

Abstract
The autoimmune process that destroys the insulin-producing pancreatic β cells in type 1 diabetes (T1D) is targeted at insulin and its precursor, proinsulin. T cells that recognize the proximal A-chain of human insulin were identified recently in the pancreatic lymph nodes of subjects who had T1D. To investigate the specificity of proinsulin-specific T cells in T1D, we isolated human CD4+ T cell clones to proinsulin from the blood of a donor who had T1D. The clones recognized a naturally processed, HLA DR4–restricted epitope within the first 13 amino acids of the A-chain (A1–13) of human insulin. T cell recognition was dependent on the formation of a vicinal disulfide bond between adjacent cysteine residues at A6 and A7, which did not alter binding of the peptide to HLA DR4. CD4+ T cell clones that recognized this epitope were isolated from an HLA DR4+ child with autoantibodies to insulin, and therefore, at risk for T1D, but not from two healthy HLA DR4+ donors. We define for the first time a novel posttranslational modification that is required for T cell recognition of the insulin A-chain in T1D.
Type 1 diabetes (T1D) is an autoimmune disease in which insulin-producing pancreatic β cells are destroyed by autoreactive T cells (1). Many genetic loci have been associated with T1D, but the highest risk is associated with the HLA complex, specifically with the class II HLA haplotypes, DR3-DQ2 and DR4-DQ8 (2, 3). Class II HLA molecules present peptide antigen to CD4+ T cells, which underlines the role of CD4+ T cells in the pathogenesis of T1D. Several lines of evidence implicate proinsulin as a target autoantigen in T cell–mediated β cell destruction in humans and the NOD mouse model of T1D (4). Proinsulin is the major product of β cells and, with the possible exception of rare self-antigen–expressing cells in lymphoid tissues (5), is the only known T1D autoantigen that is expressed exclusively in β cells. Autoantibodies to (pro)insulin are a strong risk factor for the development of T1D (6). The T1D susceptibility locus IDDM2 maps to a variable number of tandem repeats (VNTRs) upstream of the insulin gene (7); the long class III VNTR allele is associated with higher levels of proinsulin mRNA in the thymus and decreased susceptibility to T1D (8, 9). NOD mice that transgenically express proinsulin under the control of an MHC class II promoter are protected from diabetes (10, 11). NOD mice with targeted disruptions of the proinsulin I and II genes have a decreased (12) and increased (13) incidence of diabetes, respectively. Furthermore, diabetes did not develop in NOD mice that expressed a mutated proinsulin I (Y to A at B16) (14). Recently, it was reported that HLA DR4–restricted, CD4+ T cells that react to the first 15 amino acids of the insulin A-chain make up a large proportion of T cells from the pancreatic lymph nodes of two subjects with who had T1D, but not from three healthy donors (15). However, the nature of the epitope was not defined.
There is increasing evidence, mainly from animal models, that pathogenic T cells in autoimmune disease recognize epitopes that are formed by posttranslational modification of self-antigens (16). To our knowledge, there is only one report of human CD4+ T cells that recognize an epitope formed by posttranslational modification. Subjects who have rheumatoid arthritis have T cells that react to a glycosylated collagen epitope (17). Here, we sought to define the specificity of proinsulin-specific CD4+ T cells in T1D. We show for the first time that T cell recognition of the first 13 amino acids of human insulin A-chain requires posttranslational modification of adjacent cysteine residues at A6 and A7.
RESULTS AND DISCUSSION
Identification of the insulin A-chain 1–13 epitope
17 proinsulin-specific CD4+ T cell clones were isolated from the blood of a donor who had established T1D (18). The insulin A1–13 epitope was identified using an overlapping panel of 15-mer peptides. First, the clones were cultured with eight peptide pools, each containing three to four peptides, and covering the entire sequence of proinsulin. 5 of 17 clones recognized a peptide within pool 8 (Fig. 1 A); four were studied further. The three peptides in pool 8 were tested separately, and a single peptide comprising the last two amino acids of the C-peptide and the first 13 amino acids of the A-chain of insulin (KRGIVEQCCTSICSL) stimulated all four clones (Fig. 1 B). Moreover, individual peptides comprising the first 13 and 15 amino acids of the A-chain of insulin (Table S2, available at http://www.jem.org/cgi/content/full/jem.20051251/DC1) also stimulated the clones with equivalent dose response (Fig. 1 C). Furthermore, insulin and proinsulin stimulated the clones equally. The response to clinical grade recombinant insulin was HLA DR dependent (Fig. 1 D). Hence, the clones recognize a minimal epitope comprising the first 13 amino acids of the A-chain of human insulin. To confirm that the T cell clones recognized an epitope that was derived from native human insulin, their response to human islet lysate was tested. The clones proliferated in a dose-dependent manner to islet, but not spleen, lysate from the same donor (Fig. 1 E). Hence, the clones recognized a naturally processed and presented epitope in the first 13 amino acids of the A-chain of human insulin.
