

Abstract
Pre–B cells undergo apoptosis unless they are rescued by pre–B cell receptor–dependent survival signals. We previously showed that the BCR-ABL1 kinase that is expressed in pre–B lymphoblastic leukemia bypasses selection for pre–B cell receptor–dependent survival signals. Investigating possible interference of BCR-ABL1 with pre–B cell receptor signaling, we found that neither SYK nor SLP65 can be phosphorylated in response to pre–B cell receptor engagement. Instead, Bruton's tyrosine kinase (BTK) is constitutively phosphorylated by BCR-ABL1. Activated BTK is essential for survival signals that otherwise would arise from the pre–B cell receptor, including activation of PLCγ1, autonomous Ca2+ signaling, STAT5-phosphorylation, and up-regulation of BCLX L. Inhibition of BTK activity specifically induces apoptosis in BCR-ABL1 + leukemia cells to a similar extent as inhibition of BCR-ABL1 kinase activity itself. However, BCR-ABL1 cannot directly bind to full-length BTK. Instead, BCR-ABL1 induces the expression of a truncated splice variant of BTK that acts as a linker between the two kinases. As opposed to full-length BTK, truncated BTK lacks kinase activity yet can bind to BCR-ABL1 through its SRC-homology domain 3. Acting as a linker, truncated BTK enables BCR-ABL1–dependent activation of full-length BTK, which initiates downstream survival signals and mimics a constitutively active pre–B cell receptor.
The BCR-ABL1 gene rearrangement encodes a constitutively active tyrosine kinase and drives malignant transformation of pre–B cells in humans (1). In a B cell–specific mouse model, expression of BCR-ABL1 is sufficient to induce pre–B lymphoblastic leukemia and is required for maintenance of leukemic transformation (2).
During their development, human B cells have to pass multiple checkpoints at which they are selected for the expression of a functional pre–B or B cell receptor (3). However, signals from the pre–B cell receptor also may initiate apoptosis (4). Therefore, the expression of a functional pre–B cell receptor constitutes an effective means to regulate tightly the survival or elimination of individual B cell clones. In this regard, it is not surprising that in several B cell–derived malignancies, pre–B or B cell receptor function is compromised (5). For instance, Hodgkin and Reed-Sternberg cells in classic Hodgkin's disease carry “crippled” Ig variable region gene rearrangements as a result of the acquisition of deleterious somatic mutations (6). Likewise, transformation of human B cells by the EBV mimics antigen receptor–dependent survival signals through the viral oncoprotein LMP2A, and prevents the B cell receptor from signaling by excluding it from glycolipid-enriched membrane domains (7). Autonomous growth and survival of transformed B cells seem to favor a situation in which the B cell receptor is not active. In accordance with this notion, we recently observed that pre–B lymphoblastic leukemia clones harboring a BCR-ABL1 fusion gene carry only nonproductively rearranged Ig variable region genes in 9 of 12 cases (8). Even the few leukemia clones that do express a pre–B cell receptor were not responsive to pre–B cell receptor engagement (8). Analogous to the EBV-encoded LMP2A protein, the expression of the oncogenic BCR-ABL1 kinase may confer autonomous survival signals, and interfere with antigen receptor signaling.
Among others, Bruton's tyrosine kinase (BTK) represents a crucial component of the (pre)-B cell receptor signaling chain and links the (pre)-B cell receptor to Ca2+ signals in B lymphoid cells (9). In humans, deficiency of BTK leads to X-linked agammaglobulinemia which results from a differentiation block at the pre–B cell–stage (10). Of note, BTK promotes developmental progression of pre–B cells (11), and acts as a tumor suppressor to prevent pre–B lymphoblastic leukemia in mice (12, 13). To elucidate how BCR-ABL1 can bypass selection for pre–B cell receptor–dependent survival signals, the current study investigates possible interference of BCR-ABL1 with pre–B cell receptor–related signaling molecules, including spleen tyrosine kinase (SYK), SLP65, phospholipase C γ (PLCγ)2, and BTK.
RESULTS
BCR-ABL1 kinase activity induces tyrosine-phosphorylation of BTK
Normal pre–B cells can survive in human bone marrow only if they receive survival signals through their pre–B cell receptor (14). In a previous study, we observed that in the majority of cases of pre–B lymphoblastic leukemia expressing the oncogenic BCR-ABL1 kinase, the leukemia cells do not harbor a functionally rearranged IGH allele, and hence, cannot express a pre–B cell receptor on their surface (8). Therefore, we wondered how a pre–B cell–derived leukemia cell can survive and even clonally expand in the absence of pre–B cell receptor–dependent survival signals.
To determine if BCR-ABL1 interferes with signal transduction of the pre–B cell receptor, we examined whether the pre–B cell receptor–related signaling molecules SYK, SLP65, and BTK are expressed and if they can be phosphorylated upon pre–B cell receptor engagement in the leukemia cells.
Western blot using antibodies against phosphorylated SYKY323, SLP65Y96, and BTKY223 showed that neither SYK nor SLP65 was phosphorylated in response to cross-linking of the pre–B cell receptor (unpublished data). However, BTK was constitutively phosphorylated in the leukemia cells (at 77 kD; Fig. 1 A).
Figure 1.

BCR-ABL1 induces phosphorylation of BTK in pre–B lymphoblastic leukemia cells. Protein expression and tyrosine phosphorylation of the pre–B cell receptor downstream signaling molecules SYK, SLP65, and BTK was studied by Western blot in three BCR-ABL1 + pre–B lymphoblastic leukemia cell lines (VII, VIII, and IX in Table I) in the presence or absence of STI571. The BTKpY223 antibody detected full-length BTK (77 kD) as well as two additional tyrosine phosphorylated proteins of 65-kD and 52-kD size (A–C). To confirm that constitutive phosphorylation of 77-kD full-length BTK is specific for BCR-ABL1 kinase activity, BTKY223-phosphorylation was studied in four BCR-ABL1–negative B cell precursor cell lines (B, left panel; see XI, XII, XIII, and XV in Table I). Dependence of BTK on BCR-ABL1 kinase activity also was confirmed in four primary cases of BCR-ABL1 + pre–B lymphoblastic leukemia, from which matched sample pairs before and during therapy with STI571 were available (B, right panel; cases I to IV in Table I). EIF-4E was used as a loading control. To determine whether Y223 phosphorylation of BTK is mediated by transphosphorylation through BCR-ABL1 or autophosphorylation, BCR-ABL1 + leukemia cells (IX; Table I) were treated for 24 h with the BCR-ABL1 kinase inhibitor, STI571, or the BTK inhibitor, LFM-A13 (C).
Using a specific inhibitor of BCR-ABL1, termed STI571 (15), we found that phosphorylation of BTK was sensitive to inhibition of BCR-ABL1 kinase activity (77kD; Fig. 1 A). To determine whether constitutive phosphorylation of 77-kD BTK is specific for leukemia cells that carry a BCR-ABL1 gene rearrangement, BTK phosphorylation also was studied in pre–B lymphoblastic leukemias that harbored an E2A-PBX1, MLL-AF4, TEL-AML1, or TEL-PDGFRβ gene rearrangement (Fig. 1 B, Table I). Although the BTKY223-specific antibody detected smaller tyrosine-phosphorylated proteins also in other leukemias, constitutive Y223 phosphorylation of BTK (77 kD) was specific for BCR-ABL1 + pre–B cell lymphoblastic leukemia cell lines (Fig. 1 B, left).
Table I.
