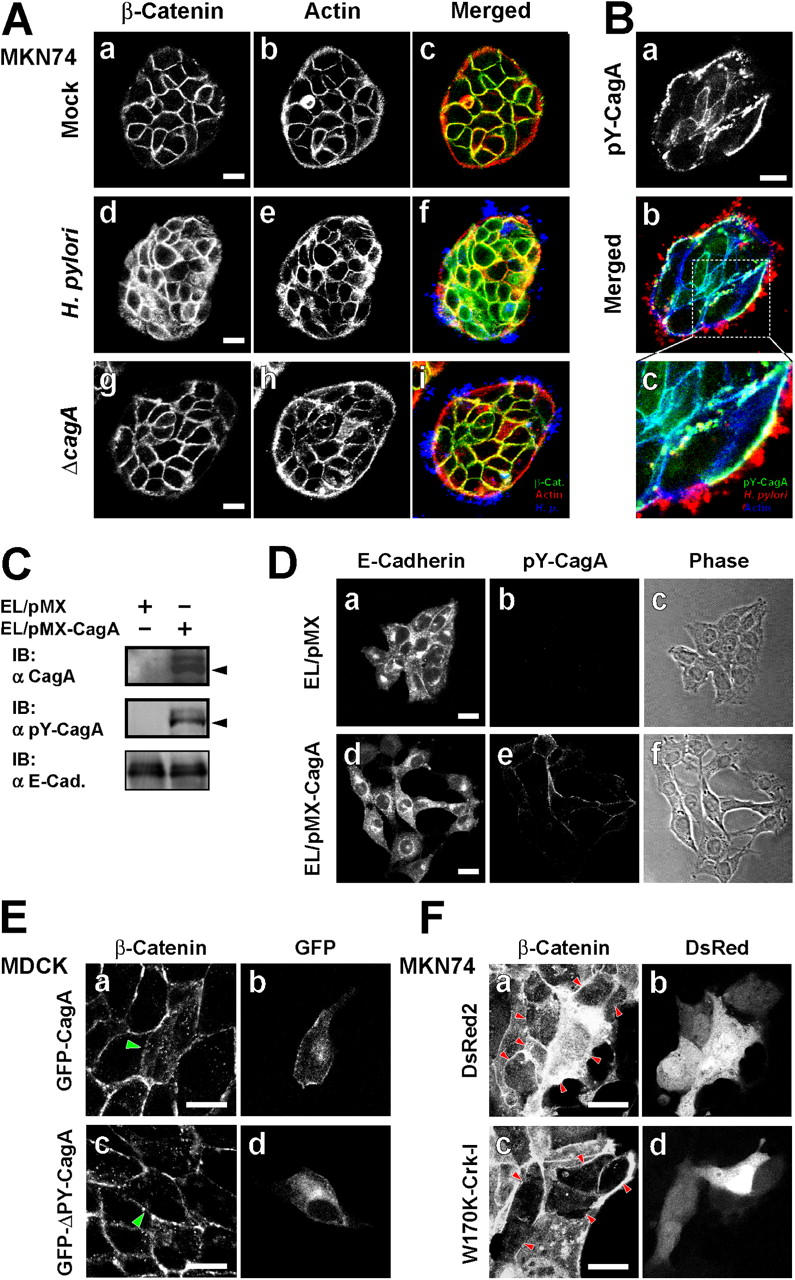
CagA disrupts epithelial AJs. (A) MKN74 cells were mock infected (a–c) and infected with H. pylori WT (d–f) or the ΔcagA strain (g–i) at an MOI of 20. The cells were fixed 48 h after infection and stained with anti–β-catenin antibody (a, c, d, f, g, and i), phalloidin (b, c, e, f, h, and i), and anti–H. pylori antibody (c, f, and i). Bar, 20 μm. (B) H. pylori–infected MKN74 cells in the same condition as A were stained with anti–pY-CagA antibody (a–c), anti–H. pylori antibody (b and c), and phalloidin (b and c). Bar, 20 μm. (C) EL cells stably expressing CagA (EL/pMX-CagA) were generated by pMX retrovirus infection (see Materials and methods), and the lysates from EL/pMX (control) and EL/pMX-CagA cells were subjected to immunoblotting with the indicated antibodies. Arrowheads indicate the CagA blot and the pY-CagA blot, respectively. (D) EL/pMX (a–c) or EL/pMX-CagA (d–f) cells were stained with anti–E-cadherin antibody (a and d) and anti–pY-CagA antibody (b and e). Phase-contrast images are also shown (c and f). Bar, 20 μm. (E) MDCK cells transiently transfected with GFP-CagA (a and b) or GFP-ΔPY-CagA plasmids (c and d) were fixed 48 h later and stained with anti–β-catenin antibody (a and c) and observed together for GFP fluorescence (b and d). Green arrowheads point to GFP-positive cells. Bar, 20 μm. (F) DsRed2 (a and b) or DsRed2-W170K-Crk-I plasmids (c and d) were microinjected into the nuclei of MKN74 cells. After incubation for 5–8 h, cells were infected with H. pylori at an MOI of 20 and fixed 48 h later. The infected cells were stained with anti–β-catenin antibody (a and c) and observed together for DsRed fluorescence (b and d). Red arrowheads point to DsRed-positive cells. Bar, 20 μm.
To further pursue the participation of Crk in CagA-dependent cell–cell dissociation, the effect of H. pylori infection on this response was investigated in MKN74 cells treated with Crk/Crk-L-RNAi (Fig. 5 A). RNAi-treated cells were infected with H. pylori ΔvacA (wild-type strain possessing an intact cagA gene) or ΔvacA/ΔcagA strain (ΔcagA mutant), and the intracellular localization of β-catenin was investigated. When the control cells were infected with the ΔvacA strain, but not with the ΔvacA/ΔcagA strain, β-catenin was dispersed within the cytoplasm (Fig. 5 B, a–d), whereas H. pylori infection hardly interfered with β-catenin localization in Crk/Crk-L knockdown cells at all (Fig. 5 B, e–h). Consistent with this observation, the cytoskeleton-associated E-cadherin (the NP-40–insoluble form) in the MKN74 cells was converted to the soluble form when infected with the wild type, but not with the ΔcagA strain (Fig. 5, C and D). The level of cytoplasmic E-cadherin actually increased 1.7-fold in the control MKN74 cells infected with the wild type, whereas it increased only 1.2-fold in the Crk/Crk-L knockdown cells infected with the wild type (Fig. 5 D). Under the same conditions, H. pylori infection slightly decreased CagA-dependent ZO-1 diffusion into the cytoplasm, suggesting less participation of Crk in the destruction of TJs than in the destruction of AJs. The β-catenin released from AJs translocates into the nucleus, where it acts as the coactivator of T cell factor (TCF)/lymphoid enhancer factor transcriptional factor (26). The β-catenin–TCF complex activates transcription of target genes, including c-myc and the cyclin D1 and matrix metalloprotease 7 genes (26), and when MKN74 cells are infected with wild-type H. pylori, β-catenin frequently translocates into the nucleus (Fig. 5 B, d, arrowheads).
