

Abstract
The genomic sequences of viruses that are highly mutable and cause chronic infection tend to diverge over time. We report that these changes represent both immune-driven selection and, in the absence of immune pressure, reversion toward an ancestral consensus. Sequence changes in hepatitis C virus (HCV) structural and nonstructural genes were studied in a cohort of women accidentally infected with HCV in a rare common-source outbreak. We compared sequences present in serum obtained 18–22 yr after infection to sequences present in the shared inoculum and found that HCV evolved along a distinct path in each woman. Amino acid substitutions in known epitopes were directed away from consensus in persons having the HLA allele associated with that epitope (immune selection), and toward consensus in those lacking the allele (reversion). These data suggest that vaccines for genetically diverse viruses may be more effective if they represent consensus sequence, rather than a human isolate.
A virus capable of genetic variation and of causing chronic infection will evolve to optimize its fitness in each host, a process which is the net sum of immune recognition (positive selection) and functional constraint on replication (negative selection). Because an estimated 1012 virions are produced each day through an error-prone, nonproofreading NS5B RNA polymerase, hepatitis C virus (HCV) is especially capable of viral evolution (1, 2). However, we previously showed that evolution is not driven by replication alone. In the acute phase of infection before adaptive immune responses (but after weeks of replication supporting a viral RNA level of more than 105 IU/ml), the same major viral variant was detected in each of a serial passage of eight chimpanzees (3). In contrast, the sequence of envelope genes, particularly HVR1, changes in virtually all humans who have been persistently infected (including the source of the inoculum passaged through this chimpanzee lineage [4]), a notable exception being persons with attenuated humoral immune responses (agammaglobulinemia), who have been shown to have reduced variability in HVR1 (5). Longitudinal studies of chimpanzees experimentally infected with HCV have revealed that amino acid replacements in immunodominant CD8+ T cell epitopes presented on MHC class I in an allele-restricted manner contribute to viral persistence (6). Thus, we hypothesized that the net evolution of HCV would demonstrate both functional constraint (reversion of sequences toward consensus) as well as positive pressure (and thus reveal immunodominant epitopes).
Although it required that persons be infected with the same inoculum, it was possible to test this hypothesis because between May 1977 and November 1978 over 500 women were inadvertently infected with HCV from a single acutely infected source, as a result of treatment with contaminated anti-D immune globulin (7). In a single amplicon, a 5.2-kb cDNA spanning 5′UTR through the NS3–NS4A junction was cloned from serum collected from 22 women 18–22 yr after infection, as well as in two specimens of frozen plasma from the inoculum donor.
Results and Discussion
HCV envelope sequence from the inoculum clustered near the base of the clade formed by sequences from the chronically infected women (Fig. 1), and the entire anti-D cohort clade was clearly distinct from all other sequences in available databases (excluding those from this outbreak), consistent with the previously reported clinical history of common-source infection from an acutely infected donor (7). Nonetheless, HCV sequences in each woman diverged along distinct paths. Recipient sequences differed from inoculum sequences at a median (range) of 51 (35–73) nucleotide sites and 23 (15–38) amino acid sites in envelope genes (E1 and E2), and 72 (12–136) nucleotide sites and 18 (8–28) amino acid sites in nonenvelope genes (Core, p7, NS2, and NS3).
Figure 1.


Phylogenetic analysis of HCV 18–22 yr after common-source outbreak. (A) Phylogenetic tree of 5.2-kb sequence alignment placing outbreak clade (*) in context with reference sequences for all major subtypes. (B) Detailed analysis of the outbreak clade, using 10 cDNA clones from each study subject to obtain the sequence of a 698-nt region spanning the E1–E2 junction. The label “inoculum” indicates 20 clones from inoculum source plasma (10 each from 2 specimens), and a full-length clone (AF313916) obtained in an independent study of this material using smaller amplicons. For both trees, numbers at nodes are bootstrap values, indicating the percentage of 1,000 permuted trees that supported the presence of that node. Bootstrap support was 100% for each of the major clades in A; in B, only bootstrap values >80% are shown, and values for nodes within a study subject's clade were omitted for clarity. Boxes highlight two subjects whose sequences were segregated into two separate clades.