Description of leukemia and lymphoma cases and cell lines studied
| Translocation
|
IGH configuration
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Case | Age | Sex | Fusion gene | IgH | Igκ/λ or SLC |
Alleles | Coding capacity | Clinical course |
| Ia | 39 | m | BCR-ABL1; p210 | – | – | (1) VH4.34-DH6.13-JH5 (2) DH5-JH |
no; out-of-frame | ALL, relapse after STI571 treatment |
| IIa | 54 | f | BCR-ABL1; p190 | – | – | (1) VH4.61-DH2.2-JH4 (2)DH2.4-JH4 |
no; out-of-frame, stop in junction |
ALL, relapse after STI571 treatment |
| IIIa | 61 | f | BCR-ABL1; p190 | ND | ND | ND | – | ALL, relapse after STI571 treatment |
| IVa | 47 | f | BCR-ABL1; p190 | ND | ND | ND | – | ALL, relapse after STI571 treatment |
| Va | 34 | m | BCR-ABL1; p190 | – | – | (1) VH4.34-DH6.13-JH5 (2) DH5-JH |
out-of-frame | ALL, complete remission after BMT |
| VIa | 32 | m | BCR-ABL1; p190 | – | – | (1) VH4.4-DH2.2-JH4 (2) DH2.4-JH4 |
out-of-frame, stop codons in junction |
ALL, died after BMT |
| VIIa | 45 | m | BCR-ABL1; p210 | – | – | (1) VH3.48-DH2.15-JH3 (2) DH2-JH |
no; out-of-frame, stop in junction |
ALL, died after relapse (BV173 cell line) |
| VIIIa | 3 | f | BCR-ABL1; p210 | μ chain | SLC | (1) DH4-JH
(2) VH3.9-DH2.21-JH3 |
no yes |
ALL, died after relapse (NALM1 cell line) |
| IXa | 9 | m | BCR-ABL1; p190 | μ chain | SLC | (1) VH3.53-DH2.8-JH6 | yesb | ALL, died after relapse (SUP-B15 cell line) |
| X | 32 | f | MLL-AF4 | – | – | (1) VH3.20-DH2.8-JH5 (2) VH6.1-DH1.7-JH4 |
no; out-of-frame no; stop in junction |
ALL, relapse after chemotherapy (RS4;11 cell line) |
| XI | 5 | f | MLL-AF4 | – | – | (1) DH3-JH
(2) IGH in germline |
no | ALL, relapse after chemotherapy (SEM cell line) |
| XII | 15 | f | TEL-AML1 | – | – | (1) VH3.15-DH3.10-JH6 (2) IGH in germline |
no; out-of-frame | ALL, relapse after chemotherapy (REH cell line) |
| XIII | 12 | m | E2A-PBX1 | μ chain | SLC | (1) VH4.34-DH3.22-JH2 (2) VH2.26-DH2.2-JH4 |
no; out-of-frame, stop in junction yes |
ALL, relapse after chemotherapy (697 cell line) |
| XIV | 15 | m | E2A-PBX1 | μ chain | SLC | (1) VH3.7-DH3.10-JH4 | yesb | ALL, cell line (KASUMI-2) established at diagnosis |
| XV | 19 | m | TEL-PDGFRβ | μ chain | SLC | (1) VH1.69-DH3.10-JH6 | yesb | ALL, relapse after chemotherapy (NALM6 cell line) |
| XVI | 5 | m | IGH-MYC | IgM | Igλ | (1) VH3.53-DH3.3-JH6 (2) VH4.34-DH2.15-JH5 |
no; stop in junction yes |
Burkitt's lymphoma cell line (MHH-PREB1) established at diagnosis |
| XVII | 73 | f | IGH-BCL2 | IgG | Igκ | (1) VH4.39-DH3.10-JH6c(2) VH3.9-DH3.22-JH6c(3) VH3.23-DH6.13-JH5c | yes no; out-of-frame no; out-of-frame |
Diffuse large B cell lymphoma cell line (KARPAS-422) established at diagnosis |
These cases were described in Klein et al. (reference 8).
The second IGH allele was not subjected to sequence analysis if a potentially functional allele was already amplified.
Three IGH VDJ gene rearrangements are compatible with three chromosome 14s in these B cell lymphoma cells. Alternatively, the cell line may comprise subclones with different IGH VDJ gene rearrangements.
ALL, acute lymphoblastic leukemia; n.d., not done; SLC, surrogate light chain.
To confirm that BTK phosphorylation is dependent on BCR-ABL1 kinase activity, we induced BCR-ABL1 expression in a murine B lymphoid cell line TONB210 (16) that carries an inducible BCR-ABL1 transgene. As opposed to BCR-ABL1 + leukemia cells, this cell line grows independently from BCR-ABL1 in the presence of IL-3. In the presence, but not in the absence, of doxycycline-induced BCR-ABL1 expression, BTK was heavily phosphorylated (see Fig. 5 C). We conclude that BTK is constitutively phosphorylated by BCR-ABL1, but not by the pre–B cell receptor in the leukemia cells.
Figure 5.

BTK links BCR-ABL1 to PLCγ1-mediated Ca2+ signals in pre–B lymphoblastic leukemia cells. Cytoplasmic Ca2+ levels were measured in single leukemia cells by confocal laser microscopy. Before Ca2+ measurement, cells were kept under control conditions or treated for 24 h with STI571 or LFM-A13 (A). Pre–B cell receptor engagement by addition of μ-heavy chain–specific antibodies (arrows) did not change cytoplasmic Ca2+ concentrations. Although PLCγ1Y783 is phosphorylated in the presence—but not in the absence (+ STI571)—of BCR-ABL1 kinase activity (B, left panels), PLCγ2 was not tyrosine phosphorylated by BCR-ABL1 (not depicted). As a control for nonspecific side effects of STI571, murine B lymphoid cells carrying a doxycycline-inducible BCR-ABL1 transgene were analyzed in the presence or absence of 1 μg/ml doxycycline (DOX). These cells remain viable in the absence of BCR-ABL1 expression if the cell culture fluid is supplemented with 2 ng/ml murine recombinant IL-3. Protein extracts from these cells were subjected to Western blot using human- and mouse-reactive antibodies (B, right). Tyrosine phosphorylation of PLCγ1 was studied in BCR-ABL1 + pre–B lymphoblastic leukemia cells in the presence or absence of STI571 or LFM-A13. EIF-4E was used as a loading control in both Western blots (B, C). Expression of full-length BTK was silenced by RNA interference in a BCR-ABL1 + pre–B lymphoblastic leukemia cell line (IX in Table I). Sorted cells carrying fluorescein-labeled siRNAs (green) were permeabilized, stained for PLCγ1pY783 (red), and analyzed by confocal laser microscopy (C, bottom). As a negative control, nontargeting siRNA duplices were used. BCR-ABL1–binding proteins were coimmunoprecipitated using an antibody against BCR. The immunoprecipitation was controlled by a Western blot using an ABL1-specific antibody (D).
BTK phosphorylation depends on BCR-ABL1 kinase activity in vivo
To confirm that phosphorylation of BTK also depends on BCR-ABL1 kinase activity in primary leukemia cells, expression of tyrosine-phosphorylated BTK was compared in matched leukemia sample pairs from four patients before and during continued therapy with STI571 (cases I to IV, Fig. 1 B, right panel). The content of BCR-ABL1 + leukemia cells was similar in the samples taken before and during STI571 treatment as assessed by semiquantitative amplification of BCR-ABL1 fusion transcripts (see Fig. 2 A). As with inhibition of BCR-ABL1 kinase by STI571 in vitro, in vivo ablation of BCR-ABL1 kinase activity resulted in the loss of BTK phosphorylation in all four patients (Fig. 1 B, right panel).
Figure 2.

BCR-ABL1 induces expression of kinase-deficient BTK isoforms. Comparing matched leukemia sample pairs taken before and during continued therapy with the BCR-ABL1 kinase-inhibitor, STI571, BTK transcripts, including splice variants (BTKp52and BTKp65), were amplified (A, top). cDNA samples were normalized for equal content of leukemia cells by amplification of BCR-ABL1 (A, bottom). To determine whether aberrant splicing of BTK is induced by BCR-ABL1, pre–B lymphoblastic leukemia cells carrying an E2A-PBX1—but no BCR-ABL1 rearrangement—were transiently transfected with a retroviral expression vector which encoded BCR-ABL1 and GFP, or GFP alone (B). GFP− and GFP+ cells were sorted after 24 h (B, top) and subjected to RT-PCR analysis. cDNA amounts were normalized by amplification of a GAPDH fragment (B, bottom). In addition to full-length BTK, sequence analysis of BTK transcripts revealed six isoforms (Table S2). Among these isoforms, two were recurrently amplified: BTKp52 and BTKp65 were detected in all nine cases of BCR-ABL1 + pre–B lymphoblastic leukemia studied (C). Both isoforms carry a deletion within their kinase domain. Skipping of exon 15 (BTKp52) leads to the loss of the reading frame and COOH-terminal truncation as the result of a premature translation stop at codon 442 (C, middle). BTKp65 lacks exons 15 and 16 and leads to an in-frame deletion of 282 bp (BTKp65).
BTKY223 can be transphosphorylated by BCR-ABL1
To clarify further BCR-ABL1–dependent phosphorylation of BTKY223, we investigated whether phosphorylation at Y223 depends only on BCR-ABL1 or also on endogenous BTK activity. In normal B cells, B cell receptor engagement leads first to LYN-mediated transphosphorylation of COOH-terminal BTKY551 (17). BTKY223 phosphorylation occurs as a secondary event by autophosphorylation through endogenous BTK activity (17). Therefore, the effect of the BTK inhibitor, LFM-A13, on BTKY223 phosphorylation in BCR-ABL1 + pre–B cell lymphoblastic leukemia cells was examined. BTKY223 phosphorylation was sensitive to BCR-ABL1 kinase inhibition by STI571, but not to BTK inhibition by LFM-A13 (Fig. 1 C). We conclude that BTKY223 is accessible to transphosphorylation by BCR-ABL1 in the absence of endogenous BTK activity.
Expression of truncated BTK splice variants in BCR-ABL1 + pre–B lymphoblastic leukemia
In the BTK Western blot analysis of BCR-ABL1 + pre–B lymphoblastic leukemia cell lines, we observed that the antibody that recognized phosphorylated BTKY223 at the expected size of 77 kD also detected two smaller tyrosine-phosphorylated proteins of ∼65 kD and 52 kD in size (Fig. 1, A–C). This prompted us to investigate splice variants of BTK at the mRNA level.
By amplifying specific BTK cDNA fragments that cover the entire coding region of BTK, we detected shorter PCR products in all three BCR-ABL1 + pre–B lymphoblastic leukemia cell lines (not depicted) and also in six primary cases of BCR-ABL1 + pre–B lymphoblastic leukemia (cases I to VI, Table I; Fig. 2 A).
In total, sequence analysis revealed six different BTK isoforms in BCR-ABL1 + pre–B lymphoblastic leukemia cells (Table S2, available at http://www.jem.org/cgi/content/full/jem.20042101/DC1; sequence data available from GenBank/EMBL/DDBJ under accession nos. AJ888376–AJ888381). All of these splice variants carry large deletions within the kinase domain (Table S2). Four of these isoforms were unique, were generated by usage of cryptic splice sites, and only were amplified from individual cases of BCR-ABL1 + pre–B lymphoblastic leukemia. However, two BTK isoforms were identified recurrently.