Figure 5.
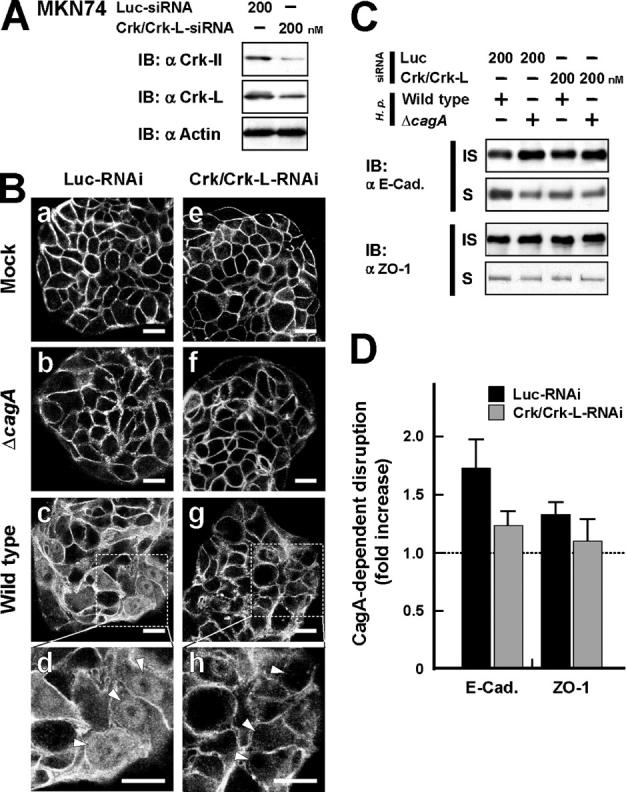
Crk stimulates CagA-dependent breakdown of cell–cell adhesion. (A) Lysates from MKN74 cells transfected for 72 h with Luc-siRNA or Crk-siRNA at 200 nM were subjected to immunoblotting with the indicated antibodies. (B) MKN74 cells treated for 72 h with Luc-RNAi (a–d) or Crk/Crk-L-RNAi (e–h) were mock infected (a and e) or infected with the H. pylori ΔvacA/ΔcagA (b and f) or ΔvacA strain (c, d, g, and h) at an MOI of 20. The cells were fixed 48 h after infection and stained with anti–β-catenin antibody. White arrowheads (d and h) point to the nuclei of the cells. Note that ΔvacA H. pylori strains were used to reduce damage to the infected cells (references 1, 60). Bar, 20 μm. (C) NP-40–soluble (S) and –insoluble fractions (IS) were prepared from H. pylori–infected cells in B, and the fractions were subjected to immunoblotting with the indicated antibodies. (D) The blots of NP-40–soluble fractions in C were quantified by densitometry. The data are presented as fold increase in amounts of ΔvacA compared with that of ΔvacA/ΔcagA under the same RNAi conditions. The data shown represent means and SDs of triplicate experiments.
Gab1, EGFR, and c-Met are not directly engaged in CagA/Crk-mediated cell scattering/hummingbird phenotype
On integrin stimulation, focal adhesion kinase (FAK) is rapidly activated and mediates the phosphorylation of focal adhesion components, such as paxillin and p130Cas (27). Crk is involved in integrin/FAK-mediated intracellular signaling via interactions between Crk-SH2 domain and paxillin/p130Cas (20, 21). In addition, Iwahara et al. have reported that Crk also functions upstream of FAK activation (28). To investigate whether FAK is involved in CagA/Crk-mediated cell scattering/hummingbird phenotype, AGS cells expressing FAK or FAK-related nonkinase (FRNK), a dominant-negative FAK mutant (29), were infected with H. pylori. As shown in Fig. 6 A, overexpression of FAK, but not FRNK, inhibited H. pylori–induced cell scattering (Fig. S1 A, d and e). Because moving cells, such as the scattered cells, need rapid reorganization of the focal adhesions, a strong adhesion to the extracellular matrix may prevent the cell scattering. On H. pylori infection, the scattered cells reduced the assembly of vinculin-containing focal contacts (unpublished data). Thus, we presume that FAK activation does not play an important role in CagA/Crk-elicited cell scattering/hummingbird phenotype.
Figure 6.

Either Gab1, EGFR, or c-Met is dispensable for CagA/Crk signaling. (A and B) AGS cells transiently transfected with the indicated plasmids were infected with H. pylori at an MOI of 100. The percentages of scattered cells were calculated 5 h after infection. (C) AGS cells were transfected for 72 h with the siRNAs as indicated. The Gab1 knockdown cells were examined by immunoblotting with anti-Gab1 antibody (top right). The arrowhead indicates the Gab1 blot. RNAi-treated cells were also infected with H. pylori at an MOI of 100. The percentages of scattered cells were calculated 5 h after infection. The black bars in A–C indicate the controls. The data shown in A–C represent means and SDs of triplicate experiments (at least 100 cells were counted in each experiment). (D) The lysates from AGS and T47D cells were subjected to immunoblotting with the indicated antibodies. (E) T47D cells were serum starved for 24 h and stimulated with H. pylori for 8 h (MOI = 100), or growth factors were dissolved in DMSO for 30 min as indicated. The cells were also pretreated with or without AG1478 30 min before stimulation. The lysates were prepare after stimulation and subjected to immunoblotting with the indicated antibodies. (F) T47D cells were pretreated for 30 min with AG1478 at 1 μM and infected with WT H. pylori (a) or the ΔvirD4 strain (b) at an MOI of 100. Phase-contrast microscopy was performed 24 h after infection. Bar, 20 μm.
Previous reports have shown that H. pylori stimulates EGFR and c-Met–mediated signaling pathways (12, 30). Gab1 is a docking protein that plays an important role in RTK activation by recruiting signaling molecules, including Grb2, SHP-2, Crk/Crk-L, and phosphatidylinositol 3–kinase (PI3K), into the vicinity of the cytoplasmic domain of the RTK (31). We therefore used two dominant-negative Gab1mutants, ΔCrk-Gab1 and ΔMet-Gab1, to investigate whether Gab1 is involved in cell scattering/hummingbird phenotype that occurs on H. pylori infection. ΔCrk-Gab1 is unable to bind Crk, but binds to SHP-2 and the PI3K p85 subunit (32), whereas ΔMet-Gab1 is unable to directly bind to c-Met (33). AGS cells expressing ΔCrk-Gab1 or ΔMet-Gab1 were infected with H. pylori; however, as shown in Fig. 6 B, neither dominant-negative Gab1 affected H. pylori–induced cell scattering (Fig. S1 A, k–m). Furthermore, knockdown of endogenous Gab1 expression in AGS cells by RNAi decreased the level of Gab1 expression to 8% of the control level (Fig. 6 C, top right), and H. pylori–induced cell scattering was completely unaffected by Gab1-RNAi (Fig. 6 C).