There was strong evidence of negative selection (sequence conservation) at some loci. Not only were there regional differences in sequence divergence in all genes, but there were also marked regional differences in the relative proportion of nonsynonymous (amino acid-changing) and synonymous (silent) changes (Fig. 2). Overall, the highest rate of nonsynonymous change was observed in the E2 gene, followed by NS2, p7, E1, NS3, and Core. Synonymous substitution rates were consistently higher than nonsynonymous rates for all genes, suggesting strong negative selection is a consistent feature of chronic HCV infection. HVR1 sequences were highly divergent at many sites, but constrained at others (8, 9).
Figure 2.

HCV divergence and convergence after a common-source outbreak. The upper panel shows a sliding-window analysis of nonsynonymous (red) and synonymous (blue) variation, calculated by comparing the mean pairwise distance between the inoculum (2 cDNA clones obtained from two inoculum source plasma specimens obtained 1 wk apart in 1977) to 44 clones (2 per study subject) obtained from chronically infected women 18–22 yr after exposure, in a sliding window 20 codons wide, moving in 1-codon increments, generated using VarPlot. Horizontal bars indicate average distance for each gene region. The lower panel is composed of sequence logos displaying the variability in HVR1 (the region indicated by dashed lines in the upper panel), with numbers indicating position relative to the H77 polyprotein. The first three rows show the subtype 1b reference sequences, inoculum sequences (20 clones from 2 plasma specimens obtained 1 wk apart), and 220 recipient sequences (10 per study subject) respectively, with the height of each single-letter amino acid code proportional to its frequency. The residue at the top of the stack in each position is the most-frequently observed, and hence the consensus residue, at that position. The fourth row shows differences between the amino acid frequencies in the recipient sequences versus the inoculum sequences as a type 2 logo, in which the height of each amino acid is determined by the log2 relative risk of observing it, with the scale indicated. Empty spaces indicate a distribution highly similar to the inoculum distribution, because the logarithm of a relative risk of one is zero.
At other loci, these data also reflect strong positive selection. For example, in HVR1when the amino acid of the inoculum matched the consensus for that viral subtype, residues either did not change or changed in an apparently “sporadic” fashion (risk of finding the residue in recipients was not different than finding the residue in the inoculum, indicated by near-zero height of the sequence logo in Fig. 2, with additional detail in Fig. S1). In contrast, when the amino acid in the inoculum differed from the consensus, there was convergent evolution toward consensus (residue was found over two times more often in the recipients than in the inoculum; Fig. 2). For example, 16 of the 22 women had replaced H in the inoculum with R at position 394, 12 replaced A with T at 396, 11 replaced L with F at 399, and 16 replaced T with S at 401. Four women had all four of these changes, not significantly different from the expected frequency of 3.2, indicating that these changes occurred independently. Interpreted in recognition of the thesis that viral sequence evolves to optimize fitness and the infinitesimal likelihood that these same amino acid substitutions occurred by chance in each of the women, these data suggest that the sequences converged to a more fit state.Further, it is likely that prior evolution of the inoculum sequence to optimize its fitness in the original host resulted in changes that diminished its fitness in the subsequent hosts.
Rather than convergent evolution, these results might have been due to shared selection and then divergence (at other sites) from a rare variant that we did not detect in the inoculum. We did detect one clone (clone #5) among 20 in the inoculum material that carried the RxTxxFxS motif at positions 394–401, but it was highly divergent from all other sequences described here, as evident from its position in the phylogenetic tree (Fig. 1 B), and therefore less likely than the other 19 sequences to represent the founder strain for these women. Although it is possible that a less divergent RxTxxFxS clone was present in the inoculum at very low frequency, shared selection of such a rare variant would support the same conclusion.