In addition to a previously described BTK isoform which lacks exons 15 and 16 (BTKp65 in Fig. 2 A; reference 18), another truncated splice variant that lacks only exon 15 was amplified from all nine cases of BCR-ABL1 + pre–B lymphoblastic leukemia (Table S2). Joint skipping of exons 15 (217 bp) and 16 (65 bp) leads to an in-frame 282-bp deletion (BTKp65; Fig. 2 C). The predicted size of the protein encoded by BTKp65 matches the 65-kD protein recognized by the BTKY223 antibody (Fig. 1, A–C). Skipping of exon 15 alone leads to loss of the reading frame and generates a premature stop codon which results in the expression of a truncated protein of 442 amino acids instead of 659 amino acids (BTKp52; Fig. 2 C). The predicted truncated BTKp52 (442 amino acids) exactly matches the size of the heavily phosphorylated 52-kD protein that we detected with the phospho-specific anti-BTKY223 antibody (Fig. 1, A–C).
By comparing matched sample pairs of leukemia cells before and during continued therapy with the BCR-ABL1 kinase inhibitor, STI571, we amplified BTKp52 (lacking exon 15 alone) or BTKp65 (lacking exons 15 and 16) in the presence (but not in the absence) of BCR-ABL1 kinase activity (Fig. 2 A). These findings suggest that aberrant splicing of BTK may be linked to BCR-ABL1 kinase activity.
BCR-ABL1 was implicated previously in altered splice-site selection and exon skipping through up-regulation of RNA-binding proteins, including FUS, hnRNP A1, and hnRNP E2 (19). In addition, a recent study shows that BCR-ABL1 induces aberrant splicing of PYK2 premRNA in human hematopoietic progenitor cells, which can be reverted by STI571 (20). These findings exemplify that BCR-ABL1 frequently interferes with normal regulation of premRNA splicing (e.g., by reduction of stringency of splice-site selection).
To assess directly whether BCR-ABL1 can interfere with splicing of BTK premRNA, we transiently transfected pre–B lymphoblastic leukemia cells that carry an E2A-PBX1, but no BCR-ABL1, gene rearrangement with an expression vector which encodes GFP only or GFP and BCR-ABL1. For both transfections, GFP+ and GFP− cells were sorted and analyzed separately for expression of BTK splice variants (Fig. 2 B). Although E2A-PBX1 + pre–B lymphoblastic leukemia cells under control conditions express BTKp65 at low levels, induced expression of BCR-ABL1 leads to the expression of multiple BTK splice variants (Table S2), including BTKp52 (Fig. 2 B). We conclude that BCR-ABL1 induces aberrant splicing of BTK and expression of BTKp52.
By comparing 19 genome-wide gene expression profiles by serial analysis of gene expression (SAGE), we identified three SAGE-tags that distinguish between full-length BTK, BTKp65, and BTKp52 (Table S3, available at http://www.jem.org/cgi/content/full/jem.20042101/DC1). A SAGE-tag represents a 14-bp cDNA sequence which immediately flanks the extreme 3′ restriction site (CATG) of the tagging enzyme (NlaIII) that is used as a unique identifier of any processed transcript. Although full-length BTK is expressed widely in normal B cell populations and BTKp65 also is expressed in non–BCR-ABL1 B lymphoid malignancies, BTKp52 seems to be expressed specifically in BCR-ABL1 + leukemia cells (Table S3).
Specific silencing of BTK splice variants by RNA interference
To investigate the function of full-length BTK and its splice variants in BCR-ABL1 + pre–B lymphoblastic leukemia cells, we established an RNA interference approach to silence full-length BTK and BTK isoforms specifically. Among the six BTK splice variants (Table S2), only BTKp52 and BTKp65 were amplified recurrently. Therefore, we focused our analysis on BTKp52 and BTKp65. To clarify the specific function of BTKp52 (lack of exon 15, loss of reading frame) and BTKp65 (in-frame deletion of exons 15 and 16), we designed three small interfering RNA (siRNA) duplices against the junction of BTK exons 14 and 16 (corresponding to BTKp52) and the junction of BTK exons 14 and 17 (corresponding to BTKp65). For each target, the three siRNA duplices were mixed and fluorescein labeled. Also for full-length BTK, three siRNA duplices were synthesized (specifically binding to BTK exon 15), mixed, and fluorescein labeled. As a control, a fluorescein-labeled nontargeting siRNA duplex was used. After two transfections, the proportion of siRNA-carrying cells ranged between 3% and 5% (Fig. 3). For each condition, 105 fluorescein+ cells were sorted and subjected to RT-PCR analysis for BTK splice variants. mRNA levels of the reference genes HPRT and COX6B remained unchanged (Fig. 3; see Fig. 8). Sorted cells exhibited specific silencing of full-length BTK, BTKp52, or BTKp65, and no cross-inhibition of BTK isoform expression was observed (Fig. 3). Because lymphoid cells typically are difficult to transfect, we conclude that double transfection of a mixture of three siRNA duplices and cell sorting of siRNA-carrying cells represents an effective strategy to silence specifically target mRNA sequences in B lymphoid leukemia cells.
Figure 3.

Specific silencing of full-length BTK or BTK splice variants by RNA interference. BCR-ABL1 + pre–B lymphoblastic leukemia cells were transfected with a nontargeting siRNA duplex in the presence or absence of STI571 (for 12 h), as well as with a mixture of siRNAs against full-length BTK, BTKp52, or BTKp65. All siRNA duplices were labeled with fluorescein before transfection. Transfection was repeated after 24 h, and 105 cells carrying fluorescein-labeled siRNAs (between 3–5% of all viable cells) were sorted by FACS 48 h after the first transfection (upper panel). Total RNA was isolated and subjected to semiquantitative RT-PCR analysis of BTK isoform expression (lower panel). mRNA levels for HPRT and COX6B (see Fig. 8 C) were stable under all conditions.
Figure 8.

BTKp52, but not BTKp65, promotes cell survival, tyrosine phosphorylation of STAT5, and up-regulation of BCLXL. BCR-ABL1 + pre–B lymphoblastic leukemia cells (IX, Table I) were transfected with fluorescein-labeled siRNA duplices against full-length BTK, BTKp52, or BTKp65 as described above (Fig. 3), and were analyzed by flow cytometry for annexin V expression (A). As a negative control, nontargeting siRNA duplices were used. Likewise, siRNA-treated leukemia cells were subjected to intracellular staining for tyrosine-phosphorylated STAT5 and analyzed by flow cytometry (B). For the analysis of mRNA levels of BCLXL, BCR-ABL1 + pre–B lymphoblastic leukemia cells were transfected with a nontargeting siRNA duplex in the presence or absence of STI571 (for 12 h), as well as with siRNAs against full-length BTK, BTKp52, or BTKp65. 105 leukemia cells carrying fluorescein-labeled siRNAs sorted by FACS and subjected to semiquantitative RT-PCR analysis for mRNA levels of BCLXL (C). mRNA levels for COX6B (C) and HPRT (Fig. 3) remained stable in all experiments.
From in vitro kinase assays of BTK mutants that were derived from BTK-deficient patients who had X-linked agammaglobulinemia, it is known that even replacement mutations in the distal portion of the kinase domain result in a complete loss of BTK activity (http://bioinf.uta.fi/BTKbase/BTKbasebrowser.html). Hence, only full-length BTK—but none of the BTK splice variants identified here—should exhibit BTK activity. Therefore, we investigated the relevance of BTK activity for BCR-ABL1–mediated survival signaling in pre–B lymphoblastic leukemia cells.
Inhibition of BTK activity induces apoptosis specifically in human BCR-ABL1 + pre–B lymphoblastic leukemia cells
To clarify the role of full-length BTK (as compared with kinase-deficient truncated BTK) in pre–B lymphoblastic leukemia cells, we analyzed the effect of BTK inhibition on the survival of BCR-ABL1 + leukemia cells in comparison with inhibition of BCR-ABL1 by STI571. As a control we used several B lymphoid cell lines that do not carry a BCR-ABL1 gene rearrangement. Neither inhibition of endogenous c-ABL1 (by STI571) nor BTK (by LFM-A13) kinase activity alone significantly reduced viability of BCR-ABL1–negative B lymphoid cells (Fig. 4 A, B). Conversely, inhibition of BCR-ABL1 rendered almost all BCR-ABL1 + pre–B lymphoblastic leukemia cells apoptotic within 5 d (Fig. 4 A). In both BCR-ABL1 + pre–B lymphoblastic leukemia cell lines that were tested, but not in BCR-ABL1–negative cell lines, inhibition of BTK had a very similar effect as inhibition of BCR-ABL1, and also induced apoptosis in ∼90 percent of the leukemia cells within 5 d (Fig. 4 A, B). These findings are in agreement with an RNA interference experiment that showed that silencing of full-length BTK expression (Fig. 3) induced apoptosis in BCR-ABL1 + pre–B lymphoblastic leukemia cells (see Fig. 8 A). Thus, BTK activity is required for sustained growth and survival of BCR-ABL1–transformed human pre–B lymphoblastic leukemia cells.
Figure 4.