Figure 1.

Identification of the A-chain 1–13 epitope. (A) Preliminary epitope mapping. Peptides, 15-mers shifted by three amino acids, comprising proinsulin, were grouped into eight pools of three to four peptides (5 μg/ml each), or proinsulin (10 μg/ml) were included as indicated. Mean of triplicates ± SEM is shown. (B) Fine epitope mapping. Three peptides from pool 8 were tested separately. (C) Responses to insulin and A-chain peptides. Insulin (open circles) and proinsulin (filled circles) were titrated from 1 μM to 0.1 nM. Peptides KR-A1-13 (filled triangles), A1–13 (filled diamonds), and A1-15 (filled squares) were titrated from 10 μM to 0.1 nM. Other conditions were the same as for A. (D) Responses to insulin. T cell clones were cultured with proinsulin (10 μg/ml), insulin (10 μg/ml), or without antigen (no antigen). Anti-HLA DR mAb (5 μg/ml) was included as indicated. (E) Responses to islet lysate. T cell clones were cultured with dilutions of human islet lysate (filled circles), spleen lysate (open circles), insulin (10 μg/ml; filled square), or without antigen (open square).
Mapping the fine HLA restriction of the insulin A1–13 epitope
Experiments with HLA isotype–specific antibodies showed that proliferation of all clones was blocked by anti-HLA DR (L243) mAb (Fig. 2 A). Next, EBV-transformed B cells from a patient who had bare lymphocyte syndrome (BLS) that were transfected with different HLA-β genes were used as APCs. Cells that were transfected with DRB1*0401, DRB1*0404, or DRB1*0405 presented the A1–13 epitope, whereas cells that were transfected with DRB4*0101 (DR53), DQB1*0201 (DQ2), or DQB1*0302 (DQ8) did not (Fig. 2 B). Hence, the A1–13 epitope can be presented by HLA DRB1*0401, 0404, and 0405. It is well established that HLA DR4 confers susceptibility to T1D (2, 3). HLA DR4 has been associated with high-affinity and high-titer insulin autoantibodies (6) that typically are found in young individuals who are at high risk for developing T1D. Achenbach et al. (6) reported that these antibodies bound to the middle of the A-chain (A8-10/13), a region that is contained within the A1–13 CD4+ T cell epitope described here. Antibodies can increase antigen uptake and processing (19). Therefore, insulin autoantibodies that bind to the A-chain of insulin could enhance the presentation of the A1–13 epitope to CD4+ T cells, and, in turn, augment autoantibody production and explain, in part, the high titers of insulin antibodies in HLA DR4+ subjects.
Figure 2.

The response to insulin A1-13 is HLA DR4 restricted. (A) Coarse HLA restriction. Antibodies specific for HLA DR (L243), HLA DQ (SPV-L3), or HLA DP (B7/21) were added to a final concentration of 5 μg/ml. (B) Fine HLA restriction. Insulin-specific T cells were incubated with irradiated, HLA-transfected BLS cells pulsed with 100 μM KR-A1-13 peptide (filled bars) or solvent (open bars). Proliferation of the BLS cells alone after irradiation (1,000–5,000 cpm) was subtracted, and results are expressed as net 3H-thymidine incorporation (Δcpm). Mean ± SEM of triplicates is shown.
