

Abstract
NFATc1 and NFATc2 are functionally redundant in the immune system, but it was suggested that NFATc1 is required exclusively for differentiation of osteoclasts in the skeletal system. Here we provide genetic evidence that NFATc1 is essential for osteoclast differentiation in vivo by adoptive transfer of NFATc1 −/− hematopoietic stem cells to osteoclast-deficient Fos −/− mice, and by Fos −/− blastocyst complementation, thus avoiding the embryonic lethality of NFATc1 −/− mice. However, in vitro osteoclastogenesis in NFATc1-deficient cells was rescued by ectopic expression of NFATc2. The discrepancy between the in vivo essential role of NFATc1 and the in vitro effect of NFATc2 was attributed to selective autoregulation of the NFATc1 gene by NFAT through its promoter region. This suggested that an epigenetic mechanism contributes to the essential function of NFATc1 in cell lineage commitment. Thus, this study establishes that NFATc1 represents a potential therapeutic target for bone disease and reveals a mechanism that underlies the essential role of NFATc1 in bone homeostasis.
The mechanism underlying irreversible cell fate determination is critical for an understanding of biologic systems in vertebrates, where the NFAT family of transcription factors plays important roles (1, 2). Mice deficient in individual NFAT proteins exhibit a relatively mild phenotype in immune cells or neurons (1, 2), which suggests that the different family members play redundant roles in these systems (1–3). Homeostasis of the skeletal system depends on a balance between bone-forming osteoblasts and bone-resorbing osteoclasts (4). The critical role of NFATc1 in the skeletal system was suggested by selective and strong induction of NFATc1 in bone marrow monocyte/macrophage precursor cells (BMMs) that were stimulated with a key cytokine for osteoclastogenesis, receptor activator of NF-κB ligand (RANKL) (5, 6). Although our previous in vitro analyses indicated that NFATc1 is the essential transcription factor for osteoclast differentiation (5), little is known about its in vivo function in osteoclastogenesis because targeted disruption of the NFATc1 gene in mice results in embryonic lethality. Furthermore, there is a contrasting report that NFATc2 also activates this process and may have a redundant role (7). Considering the close functional similarity of NFATc1 and NFATc2 (3, 7), it is important to determine whether NFATc1 is essential for osteoclast differentiation in vivo, and, if so, how it exerts an indispensable function in this cell type.
In this report, we provide in vivo genetic evidence for the essential role of NFATc1 by two novel techniques: adoptive transfer of hematopoietic stem cells to osteoclast-deficient Fos −/− mice and Fos-deficient blastocyst complementation. NFATc1-deficient osteoclast precursor cells cannot differentiate into osteoclasts. Conversely, osteoclastogenesis of NFATc1-deficient cells is rescued by forced expression of NFATc2. The discrepancy between the essential role of NFATc1 and the in vitro effect of NFATc2 is attributed to selective autoregulation of the NFATc1 gene by NFAT through its promoter region. Our analysis indicates that the essential role of NFATc1 in bone homeostasis is not determined by its distinct biochemical properties, but is determined by its unique spatiotemporal activation mechanism during osteoclastogenesis.
RESULTS
In vivo evidence for the essential role of NFATc1 in osteoclast differentiation shown by the fetal liver cell (FLC) transfer
Because in vivo analysis of the role of NFATc1 has been hampered by embryonic lethality (8, 9), one may postulate that a conditional gene targeting strategy should be appropriate to avoid the difficulty. However, there is no desirable promoter to drive cre (encoding Cre recombinase) in the very early stage of osteoclast development. Adoptive transfer of hematopoietic stem cells to irradiated lymphocyte-deficient mice (10, 11) is used as an established method to analyze the function of such a gene in the study of lymphocytes (3). Here we applied this method to the skeletal system using mice lacking c-Fos (Fos −/− mice) (12, 13) as osteoclast-deficient recipients. Hematopoietic stem cells that were derived from NFATc1 +/− and NFATc1 − / − FLCs were injected into the liver of busulfan-treated newborn mice of Fos −/− background, in which no osteoclasts form because of a cell autonomous defect (13). Osteopetrosis is ameliorated and bone marrow cavities are observed when NFATc1 +/− FLCs are transferred to Fos −/− mice (Fig. 1, A and B). Tooth eruption, which is not seen in Fos −/− mice (13), is observed in these mice (unpublished data). In contrast, when NFATc1 −/− FLCs are transferred to Fos −/− mice, they exhibit severe osteopetrosis and bone marrow cavities remain occupied with unresorbed bone (Fig. 1, A and B). Histologic analyses show that osteoclast number and parameters of bone resorption are normalized only when NFATc1 +/− cells are transferred (Fig. 1, C and D). Repopulation of hematopoietic cells was confirmed by PCR in splenic lymphocytes and monocyte/macrophage precursor cells (Fig. 1 E). Consistently, osteoclast precursor cells that were derived from the NFATc1 − / − FLC chimera could not generate osteoclasts in culture (Fig. 1 F). These results indicate that NFATc1 is indispensable for osteoclast formation in vivo, and that FLC transfer to osteoclast-deficient mice is a beneficial tool for in vivo analysis of the osteoclast lineage.
Figure 1.