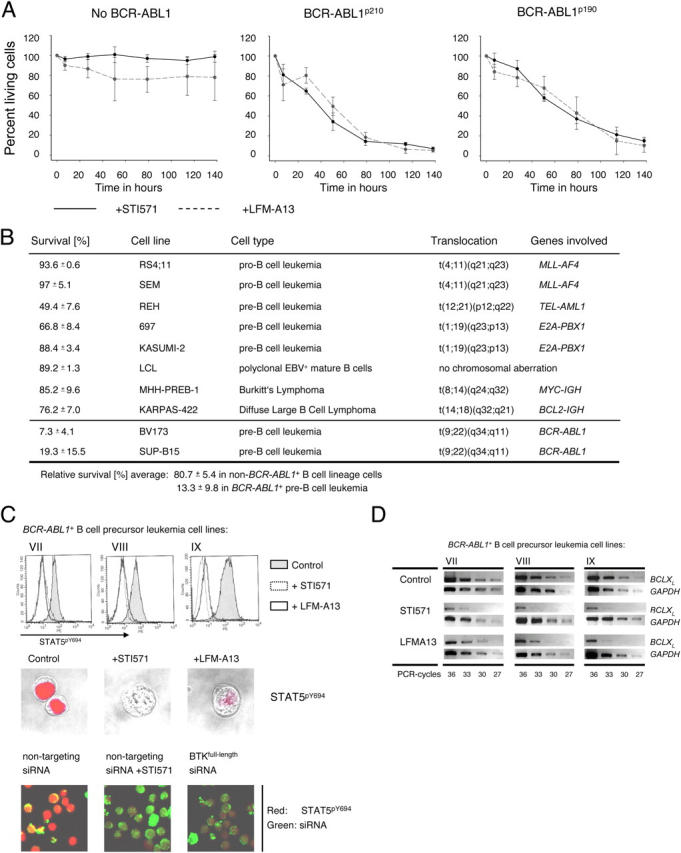
Inhibition of BTK induces apoptosis specifically in BCR-ABL1 + pre–B lymphoblastic leukemia cells. BCR-ABL1–negative B lymphoid leukemia and lymphoma cell lines (A, left panel) and two BCR-ABL + leukemia cell lines (A, middle and right panel) were incubated for the times indicated under control conditions, with STI571 (solid line) or with the BTK inhibitor, LFM-A13 (dashed line; A). Relative viability (percent living cells) was calculated as the ratio of viability in the presence of STI571 or LFM-A13 and viability in untreated cells. Two BCR-ABL1 + and eight BCR-ABL1–negative B lymphoid cell lines were incubated in the presence of LFM-A13 or under control conditions for 114 h (B). Relative survival of the treated cells as compared with control conditions is given as the mean of three independent experiments ± SD. To analyze BCR-ABL1–mediated and BTK-dependent tyrosine phosphorylation of STAT5 (C), three BCR-ABL1 + B cell precursor cell lines (VII, VIII, and IX in Table I) were treated with the BCR-ABL1 kinase inhibitor, STI571 (dotted line), with the BTK inhibitor, LFM-A13 (solid line), or incubated under control conditions (shaded areas; C). Nuclear localization of activated STAT5 in treated and untreated leukemia cells was visualized by confocal laser microscopy (C; red stain, middle). BCR-ABL1 + pre–B lymphoblastic leukemia cells were transfected with a mixture of fluorescein-labeled siRNAs against full-length BTK as shown in Fig. 3. Fluorescein+ cells carrying siRNA molecules (green) were stained for tyrosine-phosphorylated STAT5 (red) and analyzed by confocal laser microscopy (C, bottom panel). As a negative control, nontargeting siRNA duplices were used. Next, mRNA levels of BCLX L were studied in BCR-ABL1 + pre–B lymphoblastic leukemia cells in the presence or absence of STI571 or LFM-A13. Total RNA was isolated from the leukemia cells and subjected to semiquantitative RT-PCR analysis for mRNA levels of BCLX L (D). cDNA amounts were normalized by amplification of a GAPDH cDNA fragment.
BCR-ABL1–induced tyrosine phosphorylation and nuclear localization of STAT5 requires BTK activity
Besides other survival signals, STAT5 activation is critical for BCR-ABL1–mediated transformation (21, 22). Given that BTK activity can activate STAT5 through phosphorylation at tyrosine 694 (23), we investigated the impact of BCR-ABL1 or BTK inhibition on STAT5Y694 phosphorylation in three BCR-ABL1 + pre–B lymphoblastic leukemia cell lines by intracellular staining and flow cytometry. STAT5Y694 is constitutively phosphorylated in all three leukemia cell lines and exhibits nuclear localization (Fig. 4 C), which is required for STAT5 activity (21, 22). However, ablation of BCR-ABL1 kinase activity by STI571 and BTK inhibition by LFM-A13 largely reduced phosphorylation and nuclear localization of STAT5Y694 (Fig. 4 C).
In addition, three siRNA duplices against full-length BTK were mixed, fluorescein labeled, and transfected into BCR-ABL1 + pre–B lymphoblastic leukemia cells (IX in Table I) using fluorescein-labeled, nontargeting siRNAs as a control. After two transfections, fluorescein+ leukemia cells were sorted and analyzed for STAT5 phosphorylation (Fig. 4 C, lower panel). Efficiency and specificity of BTK full-length silencing was controlled by RT-PCR (Fig. 3). In agreement with BTK inhibition by LFM-A13, silencing of full-length BTK by RNA interference largely attenuated STAT5Y694 phosphorylation as shown by confocal laser microscopy (Fig. 4 C) and intracellular FACS staining (see Fig. 8 B).
BTK-mediated transcriptional activation of BCLXL in BCR-ABL1 + pre–B lymphoblastic leukemia cells
In BCR-ABL1 + leukemia cells, STAT5 mainly accomplishes its antiapoptotic effect by transcriptional activation of BCLX L (24). By comparing the frequency of SAGE-tags that distinguish between antiapoptotic BCLXL and its proapoptotic isoform, BCLXS, in 19 genome-wide SAGE-libraries, SAGE-tags specific for BCLXL were particularly frequent in BCR-ABL1 + pre–B lymphoblastic leukemia cells (Table S3).
By analyzing mRNA levels for BCLX L in BCR-ABL1 + pre–B lymphoblastic leukemia cells in the presence or absence of STI571 or LFM-A13, we found that inhibition of BCR-ABL1 or BTK leads to a considerable reduction of BCLX L expression in the leukemia cells (Fig. 4 D). This is consistent with the effect of siRNA-mediated silencing of full-length BTK on BCLXL mRNA levels (see Fig. 8 C).
A recent study identified SRC kinase activity as a requirement for BCR-ABL1–mediated transformation of murine pre–B cells (25). The findings presented here indicate that BTK activity also is required specifically for the BCR-ABL1–dependent survival signals, and thus, may represent an important target for the therapy of BCR-ABL1 + pre–B lymphoblastic leukemia.
BTK activity is required for autonomous Ca2+ signal activity in BCR-ABL1 + pre–B lymphoblastic leukemia cells
In normal pre–B cells, BTK activity induces Ca2+ release upon pre–B cell receptor engagement through BTK-dependent phosphorylation of PLCγ2. Constitutive phosphorylation of BTK by BCR-ABL1 and specific requirement of BTK activity for the survival of BCR-ABL1 + leukemia cells suggest that BCR-ABL1 also can trigger BTK-dependent Ca2+ release from cytoplasmic stores. In BCR-ABL1 + pre–B lymphoblastic leukemia cells that express a pre–B cell receptor on the surface (Table I; reference 8), BCR-ABL1 kinase activity was linked to an autonomous oscillatory Ca2+ signal, which was not changed by pre–B cell receptor engagement (arrows, left panel of Fig. 5 A). Analysis of Ca2+ release in multiple pre–B lymphoblastic leukemia cells at the single-cell level showed that inhibition of BCR-ABL1 terminated the autonomous Ca2+ signaling activity (Fig. 5 A, middle panel). Autonomous Ca2+ signals required BCR-ABL1 kinase activity and were abrogated by inhibition of BTK activity through treatment with LFM-A13 (Fig. 5 A, right panel). Comparing sorted fluorescein+ leukemia cells which were transfected with full-length BTK-specific or nontargeting siRNAs, autonomous oscillatory Ca2+ signals were diminished greatly in the leukemia cells that carried full-length BTK-specific siRNAs (see Fig. 9 B).
Figure 9.

Specific silencing of BTK full-length or BTKp52 reduces PLCγ1 phosphorylation and autonomous Ca2+ oscillations in BCR-ABL + pre–B lymphoblastic leukemia cells. BCR-ABL1 + pre–B lymphoblastic leukemia cells were transfected with nontargeting siRNAs or siRNAs against full-length BTK, BTKp52, or BTKp65, then subjected to intracellular staining for tyrosine-phosphorylated PLCγ1 and analyzed by flow-cytometry (A). Cytoplasmic Ca2+ levels [nmol/l] were measured in single siRNA-containing cells by confocal laser-scanning microscopy by continuous scanning for 6 min (B). Before analysis of Ca2+ levels, leukemia cells carrying fluorescein-labeled siRNAs were sorted by FACS. For each condition, ∼50 individual cells were recorded. As a control, cytoplasmic Ca2+ levels were measured in cells with nontargeting siRNAs (red line).