The requirement for cysteine residues in T cell recognition
The requirement for the three cysteines in the A1–13 epitope was determined by testing peptides in which serine was substituted for each cysteine (Table S2). Replacing either of the adjacent cysteines at A6 or A7 with serine completely abolished the peptide's capacity to stimulate the T cell clones (Fig. 3 A), whereas replacing the A11 cysteine with serine had no effect. The analogue peptide (S-11), with serine substituted for cysteine at position A11, was used in subsequent experiments. A peptide corresponding to the sequence of mouse insulin A1-13 (KRGIVDQCCTSICSL) was ∼10-fold less potent than was the human homologue. The absolute requirement of the adjacent cysteines at A6 and A7 for T cell recognition suggested that the epitope recognized by the T cell clones may require oxidative modification of the cysteines. To evaluate this possibility, the S-11 peptide was exposed to the disulfide-reducing agent, TCEP (Tris (2-carboxyethyl) phosphine hydrochloride). Reduction abolished the ability of S-11 to stimulate the clones (Fig. 3 B), which was consistent with oxidation-dependent modification of the adjacent cysteines required for T cell recognition. To determine if modification of the cysteines occurred spontaneously in culture medium or during antigen processing, we tested the capacity of paraformaldehyde-fixed and unfixed APCs to present the peptide to the T cell clones (Fig. 3 C). Fixation did not alter T cell reactivity. Therefore, we concluded that T cell recognition of the A1–13 epitope depended on the presence of oxidized cysteine residues at A6 and A7, a modification that did not require antigen uptake and intracellular processing.
Figure 3.
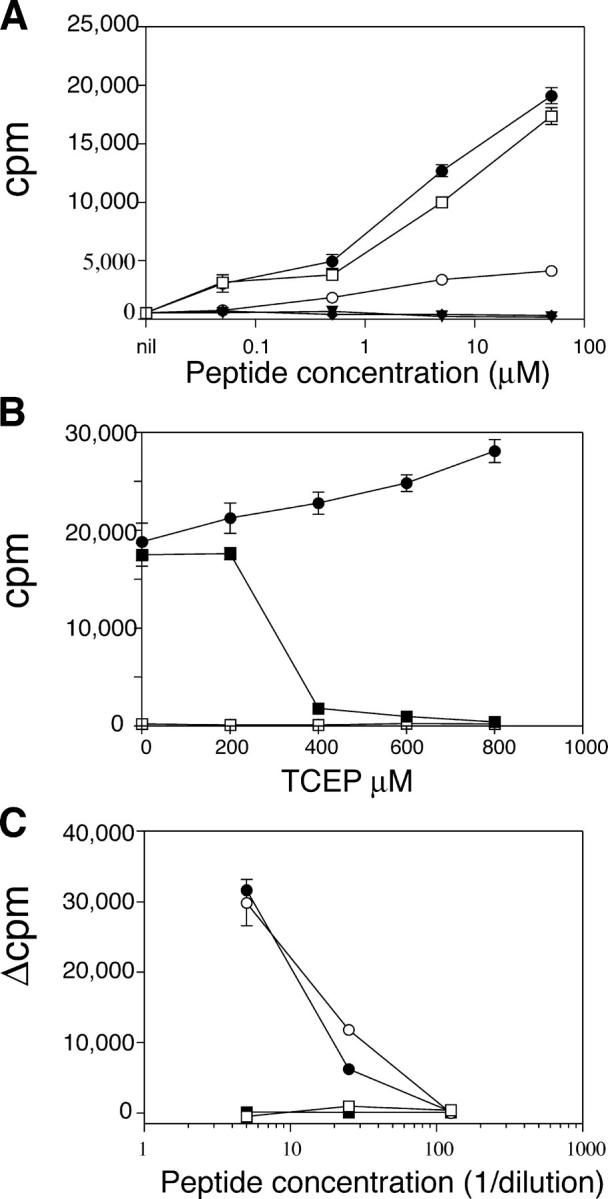
Oxidized cysteine at A6 and A7, but not antigen processing, is required to elicit responses of T cell clones. (A) Serine for cysteine substitutions. Insulin-specific T cell clones were cultured in the presence of 50—0.05 μM peptide (see Table S2). KR-A1-13 peptide (open squares), S-6 (filled triangles), S-7 (filled diamonds), S-11 (filled circles), or murine KR-A1-13 (open circles). (B) Effect of TCEP on responses to the A1-13 epitope. S-11 peptide (1 μM) was treated with TCEP at the concentrations shown (filled squares). Controls were comprised of either PHA (1.25 μg/ml) and IL-2 (2.5 U/ml; filled circles) or were without antigen (open squares). (C) Effect of APC fixation. HLA-DRB1*0404–transfected BLS cells were fixed cultured with RP-HPLC purified S-11 peptide and T cell clones (filled circles), or with unfixed BLS cells (open circles). Fixed cells with solvent alone (filled squares), or unfixed cells with solvent alone (open squares) were included. The mean ± SEM of triplicate wells is shown.