We used T47D cells to investigate whether c-Met is involved in CagA-dependent cell scattering/hummingbird phenotype, because the cells poorly express c-Met and are therefore tolerant to HGF stimulation (Fig. 6, D and E) (34). T47D cells pretreated with an EGFR inhibitor, AG1478, were infected with H. pylori, and the cell responses were investigated. Keates et al. reported that H. pylori infection induces activation of the mitogen-activated protein kinase/extracellular signal–regulated kinase (MAPK/ERK) pathway via EGFR transactivation (30). In agreement with the report, ERK activation in H. pylori-infected T47D cells was somewhat decreased by AG1478 treatment as compared with that of untreated control cells (Fig. 6 E). However, wild-type H. pylori, but not the ΔvirD4 strain (CagA secretion mutant) (14), was still able to induce cell scattering (Fig. 6 F). The results indicated that neither the EGFR/Gab1 nor c-Met/Gab1 pathway is involved in CagA/Crk signaling.
Crk downstream signaling pathways are involved in CagA/Crk-induced cell scattering/hummingbird phenotype
The Crk SH3 domain binds SoS1, C3G, and Dock180, known as GEFs for small GTPases, via their PRRs (Fig. 7 A) (20, 21), and overexpression of the PRR-peptide corresponding to each of the GEFs in the cells was expected to competitively inhibit binding of Crk to the GEFs (35, 36). We therefore introduced PRR-peptide expression vectors into AGS cells and investigated the transfectants infected with H. pylori for an effect on cell scattering/hummingbird phenotype. Although mock-transfected AGS cells scattered 75% of the cells, the cells expressing PRR-SoS1, PRR-C3G, or PRR-Dock180 scattered only 10–20% of the cells (Fig. 7 B).
Figure 7.

CagA/Crk signaling up-regulates cell proliferation and reorganization of the actin cytoskeleton. (A) Schematic representations of Crk binding GEFs. Each PRR region contains the indicated number of proline-rich (PxxP) motifs. The numbers are the amino acid positions. (B, C, F, and G) AGS cells transiently transfected with the indicated expression plasmids were infected with H. pylori at an MOI of 100. The cells in C and F were also treated with the indicated inhibitors 30 min before infection. The percentages of scattered cells were calculated 5 h after infection. At least 100 cells were counted in each experiment. The black bars indicate the controls. The data shown indicate means and SDs of triplicate experiments. (D) KATOIII cells transfecting siRNA at 400 nM for 120 h were serum starved for 2 h and infected with H. pylori at an MOI of 100 as indicated. The lysates were prepared at the indicated time points after infection and subjected to immunoblotting with the indicated antibodies. (E) The blots of phosphorylated MEK in D were quantified by densitometry. The data are presented as fold activation over the uninfected control under the same RNAi conditions. The data shown indicate means and SDs of triplicate experiments.
Because SoS1, C3G, and Dock180 are recruited to the vicinity of the cytoplasmic membrane and catalyze the activation of H-Ras, Rap1, and Rac1, respectively, we used dominant-negative mutants of the GTPases (N17 mutants) to investigate whether the GTPase activity is required for CagA/Crk-mediated cell scattering/hummingbird phenotype. Overexpression of N17-H-Ras or N17-Rap1 decreased the cell scattering to 45 and 72%, respectively, of the level in the mock control (Fig. 7 C and Fig. S1 A, f and g), suggesting that the cell scattering requires activation of H-Ras and Rap1. We previously reported that CagA-induced cell scattering/hummingbird phenotype requires activation of the MAPK/ERK pathway via the CagA/Grb2 interaction (14). Similarly, the levels of CagA-dependent scattering and MAPK/ERK-kinase (MEK) activation in AGS cells were strongly decreased by treatment with the Raf kinase inhibitors BAY43-9006 or GW5074 or with the MEK inhibitors PD98059 or U0126 (Fig. 7 C and Fig. S1, B and C), and the CagA-induced cell–cell dissociation was also almost completely blocked when treated with these Raf/MEK inhibitors (Fig. S2, C and D).
To further confirm the contribution of the CagA/Crk complex to activation of the MEK/ERK pathway, expression of all Crk proteins (Crk-II, Crk-I, and Crk-L) in KATOIII cells was knocked down by Crk/Crk-L-RNAi, and the effect on MEK activity after H. pylori infection was investigated by assaying the level of phosphorylated MEK. The results showed that the level of the CagA-dependent MEK activation in the KATOIII cells treated with Crk/Crk-L-RNAi was strongly inhibited when compared with the level in the control cells (Fig. 7, D and E). When serum-starved AGS cells were stimulated with HGF, the cells became somewhat scattered (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20051027), and the HGF/c-Met-mediated cell scattering and MEK activation were also substantially inhibited by Crk/Crk-L-RNAi (Fig. S4, B and C).
CagA-dependent cell scattering/hummingbird phenotype requires Rac1/Wiskott-Aldrich syndrome protein family verprolin homologous protein (WAVE) activation
When AGS cells expressed dominant-negative Rac1 (N17-Rac1), H. pylori–induced cell scattering decreased to 45% of the GFP mock control level (Fig. 7 F and Fig. S1 A, h). When the cells were exposed to an actin polymerization inhibitor, cytochalasin D, at 0.1 and 1 μM, cell scattering decreased to 32% and was undetectable, respectively (Fig. 7 F), whereas treatment of the cells with the specific c-Jun NH2-terminal kinase inhibitor SP600125 had no effect on the cell scattering (Fig. 7 F). The WAVE family proteins downstream of Rac1 have been shown to activate Arp2/3 complex and promote cortical actin polymerization (37). H. pylori–induced scattering of the cells transfected with dominant-negative WAVE (ΔVPH-WAVE1 or ΔVPH-WAVE2) (38), but not with dominant-negative N-WASP (neural Wiskott-Aldrich syndrome protein; ΔCof-N-WASP) (39), decreased to ∼50% of the control level (Fig. 7 G and Fig. S1 A, i and j), suggesting that CagA-mediated cell scattering/hummingbird phenotype requires Rac1/WAVE activation.