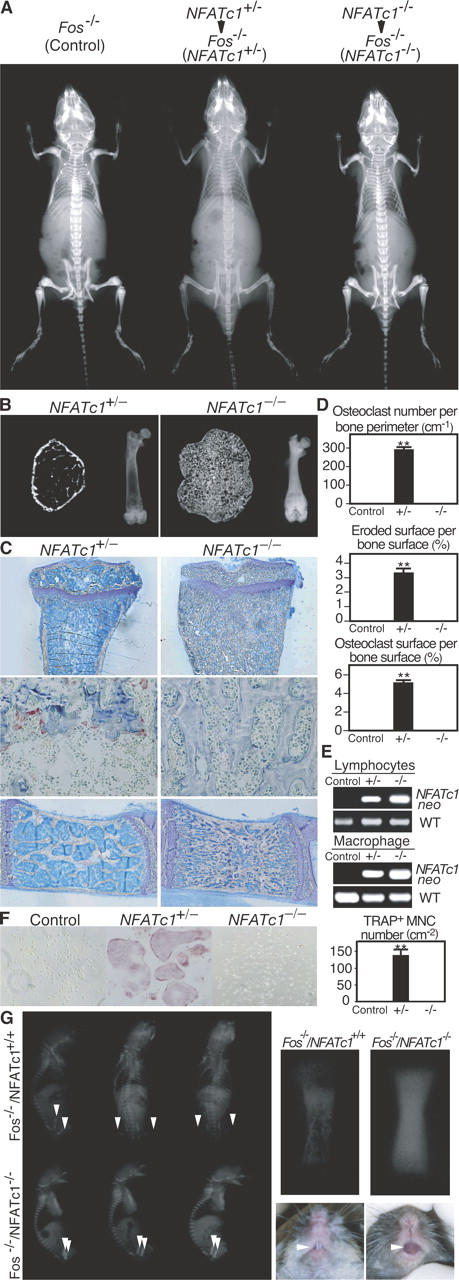
In vivo evidence for the essential role of NFATc1 in osteoclast differentiation. (A) Radiographic analysis of Fos −/− mice transferred with NFATc1 +/− or NFATc1 −/− FLCs. Severe osteopetrosis persists after NFATc1 −/− FLC transfer. (B) Microradiographic analysis of femur (left: microcomputed tomography; right: microradiograph). Bone marrow cavity is filled with unresorbed bone in NFATc1 −/− FLC chimera. (C) Histologic examination of tibia (top: toluidine blue staining; middle: tartrate-resistant acid phosphatase [TRAP] staining for detection of osteoclasts and lumbar vertebra; bottom: toluidine blue staining). No osteoclasts are observed in NFATc1 −/− FLC chimera. (D) Histomorphometric evaluation of osteoclasts in NFATc1 +/− and NFATc1 −/− FLC chimera. (E) Reconstitution of the hematopoietic system of chimeric mice by the donor cells. PCR primers specific for joint region between the NFATc1 locus and the neomycin-resistant gene (NFATc1 neo) and for wild-type (WT) NFATc1 gene are used. The similar chimeric ratios were confirmed by quantitative PCR. (F) Complete blockade of in vitro osteoclastogenesis in osteoclast precursor cells from NFATc1 −/− FLC chimera. Splenocyte-derived osteoclast precursor cells were cultured in RANKL (50 ng ml−1) and M-CSF (10 ng ml−1). We counted multinucleated (>3 nuclei) cells (MNCs) positive for TRAP staining. (G) Radiographic analysis of neonates generated by Fos − / − blastocyst complementation. Note the difference in radio-opacity at the femur (arrowheads). Right panels show the magnified view of the femur of neonates and tooth eruption of 6-wk-old mice. All of the Fos −/−/NFATc1 −/− chimeric mice exhibited osteopetrosis, but osteoclastogenesis is restored in Fos − / − /NFATc1 + / + chimeric mice when ES cells are transmitted to the hematopoietic system of recipient mice. **P < 0.001 versus control.
Fos −/− blastocyst complementation
Rag-2–deficient blastocyst complementation is an alternative method for analyzing the lymphocyte-specific functions of gene, loss of which results in embryonic lethality (14). To provide additional genetic evidence for the essential role of NFATc1 in osteoclastogenesis, we applied this method to an analysis of the osteoclast lineage using Fos − / − blastocysts (referred to as Fos-deficient blastocyst complementation). We analyzed chimeric neonates that were generated by injection of wild-type or NFATc1 − / − embryonic stem (ES) cells into Fos − / − blastocysts. The chimeric mice that result from injection of normal ES cells into Fos − / − blastocysts (Fos − / − /NFATc1 + / + chimera) should develop mature osteoclasts, which are derived from the injected ES cells. In fact, all of the chimeric mice recovered from osteopetrosis, and tooth eruption was observed when the injected ES cells were transmitted into the hematopoietic lineage (Fig. 1 G). In contrast, chimeric mice that result from injection of NFATc1 − / − ES cells into Fos − / − blastocysts (Fos − / − /NFATc1 − / − chimera) remain severely osteopetrotic, and have a defect in tooth eruption that is due to a lack of osteoclasts (Fig. 1 G and not depicted). Taken together with the results of FLC transfer, these observations provide clear evidence that NFATc1 is essential for osteoclast differentiation in vivo.
NFATc2 is dispensable for osteoclastogenesis in vivo and in vitro
NFATc2 is another NFAT protein that is expressed at a low level in osteoclast precursor cells (5, 7). In addition to the similarity of the DNA binding (Rel homology) domain, the genomic view of the NFATc1 and NFATc2 loci suggest that NFATc2 is the evolutionally closest relative of NFATc1 among the NFAT proteins (1, 2, 15, 16) (Fig. 2 A). In addition, overexpression of NFATc2 facilitates the differentiation of osteoclasts (7) (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051150/DC1). These results prompted us to investigate osteoclast differentiation in NFATc2-deficient (NFATc2 −/−) mice (17). Although NFATc2 is involved in the regulation of chondrocytes at advanced age, bone development of NFATc2 −/− mice has never been well defined (18). Skeletal development occurs normally, and we observed no increase in bone mass in NFATc2 −/− mice as shown in Fig. 2 B [see also reference 19]. We found no abnormality in the number of osteoclasts, or in the parameters of osteoclastic bone resorption (Fig. 2 C and Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20051150/DC1) in NFATc2 −/− mice. Consistently, NFATc2 −/− BMMs that are stimulated by RANKL normally differentiate into osteoclasts with bone-resorbing activity (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20051150/DC1 and not depicted). In addition, RANKL induction of NFATc1 in NFATc2 −/− cells is comparable to that in NFATc2 +/+ cells (Fig. S3). Thus, NFATc2 is dispensable for RANKL-induced NFATc1 expression and osteoclastogenesis.