Because HVR1 is a potential target of both humoral and cellular immunity and the precise recognition motifs remain difficult to identify due to the extreme variability, further examination of positive selection was focused on nonenvelope genes, and in particular, on known MHC class I–restricted epitopes. Consistent with immune selection hypothesis, the number of changes in sites of known epitopes associated with specific class I alleles was significantly greater than the number of changes in other sites (RR 1.6, P < 0.05), and greater than what was found in that same site for persons who did not possess the allele (Table I, with detail for all epitopes in Fig. S2). For HLA B*35, changes in epitopes were observed only in women having that allele, and for B*37, sequence changes were 8.5 times as likely to occur in an epitope associated with an allele in women having the allele as compared with those that lacked it (P < 0.001). An example of such an epitope is shown in a 38–amino acid region that spans an HLA A2 motif (Fig. 3, A and B). Mutations from R to K were noted at position 1397 outside the A2 epitope, and mutations from G to S were noted within the epitope in 8 and 6 of 22 women, respectively. However, whereas R to K mutation was noted in a similar percentage of A2-positive and A2-negative women (41.7% vs. 30.0%, P > 0.10), all G to S mutations were observed in A2-positive women (P = 0.015), consistent with immune escape as has been observed in the simian immunodeficiency virus macaque model (10) and chimpanzees infected with HCV (6).
Table I.
Epitope changes according to the presence of restricting HLA alleles
| Allele | Subjects with allele | Epitopes | Allele present | Allele absent | Relative risk of change (allele present versus absent) | p-value |
|---|---|---|---|---|---|---|
| Epitope changes/sites | Epitope changes/sites | |||||
| n | n | (%) | (%) | |||
| A*01 | 11 | 1 | 2/198 (1.0) | 10/198 (5.1) | 0.2 | 0.019 |
| A*02 | 13 | 13 | 39/3,198 (1.2) | 18/2,214 (0.8) | 1.5 | > 0.10 |
| A*03 | 9 | 4 | 8/648 (1.2) | 18/936 (1.9) | 0.6 | > 0.10 |
| A*11 | 3 | 3 | 6/162 (3.7) | 45/1,026 (4.4) | 0.8 | > 0.10 |
| A*24 | 1 | 2 | 0/36 (0.0) | 3/756 (0.4) | 0 | > 0.10 |
| B*07 | 7 | 3 | 4/378 (1.1) | 0/810 (0.0) | NA | 0.003 |
| B*08 | 6 | 3 | 4/312 (1.3) | 28/832 (3.4) | 0.4 | 0.057 |
| B*35 | 3 | 2 | 8/108 (7.4) | 0/684 (0.0) | NA | < 0.001 |
| B*37 | 5 | 1 | 20/80 (25.0) | 8/272 (2.9) | 8.5 | < 0.001 |
| B*44 | 7 | 1 | 0/126 (0.0) | 2/270 (0.7) | 0 | > 0.10 |
Proportion of amino acid substitutions in known epitopes located in HCV Core, p7, NS2, and NS3, according to the presence or absence of the associated HLA allele (28). Epitopes were considered in the region sequenced (corresponding to amino acid residues 1–1651 of the H77 polyprotein (GenBank/EMBL/DDBJ accession no. AF009606). Two sequences per subject were examined. P values were calculated by comparison of proportions as implemented in SigmaStat (Systat Software, Inc.). NA means not applicable due to zero cell. EpiChanges, a software program created by S.C. Ray to automate this analysis, is available from the author.
Figure 3.