Discussion
CagA/Crk interaction is involved in both H. pylori–induced cell scattering/hummingbird phenotype and cell–cell dissociation
We found that pY-CagA binds Crk adaptor proteins and that the interaction plays a critical role in promoting cell scattering/hummingbird phenotype, the most recognized hallmark of the host cell response to H. pylori infection (Figs. 1–3) (7, 12, 14, 15, 22). We also found that loss of Crk function in gastric epithelial cells considerably reduced the CagA-dependent breakdown of AJs with diffuse cytoplasmic localization of β-catenin and E-cadherin (Figs. 4 and 5 and Figs. S2 and S3). Amieva et al. reported that H. pylori infection of polarized MDCK cells promotes disruption of TJs by an interaction between nonphosphorylated CagA and ZO-1 (13). Thus, it is likely that CagA affects TJs via its ZO-1 interaction in a phosphorylation-independent manner and AJs via its Crk interaction in a phosphorylation-dependent manner, thereby leading to the morphological changes and deregulation of cell–cell adhesion in the gastric epithelium (Fig. 8).
Figure 8.
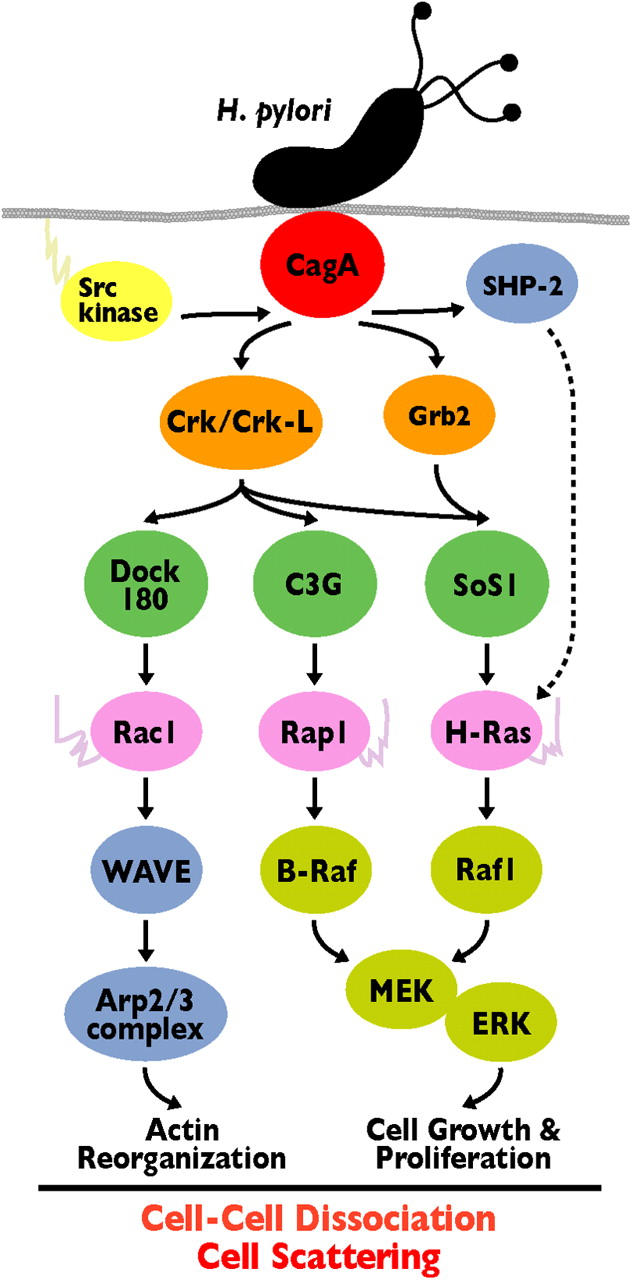
H. pylori CagA cracks the host signaling via its interaction with Crk. CagA translocated from H. pylori into the host cell is tyrosine phosphorylated by SFKs and interacts with Crk/Crk-L adaptors. Crk is involved in MAPK activation leading to cell growth and proliferation through the Crk/SoS1/H-Ras/Raf1 and Crk/C3G/Rap1/B-Raf pathways. Crk also promotes Rac1 activity through the Crk/Dock180/ELMO pathway. Activated Rac1 induces actin reorganization by stimulating downstream effectors such as WAVE family proteins. These motogenic, mitogenic, and morphologic effects downstream of CagA/Crk signaling are required for H. pylori–induced cell scattering/hummingbird phenotype and cell–cell dissociation.
MAPK signaling downstream of Crk is required for CagA-mediated cell responses
H-Ras plays an important role in mediating MEK/ERK signaling by activating Raf1 kinase (40). The H-Ras/Raf1/MEK pathway must be required for CagA activity, because cell scattering/hummingbird phenotype induced by H. pylori infection was substantially abrogated when the cells expressed dominant-negative H-Ras (N17-H-Ras) or were exposed to Raf/MEK inhibitors, such as BAY43-9006 (Fig. 7 C and Fig. S1 C). In addition, inactivation of Ras family GTPases by a farnesyltransferase inhibitor, FTI-277, also blocked the response (unpublished data). Higashi et al. recently reported, however, that CagA-induced cell scattering/hummingbird phenotype requires ERK activity but is independent of H-Ras activity (41). They only transiently transfected AGS cells with an HA-tagged CagA expression vector, which induced scattering of only ∼7–25% of the cells, whereas the infection of AGS cells with H. pylori for 5 h in the present study resulted in a very high scattering level (70–80%). Indeed, we confirmed that though the effect was marginal, a dominant-negative SHP-2 mutant inhibited CagA-dependent cell scattering/hummingbird phenotype in our bacterial infection model (unpublished data). Because Zhang et al. have demonstrated that SHP-2 promotes Ras/ERK activation by regulating SFK activation via COOH-terminal Src kinase (42). We believe that it is biologically important that MAPK activation and cell scattering/hummingbird phenotype induced by H. pylori–delivered CagA is dependent on H-Ras activity (Fig. 8).
Rap1, the H-Ras homologue, also activates MEK/ERK signaling in a cell type–specific manner that is governed by the cell type–specific expression of B-Raf kinase (35, 43, 44), and the results of several studies have actually indicated that the Crk/Rap1/B-Raf pathway contributes to sustained ERK activation (35, 43, 45). Because B-Raf mRNA is expressed in gastric cell lines, such as AGS, KATOIII, and MKN74 (unpublished data), Rap1, acting downstream of CagA/Crk/C3G signaling, may positively regulate ERK activity in gastric epithelial cells (Fig. 7, C–E). Moreover, the level of CagA- or HGF/c-Met–dependent activation of the MEK/ERK pathway was also decreased by Crk/Crk-L knockdown (Fig. 7, D and E and Fig. S4), implying that, similar to c-Met/Gab1/Crk signaling, CagA/Crk signaling may contribute to Rap1-mediated sustained ERK activation even though the signal cascade mediated by CagA/Crk is independent of c-Met and Gab1 functions (Fig. 6, B–F).
Importantly, the breakdown of epithelial cell AJs induced by CagA or HGF/c-Met signaling also requires sustained activation of MAPK (Fig. S2, C and D) (46). Lamorte et al. recently reported that the HGF/c-Met–induced cell spreading of epithelial colonies and breakdown of AJs require Crk-II and Crk-L activity (47). Thus, the sustained MAPK activation via the CagA/Crk-dependent signaling pathway is most likely an important mechanism underlying the disruption of the superficial gastric epithelium during H. pylori infection (Fig. 8).
