

Abstract
Acute infection with hepatitis C virus (HCV) rarely is identified, and hence, the determinants of spontaneous resolution versus chronicity remain incompletely understood. In particular, because of the retrospective nature and unknown source of infection in most human studies, direct evidence for emergence of escape mutations in immunodominant major histocompatibility complex class I–restricted epitopes leading to immune evasion is extremely limited. In two patients infected accidentally with an identical HCV strain but who developed divergent outcomes, the total lack of HCV-specific CD4+ T cells in conjunction with vigorous CD8+ T cells that targeted a single epitope in one patient was associated with mutational escape and viral persistence. Statistical evidence for positive Darwinian selective pressure against an immunodominant epitope is presented. Wild-type cytotoxic T lymphocytes persisted even after the cognate antigen was no longer present.
Chronic hepatitis C virus (HCV) infection is common and affects 3% of the world's population; however, the diagnosis of acute HCV rarely is made and usually is inferred based on the history of a known exposure (1, 2). Emerging data suggest that immunologic and virologic events in the early stages of infection determine the eventual outcome (3). Spontaneous resolution of acute HCV infection has been associated with robust T cell responses in chimpanzees (4, 5) and humans (6–8), and it has been suggested that a threshold frequency of CTL is required for clearance of HCV (9). Although emergence of escape mutations in class I MHC–restricted epitopes was shown to lead to immune evasion in HCV-infected chimpanzees (10, 11), direct evidence for this mechanism in humans is limited. Tsai et al. (12), by focusing on a single HLA A2–restricted HCV E1 epitope in the hypervariable region, observed variant epitope sequences with CTL antagonist activity 3 mo from onset of acute hepatitis in two patients who developed persistence. Timm et al. (13) recently described the development of CTL responses against an HLA-B8–restricted epitope (including one patient after antiviral therapy); however, mutations within this immunodominant epitope did not impact HLA class I binding or TCR recognition.
We comprehensively studied two patients who developed homologous acute HCV infection after patellar tendon transplantation from a deceased donor who was in the “window” phase of acute HCV (i.e., negative for HCV antibody but positive for HCV RNA). Antigenic and viral genetic mapping revealed striking differences in these two individuals who shared several HLA alleles but had divergent outcomes. To assess the total and specific CD4+ and CD8+ T cell response against all potential HCV epitopes in an unbiased manner, we used 750 overlapping 15-mer peptides that spanned the entire HCV polyprotein. The lack of HCV-specific CD4+ T cells, in conjunction with vigorous CD8+ T cells that targeted a single immunodominant epitope, was associated with mutational escape and viral persistence. In contrast, the patient who demonstrated vigorous and multispecific CD4+ and CD8+ T cell responses spontaneously eradicated HCV infection.
RESULTS AND DISCUSSION
Divergent outcomes after exposure to identical HCV-infected donor tissue
Two subjects with no previous history of exposure to HCV underwent elective patellar ligament (with bone) transplantation for knee reconstructive surgery in April 2002. Within 2 mo, both patients developed symptoms of fatigue and elevation in liver function tests. The first patient (PD101, a 49-yr-old white man) had elevation in his liver function tests and persistent viremia, and was treated with pegylated interferon and ribavirin. The second patient (PD102, a 51-yr-old white woman) became jaundiced with a peak total serum bilirubin of 8.1 mg/dl and spontaneously cleared HCV RNA from serum within 3 mo. Their clinical and virologic courses are shown in Fig. 1. Remarkably, both patients shared several HLA alleles, including HLA A2.
Figure 1.