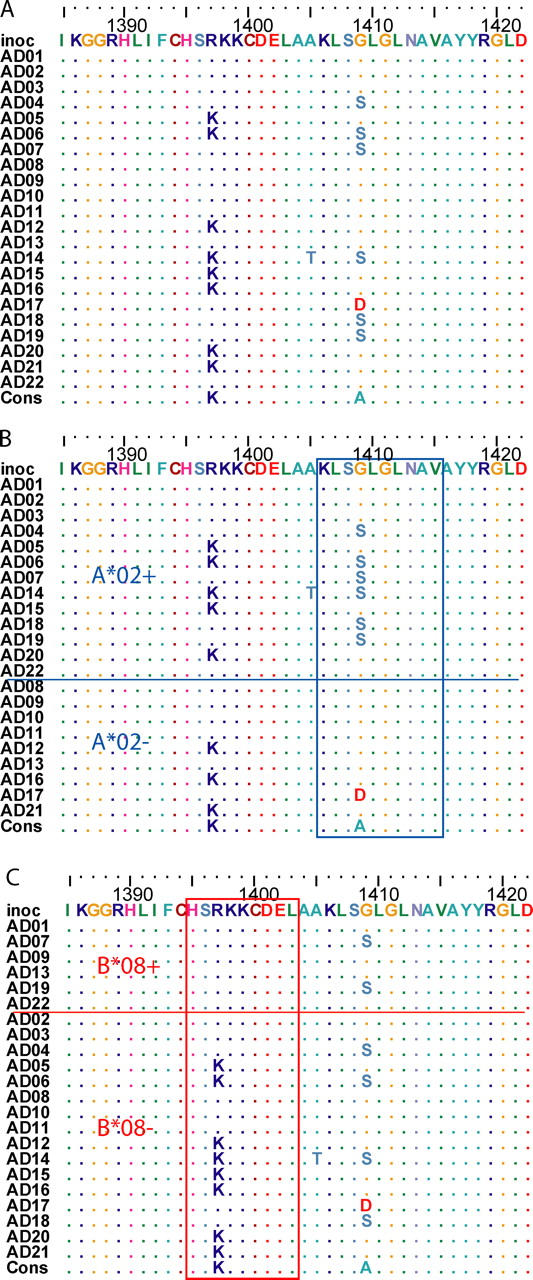
Escape versus reversion in the presence versus absence of the restricting HLA allele. (A) Amino acid alignment of a region in NS3 (positions 1388 to 1431 relative to the H77 polyprotein (GenBank/EMBL/DDBJ accession no. AF009606) showing sites with polymorphism. Study subjects are listed in arbitrary order. Identity to the inoculum sequence (“inoc”) is indicated by “.”. (B) Sorting the subjects by the presence of HLA A*02, G1409S substitution in the 4th position of a frequently recognized HLA A*02-restricted epitope at 1406–1415 is limited to subjects having the HLA A*02 allele. The subtype 1b consensus sequence for this epitope is shown below the alignment, and has been shown to be recognized as readily as the prototype (subtype 1a) KLVALGINAV sequence (27). Subject AD17 (HLA A*01, A*11) had G1409D substitution, the impact of which on recognition is unknown. (C) Variation resulting in reversion to a HLA B*08-restricted epitope. Sorting the subjects by presence of HLA B*08, R1397K substitution in the 3rd position of a frequently recognized HLA B*08- restricted epitope at 1395–1403 is limited to subjects lacking the HLA B*08 allele. The inoculum sequence differs from the prototypical epitope (HSKKKCDEL) at the 3rd position. Reversion to consensus (and the prototype epitope) occurred only in study subjects lacking the HLA B*08 allele.
As seen with envelope sequences, the opposite effect was observed in other alleles. For alleles A*01 and B*08, sequence changes were 0.2 and 0.4 times as likely to occur in an epitope restricted by an allele in women having the allele as compared with those who lack it (Table I). In fact, the R1397K substitution that was described above in both A2-positive and -negative women, only occurred in women who were not HLA B*08 positive, although the apparently A2-restricted G1409S substitution occurred in both B*08-positive and -negative women (Fig. 2 C).
Collectively, these findings indicate that HCV sequence change is a nonrandom process that reflects negative selection (change is disadvantageous) as well as positive selection. Moreover, we find evidence that positive selection represents both the direct effect of pressure applied by immune responses in the current host (in this case, HLA class I–restricted CD8+ cytotoxic T lymphocytes) as well as reversion of sequence toward a consensus, as we saw with envelope sequences.