Rac1 and its downstream effector are involved in H. pylori–induced cell responses
Cell scattering of epithelial colonies is initiated by dynamic changes in cell architecture, implying that Rho GTPases and their downstream effectors play an important role (48, 49). The cell scattering induced by HGF/c-Met requires PI3K-mediated Rac1 activation (46, 49), whereas the response induced by H. pylori is independent of PI3K activity (unpublished data) (12), suggesting that CagA/Crk/Dock180/Rac1 signaling is distinct from the pathway required for PI3K activity. In addition, a recent study has indicated that cell migration stimulated by HGF/c-Met requires WAVE, a Rac1 effector (50), and the same would also be true of CagA/Crk-mediated cell scattering/hummingbird phenotype (Fig. 7 G). Actually, neither inhibition of Cdc42 nor of N-WASP, a Cdc42 effector, affected the cell scattering (Fig. 7 G and not depicted), and even though Kodama et al. reported that SHP-2 phosphatase activity is required for HGF/c-Met-induced cell scattering through the regulation of H-Ras and RhoA activity (51), in our study H. pylori–induced cell scattering/hummingbird phenotype was not inhibited by dominant-negative RhoA or by treatment with Y-27632, a specific inhibitor of Rho-kinase/ROCK, an RhoA effector (unpublished data). Therefore, the CagA/Crk-regulated Rac1 activity may play a role in modulating the actin reorganization during H. pylori–elicited cell scattering/hummingbird phenotype (Fig. 8).
CagA/Crk signaling stimulates disruption of AJs and translocation of β-catenin into the cell nucleus
Persistent H. pylori infection results in disassembly of AJs and nuclear localization of β-catenin in the gastric epithelium through activation of the CagA/Crk signaling pathway (Figs. 4 and 5 and Figs. S2–4). When AJs are disrupted by extracellular stimuli, β-catenin is released from the AJ pool and translocates into the nucleus, where the β-catenin–TCF complex induces the transcription of genes that contribute to cancer progression (26). Loss of function of E-cadherin is common in malignant carcinomas and plays a causative role in the progression from adenoma to carcinoma (52). Germline E-cadherin mutations have been linked to an inherited gastric cancer (53), and mice lacking IQGAP1, a regulator of AJ complex formation, display gastric hyperplasia and dysplasia (54). Thus, the versatile Crk-mediated CagA activity is likely to contribute to actin reorganization, cell migration, and proliferation, thus leading to the down-regulation of AJ regulatory proteins acting as tumor suppressors. The long-term effect, therefore, may be to increase the risk of generation and proliferation of malignant gastric cells.
Materials and Methods
Cell culture and bacterial infection
H. pylori strain NCTC11637 and its isogenic mutants (ΔcagA, ΔvirD4, ΔvacA, and ΔvacA/ΔcagA; ATCC43579, 26695, and J99) have been described previously (4, 14). The ΔcagA mutant of H. pylori NCTC11637 harboring pHel-FLAG, pHel-FLAG-FL-CagA, pHel-FLAG-ΔPY-CagA, or pHel-FLAG-F5-CagA has also been described previously (14). All strains were cultured according to the standard procedure (14). AGS, MKN45, MKN74, and MDCK cells were maintained in DMEM (Sigma-Aldrich) containing 10% FBS. KATOIII, T47D, and NCI-N87 cells were maintained in RPMI 1640 (Sigma-Aldrich) with 10% FBS.
Culture cells were infected with H. pylori at a multiplicity of infection (MOI) of 20–100. H. pylori infection of AGS cells induces the dispersal of epithelial colonies after cell–cell dissociation. The compact round cells then begin to elongate and move from one place to another with protruding lamellipodial structures. We defined cell scattering/hummingbird phenotype as an elongated cell showing 40–200 μm of spindle-like structures with some lamellipodial protrusions for a counting experiment of the cells.
Plasmids, antibodies, and reagents
The cDNAs of crk-II, crk-I, crk-L, PRR-SoS1 (residues 1020–1133 of SoS1), PRR-C3G (residues 279–660 of C3G), and PRR-Dock180 (residues 1650–1865 of Dock180) were amplified by RT-PCR from the total RNA of AGS cells. The crk-I, PRR-SoS1, PRR-C3G, and PRR-Dock180 genes were cloned into pcDL-SRα-myc (14) for NH2-terminal Myc-tagged protein expression. The R38K, W170K, and R38K/W170K Crk-I mutants were generated with the QuickChange site-directed mutagenesis kit (Stratagene). The crk-I and W170K-Crk-I genes were also cloned into pEGFP-C1 and pDsRed2-C1 (CLONTECH Laboratories, Inc.), respectively, for fluorescent protein expression. crk-II, crk-I, crk-L, SH2-Crk (residues 1–121 of crk-I), SH3-Crk (residues 122–204 of crk-I), and R38K-Crk-I genes were cloned into pGEX-4T1 (GE Healthcare) for preparation of GST fusion protein. The crkR gene (adenine residues 369 and 372 of the crk gene substituted by thymine) was generated by site-directed mutagenesis to ensure the specificity of Crk-siRNA inhibition.
H-ras cDNA was obtained from the pCMV-Ras vector (CLONTECH Laboratories, Inc.). rap1 cDNA was amplified by RT-PCR from the total RNA of AGS cells. The H-ras gene and rap1 gene were cloned into pEGFP-C1, and the N17-H-Ras mutant (S17N) and N17-Rap1 mutant (S17N) were each generated by site-directed mutagenesis. The exoT gene was amplified by PCR from the P. aeruginosa PAO1 genome and cloned into pEGFP-C1. The ADPRT-ExoT (R149K) and EEDD-ADPRT mutants (R149K, E383D, and E385D) were generated by site-directed mutagenesis. pMX-puro was provided by T. Kitamura (University of Tokyo, Tokyo, Japan) (55). The cagA gene was cloned into pMX-puro to produce retrovirus and virus infection. All other full-length, ΔPY, and F5 CagA constructs were prepared as described previously (14). FAK cDNA was provided by T. Yamamoto (56). The FRNK gene was amplified by PCR from residues 691–1053 of the FAK gene. pDNA1.1-Gab1 and pDNA1.1-Gab1-ΔCrk were provided by M. Park (32). The ΔMet-Gab1 mutant (V490A) was produced by site-directed mutagenesis. The ΔCof-N-WASP and N17-Rac1 constructs were prepared as described previously (39, 57). The ΔVPH-WAVE1 and ΔVPH-WAVE2 constructs were provided by T. Takenawa (University of Tokyo, Tokyo, Japan) (38).