Figure 2.
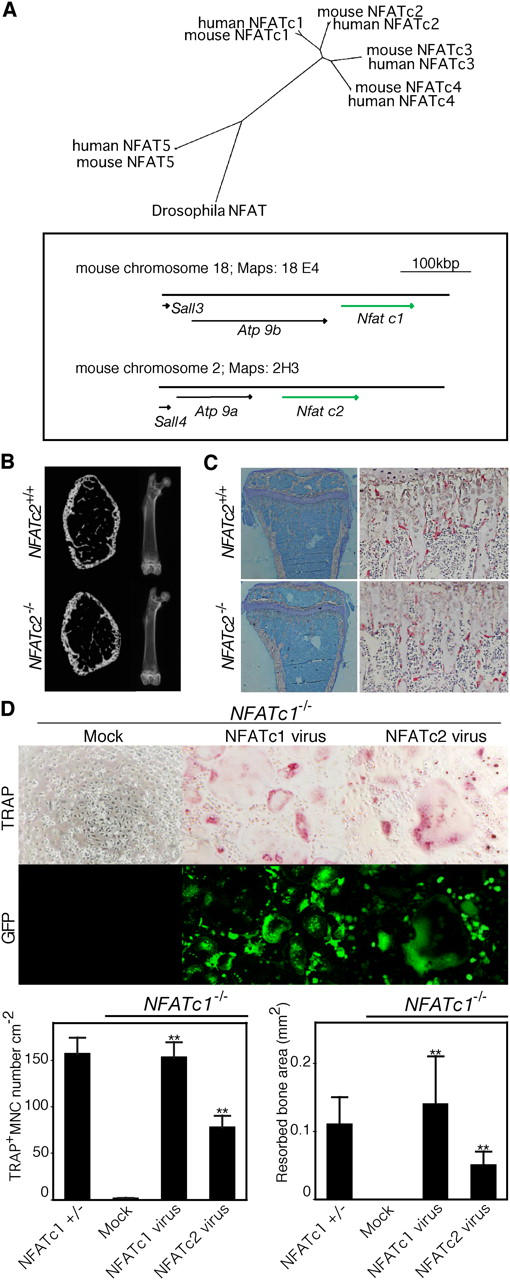
In vitro compensation of NFATc1 deficiency by forced expression of NFATc2. (A) Phylogenetic tree analysis for the DNA binding (Rel homology) region of NFAT family proteins (top). A wide-range genomic view of NFATc1 and NFATc2 genes suggests that these genes were generated by chromosomal gene duplication (bottom). (B) Loss-of-function analyses of NFATc2 in osteoclast differentiation. Microradiographic analysis of femur derived from NFATc2 −/− mice at 5 wk of age (see Fig. 1 B). (C) Histologic examination of tibia from NFATc2 −/− mice (left: toluidine blue staining; right: tartrate-resistant acid phosphatase staining [TRAP]). (D) Recovery of osteoclastogenesis in NFATc1 −/− FLCs by retrovirus-mediated expression of NFATc1 or NFATc2. Forced expression of NFATc2 as well as that of NFATc1 induces formation of osteoclasts with pit-forming activity on dentin slices (RANKL, 50 ng ml−1). Infection efficiency was monitored by GFP, which is expressed bicistronically. **P < 0.001 versus mock.
Osteoclast differentiation of NFATc1-deficient cells is rescued by forced expression of NFATc2
Because NFATc1 and NFATc2 have a similar ability to activate gene expression in T cells (1–3), it has been unexpected that NFATc1 plays a nonredundant role in the skeletal system. NFATc1 +/− cells differentiate into osteoclasts with bone-resorbing activity when they are stimulated by RANKL in the presence of M-CSF, whereas NFATc1 −/− cells are unable to generate osteoclasts (Fig. 2 D), despite normal development into the monocyte/macrophage lineage (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20051150/DC1). To investigate the function of NFATc1 and NFATc2 further, we ectopically expressed NFATc1 and NFATc2 under the control of the LTR promoter using retrovirus-mediated gene transfer (20) in NFATc1 −/− osteoclast precursor cells. As expected, the formation of bone-resorbing osteoclasts was rescued by retroviral expression of NFATc1. Surprisingly, osteoclast formation recovered, albeit at a reduced efficiency, as the result of forced expression of NFATc2. This demonstrated that forced expression of NFATc2 compensates for the loss of the NFATc1 gene (Fig. 2 D).
Selective autoamplification of NFATc1 during osteoclastogenesis
How can we reconcile the in vivo essential role of NFATc1 in osteoclastogenesis with the observation that NFATc1 deficiency is compensated for by forced expression of NFATc2? Whereas NFATc2 is expressed constitutively in BMMs at a low level, mRNA of NFATc1 is induced selectively and potently by RANKL (5) (Fig. 3 A). The induction of NFATc1, but not NFATc2, is down-regulated by the calcineurin inhibitor, FK506, which suppresses the activity of NFAT (Fig. 3 A). This suggests that NFATc1 is autoregulated selectively by NFAT when BMMs are stimulated with RANKL, and that the resulting expression pattern may explain the specificity of NFATc1. Although NFATc1 also is autoregulated in T cells (20, 21), the expression pattern during osteoclastogenesis (autoamplification) is characterized by a monotone increase and magnitude of amplification. Three isoforms (A, B, and C) of NFATc1 are well documented in T cells (20, 22), but it remains to be clarified which isoform is involved in osteoclastogenesis. Using specific PCR primers, we found that mRNA expression of the shortest isoform, NFATc1/A, is induced selectively in RANKL-stimulated BMMs (Fig. 3 B); immunoblot analysis yielded consistent results (Fig. 3 C). Therefore, we investigated the regulatory mechanism of the P1 promoter of the NFATc1 gene, which regulates NFATc1/A induction (20), in comparison with the NFATc2 promoter. To determine the transcriptional start site of mouse NFATc2 gene, a 5′ rapid amplification of cDNA ends experiment was performed. The putative transcription factor binding sites in the 5′ flanking region of the NFATc1 and NFATc2 gene are shown in Fig. 3 D. Unexpectedly, these results suggest that not only the NFATc1 promoter but also the NFATc2 promoter contain multiple NFAT binding sites.