To independently evaluate this paradigm, we compared the amino acid sequences of these women with an HCV 1b consensus sequence. For the epitopes that showed evidence of HLA class I–restricted positive selection (a significantly increased risk of mutations from the inoculum occurred when the restricting allele was present), there was also an increased number of changes away from the 1b prototype consensus in women with one of these alleles, but not those without (Table II). In addition, for epitopes that showed the converse effect, i.e., evidence of positive selection when the allele was absent (a significantly lower risk of mutations from the inoculum when the restricting allele was present), there was also an increased number of changes toward consensus in those who lacked the allele versus those who had the allele, suggesting reversion (Table II). These findings are supported by an accompanying report, Cox et al. (11), which shows that amino acid substitutions in CD8+ T cell epitopes are associated with a loss of T cell recognition during acute infection, whereas nonepitope changes revert toward consensus at a rate much higher than expected by chance.
Table II.
Classification of amino acid substitutions in MHC class I–restricted epitopes as being toward or away from a 1b consensus sequence in 22 women exposed to a common HCV subtype 1b inoculum
| Effect of allele presence on likelihood of amino acid replacement | Changes in epitopes relative to 1b consensus per 1,000 sites | |||
|---|---|---|---|---|
| Allele present
|
Allele absent
|
|||
| Toward consensus | Away from consensus | Toward consensus | Away from consensus | |
| 566 sites | 1,766 sites | |||
| Increased relative risk (B*07, B*35, B*37) |
0.0 | 56.5a | 0.0 | 4.5 |
| 510 sites | 1,030 sites | |||
| Decreased relative risk (A*01, B*08) |
3.8 | 4.7 | 22.3a | 4.8 |
Results are stratified (in columns) by the presence or the absence of the relevant allele, and grouped (rows) according to relative risk for change shown in Table I. Changes are per 1000 epitope sites, based on whether the resulting residue matches HCV 1b consensus.
P < 0.001 for comparison of toward versus away.
The persistence of R at position 1397 in half of the B*08-negative women suggests an alternative hypothesis, namely that reversion is a neutral process. If that were true, then R at position 1397 would be expected in about half of all B*08-negative HCV-infected persons; however, 82 of 83 subtype 1b reference sequences have K at position 1397. It is highly unlikely that HLA B*08 was present in more than half of the persons from whom the reference sequences were obtained; therefore, the available evidence does not support the hypothesis that R versus K at position 1397 is neutral (random). A similar phenomenon of delayed reversion has been observed in the setting of primary infection with drug-resistant HIV-1, suggesting that this is a complex process, and compensatory mutations may play a role. (12)
Indirectly, these results suggest that immune escape is costly to the virus. Fitness is conventionally measured as a competition among genetic variants, and when the viral population size is large the most successful variants present at any one time in a host are by definition the most fit under the overall selection pressures. We assume that over very long periods of time (relative to the viral life cycle) the residues in the viral genome have the opportunity to vary substantially, and independently to a first approximation (this is a fundamental assumption of phylogenetic analysis). Therefore, although there may be some covariation, analytically the positions in the sequence can be considered independent variables. Finding a strong positive correlation between the presence of HLA alleles and substitutions in allele-associated epitopes suggests that those changes increase viral fitness in the presence of the associated immune response. Likewise, a strong association between the absence of alleles and substitutions in allele-associated epitopes, particularly when found to represent reversion to consensus, suggests that positive selection in a previous host carried a fitness cost in terms of viral replicative capacity.
Prior studies have demonstrated reversion of CTL escape–variant sequences in macaques experimentally infected with SIV (13), reversion of one epitope each of HIV-1 and HCV in humans (14, 15), and evidence of HIV-1 adaptation to common HLA alleles (16). This is the first report of viral adaptation to multiple HLA alleles across multiple genes in HCV, and provides additional support for the suggestion, based on minimizing differences between vaccine and circulating strains, that vaccine effectiveness may be enhanced by using a consensus (17) or ancestral (18) sequence.