Rabbit anti-CagA polyclonal antibody (pAb), rabbit anti–pY-CagA pAb, and rabbit anti-UreA pAb were prepared as described previously (14). Mouse anti–H. pylori pAb was obtained from Monosan. Rabbit anti–H. pylori pAb was from Biomeda. Mouse anti-Crk mAb, mouse anti–E-cadherin mAb, mouse anti–β-catenin mAb, and mouse anti–Myc tag mAb (9B11) were obtained from BD Transduction Laboratories. Rabbit anti–Crk-II pAb, rabbit anti–Crk-L pAb, rabbit anti-EGFR pAb, rabbit anti–c-Met pAb, and mouse anti-FLAG mAb (M5) were purchased from Santa Cruz Biotechnology, Inc. Mouse antiactin mAb was obtained from Chemicon International. Rabbit anti-GFP pAb was purchased from MBL International Corporation. Mouse anti–ZO-1 mAb and mouse anti-occludin mAb were obtained from Zymed Laboratories. Rabbit anti-Gab1 pAb was purchased from Upstate Biotechnology. Rabbit anti-pMEK1/2 pAb, rabbit anti-MEK1/2 pAb, mouse anti-pERK1/2 mAb, and rabbit anti-ERK1/2 pAb were obtained from Cell Signaling Technology. Rhodamine phalloidin and AlexaFlour 633 phalloidin were purchased from Invitrogen. Human EGF was obtained from PeproTech. Human HGF was purchased from Sigma-Aldrich. PD98059, U0126, BAY43-9006, GW5074, SP600125, AG1478, and cytochalasin D were obtained from Calbiochem.
Transfection and microinjection
Transient transfection with the appropriate DNA constructs was performed using Fugene6 (Roche) or GenomONE-Neo (Ishihara Sangyo) for 18–20 h according to the manufacturer's instructions. Microinjection into the nuclei of MKN74 cells with the appropriate DNA constructs was performed as described previously (58), and the microinjected cells were incubated for 5–8 h before bacterial infection.
Retrovirus construction and infection
Production of an ecotropic retrovirus by transient transfection of PLAT-E packaging cells with pMX vector was performed as described elsewhere (55). In brief, 48 h after transfection of PLAT-E cells with pMX-puro (empty vector) or pMX-puro-CagA, culture supernatants were harvested, and EL cells (mouse L fibroblasts stably expressing E-cadherin [59]) were infected with the supernatants by using DOTAP (Roche). Stable transformants were isolated by resistance to 5 μg/ml puromycin, and the resulting clones were named EL/pMX and EL/pMX-CagA, respectively.
RNAi
siRNA corresponding to bases 264–284 of the human crk coding sequence, bases 409–429 of the human crk-L coding sequence, and bases 153–173 of the luciferase GL2 coding sequence were synthesized, purified, and duplexed by Dharmacon Research. The heterogeneous diced siRNA targeting bases 401–900 of the human Gab1 coding sequence were generated with a complete dicer RNAi kit (BLOCK-iT; Invitrogen) according to the manufacturer's instructions. Culture cells were transiently transfected with the appropriate siRNA by using Lipofectamine 2000 or Oligofectamine (Invitrogen).
Fluorescence microscopy
Fluorescence staining with appropriate antibodies and reagents was performed as described previously (14, 39).
Time-lapse imaging
AGS cells were grown in 35-mm glassbottom dishes and infected with H. pylori at an MOI of 100. The cells were washed twice 60 min after infection, and the medium was replaced with fresh DMEM. Phase-contrast images were taken every 10 min from 90 min after infection until 360 min after infection by fluorescent microscopy (Axiovert 135-SENSYS; Carl Zeiss MicroImaging, Inc.).
Immunoprecipitation, GST-pulldown assay, and immunoblotting
Immunoprecipitation, GST pull-down assay, and immunoblotting with the appropriate antibodies or GST fusion proteins were performed as described previously (14). The blots were quantified by measuring mean intensity with image software (version 1.63; National Institutes of Health).
Cell fractionation
1% NP-40–soluble and –insoluble fractions were prepared by lysing cells in NP-40 buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 4 mM EDTA, 25 mM NaF, 1% NP-40, 1 mM Na3VO4, and complete protease inhibitors; Roche) by 10 passages through a 27-gauge syringe and allowing to then stand at 4°C for 30 min. The lysates were then centrifuged at 10,000 g for 30 min, and the supernatant was collected as the NP-40–soluble fraction. The pellet was resuspended in 50 ml SDS buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 4 mM EDTA, 25 mM NaF, 1% SDS, 1 mM Na3VO4), and after adding 450 ml of NP-40 buffer, the lysate was homogenized by 10 passages through a 27-gauge syringe and placed on a rotating wheel at 4°C for 30 min. The lysates were then centrifuged at 10,000 g for 30 min, and the supernatant was collected as the NP-40–insoluble fraction.
Online supplemental material
Fig. S1 shows distinct effects on CagA-mediated scattering of AGS cells after transfection of various constructs or exposure to various inhibitors. Fig. S2 describes a difference in cell–cell junctions between AGS cells and MKN74 cells, and Fig. S3 shows CagA-mediated breakdown of E-cadherin–containing AJs in breast T47D and gastric NCI-N87 cells. Fig. S4 shows the contribution of Crk adaptors to HGF/c-Met signaling, including cell motility and proliferation, in gastric epithelial cells. Videos S1 and S2 show time-lapse microscopy analysis of H. pylori–infected AGS cells that were expressed as GFP-CrkR after Luc-RNAi and Crk/Crk-L-RNAi, respectively. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051027/DC1.
Acknowledgments
We thank T. Kitamura for providing pMX-puro vector and PLAT-E cells and T. Takenawa for providing expression vectors for WAVE. We are also grateful to members of our laboratory for helpful discussions and suggestions.
This work was supported by grants-in-aid for Scientific Research (155490141 and 17559127) on Priority Area (14021011), the Special Coordination Funds for Promoting Science and Technology from the Japanese Ministry of Education, Culture, Sports, Science and Technology and Core Research for Evolutional Science and Technology from Japan Science and Technology Agency.
The authors have no conflicting financial interests.
Abbreviations used: ADPRT, ADP-ribosyl transferase; AJ, adherens junction; CrkR, RNAi-resistant Crk; EGFR, epidermal growth factor; EL, E-cadherin–expressing L; ERK, extracellular signal–regulated kinase; FAK, focal adhesion kinase; FRNK, FAK-related nonkinase; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; GST, glutathione S-transferase; HGF, hepatocyte growth factor; MAPK, mitogen-activated protein kinase; MDCK, Madin-Darby canine kidney; MEK, MAPK/ERK-kinase; MOI, multiplicity of infection; N-WASP, neural Wiskott- Aldrich syndrome protein; pAb, polyclonal antibody; PI3K, phosphatidylinositol 3–kinase; PRR, proline-rich region; pY, tyrosine phosphorylated; RNAi, RNA interference; RTK, receptor tyrosine kinase; SFK, Src family kinase; SH, Src homology; SHP-2, SH2-containing protein tyrosine phosphatase–2; siRNA, small interfering RNA; TCF, T cell factor; TJ, tight junction; WAVE, Wiskott- Aldrich syndrome protein family verprolin homologous protein; ZO-1, zonula occludens–1.