Analysis of oxidized cysteine residues at A6 and A7
To determine the nature of the oxidized cysteine residues, the S-11 peptide was incubated in culture medium and the components were separated by reversed-phase (RP)–HPLC. A single fraction stimulated proliferation of the T cell clones (Fig. 4 A). No fractions stimulated proliferation when the S-6 peptide was used in similar experiments (unpublished data). Analysis of the active fraction by mass spectrometry (MS) showed that it contained a single species that was 2 Dltons smaller than predicted for the parental S-11 peptide (Fig. 4, B–E). Tandem MS/MS analysis revealed that this loss of 2 Dltons arose at the adjacent cysteines (Table S3, available at http://www.jem.org/cgi/content/full/jem.20051251/DC1). This indicated that the epitope contained a vicinal disulfide bond (i.e., a disulfide bond between the adjacent A6 and A7 cysteines). This is the only modification that would eliminate two protons to result in a 2-Dlton decrease in mass. The vicinal disulfide bond formed within minutes, and treatment of the modified S-11 peptide with dithiothreitol reduced the vicinal disulfide bond and increased the mass by 2 Dltons (unpublished data). Using MS, we found no evidence of cysteinylation, sulphation, or S-nitrosylation of these residues. There was no evidence, by MS, for a vicinal disulfide bond in the recombinant proinsulin that was used to stimulate T cells.
Figure 4.

The A1–13 epitope contains a vicinal disulfide bond between cysteine A6 and A7. (A) Isolation of the modified peptide. The absorbance at 214 nm (line) and proliferation of an insulin-specific T cell clone (bars) in response to each fraction (1/400 dilution) is shown. MALDI-QTOF mass spectrometry was used to analyze the parental S-11 peptide at low (B) and high (C) resolution. The active fraction (fraction 7) from the serum-modification experiment at low (D) and high (E) resolution is shown.
Previous studies suggested that the oxidation state of insulin-derived peptides may play a role in immunogenicity (20). The first 14 amino acids of the A-chain of bovine insulin (GIVEQCCASVCSLY) are recognized by murine (20) and human T cells (21) after immunization. Nonetheless, T cell epitopes containing vicinal disulfide bonds have not been reported previously. The vicinal disulfide bond between A6 and A7 occurs during refolding of insulin in vitro (22). This suggests that the nonnative vicinal disulfide bond that formed between cysteines A6 and A7 predominates under oxidatively permissive conditions in vitro for the synthetic peptide, and after presentation of exogenous recombinant and native (pro)insulin.
Binding and modeling of modified and unmodified insulin A1-13 peptide to HLA DR4
The S-11 peptide containing a vicinal disulfide bond (KRGIVEQC-CTSISSL) and the serine-substituted homologue (KRGIVEQSSTSISSL) had similar binding affinities for HLA DRB1*0404 (0.024 μM and 0.030 μM, respectively, Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051251/DC1). The serine-substituted peptide was used to avoid spontaneous oxidation of cysteine during the HLA binding assay. Both peptides also bound equally well to HLA DRB1*0401 (unpublished data). Thus, the vicinal disulfide bond did not alter binding to HLA DR4. The binding register of the insulin A1–13 epitope was determined to be the core nonamer VEQCCTSIS, using the ProPred algorithm (23). The formation of a vicinal disulfide bond between cysteine residues at A6 and A7 impacts on the conformation and solvent accessibility of the central portion of the epitope (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20051251/DC1). The vicinal disulfide bond creates a structure that is unable to be mimicked by other naturally occurring amino acids; this explains the failure of serine substitution to stimulate, even in part, the T cell clones.