Pre–B cell receptor–dependent Ca2+ signals typically are initiated by BTK-mediated phosphorylation of PLCγ2 (9). Assuming that BCR-ABL1 targets downstream molecules of the pre–B cell receptor signal cascade in BCR-ABL1 + pre–B lymphoblastic leukemia cells, we investigated whether BCR-ABL1–dependent autonomous Ca2+ signals also are transduced by BTK and PLCγ2. Unexpectedly, PLCγ1—but not PLCγ2 (not depicted)—is phosphorylated by BCR-ABL1 in human leukemia cells (Fig. 5, B and C) and in mouse B lymphoid cells that carry an inducible BCR-ABL1 transgene (Fig. 5 B). As shown by coimmunoprecipitation, BCR-ABL1 does not bind directly to PLCγ1 (Fig. 5 D). Phosphorylation of PLCγ1 was sensitive to inhibition of BTK by LFM-A13 (Fig. 5 C) which indicates that PLCγ1 is activated through BTK as in normal lymphocytes and not directly by BCR-ABL1. Silencing of full-length BTK by RNA interference using fluorescein-labeled full-length BTK-specific siRNAs also results in a marked reduction of PLCγ1Y783 phosphorylation as assessed by confocal laser microscopy (Fig. 5 C) and intracellular FACS staining (see Fig. 9 A).
PLCγ1 contributes to survival signals in BCR-ABL1 + pre–B lymphoblastic leukemia cells
Whereas the role of STAT5 in BCR-ABL1–mediated survival signaling is well-established (21, 22, 24), a possible role of PLCγ1 in the promotion of survival of BCR-ABL1 + pre–B lymphoblastic leukemia cells has not been investigated. To determine whether BCR-ABL1– and BTK-dependent activation of PLCγ1 and PLCγ1-mediated Ca2+ release contributes to BCR-ABL1–mediated survival signals, we silenced PLCγ1 mRNA expression by RNA interference in three pre–B lymphoblastic leukemia cell lines that carry a BCR-ABL1 gene rearrangement (VII to IX, Table I) and four cell lines that carry other chromosomal translocations (MLL-AF4, X; TEL-AML1, XII; E2A-PBX1, XIII; TEL-PDGFRB, XV; Table I). As a control, nontargeting siRNAs were used. Cells were transfected twice with fluorescein-labeled siRNAs. To assess viability, fluorescein+ cells were analyzed for propidium iodide uptake and annexin V staining after 48 h. Among the three BCR-ABL1 + pre–B lymphoblastic leukemia cell lines, viability (mean of three experiments ± SD) was 90.3% ± 2.7% with nontargeting siRNAs and 18.6% ± 13.7% with PLCγ1-specific siRNAs. Among four leukemia cell lines that carry other gene rearrangements, viability was 84.3% ± 12.2% with nontargeting siRNAs and 67.6% ± 16.9% with PLCγ1-specific siRNAs. Unlike BCR-ABL1–negative leukemia cells, silencing of PLCγ1 in BCR-ABL1 + pre–B lymphoblastic leukemia cells results in a fivefold reduction of viability. Thus, PLCγ1 contributes to survival signaling specifically in BCR-ABL1 + pre–B lymphoblastic leukemia cell lines.
COOH-terminally truncated BTK functions as a link between full-length BTK and BCR-ABL1
BCR-ABL1–dependent phosphorylation of BTK suggests that BTK represents a substrate of the BCR-ABL1 kinase. However, previous work demonstrated that BCR-ABL1 cannot phosphorylate full-length BTK directly, but the NH2-terminal BTK-SH3 domain fused to GST (26).
To determine how BCR-ABL1 can induce phosphorylation of full-length BTK, we studied whether full-length BTK or BTK splice variants are part of the BCR-ABL1-signalosome. Using protein extracts of two BCR-ABL1 + pre–B lymphoblastic leukemia cell lines, BCR-ABL1 signaling complexes were immunoprecipitated with a BCR-specific antibody. Immunoprecipitation was controlled using an anti-ABL1 antibody. Western blot showed that low amounts of full-length BTK coimmunoprecipitates with BCR-ABL1 (Fig. 6 A). As a control for quantitative distribution of full-length BTK, BTKp65, and BTKp52 before coimmunoprecipitation with BCR-ABL1, whole cell lysates (WCLs) from two BCR-ABL1 + leukemia cell lines were subjected to Western blot (Fig. 6 A). Although full-length BTK, BTKp65, and BTKp52 are expressed at similar levels in the leukemia cells (WCL, Fig. 6 A), the amount of full-length BTK that coimmunoprecipitated with BCR-ABL1 was ∼20-fold less than the amount of BTKp52 that coimmunoprecipitated with BCR-ABL1. Although BTKp65 is expressed clearly in the leukemia cells (WCL, Fig. 6 A), coimmunoprecipitation of BTKp65 with BCR-ABL1 could not be detected (Fig. 6 A).
Figure 6.
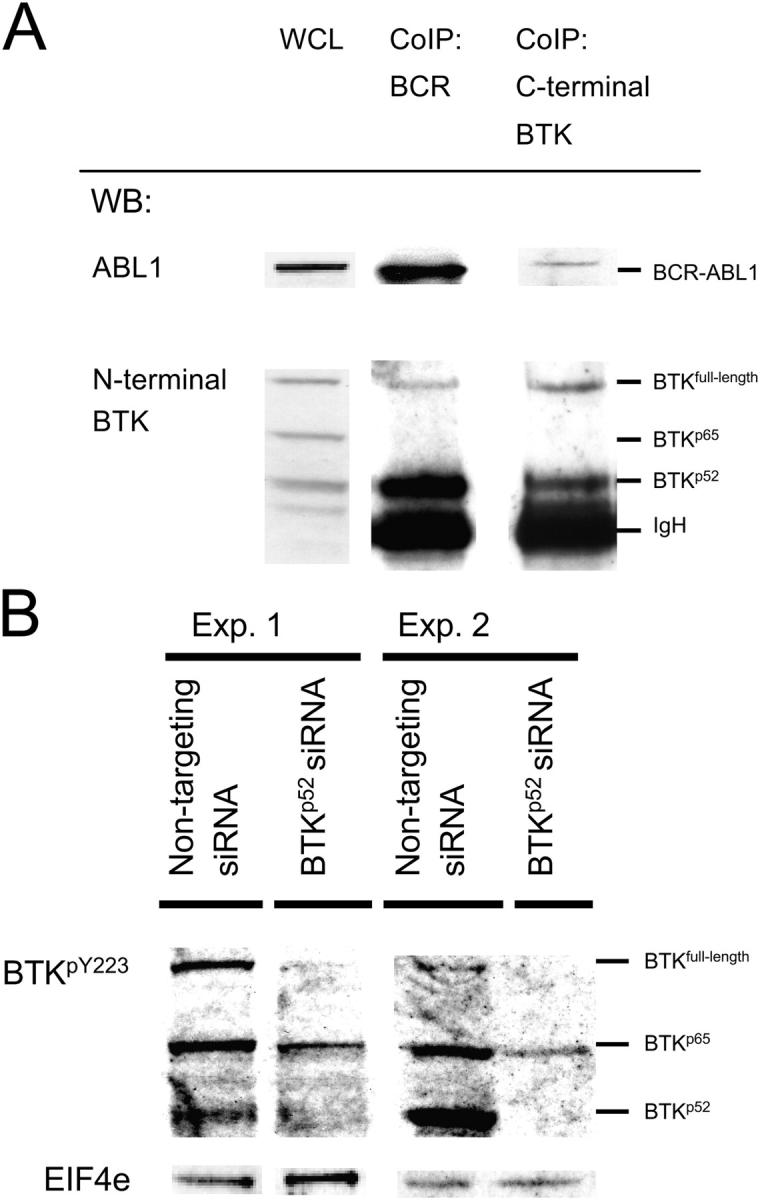
COOH-terminally truncated BTK functions as a linker between full-length BTK and BCR-ABL1. Proteins binding to BCR-ABL1 were coimmunoprecipitated with an anti–BCR antibody (A). Immunoprecipitation was controlled by an anti-ABL1–specific Western blot (WB) showing the characteristic BCR-ABL1 fusion protein expressed by the leukemia cell line. Proteins binding to full-length BTK were coimmunoprecipitated with an antibody against COOH-terminal BTK. Immunoprecipitation was controlled by Western blot using an antibody against NH2-terminal BTK. Full-length BTK and BTKp52 proteins coimmunoprecipitating with BCR-ABL1 or full-length BTK were visualized by Western blot using an antibody against NH2-terminal BTK (A). As a control for quantitative distribution of full-length BTK and BTKp52 before coimmunoprecipitation with BCR-ABL1 or full-length BTK, WCLs were used (A). To analyze the effect of BTKp52 on BCR-ABL1–dependent phosphorylation of full-length BTK, BTKp52 expression was inhibited by RNA interference as described in Fig. 3. As a control, nontargeting siRNA duplices were used. siRNAs were fluorescein-labeled and fluorescein+ cells were sorted and subjected to Western blot using antibodies specific for tyrosine-phosphorylated BTKY223 (B). Because patterns of tyrosine phosphorylation varied between replicate experiments, Western blots of two representative experiments (Exp. 1 and 2) are shown. EIF4e was used as a loading control.