The ability of viruses to restrict adaptive immune responses and evade those that are formed contributes to persistence and is a major barrier to vaccine development. These data suggest that escape variants have greater fitness in the presence of an individual host's immune response, and that immune evasion contributes to the sequence divergence observed in a each persistently infected host. Nonetheless, this divergence may actually reduce the fitness of the virus in the population (that is, in other hosts). From an evolutionary perspective, these forces maintain the virus as a distinct pathogen. However, the data also suggest that immune responses to consensus sequences (rather than a product based on the sequence in a given host) may establish the highest barrier to viral escape and consequently the most effective protection against chronic infection.
Materials and methods
Study subjects
22 women from this outbreak were studied because specimens were available, they provided consent, their HLA class I genotyping was complete, and they had at least one of the three most common A gene alleles (A*01, A*02, or A*03; reference 19). Informed consent was obtained from the subjects studied and the research protocol was approved by the Cork University Hospital Ethics Committee. Work performed in Baltimore was approved by the Johns Hopkins Medicine Institutional Review Board.
Hemigenomic cDNA cloning.
The region encoding Core, E1, E2, p7, NS2, and NS3 was amplified and cloned as previously described (20), and 40 clones per specimen were stored. For each specimen, envelope sequences from 10 random randomly selected clones were determined using primer H77-1868a21 (20) on a PRISM version 3100 sequencer (ABI). These sequence data are available from GenBank/EMBL/DDBJ under accession nos. DQ061331 through DQ061378.
Estimation of consensus sequence.
An alignment of full-length HCV subtype 1b sequences was obtained from the Los Alamos National Laboratories HCV database (http://hcv.lanl.gov). The alignment was edited by hand to remove gaps introduced for alignment to other genotypes, and to remove duplicate sequences from the same human source and those obtained from nonhuman sources. The resulting alignment included 83 sequences. A majority-rule consensus sequence was formed, with residues occurring in less than 42 sequences flagged as nonconsensus. Changes in the anti-D recipients were then classified as “toward” (change results in a residue matching the consensus) or “away” (change results in a residue not matching the consensus, or residue is nonconsensus).
Estimation of the likelihood of convergence.
The expected frequency of covariation assuming independence was calculated as the product of the marginal frequencies, and compared with the observed value using the Chi-squared distribution with three degrees of freedom.
If we assume that all amino acid replacements are equally likely over a time period that is very long with respect to the rate of mutation, then sharing of amino acids at four variable sites in just two study subjects would be expected to occur at a frequency of 1/204 or 0.00000625. Of course, all amino acid replacements are not equally likely, even in the highly variable HVR1 (8); therefore, the likelihood of sharing of four variable sites by two subjects would be higher, e.g., 1/34 or 0.012 if each site is equally likely to have one of three residues. Because the likelihood of shared residues at variable sites in multiple study subjects is the product of such probabilities, the observed findings in this study are clearly incompatible with random substitution and most consistent with convergent evolution.
Phylogenetic analysis
Sequences were aligned using ClustalX (21), codon boundaries were restored by hand in BioEdit (22), and phylogenetic analysis was performed using PAUP* version 4b10 (Sinauer Associates) using a HKY85+G model and parameters (Ti/Tv 2.78, γ = 0.37) selected with the aid of ModelTest (23). Initial results from one specimen were consistent with subtype 1a, and that specimen was not examined further. Reference sequences included 3 from subtype 1a, 83 from subtype 1b (including AF313916), 2 from subtype 1c, 6 from subtype 2a, 8 from subtype 2b, 1 each from subtypes 2c and 2k, 4 from subtype 3a, and 1 each from subtypes 3b, 3k, 4a, 5a, 6a, 6b, 6d, 6g, 6h, and 6k (obtained from http://hcv.lanl.gov).
VarPlot was used to calculate nonsynonymous and synonymous distances using the method of Nei and Gojobori, in a sliding window 20 codons wide, moving in 1 codon steps, as previously described (24).
Sequence logos.