Figure 3.

Autoamplification of NFATc1 during osteoclastogenesis. (A) GeneChip analysis of mRNA expression of NFATc1 and NFATc2 in RANKL-stimulated BMMs. Strong induction of NFATc1 by RANKL is inhibited by FK506 (2.5 μg ml−1), which inhibits NFAT activity. NFATc2 expression is constitutive and is not affected by FK506. (B) Semiquantitative RT-PCR analysis of the mRNA of NFATc1 isoforms in BMMs. NFATc1/A is induced ∼10-fold by RANKL. (C) Immunoblot analysis of NFATc1 isoforms during RANKL-induced osteoclastogenesis in BMMs. A Daudi lymphoma cell line is shown as a positive control for all three isoforms (A, B, and C) of NFATc1 (22). (D) A schematic illustration of putative transcription factor binding sites in the 5′ flanking region of the NFATc1 (20) and NFATc2 genes. Binding sites in the 5′ flanking region of the NFATc2 gene were determined by the TRANSFAC retrieval program. Arrows indicate primers for ChIP experiments. (E) ChIP assay of NFATc1 and NFATc2 promoters in RANKL-stimulated BMMs. (F) Luciferase assay of NFATc1 and NFATc2 promoters in HEK293T cells.
Recruitment of NFATc1 and other transcription factors to the NFAT promoters
Chromatin immunoprecipitation (ChIP) analysis shows that NFATc2 is recruited to the NFATc1 promoter at the earliest phase of osteoclast differentiation (Fig. 3 E). 24 h after RANKL stimulation, NFATc1 mainly occupies the promoter instead of NFATc2, and c-Fos is concomitantly recruited to the promoter (Fig. 3 E); this is consistent with the strong induction of NFATc1 at this time point. This occupancy persists during terminal differentiation of osteoclasts, and indicates that autoamplification of NFATc1 occurs during this process and that c-Fos may contribute to it. This is congruent with the critical role of c-Fos in RANKL-mediated induction of NFATc1, which was reported previously (5, 23). NF-κB is activated rapidly by RANKL (24), and a recent study suggests that NF-κB activity is important for RANKL-mediated induction of NFATc1 at the early phase of osteoclastogenesis (25). Consistent with this, NF-κB components p50 and p65 are recruited to the NFATc1 promoter 1 h after RANKL stimulation. However, this recruitment is not observed after 24 h (Fig. 3 E), which suggests that NF-κB is important for the initial induction of NFATc1 in cooperation with NFATc2. Although NFATc2 is detected on the NFATc1 promoter without RANKL stimulation, NFATc1 is not induced under this condition, which suggests that additional stimulation, such as NF-κB activation, is important for the efficient triggering of this promoter. This is consistent with the observation in the luciferase assay that the NFATc1 promoter is not activated fully by NFATc2 alone, but is activated markedly by the coexpression of NFATc2 and NF-κB (Fig. 3 F). The basal calcineurin activity in osteoclast precursor cells may contribute to the nuclear localization of NFATc2 without RANKL addition (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20051150/DC1). Because osteoclast differentiation was not affected in NFATc2 − / − mice, the function of NFATc2 evidently can be compensated by other factors, although it may contribute to physiologic regulation of the initial induction of NFATc1. A reporter plasmid that is driven by the NFATc1-P1 promoter, but not by the NFATc2 promoter, is activated markedly by NFAT in cooperation with NF-κB (Fig. 3 F). These results indicate that the NFATc1 promoter is distinct from the NFATc2 promoter in that the former is exclusively under autoregulation by NFAT; this further suggests that an autoregulatory mechanism contributes to the essential role of NFATc1.
Epigenetic regulation of NFATc1 gene determines its unique spatiotemporal induction pattern during osteoclastogenesis
To gain insight into the selective recruitment of NFATc1 to the NFATc1 promoter, we examined the association of transcriptional cofactors with these promoters. NFATc1 promoter is associated increasingly with transcriptional coactivators with histone acetylase activity, such as CREB-binding protein (CBP) and p300/CBP-associated factor (PCAF), but is dissociated with histone deacetylase 1 (HDAC1; Fig. 4 A). It is notable that histone acetylation in the NFATc1 promoter increased gradually after RANKL stimulation and that the high acetylation status was sustained, but this was not observed in the NFATc2 promoter (Fig. 4 B). Methylation of histone H3 lysine 4, which is characteristic of the transcriptionally active locus, is up-regulated exclusively in the NFATc1 promoter (Fig. 4 B). Conversely, the NFATc2 promoter is associated constantly with methylated DNA-binding proteins, such as the methyl-CpG binding protein 2 (MeCP2) (Fig. 4 C), which suggests that the NFATc2 promoter specifically is silenced during osteoclast differentiation. Consistent with this notion, we detected high levels of methylated histone H3 lysine 9 only in the NFATc2 promoter (Fig. 4 C). These results suggest that epigenetic regulation underlies the specific autoamplification of NFATc1.
Figure 4.