References
- 1.Rieder, G., W. Fischer, and R. Haas. 2005. Interaction of Helicobacter pylori with host cells: function of secreted and translocated molecules. Curr. Opin. Microbiol. 8:67–73. [DOI] [PubMed] [Google Scholar]
- 2.Suerbaum, S., and P. Michetti. 2002. Helicobacter pylori infection. N. Engl. J. Med. 347:1175–1186. [DOI] [PubMed] [Google Scholar]
- 3.Montecucco, C., and R. Rappuoli. 2001. Living dangerously: how Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2:457–466. [DOI] [PubMed] [Google Scholar]
- 4.Asahi, M., T. Azuma, S. Ito, Y. Ito, H. Suto, Y. Nagai, M. Tsubokawa, Y. Tohyama, S. Maeda, M. Omata, et al. 2000. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med. 191:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Odenbreit, S., J. Puls, B. Sedlmaier, E. Gerland, W. Fischer, and R. Haas. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 287:1497–1500. [DOI] [PubMed] [Google Scholar]
- 6.Ogura, K., S. Maeda, M. Nakao, T. Watanabe, M. Tada, T. Kyutoku, H. Yoshida, Y. Shiratori, and M. Omata. 2000. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J. Exp. Med. 192:1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal, E.D., J. Cha, J. Lo, S. Falkow, and L.S. Tompkins. 1999. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc. Natl. Acad. Sci. USA. 96:14559–14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaser, M.J., G.I. Perez-Perez, H. Kleanthous, T.L. Cover, R.M. Peek, P.H. Chyou, G.N. Stemmermann, and A. Nomura. 1995. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 55:2111–2115. [PubMed] [Google Scholar]
- 9.Selbach, M., S. Moese, C.R. Hauck, T.F. Meyer, and S. Backert. 2002. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J. Biol. Chem. 277:6775–6778. [DOI] [PubMed] [Google Scholar]
- 10.Stein, M., F. Bagnoli, R. Halenbeck, R. Rappuoli, W.J. Fantl, and A. Covacci. 2002. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 43:971–980. [DOI] [PubMed] [Google Scholar]
- 11.Tsutsumi, R., H. Higashi, M. Higuchi, M. Okada, and M. Hatakeyama. 2003. Attenuation of Helicobacter pylori CagA × SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J. Biol. Chem. 278:3664–3670. [DOI] [PubMed] [Google Scholar]
- 12.Churin, Y., L. Al-Ghoul, O. Kepp, T.F. Meyer, W. Birchmeier, and M. Naumann. 2003. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J. Cell Biol. 161:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amieva, M.R., R. Vogelmann, A. Covacci, L.S. Tompkins, W.J. Nelson, and S. Falkow. 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 300:1430–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mimuro, H., T. Suzuki, J. Tanaka, M. Asahi, R. Haas, and C. Sasakawa. 2002. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol. Cell. 10:745–755. [DOI] [PubMed] [Google Scholar]
- 15.Higashi, H., R. Tsutsumi, S. Muto, T. Sugiyama, T. Azuma, M. Asaka, and M. Hatakeyama. 2002. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 295:683–686. [DOI] [PubMed] [Google Scholar]
- 16.Hirata, Y., S. Maeda, Y. Mitsuno, K. Tateishi, A. Yanai, M. Akanuma, H. Yoshida, T. Kawabe, Y. Shiratori, and M. Omata. 2002. Helicobacter pylori CagA protein activates serum response element-driven transcription independently of tyrosine phosphorylation. Gastroenterology. 123:1962–1971. [DOI] [PubMed] [Google Scholar]
- 17.Mayer, B.J., M. Hamaguchi, and H. Hanafusa. 1988. A novel viral oncogene with structural similarity to phospholipase C. Nature. 332:272–275. [DOI] [PubMed] [Google Scholar]
- 18.Matsuda, M., S. Tanaka, S. Nagata, A. Kojima, T. Kurata, and M. Shibuya. 1992. Two species of human CRK cDNA encode proteins with distinct biological activities. Mol. Cell. Biol. 12:3482–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.ten Hoeve, J., C. Morris, N. Heisterkamp, and J. Groffen. 1993. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene. 8:2469–2474. [PubMed] [Google Scholar]
- 20.Feller, S.M. 2001. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 20:6348–6371. [DOI] [PubMed] [Google Scholar]
- 21.Chodniewicz, D., and R.L. Klemke. 2004. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim. Biophys. Acta. 1692:63–76. [DOI] [PubMed] [Google Scholar]
- 22.Selbach, M., S. Moese, R. Hurwitz, C.R. Hauck, T.F. Meyer, and S. Backert. 2003. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 22:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun, J., and J.T. Barbieri. 2003. Pseudomonas aeruginosa ExoT ADP-ribosylates CT10 regulator of kinase (Crk) proteins. J. Biol. Chem. 278:32794–32800. [DOI] [PubMed] [Google Scholar]
- 24.Conlin, V.S., S.B. Curtis, Y. Zhao, E.D. Moore, V.C. Smith, R.M. Meloche, B.B. Finlay, and A.M. Buchan. 2004. Helicobacter pylori infection targets adherens junction regulatory proteins and results in increased rates of migration in human gastric epithelial cells. Infect. Immun. 72:5181–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heczko, U., V.C. Smith, R. Mark Meloche, A.M. Buchan, and B.B. Finlay. 2000. Characteristics of Helicobacter pylori attachment to human primary antral epithelial cells. Microbes Infect. 2:1669–1676. [DOI] [PubMed] [Google Scholar]
- 26.Conacci-Sorrell, M., J. Zhurinsky, and A. Ben-Ze'ev. 2002. The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Invest. 109:987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitra, S.K., D.A. Hanson, and D.D. Schlaepfer. 2005. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6:56–68. [DOI] [PubMed] [Google Scholar]
- 28.Iwahara, T., T. Akagi, Y. Fujitsuka, and H. Hanafusa. 2004. CrkII regulates focal adhesion kinase activation by making a complex with Crk-associated substrate, p130Cas. Proc. Natl. Acad. Sci. USA. 101:17693–17698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richardson, A., and T. Parsons. 1996. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature. 380:538–540. [DOI] [PubMed] [Google Scholar]
- 30.Keates, S., S. Sougioultzis, A.C. Keates, D. Zhao, R.M. Peek Jr., L.M. Shaw, and C.P. Kelly. 2001. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 276:48127–48134. [DOI] [PubMed] [Google Scholar]
- 31.Liu, Y., and L.R. Rohrschneider. 2002. The gift of Gab. FEBS Lett. 515:1–7. [DOI] [PubMed] [Google Scholar]