Analysis of T cell clones from an individual at risk for T1D and healthy donors
To determine whether T cells specific for the cysteine-modified epitope are present before the onset of clinical T1D, we isolated 11 proinsulin-specific CD4+ T cell clones from an asymptomatic HLA DRB1*0404+ 12-yr-old with autoantibodies to insulin. Three of the clones responded to proinsulin, insulin, KR-A1-13, and S-11 peptides, but not to S-6 or S-7 peptides (Fig. 5 A). They recognized S-11 peptide in association with HLA DRB1*0404-0405 (Fig. 5 B). Furthermore, the RP-HPLC fraction containing the A1-13 epitope with the vicinal disulfide bond stimulated these clones (Fig. 5 C). In contrast, none of the 13 proinsulin-specific CD4+ T cell clones that were isolated from two healthy HLA DR4+ donors (6 and 7 clones, respectively) proliferated in response to KR-A1-13 peptide (unpublished data). Hence, T cells specific for the A1–13 epitope were not detected in healthy subjects, but were present in the blood of an individual with insulin autoantibodies.
Figure 5.
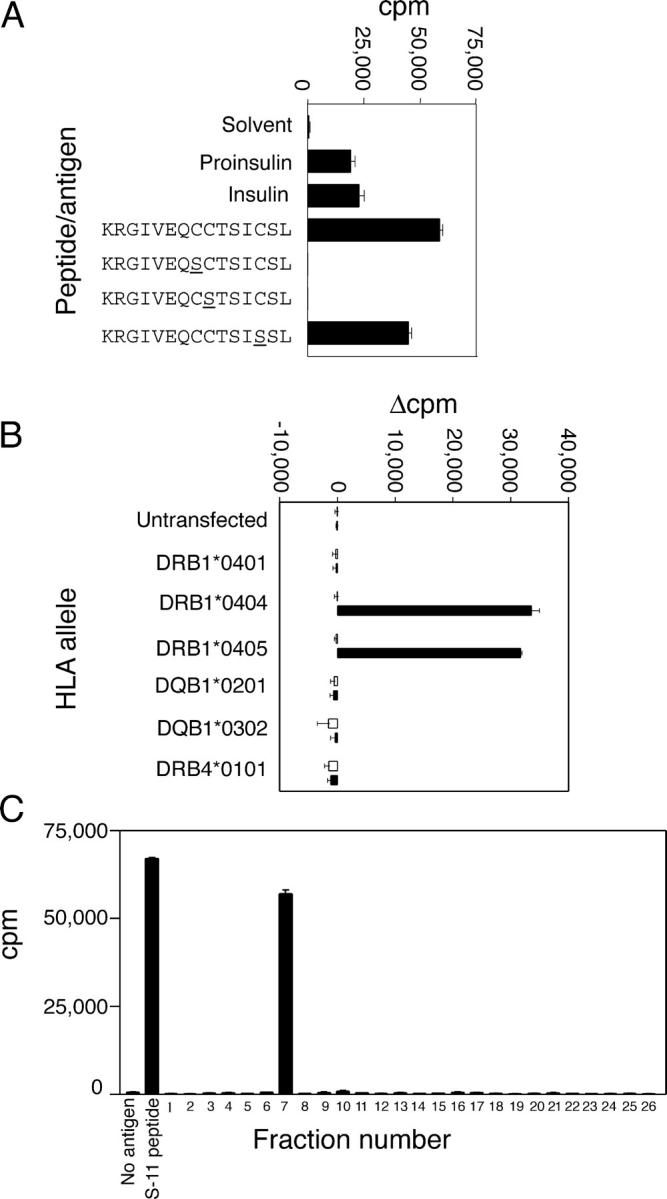
T cell clones from a donor at high risk for T1D recognize the A1–13 epitope. (A) Isolation of T cell clones. T cell clones were cultured with proinsulin (10 μg/ml), insulin (10 μg/ml), or the KR-A1-13, S-6, S-7, or S-11 peptides (10 μM each), or without antigen. (B) HLA restriction. Irradiated BLS cells that were transfected with the HLA genes shown were pulsed with 100 μM S-11 peptide (filled bars) or solvent alone (open bars). The results are expressed as net cpm proliferation (Δcpm) after subtracting the proliferation of BLS cells alone. (C) Peptide modification in culture medium. Fixed BLS cells that were transfected with HLA DRB1*0404 were cultured with RP-HPLC fractions from Fig. 4. MS analysis of fraction 7 is shown in Fig. 4, D and E. Each point is the mean ± SEM of triplicate wells.