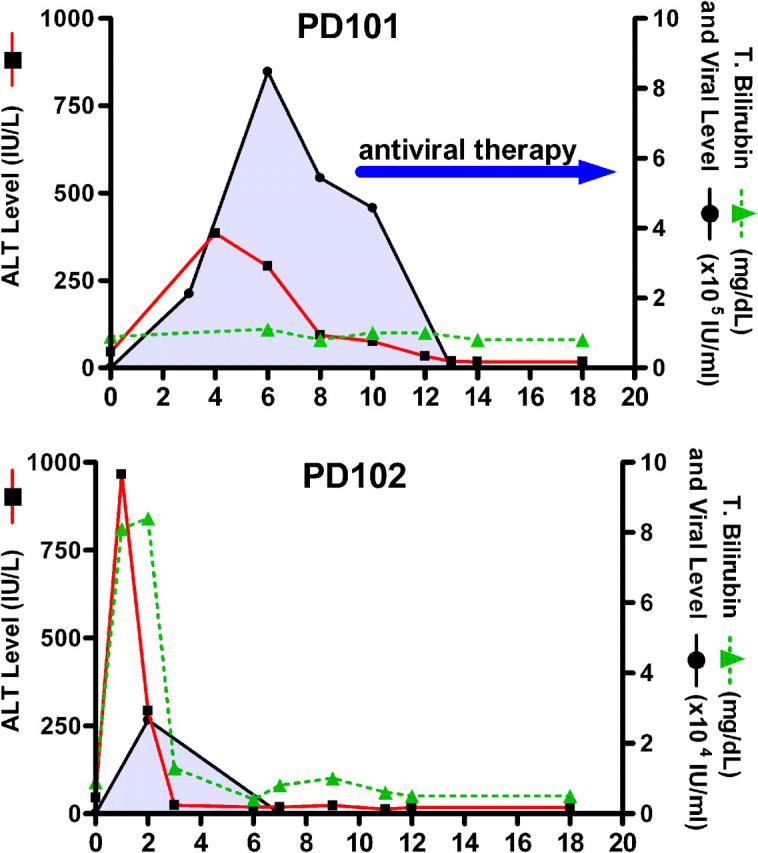
HCV infection in two subjects who were exposed accidentally on the same day to HCV genotype 1a–infected patellar tendon that resulted in divergent outcomes. PD101 (top) developed elevation in serum alanine aminotransferase (ALT) and HCV RNA viremia that persisted until initiation of antiviral therapy with pegylated interferon and ribavirin. PD102 developed marked elevations in serum alanine aminotransferase and bilirubin; viremia became undetectable spontaneously without antiviral therapy.
HCV genome-wide analysis of CD4 + and CD8 + T cell responses
To assess CD4 + and CD8 + T cell responses to the whole HCV polyprotein comprehensively, 15 mer-peptides, overlapping by 11 amino acids, were pooled and used in an IFN-γ ELISPOT assay. In brief, autologous dendritic cells were used as APCs to stimulate bead-purified CD8 + T cells with 32 peptide pools that spanned subgenomic regions; the remaining “CD4+ others” were stimulated on the same day (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20042284/DC1).
Longitudinal direct ex vivo analysis of HCV-genome wide CD4+ and CD8+ T cell responses revealed striking differences after infection in the two individuals (Fig. 2). None of the overlapping HCV peptides nor recombinant HCV proteins (not depicted) elicited CD4+ T cell responses from PD101; only one pool (NS3-5H, amino acids 1369–1479) demonstrated significant CD8+ T cell responses 6 mo after infection (Fig. 2 A). Conversely, PD102 demonstrated vigorous and multispecific CD4+ and CD8+ T cell responses (Fig. 2 B). Therefore, these results were in accord with, and extend, those reported in acute resolving HCV infection, and demonstrated an association between a strong and broad T cell response with recovery (6–8). 6 mo later, despite the fact that serum HCV RNA remained negative during that span, PD102 maintained vigorous HCV-specific CD4+ T cell responses, with 40% of the polypeptide (13 of 32 peptide pools) eliciting responses (Fig. 2 D). Because PD101 developed persistent viremia, he was treated with pegylated interferon and ribavirin starting 9.5 mo after infection. The patient's CD8+ T cells targeted determinants limited to the NS3-5H pool throughout follow-up, including 24 mo after infection (Fig. 2 E). Of the individual peptides that made up the latter pool, only one 15-mer peptide (amino acids 1401–1415) stimulated CD8+ T cell responses. Therefore, only one of the 750 peptides screened elicited CD8+ T cell responses in PD101. Mapping identified peptide 1406–1415 as the minimal epitope, previously reported to be detectable by tetramer analysis in almost one third of HLA A2-positive, HCV-exposed individuals (14).
Figure 2.