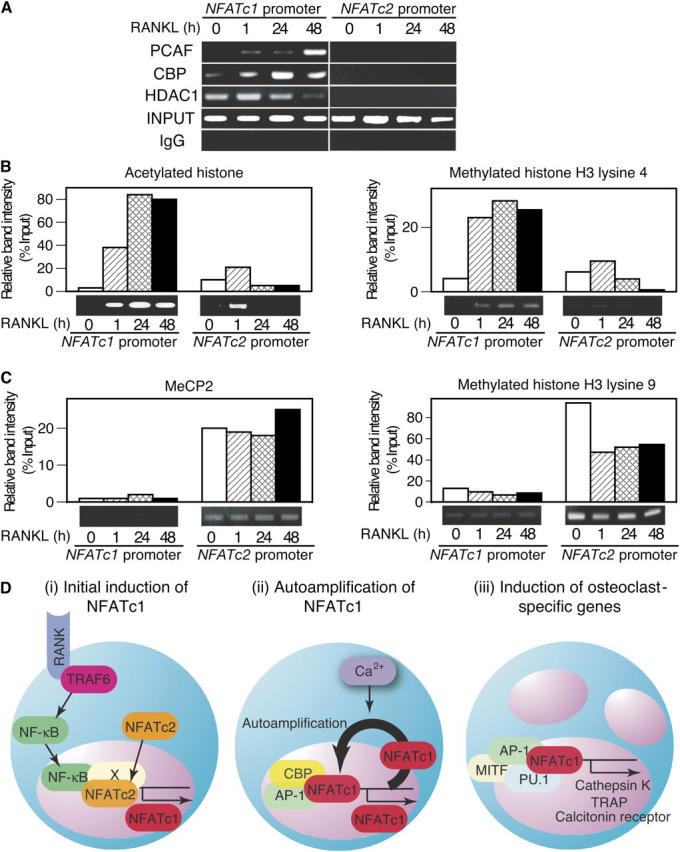
Epigenetic regulation of NFATc1 promoter underlies the selective autoamplification of NFATc1. (A) Analysis of chromatin modification–related factors in NFATc1 and NFATc2 promoters by ChIP in RANKL-stimulated BMMs during osteoclastogenesis. (B) ChIP assay for acetylated histone and methylated histone H3 lysine 4. (C) ChIP assay for MeCP2 and methylated histone H3 lysine 9. (D) A schematic diagram of three stages of osteoclast differentiation that are governed by NFATc1. (i) NFATc2 is recruited to the NFATc1 promoter at the very early phase, but this is not enough to activate the NFATc1 promoter. The binding of RANKL to receptor activator of NF-κB (RANK) results in the recruitment of TNF receptor–associated factor (TRAF) 6, and leads to the activation of downstream molecules, such as NF-κB (25, 34). Cooperation of NFATc2 and NF-κB activates the initial induction of NFATc1, but because NFATc2-deficient mice have no obvious defect in osteoclast differentiation, unknown factor(s) (shown as X) may compensate for the loss of NFATc2 in these mice. (ii) RANKL–RANK interaction cooperates with immunoreceptors to activate the calcium signals (28), which stimulate the NFATc1 activation by way of calcineurin (5). NFATc1 bind to its own promoter, which leads to the robust induction of NFATc1; AP-1 (containing c-Fos) is critical for this autoamplification. Selective recruitment of NFATc1 to the promoter of NFATc1, but not NFATc2, is explained in part, by epigenetic regulation. (iii) Several osteoclast-specific genes, such as cathepsin K, TRAP, and calcitonin receptor, are activated by a transcriptional complex that contains NFATc1 and other cooperators, such as AP-1, PU.1, and microphthalmia-associated transcription factor (MITF).
DISCUSSION
Our results illustrate that the essential importance of a gene is not only attributable to the specific biochemical function of the coded protein, but also is determined epigenetically by the distinct regulatory mechanism of gene expression. The role of GATA-1 and GATA-3 in erythropoiesis and Th2 cell differentiation are examples in which the essential importance of one member of a group of structurally related proteins of interchangeable function is based on an autoregulatory mechanism (26, 27). These studies suggest that autoregulation of transcription factors is one of the critical mechanisms for cell fate determination (2). In a future study, generation of mice with an NFATc2 knock-in into the NFATc1 locus will be an intriguing strategy to provide the conclusive in vivo evidence.
As depicted in Fig. 4 D, in the differentiation of osteoclasts, preexisting NFATc2 cooperates with other RANKL-stimulated transcription factors, such as NF-κB, to activate the initial induction of NFATc1, followed by an autoamplification phase of NFATc1. It was reported that the activator protein (AP)–1 component, c-Fos, is critical for RANKL-mediated induction of NFATc1 (5, 23). Consistent with this, c-Fos is recruited selectively to the NFATc1 promoter at the autoamplification phase (Fig. 3 E). The composite NFAT/AP-1 site is observed exclusively in the NFATc1 promoter (Fig. 3 D) (20). This lends further support to the notion that the cooperation of NFAT and AP-1 is responsible for the specific activation of the NFATc1 promoter and its autoamplification.
Calcium signal-mediated activation of NFATc1 also triggers the autoamplification loop of NFATc1 and ensures a sustained NFATc1-dependent transcriptional program (5, 28) in which osteoclast-specific genes are activated by a transcriptional complex that involves NFATc1, AP-1, and other cooperators (5, 29). Accumulating evidence indicates that NFATc1 regulates many osteoclast-specific genes, such as cathepsin K (29), TRAP (5, 30), calcitonin receptor (5, 30), and osteoclast-associated receptor (30), in cooperation with other transcription factors (e.g., PU.1 and microphthalmia-associated transcription factor), although the components of the transcriptional complex are not always the same (30) (Fig. 4 D, iii). A distinct pattern of calcium oscillation, which efficiently keeps NFATc1 transcriptionally active in the nucleus (31), may explain the specific spaciotemporal expression of NFATc1 in the osteoclast lineage (5), in contrast to T cells. FK506 inhibited osteoclast formation, even when it was added at the late phase of osteoclastogenesis (48–72 h after RANKL stimulation, unpublished data); this suggests that calcium signaling is also critical for the autoamplification of NFATc1.