A sequence logo is a graphical representation of a group of aligned sequences, at each position of which the frequency of each residue is represented by the height of the single-letter representation of that residue (25). The sequence logos in Fig. 2 were generated using a novel software program, VisSPA (Visual Sequence Pattern Analysis, available on request from the author S.C. Ray). The algorithm is identical to that described for type 2 logos by Gorodkin et al. (26), except that the a priori distributions for the logo are calculated empirically from input sequences, and missing values in the a priori distribution are assigned the lowest frequency of residues at that site (if more than one state is represented) or 1/20 if the a priori distribution has only one residue at that site.
Online supplemental material
Fig. S1 shows the variability of HVR1 in each study subject. Fig. S2 shows the sequences at the sites of known MHC class I–restricted alleles in each study subject. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050122/DC1.
Acknowledgments
The authors are particularly grateful to the women whose generosity was crucial to the success of this study.
This research was funded by National Institutes of Health grants U19 AI40035 and R01 DA016078, and Irish Health Research Board grant HC08/97.
The authors have no conflicting financial interests.
Abbreviation used: HCV, hepatitis C virus.
References
- 1.Neumann, A.U., N.P. Lam, H. Dahari, D.R. Gretch, T.E. Wiley, T.J. Layden, and A.S. Perelson. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 282:103–107. [DOI] [PubMed] [Google Scholar]
- 2.Martell, M., J.I. Esteban, J. Quer, J. Genesca, A. Weiner, R. Esteban, Guardia, and J. Gomez. 1992. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J.Virol. 66:3225–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ray, S.C., Q. Mao, R.E. Lanford, S. Bassett, O. Laeyendecker, Y.M. Wang, and D.L. Thomas. 2000. Hypervariable region 1 sequence stability during hepatitis C virus replication in chimpanzees. J. Virol. 74:3058–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogata, N., H.J. Alter, R.H. Miller, and R.H. Purcell. 1991. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA. 88:3392–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaud, U., B. Langer, T. Petropoulou, H.C. Thomas, and P. Karayiannis. 2003. Changes in hypervariable region 1 of the envelope 2 glycoprotein of hepatitis C virus in children and adults with humoral immune defects. J. Med. Virol. 69:350–356. [DOI] [PubMed] [Google Scholar]
- 6.Erickson, A.L., Y. Kimura, S. Igarashi, J. Eichelberger, M. Houghton, J. Sidney, D. McKinney, A. Sette, A.L. Hughes, and C.M. Walker. 2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 15:883–895. [DOI] [PubMed] [Google Scholar]
- 7.Kenny-Walsh, E. 1999. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. Irish Hepatology Research Group. N. Engl. J. Med. 340:1228–1233. [DOI] [PubMed] [Google Scholar]
- 8.McAllister, J., C. Casino, F. Davidson, J. Power, E. Lawlor, P.L. Yap, P. Simmonds, and D.B. Smith. 1998. Long-term evolution of the hypervariable region of hepatitis C virus in a common-source-infected cohort. J. Virol. 72:4893–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penin, F., C. Combet, G. Germanidis, P.O. Frainais, G. Deleage, and J.M. Pawlotsky. 2001. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol. 75:5703–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen, T.M., D.H. O'Connor, P. Jing, J.L. Dzuris, B.R. Mothe, T.U. Vogel, E. Dunphy, M.E. Liebl, C. Emerson, N. Wilson, et al. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 407:386–390. [DOI] [PubMed] [Google Scholar]
- 11.Cox, A.L., T. Mosbruger, Q. Mao, Z. Liu, X.-H. Wang, H.-C. Yang, J. Sidney, A. Sette, D. Pardoll, D.L. Thomas, and S.C. Ray. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 201:1741–1752. [DOI] [PMC free article] [PubMed]