HCV–genome-wide analysis of CD4+ and CD8+ T cell responses in PD101 and PD102 at 6, 12, and 24 mo after infection. Overlapping 15 mer-peptides (n = 750) were synthesized to span the complete HCV polyprotein derived from HCV-1a (the sequence data are available from GenBank/EMBL/DDBJ under accession no. M62321) and divided into 32 peptide pools. IFN-γ production was detected using an established ELISPOT protocol. CD8+ cells were isolated from PBMCs by magnetic-activated cell sorting magnetic bead separation (Miltenyi Biotec) and the remaining CD4+ others were used. Negative controls in the ELISPOT assay were wells with purified T cells but no peptide (n = 8). After plates were dry, spots were quantified by a Zeiss microscope using KS ELISPOT software. Responses were considered positive if greater than the mean plus three SD of the control wells (DMSO only), if there were at least 10 spots above background, and/or were significant by Student's t test (P < 0.05). Shown are the mean ± SEM. (A and B) CD4+ and CD8+ T cell responses for PD101 and PD102 at 6 mo after infection. The difference in total immune response to the HCV peptides between patients was statistically significant (P = 0.0005, Fisher's two-tail test). (C and D) CD4+ and CD8+ T cell responses for PD101 and PD102 at 12 mo after infection. (E and F) T cell responses for PD101 and PD102 at 24 mo after infection.
Tetramer frequencies for this epitope ranged from 1.43% (8 mo) to 2.24% (22 mo after infection; Table S1, available at http://www.jem.org/cgi/content/full/jem.20042284/DC1). Direct ex vivo phenotypic analysis of NS3 1406 tetramer-positive cells revealed intermediate memory cells, consistently high expression of the costimulatory molecule CD28, and decreasing levels of HLA-DR after viral clearance with therapy. Over time, CCR7 expression increased, which indicated the presence of early (central)—rather than mature—effector memory CTLs (reference 15 and Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20042284/DC1). To investigate the proliferative capacity of these NS4 1406–specific CD8+ T cells at the single cell level, PBMCs were cultured short-term with HCV peptides and low-dose IL-2 and stained with carboxyfluorescein succinimidyl ester (CFSE), as described previously (14). As shown in Fig. 3, PD101 demonstrated a strikingly high frequency of proliferating NS3 1406 tetramer+ cells.
Figure 3.

Proliferation of HCV-specific tetramer+ CD8+ T cells. On day 8 of culture in HCV peptide–stimulated T cell lines derived from PD101 and PD102, CFSE-labeled tetramer+ cells were detected. In brief, 107 PBMCs were cultured in the presence of four HCV epitope peptides (NS3 1406–1415 [KLVALGINAV], NS3 1073–1081 [CINGVWCTV], and core 132–140 [DLMGYIPLV]) with IL-2 (0.5 ng/ml) added on days 0 and 3. Because the CFSE signal is diluted with each cell division, signals in the left upper quadrant represent viral-specific CD8+ T cells that have proliferated in culture. Those in right upper quadrant represent tetramer+ T cells that have not divided (reference 14). (A) In PD101, the NS3 1406–specific CD8+ T cells expanded after peptide stimulation (representing 29% of gated CD8+ T cells) and NS3 1073–specific CD8+ T cells demonstrated modest proliferation, whereas the other epitope tetramer+ cells were viable but failed to expand, as indicated by the tetramer+ cells in the upper right quadrants of the dot plots. Analysis from two other time points after infection revealed similar results. (B) For PD102, in vitro culture for 8 d led to proliferation of NS3 1406– and NS3 1073–specific CDD8+ T cell responses. In contrast, neither core-specific nor NS5B 2594–specific responses expanded in culture. Analysis from two other time points revealed similar results.
Evidence for immune escape of a single dominant epitope
Sequencing of a contiguous region encoding the NS3-5H pool revealed an amino acid substitution at amino acid position 4 of the epitope (A→V) at 4.5, 6, 8, and 10 mo after infection in PD101 (Fig. 4 A). Sequencing of the dominant virus from the donor and PD102 at 6 wk after infection revealed identity with the prototype sequence; this indicated that the alanine to valine change in PD101 represented a new mutation that arose after transmission from the donor organ. This de novo nonsynonymous mutation was propagated stably until antiviral therapy was initiated and was compatible with virus replication; serum viral load increased between the third and sixth month after infection (Fig. 1). Therefore, in accord with a recent HIV model, one could speculate that this rapidly emerging viral variant likely faced a low “genetic barrier” (requiring only one or a few nucleotide changes) that carried a low fitness cost to replication (16).