Preferential binding of BCR-ABL1 to BTKp52 that lacked the COOH terminus but retained the NH2-terminal BTK-SH3 domain is consistent with previous findings that demonstrated that BCR-ABL1 cannot directly phosphorylate full-length BTK but can phosphorylate a BTK-SH3 domain-GST fusion molecule (26). However, binding of BTKp52 to BCR-ABL1 also may be indirect in this case. That the NH2-terminal BTK SH3 domain, but not full-length BTK, can be phosphorylated readily by BCR-ABL1 suggests that the COOH terminus of BTK may interfere with binding of BCR-ABL1 to BTK. Likewise, BTKp65 molecules that harbor an in-frame deletion but retain the BTK COOH terminus do not immunoprecipitate with BCR-ABL1. In this regard, the expression of truncated BTK splice variants that lack the COOH terminus of BTK may represent a mechanism to facilitate phosphorylation of full-length BTK by BCR-ABL1. Because BTK molecules also can self-associate through NH2-terminal SH3 domains (27), BTKp52 may bind to BCR-ABL1 as well as to full-length BTK and acts as link between the two kinases.
We next investigated whether full-length BTK can bind to BTKp52. Using an antibody specific for COOH-terminal BTK (only recognizes full-length BTK) for immunoprecipitation and an NH2-terminal BTK antibody (recognizes full-length BTK and BTKp52) for subsequent Western blotting, we observed that BTKp52 coimmunoprecipitates with full-length BTK (Fig. 6 A). These findings suggest a role for BTKp52 as a link between BCR-ABL1 and full-length BTK.
In this case, BTKp52 also would facilitate tyrosine phosphorylation of full-length BTK by BCR-ABL1. Therefore, we tested whether silencing of BTKp52 in BCR-ABL1 + pre–B lymphoblastic leukemia cells results in decreased tyrosine phosphorylation of full-length BTK. BCR-ABL1 + pre–B lymphoblastic leukemia cells were transfected with nontargeting or BTKp52-specific fluorescein-labeled siRNAs (Fig. 3). Fluorescein+ cells were sorted and analyzed for expression of tyrosine-phosphorylated BTK by Western blot (Fig. 6 B). Silencing of BTKp52 mRNA expression by siRNAs did not interfere with mRNA expression of full-length BTK (Fig. 3), yet tyrosine phosphorylation of full-length BTK was decreased by >90% (Fig. 6 B).
Given that BCR-ABL1 and full-length BTK cannot bind directly to each other; BTKp52 can bind to BCR-ABL1 and full-length BTK; and expression of BTKp52 is required for efficient tyrosine phosphorylation of full-length BTK, we conclude that BTKp52 can act as a link between BCR-ABL1 and full-length BTK.
This is in agreement with previous work which suggested that kinase-deficient BTK may act as a linker molecule between upstream kinases (e.g., SYK) and downstream effector molecules (e.g., PLCγ2; reference 11).
COOH-terminally truncated BTK can link full-length BTK to BCR-ABL1 through its SH3 domain
Based on this assumption, one would predict that the coexpression of BTKp52 with full-length BTK can facilitate the interaction between BTK and BCR-ABL1, and hence, the phosphorylation of downstream signaling molecules. To test this possibility, BCR-ABL1, BTK, and its truncated variant, BTKp52 were expressed in 293T embryonic kidney cells alone, in various combinations, or in the presence of BTK (LFM-A13) or BCR-ABL1 (STI571) kinase inhibitors. As a readout of this experiment, tyrosine phosphorylation of PLCγ1 and STAT5 was measured. According to a previous study, 293T cells do not express BTK but do express PLCγ1 and STAT5 (28).
The expression of full-length BTK, BTKp52, or BCR-ABL1 alone had no effect on tyrosine phosphorylation of downstream molecules in general. However, to some extent, single transfection with full-length BTK induced activation of PLCγ1. Likewise, to some extent, BCR-ABL1 alone induced tyrosine phosphorylation of STAT5 (Fig. 7, A and B); this indicates that BCR-ABL1 can activate STAT5 partially in the absence of BTK. Coexpression of BTKp52 which lacked a functional kinase domain and BCR-ABL1 did not increase tyrosine phosphorylation as compared with expression of BCR-ABL1 alone. However, coexpression of BTKp52 with full-length BTK and BCR-ABL1 drastically up-regulated PLCγ1 and STAT5 phosphorylation (Fig. 7 A). As assessed by intracellular staining, phosphorylated PLCγ1 and STAT5 were distributed in the cytoplasm and the nucleus, respectively (Fig. 7 B).
Figure 7.

BTKp52 facilitates BTK- and BCR-ABL1–dependent activation of PLCγ1 and STAT5 through its SH3 domain. BCR-ABL1, full-length BTK, BTKp52, and the SH3-domain of BTK were expressed in 293T embryonic kidney cells alone or in various combinations in the presence or absence of STI571 or LFM-A13. As a readout, cells were harvested, subjected to intracellular staining for tyrosine-phosphorylated PLCγ1 or STAT5, and analyzed by flow cytometry (A). To visualize cytoplasmic localization of tyrosine-phosphorylated PLCγ1 and nuclear localization of activated STAT5, the stained cells also were subjected to analysis by confocal laser microscopy (B). 106 293T cells were transfected transiently with full-length BTK for 24 h and subjected to immunoprecipitation of full-length BTK. Immunoprecipitation (IP) was controlled by a BTK-specific Western blot (C). Kinase activity of immunoprecipitated BTK was analyzed in an in vitro kinase assay using 150 ng of a PLCγ1 fragment (amino acids 530 to 850) as substrate. In parallel, 25 ng of recombinant active BTK and 100 ng of kinase-deficient SH3-domain of BTK were used in kinase assays as positive and negative controls, respectively.
Activation of PLCγ1 and STAT5 upon triple transfection with full-length BTK, BTKp52, and BCR-ABL1 largely was sensitive to BTK (LFM-A13) or BCR-ABL1 (STI571) kinase inhibition. Hence, enzymatic activity of both kinases is required for activation of PLCγ1 and STAT5. As a control, the enzymatic activity of BTK expressed in transfected 293T cells was confirmed in an in vitro kinase assay as described in the Materials and methods section. The kinase activity of BTK which was immunoprecipitated from 105 transfected 293T cells roughly was comparable to that of 25-ng (0.008 U) recombinant BTK (Fig. 7 C).
Because BTK-SH3 domains can bind directly to BCR-ABL1 (26) and also bind to proline-rich regions of other BTK molecules (27), we tested whether the BTK-SH3 domain is sufficient to link full-length BTK to BCR-ABL1. In fact, transfection of 293T cells with a vector encoding only the BTK-SH3 domain had a similar effect compared with transfection with BTKp52 comprising PH, TH, SH3, SH2 and a truncated kinase domain (Fig. 2 C). The SH3 domain was sufficient to enable BCR-ABL1-driven activation of PLCγ1 and STAT5 in the presence of active full-length BTK (Fig. 7 A). We conclude that BTKp52, mainly through its SH3 domain, can act as a linker between full-length BTK and BCR-ABL1.
BTKp52 but not BTKp65 cooperates with full-length BTK to transduce BCR-ABL1–dependent survival signals
To clarify the specific contribution of BTKp52 and BTKp65 to BCR-ABL1 downstream survival signaling, annexin V expression, STAT5 phosphorylation, and BCLXL mRNA levels were measured in leukemia cells after transfection with fluorescein-labeled siRNAs against BTKp52 and BTKp65 and nontargeting siRNAs (Fig. 8, A–C). Transfection of leukemia cells with nontargeting siRNAs or siRNAs against BTKp65 had no effect on viability as assessed by annexin V staining (Fig. 8 A), tyrosine phosphorylation of STAT5 (Fig. 8 B), and BCLXL mRNA levels (Fig. 8 C). In contrast, siRNA-mediated silencing of BTKp52 induced apoptosis in BCR-ABL1 + pre–B lymphoblastic leukemia cells to a similar extent as full-length BTK (Fig. 8 A). Likewise, silencing of full-length BTK or BTKp52 decreased tyrosine phosphorylation of STAT5 (Fig. 8 B) and reduced mRNA levels of BCLXL in the leukemia cells (Fig. 8 C). Collectively, these findings indicate that survival signals in BCR-ABL1 + pre–B lymphoblastic leukemia cells require BTK activity, and BTKp52 to act as a link between full-length BTK and BCR-ABL1.
Requirement of BTKp52 for BCR-ABL1–driven autonomous Ca2+ signaling activity
Autonomous oscillatory Ca2+ signals in BCR-ABL1 + pre–B lymphoblastic leukemia cells require kinase activity of BCR-ABL1 and BTK (Fig. 5 A), as well as PLCγ activity (9). Therefore, we investigated the impact of specific silencing of full-length BTK, BTKp52, and BTKp65 on PLCγ1 activation (Fig. 9 A). siRNA-mediated knockdown of full-length BTK and BTKp52 expression similarly reduced tyrosine phosphorylation of PLCγ1, whereas siRNAs against BTKp65 had no effect (Fig. 9 A). In agreement with this, siRNA-mediated inhibition of full-length BTK or BTKp52 expression—but not expression of BTKp65—largely reduced the amplitude of autonomous Ca2+ oscillations in BCR-ABL1 + pre–B lymphoblastic leukemia cells (Fig. 9 B). We could not identify a specific function of BTKp65 with respect to BCR-ABL1–mediated transformation in pre–B lymphoblastic leukemia cells.