Successful application of FLC transfer and blastocyst complementation, first established in the immune system, provided genetic evidence that NFATc1 is essential for osteoclast differentiation and maintenance of the skeletal system. Modulation of the NFATc1 autoamplification pathway has promise as a strategy for suppressing the excessive osteoclast formation that is characteristic of a variety of bone diseases.
MATERIALS AND METHODS
Mice and analysis of bone phenotype.
Generation of Fos −/− and NFATc1 − / − mice has been described previously (8, 13). NFATc2 mice (17) were provided by L.H. Glimcher (Harvard Medical School, Boston, MA). Histologic, histomorphometric, and microradiographic examinations were performed using essentially the same methods as described (32). Statistical analysis was performed using Student's t test. All mice were kept in a specific pathogen-free environment, and all animal experiments were performed with the approval of the institutional committee of Tokyo Medical and Dental University.
In vitro osteoclastogenesis and retroviral gene transfer.
Bone marrow cells or FLCs were cultured in α-MEM (GIBCO BRL) containing 10% FBS (Sigma-Aldrich) and 10 ng ml−1 M-CSF (R&D Systems). After 2 d, adherent cells were used as osteoclast precursor cells. In osteopetrotic mice, osteoclast precursor cells were obtained similarly from splenocytes. We described the method of RANKL-induced in vitro osteoclastogenesis, retroviral gene transfer, characterization of osteoclasts, and immunostaining (5, 24, 32). All data are expressed as mean ± SEM (n = 6). Retroviral vectors, pEGZ-NFATc1/A (NFATc1 virus) and pEGZ-NFATc2 (NFATc2 virus), were described previously (20), and infection efficiencies of both retrovirus vectors into osteoclast precursor cells were ∼40%, as described previously (24).
Fetal liver cell transfer.
NFATc1 − / − and NFATc1 + / − embryos were obtained by crossing NFATc1 + / − parental mice. Total liver cells (107 cells) from embryonic day (E) 13.5 embryos were injected into the liver of recipient Fos − / − newborn (2 d) mice, which were analyzed at 6 wk of age. The recipient mice were treated with busulfan (Sigma-Aldrich) in utero by i.p. administration to pregnant mice (12.5 mg kg−1) twice at 17.5 and 18.5 d postcoitum.
Fos −/− blastocyst complementation.
Establishment of NFATc1 − / − ES cell lines has been described (33). NFATc1 − / − ES cells or wild-type ES cells (E14K) were injected into E3.5 Fos − / − blastocysts that were obtained by fertilizing Fos − / − gametes from Fos − / − paternal and maternal mice using in vitro fertilization technique. Resultant blastocysts were transferred to pseudopregnant foster mothers to generate chimeric mice.
RT-PCR and GeneChip analysis.
RNA extraction, semiquantitative RT-PCR, and GeneChip analysis were performed as described previously (5, 32). The following PCR primers were used to detect NFAT isoforms. NFATc1/A: 5′-GGTAACTCTGTCTTTCTAACCTTAAGCTC-3′ (sense) and 5′-GTGATGACCCCAGCATGCACCAGTCACAG-3′ (antisense); NFATc1/B: 5′-CCCATCCGCCAGGCTACAGCCGCAGTAA-3′ (sense) and 5′-TTCGGTAAGTTGGGATTTCTGAGTGGTACC-3′ (antisense); NFATc1/C: 5′-CCCATCCGCCAGGCTACAGCCGCAGTAA-3′ (sense) and 5′-TGAGTGGTACCAGATGTGGGTCCAGTTTAT-3′ (antisense).
Chromatin immunoprecipitation.
ChIP was performed with the ChIP Assay Kit (Upstate Biotechnology) according to the manufacturer's instructions, using antibodies against NFATc1, NFATc2, p300/CBP-associated factor, histone deacetylase 1, p50, CBP, MeCP2 (Santa Cruz Biotechnology, Inc.), p65, acetylated histone, mono/di/trimethyl-histone H3 lysine 4, dimethyl-histone H3 lysine 9 (Upstate Biotechnology), c-Fos (Calbiochem), and normal IgG (Santa Cruz Biotechnology, Inc.). The purified DNA was analyzed by PCR using primers that detect sequences containing the NFATc1-P1 promoter: 5′-CCGGGACGCCCATGCAATCTGTTAGTAATT-3′ (sense) and 5′-GCGGGTGCCCTGAGAAAGCTACTCTCCCTT-3′ (antisense) and the NFATc2 promoter: 5′-TTATCAGGGAGCACTGCCCATCTCCGCTTT-3′ (sense) and 5′-CGGTCTGGCCTGAGCGACAGGCCCAGACAA-3′ (antisense).
Luciferase reporter gene assay.
The reporter plasmid pNFATc1P1-0.8 kb-luc (pNFATc1-pr-luc) was described previously (20). pNFATc2-0.8 kb-luc (pNFATc2-pr-luc) was constructed by inserting a 0.8-kb NheI–Xho fragment of the mouse NFATc2 promoter into the same sites of the pGL3 basic promoter vector (Promega). The mouse NFATc2 promoter region was amplified by PCR using following primers: 5′-GCTAGCTGTTTTGGTGACTGTTATCATGCTGGG-3′ (sense) and 5′-CTCGAGCTTCCTGCTCAAGGCACCTGTTGCAG-3′ (antisense). Transfection into HEK293T cells and measurement of luciferase activity were performed as described (5) using the Dual-luciferase reporter assay system (Promega).
Online supplemental material.