Figure 4.

Mutant viral sequence, recognition, and binding of mutant peptide. (A) Viral sequences corresponding to the single peptide that elicited CD8+ T cells from PD101 who developed viral persistence. The prototype HCV-1 peptide sequence is shown; dashes indicate identity with the prototype and substitutions are represented by standard letter codes at different time points after infection. The donor sequence is identical to the prototype sequence and the sequence derived from PD102 who spontaneously cleared HCV RNA. In contrast, PD101 developed a mutation within the NS3 1406–1415 epitope (A1409V) by 4.5 mo after infection that was propagated until antiviral therapy was initiated. (B) CD8+ T cells from PD101 (from 8 mo after infection) recognize wild-type NS3 1406 peptide, but not mutant 1406 peptide. Dose titrations of wild-type, mutant, and combination of both peptides reveal high frequency of IFN-γ–producing effector cells after pulsing of autologous DCs with wild-type peptide and culture with bead-purified CD8+ T cells without antagonism by the mutant peptide. Wild-type and nonwild-type peptides were screened at multiple concentrations. (C) MHC class I binding affinity. Transporter associtated with antigen-processing–deficient T2 cells (105) were cultured for 16 h at 26°C to increase expression of peptide-receptive cell surface molecules, and then were incubated with various concentrations of individual peptides (wild-type NS3 1406, mutant NS3 1406, or DMSO) at 37°C for 1 h, washed, and stained with FITC-labeled anti–HLA-A2 antibody (BD Biosciences), as described previously (24). The variant peptide bound to the HLA-A2 with decreased affinity (P = 0.0014), comparable to DMSO (no peptide) control. Data are expressed as the mean fluorescence intensity.
To determine if escape from T cell surveillance and chronic infection might have resulted from a mutation in this immunodominant NS3 1406 epitope, the mutant peptide was loaded on autologous DCs, and CD8+ T cell responses were tested directly ex vivo in an ELISPOT assay. As shown in Fig. 4 B, the mutant epitope failed to elicit IFN-γ CD8+ T cell responses; this supported the concept that virus variation which evolved in vivo outstripped the capacity of T cells to control the infection (i.e., circulating T cells did not react to the infecting sequence). Furthermore, because peptide variants of class I–restricted epitopes potentially could antagonize naturally occurring epitopes (17), we explored this possibility by using different ratios of wild-type and mutant peptide concentrations. Mutant peptide did not inhibit IFN-γ production after stimulation with the wild-type peptide (Fig. 4 B). These results may be due to the fact that the variant peptide bound to the HLA-A2 molecule with decreased affinity (Fig. 4 C).
To assess whether the emergence of the viral variant was the result of genetic drift or selective immune pressure, nucleotide sequences that contained the first 2741 codons of the polyprotein open reading frame were determined for the dominant quasispecies for the donor, PD101, and PD102 by directly sequencing the RT-PCR products. First, the homology between the amino acid sequence for PD101 and the peptides that we synthesized was 96.64%; thus, the peptides that were used to screen responses closely matched the infecting virus (Fig. 5). Second, the sequences from PD101 and PD102 differed by only 11 nucleotides in the open reading frame (99.9% identity) and led to 8 amino acid differences (99.7% identity). Third, the nucleotide sequences for the donor and PD102 were identical except for 1 nucleotide in the 5′UTR; their polyproteins from amino acid 1–2741 were identical. The amino acid differences between the sequences were (donor/PD102:PD101): F399L, A937V, V1384A, A1409V, T1474I, A2169T, P2341S, and D2597E. As shown in Fig. 5 B, these nonsynonymous mutations occurred throughout the HCV genome, including two HLA-A2–restricted epitopes. Therefore, in PD101, the rate of amino acid substitution within the immunodominant NS3 1406 CTL epitope (the only peptide that elicited CD8 responses) was 1 of 10 amino acids (10%) versus 4 of 2731 (0.15%) in the remaining flanking or non-HLA-A2–restricted regions. An exact Fisher test indicates that the number of substitutions is not independent of the amino acid position (P = 0.018). These data support a role for CD8+ T cell selective pressure as the cause of amino acid substitution because it is unlikely that this mutation arose randomly in this epitope.