DISCUSSION
The leukemogenic BCR-ABL1 kinase mimics a constitutively active pre–B cell receptor in pre–B lymphoblastic leukemia cells. Although the leukemia cells frequently carry only nonfunctional IGH-alleles (8), pre–B cell receptor signaling is compromised even in the few cases in which the leukemia cells express a pre–B cell receptor on their surface. Important components of the pre–B cell receptor signaling cascade, including SYK, SLP65, and BTK, are not phosphorylated in response to pre–B cell receptor–engagement, and BTK is constitutively phosphorylated by BCR-ABL1. BCR-ABL1–dependent activation of BTK is critical for autonomous survival signals that otherwise would arise from the pre–B cell receptor. We conclude that BTK activity is no longer responsive to pre–B cell receptor–dependent signals, but contributes to multiple aspects of BCR-ABL1–driven survival signaling in pre–B lymphoblastic leukemia cells.
Although BCR-ABL1 cannot interact with full-length BTK directly (26), BCR-ABL1–induced aberrant splicing of BTK premRNA leads to the expression of a truncated splice variant of BTK that acts as a linker molecule between the two kinases. Acting as a linker molecule, not only can truncated BTKp52 facilitate BCR-ABL1–dependent activation of full-length BTK but it can also facilitate BCR-ABL1–dependent activation of downstream molecules, including PLCγ1, STAT5, and BCLXL.
The interaction between BCR-ABL1 and BTK is critical because inhibition of BCR-ABL1 by STI571 and interference with BTK activity specifically induces apoptosis in BCR-ABL1 + B lymphoid leukemia cells. Because resistance to STI571 is frequent in the therapy of this leukemia entity (15), inhibition of BTK or its truncated splice variant potentially represents a therapeutic approach to circumvent STI571 resistance of BCR-ABL1 + pre–B lymphoblastic leukemia cells.
MATERIALS AND METHODS
Patient samples and cell lines.
Clinical data on patient samples and cytogenetic data on cell lines used are summarized in Table I and are described in detail in supplemental Materials and methods (available at http://www.jem.org/cgi/content/full/jem.20042101/DC1). All studies on human materials were performed on samples provided in compliance with Institutional Review Board regulations (Department of Hematology, University of Frankfurt).
Sequence analysis of BTK and semiquantitative RT-PCR.
In a search for BTK isoforms, fragments of the BTK cDNA were amplified covering the entire coding region using the PCR primer pairs listed in Table S1 (available at http://www.jem.org/cgi/content/full/jem.20042101/DC1. BTK amplification products were sequenced as described previously (29). Primers used for semiquantitative RT-PCR analysis of human BCR-ABL1, BCLX L, GAPDH, HPRT, and COX6B transcripts are listed in Table S1.
Transient expression of BCR-ABL1 in pre–B lymphoblastic leukemia cells.
Pre–B lymphoblastic leukemia cells (697 cell line) that carry an E2A-PBX1, but no BCR-ABL1, gene rearrangement were transfected transiently by electroporation (250 V and 950 μF) with pMIG-GFP or pMIG-GFP BCR-ABL1 vectors that encoded GFP or GFP plus BCR-ABL1 as described previously (30). For both transfections, GFP+ and GFP− cells were sorted after 24 h and subjected to mRNA isolation.
RNA interference with BTK and PLCγ1 expression.
For each target, three different siRNA duplices were synthesized (MWG Biotech). Sequences of siRNA duplices are listed in Table S1. For knockdown of PLCγ1 mRNA expression, a pool of three validated siRNAs which target different PLCγ1 exons was used (Upstate). As a control, a nontargeting siRNA duplex was used that does not match a known mRNA sequence. All siRNA duplices were labeled with fluorescein using an siRNA labeling kit (Ambion) according to the manufacturer's protocol. BCR-ABL1 + pre–B lymphoblastic leukemia cells were transfected with a mixture of three fluorescein-labeled siRNAs for each target sequence at a concentration of 100 nmol/l using oligofectamine in Opti-MEM1 medium (Invitrogen). After 24 h, leukemia cells were retransfected with labeled siRNAs and incubated for an additional 24 h. Transfection efficiency was controlled by fluorescence microscopy and by FACS. The silencing effect of siRNAs for specific BTK isoforms was controlled by RT-PCR analysis of BTK splice variants in sorted of fluorescein+ cells. siRNA-containing fluorescein+ cells were sorted using a FACStar 440 cell sorter. For each condition, 5 × 105 cells were sorted and subjected to RNA isolation and cDNA synthesis.
Western blotting.
For the detection of signaling molecules by Western blot, antibodies against BTK, PLCγ1, PLCγ2, SLP65, and SYK (Cell Signaling Technology), and phosphotyrosine-specific antibodies against BTKY223, PLCγ1Y783, PLCγ2Y1217, SLP65Y96, and SYKY323 (Cell Signaling Technology) were used with the WesternBreeze immunodetection system (Invitrogen). Mouse monoclonal antibodies against tyrosine-phosphorylated BTKY223 and BTKY551 were provided by O.N. Witte (University of California, Los Angeles, CA).
Coimmunoprecipitation.
For immunoprecipitation of BCR-ABL1 or BTK, 1.7 mg protein lysate in a volume of 800 μl was incubated with 30 μl rProtein G Agarose beads (Invitrogen) for 2 h at 4°C on a shaker. The supernatant was separated from the agarose beads and incubated with 6 μg of an antibody against BCR (Santa Cruz Biotechnology) or with 15 μg of an antibody against COOH-terminal BTK (provided by Owen N. Witte). After 8 h, 30 μl of agarose beads were added and incubation was continued overnight. The agarose beads were washed twice with lysis buffer, resuspended in 35 μl of lysis buffer, and subjected to Western blotting.
Flow cytometry.
For FACS analysis of BCR-ABL1 + pre–B lymphoblastic leukemia cells, antibodies against tyrosine-phosphorylated BTKY223, tyrosine-phosphorylated PLCγ1Y783, and tyrosine-phosphorylated STAT5Y694 (Cell Signaling Technologies) were used. Apoptotic or dead cells were identified by propidium iodide or PE-labeled annexin V (BD Biosciences). As secondary antibody, anti–rabbit or anti–mouse IgG-Cy3 (Jackson ImmunoResearch Laboratories) was used.
Immunofluorescence and confocal laser microscopy.
Nuclear, cytoplasmic, or membrane localization of phosphorylated BTKY223, PLCγ1Y783, and STAT5Y694 was analyzed using primary antibodies from Cell Signaling Technology together with anti–rabbit IgG-Cy5 (Jackson ImmunoResearch Laboratories) as secondary antibody. Cells were fixed with 0.4% paraformaldehyde and incubated for 10 min in 90% methanol on ice and subjected to confocal laser-scanning microscopy as described previously (14).
Expression of BCR-ABL1, full-length BTK, BTKp52, and the BTK-SH3 domain in 293T cells.
106 293T embryonic kidney cells were plated on a 24-well plate 24 h before transfection. Different combinations of expression vectors were prepared as indicated and 5 μg of each vector was incubated with 8 μl of FuGENE6 (Roche) and 50 μl of serum- and antibiotic-free RPMI 1640 medium for 15 min at room temperature. The expression vector pMIG-bcr/abl for BCR-ABL1 p210 (provided by D. Baltimore, California Institute of Technology, La Jolla, CA) was used as described previously (30). The expression vector pMIG-flagBtk for human full-length BTK was generated by cloning of the human BTK cDNA into the pMIG-R vector (30). For expression of BTK p52 (GenBank/EMBL/DDBJ accession no. AJ888378), the expression vector pcDNA3.1 was used with the directional TOPO Expression Kit (Invitrogen). For expression of the BTK-SH3 domain, the pEBG BTK-SH3 vector (a gift from M. Bäckesjø, Karolinska Institute, Huddinge, Sweden) was used as described previously (26).
Measurement of Ca2+ signals in BCR-ABL1+ pre–B lymphoblastic leukemia cells.
Pre–B lymphoblastic leukemia cells that carry a BCR-ABL1 gene rearrangement were cultured in the presence or absence of the BCR-ABL1 inhibitor, STI571 (Novartis), or the BTK inhibitor, LFM-A13 (Calbiochem), for the times indicated. In a different set of experiments, cells were transfected with fluorescein-labeled siRNA duplices and fluorescein+ cells were sorted and conditioned in RPMI medium at 37°C before measurement of cytoplasmic Ca2+ concentrations. Cells were washed and stained with Fluo-3 dye (Calbiochem) for 30 min. Changes of cytosolic Ca2+ were measured by confocal laser microscopy (31). For each condition, Ca2+ release of ∼50 individual cells was recorded.
In vitro kinase assay.
BTK that was immunoprecipitated from transfected 293T cells was used in a kinase assay, including a Mg2+/ATP cocktail (100 μmol/l) and a 61-kD fragment of PLCγ1 (Santa Cruz Biotechnology) as a substrate of BTK. Equal amounts of PLCγ1 peptide (150 ng) and ATP were incubated for 10 min at 30°C with BTK immunoprecipitates from 105 transfected 293T cells; 25-ng active human recombinant BTK (Upstate) was used as a positive control or 100 ng of a peptide corresponding to the BTK-SH3 domain (Labvision; lacking kinase activity) was used as a negative control. The kinase activity of BTK that was immunoprecipitated from 5 × 105 transfected 293T cells was roughly comparable to that of 25-ng (0.008 U) recombinant BTK (Fig. 7 C).
Online supplemental material.