- 12.Gandhi, R.T., A. Wurcel, E.S. Rosenberg, M.N. Johnston, N. Hellmann, M. Bates, M.S. Hirsch, and B.D. Walker. 2003. Progressive reversion of human immunodeficiency virus type 1 resistance mutations in vivo after transmission of a multiply drug-resistant virus. Clin. Infect. Dis. 37:1693–1698. [DOI] [PubMed] [Google Scholar]
- 13.Friedrich, T.C., E.J. Dodds, L.J. Yant, L. Vojnov, R. Rudersdorf, C. Cullen, D.T. Evans, R.C. Desrosiers, B.R. Mothe, J. Sidney, et al. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat. Med. 10:275–281. [DOI] [PubMed] [Google Scholar]
- 14.Leslie, A.J., K.J. Pfafferott, P. Chetty, R. Draenert, M.M. Addo, M. Feeney, Y. Tang, E.C. Holmes, T. Allen, J.G. Prado, et al. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282–289. [DOI] [PubMed] [Google Scholar]
- 15.Timm, J., G.M. Lauer, D.G. Kavanagh, I. Sheridan, A.Y. Kim, M. Lucas, T. Pillay, K. Ouchi, L.L. Reyor, J.S. Zur Wiesch, et al. 2004. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 200:1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore, C.B., M. John, I.R. James, F.T. Christiansen, C.S. Witt, and S.A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 296:1439–1443. [DOI] [PubMed] [Google Scholar]
- 17.Gaschen, B., J. Taylor, K. Yusim, B. Foley, F. Gao, D. Lang, V. Novitsky, B. Haynes, B.H. Hahn, T. Bhattacharya, and B. Korber. 2002. Diversity considerations in HIV-1 vaccine selection. Science. 296:2354–2360. [DOI] [PubMed] [Google Scholar]
- 18.Nickle, D.C., M.A. Jensen, G.S. Gottlieb, D. Shriner, G.H. Learn, A.G. Rodrigo, and J.I. Mullins. 2003. Consensus and ancestral state HIV vaccines. Science. 299:1515–1518. [DOI] [PubMed] [Google Scholar]
- 19.Fanning, L.J., E. Kenny-Walsh, and F. Shanahan. 2004. Persistence of hepatitis C virus in a white population: associations with human leukocyte antigen class 1. Hum. Immunol. 65:745–751. [DOI] [PubMed] [Google Scholar]
- 20.Liu, Z., D.M. Netski, Q. Mao, O. Laeyendecker, J.R. Ticehurst, X.H. Wang, D.L. Thomas, and S.C. Ray. 2004. Accurate representation of the hepatitis C virus quasispecies in 5.2-kilobase amplicons. J. Clin. Microbiol. 42:4223–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeanmougin, F., J.D. Thompson, M. Gouy, D.G. Higgins, and T.J. Gibson. 1998. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 23:403–405. [DOI] [PubMed] [Google Scholar]
- 22.Hall, T.A. 2001. BioEdit: Biological sequence alignment editor for Windows 95/98/NT version 5.0.7. software. http://www.mbio.ncsu.edu/RNaseP/info/programs/BIOEDIT/bioedit.html (accessed March 5, 2003).
- 23.Posada, D., and K.A. Crandall. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics. 14:817–818. [DOI] [PubMed] [Google Scholar]
- 24.Ray, S.C., Y.M. Wang, O. Laeyendecker, J. Ticehurst, S.A. Villano, and D.L. Thomas. 1998. Acute hepatitis C virus structural gene sequences as predictors of persistent viremia: hypervariable region 1 as decoy. J. Virol. 73:2938–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider, T.D., and R.M. Stephens. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18:6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorodkin, J., L.J. Heyer, S. Brunak, and G.D. Stormo. 1997. Displaying the information contents of structural RNA alignments: the structure logos. Comput. Appl. Biosci. 13:583–586. [DOI] [PubMed] [Google Scholar]
- 27.Chang, K.M., B. Rehermann, J.G. McHutchison, C. Pasquinelli, S. Southwood, A. Sette, and F.V. Chisari. 1997. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J. Clin. Invest. 100:2376–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward, S., G. Lauer, R. Isba, B. Walker, and P. Klenerman. 2002. Cellular immune responses against hepatitis C virus: the evidence base 2002. Clin. Exp. Immunol. 128:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]