Fig. S1 shows the gain-of-function analyses of NFATc1 and NFATc2 in osteoclasts. Fig. S2 shows the histomorphometric evaluation of osteoclasts in NFATc2 −/− mice. Fig. S3 shows the normal osteoclast differentiation and expression of NFATc1 in NFATc2 −/− monocyte/macrophage precursor cells that were stimulated with RANKL/M-CSF. Fig. S4 shows the development into the monocyte/macrophage lineage from NFATc1 −/− FLCs. Fig. S5 shows the effect of FK506 on the recruitment of NFATc2 to the NFATc1 promoter without RANKL stimulation. Also, details of the methods for phylogenetic tree analysis and 5′ rapid amplification of cDNA ends analysis are available. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051150/DC1.
Acknowledgments
We are grateful to L.H. Glimcher for the mice. We thank T. Taniguchi, S. Mori, H. Endo, T. Watanabe, N. Yoshida, K. Aoki, T. Ishida, H. Murayama, M. Tsujimoto, M. Isobe, S. Kamano, T. Honda, T. Koga, S. Harumiya, J. Hirooka, A. Suematsu, Y. Kim, K. Nishikawa, H.J. Gober, N. Kumasaki, H. Takatsuna, Y. Morishita, T. Yokochi, M.A. Hayashi, and I. Kawai for discussion and technical assistance.
This work was supported in part by the Precursory Research for Embryonic Science and Technology and Solution Oriented Research for Science and Technology programs of the Japan Science and Technology Agency; grants for the Genome Network Project from Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT); grants for the 21st century Center of Excellence program; grants-in-aid for Scientific Research from the Japan Society for the Promotion of Science and MEXT; Health Sciences Research Grants from the Ministry of Health, Labor and Welfare of Japan; and grants from the Mitsubishi Foundation, The Kato Trust for Nambyo Research, Takeda Science Foundation, Daiwa Securities Health Foundation, The Naito Foundation, Kowa Life Science Foundation, Suzuken Memorial Foundation, Kato Memorial Bioscience Foundation, Cell Science Research Foundation, and Uehara Memorial Foundation. The work in E. Serfling's laboratory was supported by the German Research Foundation, the Wilhelm-Sander Foundation, and the Scheel Foundation for Cancer Research.
The authors have no conflicting financial interests.
Abbreviations used: AP, activator protein; BMM, bone marrow monocyte/macrophage precursor cell; CBP, CREB-binding protein; ChIP, chromatin immunoprecipitation; ES, embryonic stem; FLC, fetal liver cell; MeCP2, methyl-CpG binding protein 2; RANKL, receptor activator of NF-κB ligand.
References
- 1.Crabtree, G.R., and E.N. Olson. 2002. NFAT signaling: choreographing the social lives of cells. Cell. 109(Suppl):S67–79. [DOI] [PubMed] [Google Scholar]
- 2.Hogan, P.G., L. Chen, J. Nardone, and A. Rao. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17:2205–2232. [DOI] [PubMed] [Google Scholar]
- 3.Peng, S.L., A.J. Gerth, A.M. Ranger, and L.H. Glimcher. 2001. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity. 14:13–20. [DOI] [PubMed] [Google Scholar]
- 4.Teitelbaum, S.L., and F.P. Ross. 2003. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 4:638–649. [DOI] [PubMed] [Google Scholar]
- 5.Takayanagi, H., S. Kim, T. Koga, H. Nishina, M. Isshiki, H. Yoshida, A. Saiura, M. Isobe, T. Yokochi, J. Inoue, et al. 2002. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell. 3:889–901. [DOI] [PubMed] [Google Scholar]
- 6.Theill, L.E., W.J. Boyle, and J.M. Penninger. 2002. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu. Rev. Immunol. 20:795–823. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda, F., R. Nishimura, T. Matsubara, S. Tanaka, J. Inoue, S.V. Reddy, K. Hata, K. Yamashita, T. Hiraga, T. Watanabe, et al. 2004. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J. Clin. Invest. 114:475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Pompa, J.L., L.A. Timmerman, H. Takimoto, H. Yoshida, A.J. Elia, E. Samper, J. Potter, A. Wakeham, L. Marengere, B.L. Langille, et al. 1998. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 392:182–186. [DOI] [PubMed] [Google Scholar]
- 9.Ranger, A.M., M.J. Grusby, M.R. Hodge, E.M. Gravallese, F.C. de la Brousse, T. Hoey, C. Mickanin, H.S. Baldwin, and L.H. Glimcher. 1998. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 392:186–190. [DOI] [PubMed] [Google Scholar]
- 10.Shinkai, Y., G. Rathbun, K.P. Lam, E.M. Oltz, V. Stewart, M. Mendelsohn, J. Charron, M. Datta, F. Young, A.M. Stall, and F.W. Alt. 1992. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 68:855–867. [DOI] [PubMed] [Google Scholar]
- 11.Mombaerts, P., J. Iacomini, R.S. Johnson, K. Herrup, S. Tonegawa, and V.E. Papaioannou. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 68:869–877. [DOI] [PubMed] [Google Scholar]
- 12.Wang, Z.Q., C. Ovitt, A.E. Grigoriadis, U. Mohle-Steinlein, U. Ruther, and E.F. Wagner. 1992. Bone and haematopoietic defects in mice lacking c-fos. Nature. 360:741–745. [DOI] [PubMed] [Google Scholar]
- 13.Grigoriadis, A.E., Z.Q. Wang, M.G. Cecchini, W. Hofstetter, R. Felix, H.A. Fleisch, and E.F. Wagner. 1994. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 266:443–448. [DOI] [PubMed] [Google Scholar]