A detailed description of patient samples and cell lines that were used in this paper is available as supplemental Materials and methods. Table S1 lists all PCR primers and siRNA duplices used. Table S2 describes all aberrant splice variants of BTK amplified from BCR-ABL1 + pre–B lymphoblastic leukemia cells. Table S3 depicts SAGE-data on mRNA levels of BCLX and BTK isoforms in normal B cell subsets and BCR-ABL1 + leukemia cells. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20042101/DC1.
Acknowledgments
We would like to thank M. Krönke and K. Rajewsky for continuous support and discussions and S. Jauch and P. Wurst for excellent technical assistance.
N. Feldhahn is supported by a fellowship of the José-Carreras-Leukemia Foundation. F. Klein is supported by the Studienstiftung des Deutschen Volkes. M. Müschen is supported by the Deutsche Forschungsgemeinschaft through the Emmy-Noether-Programm and through grant nos. MU1616/2-1 and MU1616/3-1, the German José-Carreras-Leukemia-Foundation, the Deutsche Krebshilfe through Molecular Mechanisms of Malignant Lymphoma, and the Ministry of Science and Research for North Rhine-Westphalia through the Stem Cell Network NRW.
The authors have no conflicting financial interests.
Abbreviations used: BTK, Bruton's tyrosine kinase; PLCγ, phospholipase C γ; siRNA, small interfering RNA; SAGE, serial analysis of gene expression; SH3, SRC-homology domain 3; SYK, spleen tyrosine kinase; WCL, whole cell lysate.
References
- 1.Look, A.T. 1997. Oncogenic transcription factors in the human acute leukemias. Science. 278:1059–1064. [DOI] [PubMed] [Google Scholar]
- 2.Huettner, C.S., P. Zhang, R.A. Van Etten, and D.G. Tenen. 2000. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat. Genet. 24:57–60. [DOI] [PubMed] [Google Scholar]
- 3.Rajewsky, K. 1996. Clonal selection and learning in the antibody system. Nature. 381:751–758. [DOI] [PubMed] [Google Scholar]
- 4.Kato, I., T. Takai, and A. Kudo. 2002. The pre-B cell receptor signaling for apoptosis is negatively regulated by Fc gamma RIIB. J. Immunol. 168:534–629. [DOI] [PubMed] [Google Scholar]
- 5.Küppers, R., U. Klein, M.L. Hansmann, and K. Rajewsky. 1999. Cellular origin of human B-cell lymphomas. N. Engl. J. Med. 341:1520–1529. [DOI] [PubMed] [Google Scholar]
- 6.Kanzler, H., R. Küppers, and M.L. Hansmann, and K. Rajewsky. 1996. Hodgkin and Reed-Sternberg cells in Hodgkin's disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 184:1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dykstra, M.L., R. Longnecker, and S.K. Pierce. 2001. Epstein-Barr virus coopts lipid rafts to block the signaling and antigen transport functions of the BCR. Immunity. 14:57–67. [DOI] [PubMed] [Google Scholar]
- 8.Klein, F., N. Feldhahn, L. Harder, H. Wang, M. Wartenberg, W.K. Hofmann, P. Wernet, R. Siebert, and M. Müschen. 2004. The BCR-ABL1 Kinase Bypasses Selection for the expression of a pre-B cell receptor in pre-B acute lymphoblastic leukemia cells. J. Exp. Med. 199:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fluckiger, A.C., Z. Li, R.M. Kato, M.I. Wahl, H.D. Ochs, R. Longnecker, J.P. Kinet, O.N. Witte, A.M. Scharenberg, and D.J. Rawlings. 1998. Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J. 17:1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satterthwaite, A.B., and O.N. Witte. 2000. The role of Bruton's tyrosine kinase in B-cell development and function: a genetic perspective. Immunol. Rev. 175:120–127. [PubMed] [Google Scholar]
- 11.Middendorp, S., G.M. Dingjan, A. Maas, K. Dahlenborg, and R.W. Hendriks. 2003. Function of Bruton's tyrosine kinase during B cell development is partially independent of its catalytic activity. J. Immunol. 171:5988–5996. [DOI] [PubMed] [Google Scholar]
- 12.Kersseboom, R., S. Middendorp, G.M. Dingjan, K. Dahlenborg, M. Reth, H. Jumaa, and R.W. Hendriks. 2003. Bruton's tyrosine kinase cooperates with the B cell linker protein SLP-65 as a tumor suppressor in pre-B cells. J. Exp. Med. 198:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Middendorp, S., A.J.E. Zijstra, R. Kersseboom, G.M. Dingjan, H. Jumaa, and R. Hendriks. 2005. Tumor suppressor function of Bruton's tyrosine kinase is independent of its catalytic activity. Blood. 105:259–265. [DOI] [PubMed] [Google Scholar]
- 14.Kitamura, D., J. Roes, R. Kuhn, and K. Rajewsky. 1991. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 350:423–426. [DOI] [PubMed] [Google Scholar]
- 15.Druker, B.J., C.L. Sawyers, H. Kantarjian, D.J. Resta, S.F. Reese, J.M. Ford, R. Capdeville, and M. Talpaz. 2001. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 344:1038–1042. [DOI] [PubMed] [Google Scholar]
- 16.Klucher, K.M., D.V. Lopez, and G.Q. Daley. 1998. Secondary mutation maintains the transformed state in BaF3 cells with inducible BCR/ABL expression. Blood. 91:3927–3934. [PubMed] [Google Scholar]
- 17.Wahl, M.I., A.C. Fluckiger, R.M. Kato, H. Park, O.N. Witte, and D.J. Rawlings. 1997. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton's tyrosine kinase via alternative receptors. Proc. Natl. Acad. Sci. USA. 94:11526–11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman, P.A., C.M. Wood, A.O. Vassilev, C. Mao, and F.M. Uckun. 2003. Defective expression of Bruton's tyrosine kinase in acute lymphoblastic leukemia. Leuk. Lymphoma. 44:1008–1011. [DOI] [PubMed] [Google Scholar]
- 19.Perrotti, D., and B. Calabretta. 2002. Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene. 21:8577–8583. [DOI] [PubMed] [Google Scholar]
- 20.Salesse, S., S.J. Dylla, and C.M. Verfaillie. 2004. p210BCR/ABL-induced alteration of pre-mRNA splicing in primary human CD34+ hematopoietic progenitor cells. Leukemia. 18:727–733. [DOI] [PubMed] [Google Scholar]
- 21.Nieborowska-Skorska, M. M.A. Wasik, A. Slupianek, P. Salomoni, T. Kitamura, B. Calabretta, and T. Skorski. 1999. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J. Exp. Med. 189:1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sillaber, C., F. Gesbert, D.A. Frank, and M. Sattler, and J.D. Griffin 2000. STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood. 95:2118–2125. [PubMed] [Google Scholar]
- 23.Mahajan, S., A. Vassilev, N. Sun, Z. Ozer, C. Mao, and F.M. Uckun. 2001. Transcription factor STAT5A is a substrate of Bruton's tyrosine kinase in B cells. J. Biol. Chem. 276:31216–31228. [DOI] [PubMed] [Google Scholar]
- 24.Gesbert, F., and J.D. Griffin. 2000. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood. 96:2269–2276. [PubMed] [Google Scholar]
- 25.Hu, Y., Y. Liu, S. Pelletier, E. Buchdunger, M. Warmuth, D. Fabbro, M. Hallek, R.A. Van Etten, and S. Li. 2004. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat. Genet. 36:453–461. [DOI] [PubMed] [Google Scholar]
- 26.Backesjo, C.M., L. Vargas, G. Superti-Furga, and C.I. Smith. 2002. Phosphorylation of Bruton's tyrosine kinase by c-Abl. Biochem. Biophys. Res. Commun. 299:505–510. [DOI] [PubMed] [Google Scholar]
- 27.Laederach, A., K.W. Cradic, K.N. Brazin, J. Zamoon, B.D. Fulton, X.Y. Huang, and A.H. Andreotti. 2002. Competing modes of self-association in the regulatory domains of Bruton's tyrosine kinase: intramolecular contact versus asymmetric homodimerization. Protein Sci. 11:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohya, K., S. Kajigaya, A. Kitanaka, K. Yoshida, A. Miyazato, Y. Yamashita, T. Yamanaka, U. Ikeda, K. Shimada, K. Ozawa, and H. Mano. 1999. Molecular cloning of a docking protein, BRDG1, that acts downstream of the Tec tyrosine kinase. Proc. Natl. Acad. Sci. USA. 96:11976–11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein, F., N. Feldhahn, S. Lee, H. Wang, F. Ciuffi, M. von Elstermann, M.L. Toribio, H. Sauer, M. Wartenberg, V.S. Barath, et al. 2003. T lymphoid differentiation in human bone marrow. Proc. Natl. Acad. Sci. USA. 100:6747–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pear, W.S., J.P. Miller, L. Xu, J.C. Pui, B. Soffer, R.C. Quackenbush, A.M. Pendergast, R. Bronson, J.C. Aster, and M.L. Scott, and D. Baltimore. 1998. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 92:3780–3792. [PubMed] [Google Scholar]
- 31.Feldhahn, N., I. Schwering, S. Lee, M. Wartenberg, F. Klein, H. Wang, G. Zhou, S.M. Wang, J.D. Rowley, J. Hescheler, et al. 2002. Silencing of B cell receptor signals in human naive B cells. J. Exp. Med. 196:1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]