Kent et al. (15) recently reported that CD4+ T cells that are specific for the first 15 amino acids of the A-chain of insulin were overrepresented in the pancreatic lymph nodes from two HLA DR4+ diabetic subjects, but not in the pancreatic lymph nodes from one HLA DR4+ subject and two other control subjects. We isolated proinsulin-specific CD4+ T cell clones from blood, the only tissue that is available for routine analysis in humans. T cell clones that recognized the first 13 amino acids of the A-chain of insulin also responded to islet cell lysate, which demonstrated that the A-chain epitope is naturally processed and presented. The epitope requires a vicinal disulfide bond between cysteines A6 and A7, which is essential for T cell recognition, but not for binding to HLA DR4. T cell responses to A1–13 were not a result of injecting insulin, because CD4+ T cells that recognized the A1–13 epitope were isolated from a donor who had never injected insulin, but had evidence of insulin autoimmunity. Our findings extend to T1D the paradigm that autoimmune responses can target epitopes that are formed by posttranslational modification.
MATERIALS AND METHODS
Human subjects and peripheral blood mononuclear cell isolation.
Blood was obtained by venepuncture with informed consent and ethics committee approval. The HLA typing and clinical status of each donor are shown in Table S1 (available at http://www.jem.org/cgi/content/full/jem.20051251/DC1). PBMCs were cultured in IMDM (GIBCO BRL) supplemented with 5% pooled male human serum, 2 mM glutamine (Glutamax; GIBCO BRL), 5 × 10−5 M 2-mercaptoethanol (Sigma-Aldrich), penicillin (100 U/ml), streptomycin (100 μg/ml; GIBCO BRL), and 100 μM nonessential amino acids (GIBCO BRL), hereafter referred to as culture medium. Islets and spleen samples were obtained from the Tom Mandel Islet Transplant Program with the approval of the St Vincent's Hospital Human Ethics Committee.
Antigens.
Synthetic peptides were purchased from Mimotopes, and reconstituted to 5 mM in 0.5% acetic acid, 40% acetonitrile in water. A library of 15-mer peptides overlapping by 12 amino acids, comprising the entire sequence of human proinsulin, was used for initial epitope-mapping experiments. All single peptides were ≥80% pure, and are summarized in Table S2. The vicinal disulfide bond (represented by C-C) in peptide S-11 was generated by incubating peptide in serum-free IMDM for 1 h. Modified peptide was purified by RP-HPLC, and analyzed as described below. Recombinant human proinsulin was produced in-house using a published protocol (24), and was shown to be folded correctly and free of endotoxin. Human islet lysate was prepared from >100 hand-picked islets resuspended in 0.5 ml of serum-free IMDM, then frozen, thawed, and solicited three times. Spleen lysate from the same donor was prepared similarly.
CFSE staining and T cell cloning.
Staining with the dye, CFSE (5,6-carboxylfluorescein diacetate succinimidyl ester; Invitrogen), T cell cloning, and testing for antigen specificity was performed as described (18).
3H-thymidine incorporation assays.
Assays were performed in 96-well round-bottom plates in 5% pooled human serum/IMDM. APCs were (a) irradiated (20 Gy) autologous or HLA-matched PBMCs (fresh or thawed); (b) HLA-typed EBV-transformed B cell lines from the 9th International HLA Typing Workshop; or (c) EBV-transformed B cells, from a donor who had bare lymphocyte syndrome (BLS), that were transfected with different HLA genes. EBV lines were irradiated at 50 Gy. In some experiments, APCs were fixed with 1% paraformaldehyde for 20 min at room temperature, and washed twice in PBS and once in culture medium before use in proliferation assays. In other experiments, freshly prepared TCEP (Tris (2-carboxyethyl) phosphine hydrochloride (Pierce Chemical Co.) was added to the antigen solution, before dilution by medium and addition of APCs and T cells to the final concentration shown in the figures. For all experiments, T cells and APCs were cultured in triplicate in 96-well round-bottom plates. After 2 d, 3H-thymidine (0.5 μCi/well) was added for 18 h, after which the cells were harvested and incorporated radioactivity was measured by β-scintillation counting. Results are expressed as the mean ± SEM of triplicate wells. All experiments were done at least twice with at least two clones.
HPLC fractionation and mass spectrometry.