- 14.Chen, J., R. Lansford, V. Stewart, F. Young, and F.W. Alt. 1993. RAG-2–deficient blastocyst complementation: an assay of gene function in lymphocyte development. Proc. Natl. Acad. Sci. USA. 90:4528–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho, S.N., D.J. Thomas, L.A. Timmerman, X. Li, U. Francke, and G.R. Crabtree. 1995. NFATc3, a lymphoid-specific NFATc family member that is calcium-regulated and exhibits distinct DNA binding specificity. J. Biol. Chem. 270:19898–19907. [DOI] [PubMed] [Google Scholar]
- 16.Graef, I.A., J.M. Gastier, U. Francke, and G.R. Crabtree. 2001. Evolutionary relationships among Rel domains indicate functional diversification by recombination. Proc. Natl. Acad. Sci. USA. 98:5740–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodge, M.R., A.M. Ranger, F. Charles de la Brousse, T. Hoey, M.J. Grusby, and L.H. Glimcher. 1996. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp–deficient mice. Immunity. 4:397–405. [DOI] [PubMed] [Google Scholar]
- 18.Ranger, A.M., L.C. Gerstenfeld, J. Wang, T. Kon, H. Bae, E.M. Gravallese, M.J. Glimcher, and L.H. Glimcher. 2000. The nuclear factor of activated T cells (NFAT) transcription factor NFATp (NFATc2) is a repressor of chondrogenesis. J. Exp. Med. 191:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koga, T., Y. Matsui, M. Asagiri, T. Kodama, B. de Crombrugghe, K. Nakashima, and H. Takayanagi. 2005. NFAT and Osterix cooperatively regulate bone formation. Nat. Med. 11:880–885. [DOI] [PubMed] [Google Scholar]
- 20.Chuvpilo, S., E. Jankevics, D. Tyrsin, A. Akimzhanov, D. Moroz, M.K. Jha, J. Schulze-Luehrmann, B. Santner-Nanan, E. Feoktistova, T. Konig, et al. 2002. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity. 16:881–895. [DOI] [PubMed] [Google Scholar]
- 21.Zhou, B., R.Q. Cron, B. Wu, A. Genin, Z. Wang, S. Liu, P. Robson, and H.S. Baldwin. 2002. Regulation of the murine Nfatc1 gene by NFATc2. J. Biol. Chem. 277:10704–10711. [DOI] [PubMed] [Google Scholar]
- 22.Chuvpilo, S., M. Zimmer, A. Kerstan, J. Glockner, A. Avots, C. Escher, C. Fischer, I. Inashkina, E. Jankevics, F. Berberich-Siebelt, et al. 1999. Alternative polyadenylation events contribute to the induction of NF-ATc in effector T cells. Immunity. 10:261–269. [DOI] [PubMed] [Google Scholar]
- 23.Matsuo, K., D.L. Galson, C. Zhao, L. Peng, C. Laplace, K.Z. Wang, M.A. Bachler, H. Amano, H. Aburatani, H. Ishikawa, and E.F. Wagner. 2004. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J. Biol. Chem. 279:26475–26480. [DOI] [PubMed] [Google Scholar]
- 24.Takayanagi, H., K. Ogasawara, S. Hida, T. Chiba, S. Murata, K. Sato, A. Takaoka, T. Yokochi, H. Oda, K. Tanaka, et al. 2000. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature. 408:600–605. [DOI] [PubMed] [Google Scholar]
- 25.Takatsuna, H., M. Asagiri, T. Kubota, K. Oka, T. Osada, C. Sugiyama, H. Saito, K. Aoki, K. Ohya, H. Takayanagi, and K. Umezawa. 2005. Inhibition of RANKL-induced osteoclastogenesis by (-)-DHMEQ, a novel NF-κB Inhibitor, through downregulation of NFATc1. J. Bone Miner. Res. 20:653–662. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi, S., R. Shimizu, N. Suwabe, T. Kuroha, K. Yoh, J. Ohta, S. Nishimura, K.C. Lim, J.D. Engel, and M. Yamamoto. 2000. GATA factor transgenes under GATA-1 locus control rescue germline GATA-1 mutant deficiencies. Blood. 96:910–916. [PubMed] [Google Scholar]
- 27.Ouyang, W., M. Lohning, Z. Gao, M. Assenmacher, S. Ranganath, A. Radbruch, and K.M. Murphy. 2000. Stat6-independent GATA-3 autoactivation directs IL-4–independent Th2 development and commitment. Immunity. 12:27–37. [DOI] [PubMed] [Google Scholar]
- 28.Koga, T., M. Inui, K. Inoue, S. Kim, A. Suematsu, E. Kobayashi, T. Iwata, H. Ohnishi, T. Matozaki, T. Kodama, et al. 2004. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 428:758–763. [DOI] [PubMed] [Google Scholar]
- 29.Matsumoto, M., M. Kogawa, S. Wada, H. Takayanagi, M. Tsujimoto, S. Katayama, K. Hisatake, and Y. Nogi. 2004. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J. Biol. Chem. 279:45969–45979. [DOI] [PubMed] [Google Scholar]
- 30.Kim, Y., K. Sato, M. Asagiri, I. Morita, K. Soma, and H. Takayanagi. 2005. Contribution of NFATc1 to the transcriptional control of immunoreceptor OSCAR but not TREM-2 during osteoclastogenesis. J. Biol. Chem. 280:32905–32913. [DOI] [PubMed] [Google Scholar]
- 31.Tomida, T., K. Hirose, A. Takizawa, F. Shibasaki, and M. Iino. 2003. NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J. 22:3825–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takayanagi, H., S. Kim, K. Matsuo, H. Suzuki, T. Suzuki, K. Sato, T. Yokochi, H. Oda, K. Nakamura, N. Ida, et al. 2002. RANKL maintains bone homeostasis through c-Fos–dependent induction of interferon-β. Nature. 416:744–749. [DOI] [PubMed] [Google Scholar]
- 33.Yoshida, H., H. Nishina, H. Takimoto, L.E. Marengere, A.C. Wakeham, D. Bouchard, Y.Y. Kong, T. Ohteki, A. Shahinian, M. Bachmann, et al. 1998. The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity. 8:115–124. [DOI] [PubMed] [Google Scholar]
- 34.Takayanagi, H. 2005. Mechanistic insight into osteoclast differentiation in osteoimmunology. J. Mol. Med. 83:170–179. [DOI] [PubMed] [Google Scholar]