- 32.Lamorte, L., D.M. Kamikura, and M. Park. 2000. A switch from p130Cas/Crk to Gab1/Crk signaling correlates with anchorage-independent growth and JNK activation in cells transformed by the Met receptor oncoprotein. Oncogene. 19:5973–5981. [DOI] [PubMed] [Google Scholar]
- 33.Schaeper, U., N.H. Gehring, K.P. Fuchs, M. Sachs, B. Kempkes, and W. Birchmeier. 2000. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J. Cell Biol. 149:1419–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beviglia, L., K. Matsumoto, C.S. Lin, B.L. Ziober, and R.H. Kramer. 1997. Expression of the c-Met/HGF receptor in human breast carcinoma: correlation with tumor progression. Int. J. Cancer. 74:301–309. [DOI] [PubMed] [Google Scholar]
- 35.York, R.D., H. Yao, T. Dillon, C.L. Ellig, S.P. Eckert, E.W. McCleskey, and P.J. Stork. 1998. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 392:622–626. [DOI] [PubMed] [Google Scholar]
- 36.Posern, G., J. Zheng, B.S. Knudsen, C. Kardinal, K.B. Muller, J. Voss, T. Shishido, D. Cowburn, G. Cheng, B. Wang, et al. 1998. Development of highly selective SH3 binding peptides for Crk and CRKL which disrupt Crk-complexes with DOCK180, SoS and C3G. Oncogene. 16:1903–1912. [DOI] [PubMed] [Google Scholar]
- 37.Takenawa, T., and H. Miki. 2001. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 114:1801–1809. [DOI] [PubMed] [Google Scholar]
- 38.Suetsugu, S., H. Miki, and T. Takenawa. 1999. Identification of two human WAVE/SCAR homologues as general actin regulatory molecules which associate with the Arp2/3 complex. Biochem. Biophys. Res. Commun. 260:296–302. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki, T., H. Miki, T. Takenawa, and C. Sasakawa. 1998. Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 17:2767–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Downward, J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 3:11–22. [DOI] [PubMed] [Google Scholar]
- 41.Higashi, H., A. Nakaya, R. Tsutsumi, K. Yokoyama, Y. Fujii, S. Ishikawa, M. Higuchi, A. Takahashi, Y. Kurashima, Y. Teishikata, et al. 2004. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J. Biol. Chem. 279:17205–17216. [DOI] [PubMed] [Google Scholar]
- 42.Zhang, S.Q., W. Yang, M.I. Kontaridis, T.G. Bivona, G. Wen, T. Araki, J. Luo, J.A. Thompson, B.L. Schraven, M.R. Philips, and B.G. Neel. 2004. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol. Cell. 13:341–355. [DOI] [PubMed] [Google Scholar]
- 43.Stork, P.J. 2003. Does Rap1 deserve a bad Rap? Trends Biochem. Sci. 28:267–275. [DOI] [PubMed] [Google Scholar]
- 44.Wellbrock, C., M. Karasarides, and R. Marais. 2004. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5:875–885. [DOI] [PubMed] [Google Scholar]
- 45.Sasagawa, S., Y. Ozaki, K. Fujita, and S. Kuroda. 2005. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol. 7:365–373. [DOI] [PubMed] [Google Scholar]
- 46.Potempa, S., and A.J. Ridley. 1998. Activation of both MAP kinase and phosphatidylinositide 3-kinase by Ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol. Biol. Cell. 9:2185–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamorte, L., I. Royal, M. Naujokas, and M. Park. 2002. Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol. Biol. Cell. 13:1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ridley, A.J., P.M. Comoglio, and A. Hall. 1995. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol. Cell. Biol. 15:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Royal, I., N. Lamarche-Vane, L. Lamorte, K. Kaibuchi, and M. Park. 2000. Activation of cdc42, rac, PAK, and rho-kinase in response to hepatocyte growth factor differentially regulates epithelial cell colony spreading and dissociation. Mol. Biol. Cell. 11:1709–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawamura, K., K. Takano, S. Suetsugu, S. Kurisu, D. Yamazaki, H. Miki, T. Takenawa, and T. Endo. 2004. N-WASP and WAVE2 acting downstream of phosphatidylinositol 3-kinase are required for myogenic cell migration induced by hepatocyte growth factor. J. Biol. Chem. 279:54862–54871. [DOI] [PubMed] [Google Scholar]
- 51.Kodama, A., T. Matozaki, A. Fukuhara, M. Kikyo, M. Ichihashi, and Y. Takai. 2000. Involvement of an SHP-2-Rho small G protein pathway in hepatocyte growth factor/scatter factor-induced cell scattering. Mol. Biol. Cell. 11:2565–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perl, A.K., P. Wilgenbus, U. Dahl, H. Semb, and G. Christofori. 1998. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 392:190–193. [DOI] [PubMed] [Google Scholar]
- 53.Guilford, P., J. Hopkins, J. Harraway, M. McLeod, N. McLeod, P. Harawira, H. Taite, R. Scoular, A. Miller, and A.E. Reeve. 1998. E-cadherin germline mutations in familial gastric cancer. Nature. 392:402–405. [DOI] [PubMed] [Google Scholar]
- 54.Li, S., Q. Wang, A. Chakladar, R.T. Bronson, and A. Bernards. 2000. Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Mol. Cell. Biol. 20:697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morita, S., T. Kojima, and T. Kitamura. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063–1066. [DOI] [PubMed] [Google Scholar]
- 56.Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, and T. Yamamoto. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 377:539–544. [DOI] [PubMed] [Google Scholar]
- 57.Yoshida, S., E. Katayama, A. Kuwae, H. Mimuro, T. Suzuki, and C. Sasakawa. 2002. Shigella deliver an effector protein to trigger host microtubule destabilization, which promotes Rac1 activity and efficient bacterial internalization. EMBO J. 21:2923–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki, T., H. Mimuro, H. Miki, T. Takenawa, T. Sasaki, H. Nakanishi, Y. Takai, and C. Sasakawa. 2000. Rho family GTPase Cdc42 is essential for the actin-based motility of Shigella in mammalian cells. J. Exp. Med. 191:1905–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagafuchi, A., Y. Shirayoshi, K. Okazaki, K. Yasuda, and M. Takeichi. 1987. Transformation of cell adhesion properties by exogenously introduced E-cadherin cDNA. Nature. 329:341–343. [DOI] [PubMed] [Google Scholar]
- 60.Cover, T.L., and S.R. Blanke. 2005. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 3:320–332. [DOI] [PubMed] [Google Scholar]