Modifications of KR-A1-13 peptide in culture media were investigated after incubating 1.8 mg of peptide with 1.8 ml of culture media for 1 h at 37°C. The incubate was fractionated by RP-HPLC using an AKTA Basic HPLC equipped with a multi-wavelength tunable UV detector and a Frac 950 fraction collector (GE Healthcare). Proteins were separated on a Vydac C18 column (4.6 i.d. × 250 mm, 300 Å pore size, 5-μm nominal particle size), using a linear gradient of buffer A (0.1% TFA) to 60% B (acetonitrile/0.09% TFA; 0.86%/min), at a flow rate of 1 ml/min. Fractions (500 μl) were collected and 1-μl aliquots were mixed with 1 μl of 2,5-dihydroxybenzoic acid (Agilent Technologies) and dried onto a sample stage for analysis by MALDI-QqTOF MS (QSTAR pulsar i Applied Biosystems). Selected ions were subjected to further analysis using MS/MS analysis. Fragment ions that were generated in this way were assigned manually, based on the known sequence of the parental peptide and modified amino acid residues identified within the sequence, as described (25).
Online supplemental material.
Table S1 shows subjects defined by their clinical status and HLA type. Table S2 shows that insulin A-chain peptides used in this study. Table S3 shows the assignment of the b-series ions from MS/MS analysis of S-11 peptide with a vicinal disulfide bond between cysteines at A6 and A7. Fig. S1 shows HLA DR binding. Fig. S2 shows the modeling of modified and unmodified insulin A1-13 peptide complexed with HLA DR*0404. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051251/DC1.
Acknowledgments
We thank J. McCluskey, Y. Zhan, and A. Lew for reading the manuscript.
L.C. Harrison is a Senior Principal Research Fellow of the National Health and Medical Research Council (NHMRC), and A.W. Purcell is the Russell Grimwade Fellow at the University of Melbourne. This work was funded by a Center Program grant from the Juvenile Diabetes Research Foundation and by the NHMRC of Australia.
The authors have no conflicting financial interests.
References
- 1.Eisenbarth, G.S. 1986. Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med. 314:1360–1368. [DOI] [PubMed] [Google Scholar]
- 2.Noble, J.A., A.M. Valdes, M. Cook, W. Klitz, G. Thomson, and H.A. Erlich. 1996. The role of HLA class II genes in insulin-dependent diabetes mellitus: molecular analysis of 180 Caucasian, multiplex families. Am. J. Hum. Genet. 59:1134–1148. [PMC free article] [PubMed] [Google Scholar]
- 3.Pugliese, A., and G.S. Eisenbarth. 1996. Human type 1 diabetes mellitus: genetic susceptiblity and resistance. Type 1 Diabetes. Molecular, Cellular, and Clinical Immunology. G.S. Eisenbarth and K.J. Lafferty, editors. Oxford University Press, New York. 134–152.
- 4.Narendran, P., S.I. Mannering, and L.C. Harrison. 2003. Proinsulin–a pathogenic autoantigen in type 1 diabetes. Autoimmun. Rev. 2:204–210. [DOI] [PubMed] [Google Scholar]
- 5.Pugliese, A., D. Brown, D. Garza, D. Murchison, M. Zeller, M. Redondo, J. Diez, G.S. Eisenbarth, D.D. Patel, and C. Ricordi. 2001. Self-antigen-presenting cells expressing diabetes-associated autoantigens exist in both thymus and peripheral lymphoid organs. J. Clin. Invest. 107:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Achenbach, P., K. Koczwara, A. Knopff, H. Naserke, A.G. Ziegler, and E. Bonifacio. 2004. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J. Clin. Invest. 114:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett, S.T., A.M. Lucassen, S.C. Gough, E.E. Powell, D.E. Undlien, L.E. Pritchard, M.E. Merriman, Y. Kawaguchi, M.J. Dronsfield, F. Pociot, et al. 1995. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat. Genet. 9:284–292. [DOI] [PubMed] [Google Scholar]
- 8.Vafiadis, P., S.T. Bennett, J.A. Todd, J. Nadeau, R. Grabs, C.G. Goodyer, S. Wickramasinghe, E. Colle, and C. Polychronakos. 1997. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 15:289–292. [DOI] [PubMed] [Google Scholar]
- 9.Pugliese, A., M. Zeller, A. Fernandez Jr., L.J. Zalcberg, R.J. Bartlett, C. Ricordi, M. Pietropaolo, G.S. Eisenbarth, S.T. Bennett, and D.D. Patel. 1997. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 15:293–297. [DOI] [PubMed] [Google Scholar]
- 10.French, M.B., J. Allison, D.S. Cram, H.E. Thomas, M. Dempsey-Collier, A. Silva, H.M. Georgiou, T.W. Kay, L.C. Harrison, and A.M. Lew. 1997. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes. 46:34–39. [DOI] [PubMed] [Google Scholar]
- 11.Steptoe, R.J., J.M. Ritchie, and L.C. Harrison. 2003. Transfer of hematopoietic stem cells encoding autoantigen prevents autoimmune diabetes. J. Clin. Invest. 111:1357–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moriyama, H., N. Abiru, J. Paronen, K. Sikora, E. Liu, D. Miao, D. Devendra, J. Beilke, R. Gianani, R.G. Gill, and G.S. Eisenbarth. 2003. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc. Natl. Acad. Sci. USA. 100:10376–10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thebault-Baumont, K., D. Dubois-Laforgue, P. Krief, J.P. Briand, P. Halbout, K. Vallon-Geoffroy, J. Morin, V. Laloux, A. Lehuen, J.C. Carel, et al. 2003. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J. Clin. Invest. 111:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama, M., N. Abiru, H. Moriyama, N. Babaya, E. Liu, D. Miao, L. Yu, D.R. Wegmann, J.C. Hutton, J.F. Elliott, and G.S. Eisenbarth. 2005. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 435:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kent, S.C., Y. Chen, L. Bregoli, S.M. Clemmings, N.S. Kenyon, C. Ricordi, B.J. Hering, and D.A. Hafler. 2005. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 435:224–228. [DOI] [PubMed] [Google Scholar]
- 16.Doyle, H.A., and M.J. Mamula. 2001. Post-translational protein modifications in antigen recognition and autoimmunity. Trends Immunol. 22:443–449. [DOI] [PubMed] [Google Scholar]
- 17.Backlund, J., S. Carlsen, T. Hoger, B. Holm, L. Fugger, J. Kihlberg, H. Burkhardt, and R. Holmdahl. 2002. Predominant selection of T cells specific for the glycosylated collagen type II epitope (263-270) in humanized transgenic mice and in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 99:9960–9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mannering, S.I., J.A. Dromey, J.S. Morris, D.J. Thearle, K.P. Jensen, and L.C. Harrison. 2005. An efficient method for cloning human autoantigen-specific T cells. J. Immunol. Methods. 298:83–92. [DOI] [PubMed] [Google Scholar]
- 19.Banga, J.P., J.K. Moore, N. Duhindan, A.M. Madec, P.M. van Endert, J. Orgiazzi, and J. Endl. 2004. Modulation of antigen presentation by autoreactive B cell clones specific for GAD65 from a type I diabetic patient. Clin. Exp. Immunol. 135:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen, P.E. 1991. Reduction of disulfide bonds during antigen processing: evidence from a thiol-dependent insulin determinant. J. Exp. Med. 174:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller, G.G., J.F. Hoy, L.J. Nell, and J.W. Thomas. 1988. Antigen processing and the human T cell receptor repertoire for insulin. J. Immunol. 141:3293–3298. [PubMed] [Google Scholar]
- 22.Hua, Q.X., W. Jia, B.H. Frank, N.F. Phillips, and M.A. Weiss. 2002. A protein caught in a kinetic trap: structures and stabilities of insulin disulfide isomers. Biochemistry. 41:14700–14715. [DOI] [PubMed] [Google Scholar]
- 23.Singh, H., and G.P. Raghava. 2001. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 17:1236–1237. [DOI] [PubMed] [Google Scholar]
- 24.Cowley, D.J., and R.B. Mackin. 1997. Expression, purification and characterization of recombinant human proinsulin. FEBS Lett. 402:124–130. [DOI] [PubMed] [Google Scholar]
- 25.Purcell, A.W., and J.J. Gorman. 2004. Immunoproteomics: mass spectrometry-based methods to study the targets of the immune response. Mol. Cell. Proteomics. 3:193–208. [DOI] [PubMed] [Google Scholar]