Figure 5.

Comparison of viral genetic sequence of donor, PD101, and PD102. (A) Dendrogram showing genetic relatedness of donor, PD101, and PD102 relative to other HCV isolates. Donor, PD101 (the sequence data are available from GenBank/EMBL/DDBJ under accession no. AY695436), and PD102 PD102 (the sequence data are available from GenBank/EMBL/DDBJ under accession no. AY695437) amino acid sequences were aligned against the corresponding sequences from the reference 1a isolate from which the peptides were derived (M62321), and a genotype 1b isolate (J4). The length of the horizontal lines connecting two isolates by the shortest route (right to left and left to right) is proportional to the genetic distance between the isolates. (B) Viral sequences for the prototype HCV-1 peptide sequence, donor, PD101 (6 mo after infection), and PD102 (1.5 mo) are shown; dashes indicate identity with the prototype and substitutions are represented by standard letter codes at different time points after infection. Known class I restricted epitopes are indicated in brackets [HLA A2 and B35].
The other HLA A2–restricted epitope (NS5B) demonstrated an amino acid substitution in PD101 (D2597E) that was not present in the donor or PD102. Specific stimulation with this mutant peptide (D2597E, T2600S), prototype 1a, and donor (T2600S) peptide demonstrated proliferation to the peptide derived from the initial infecting sequence (T2600S), but not the mutant peptide (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20042284/DC1).
Reports of individuals who were exposed to a single source HCV infection have been limited to subjects who acquired the infection in the remote past (18), and consequently, immunologic and virologic analyses were performed long after the outcome of infection was determined. In this study, we prospectively tracked two patients early after they acquired HCV infection from the same acutely infected, “window period” donor. Comprehensive mapping of CD4+ and CD8+ T cell responses with overlapping 15 mer-peptides spanning the entire HCV polyprotein revealed statistically significant differences in the breadth and vigor of T cell responses between both individuals. These results are even more compelling when one considers that the autologous, infecting viral sequences and the peptides that were used to screen responses were nearly identical (presumably facilitating immune recognition), and that the subjects shared several HLA alleles, including the highly prevalent HLA A2 (also present in the donor).
Our results are consistent with the concept that the variant HCV carrying a substitution within an immunodominant CD8+ T cell epitope was selected by the monospecific antiviral CTL response, and the variant epitope (perhaps because of lack of HCV-specific CD4+ T cell help [11]) failed to generate new variant-specific CTL populations. The only peptide region that elicited IFN-γ response in PD101 was the class I–restricted epitope region where a nonsynonymous mutation arose and was maintained. In contrast, the likelihood of CTL escape is low when the CTL response is directed against multiple viral epitopes simultaneously, as in the case of PD102. We acknowledge the possibility that T cell responses were broader at time points earlier than the 6 mo when samples first became available.
One of the most striking findings in our study was that the CTL effector response (IFN-γ production and proliferative capacity) in PD101 was maintained even when the cognate antigen that it recognizes was no longer present (after viral escape and after therapeutic viral eradication). This apparent paradox was described previously in a human study of HBV infection (19) and in HCV-infected chimpanzees (9); it could be related to recurrent stimulation from an undefined reservoir of wild-type virus (20), as suggested by the recent demonstration of low-level HCV replication in PBMCs of patients with therapy-induced or spontaneous resolution of HCV infection (20). Alternatively, mutant epitopes could stimulate expansion of the wild-type–specific CTL—a phenomenon known as “original antigenic sin”—that initially was described for antibodies, but also extended to class I HLA-restricted responses (9, 21). Direct enumeration of the frequency of mutant-specific CTL with tetramers was not possible because attempts to synthesize them failed on two occasions, possibly a reflection of the fact the mutant peptide bound to the HLA-A2 molecule with lower affinity than the wild-type peptide (which also might explain the lack of antagonistic activity of the mutant peptide; Fig. 4 B).
In summary, our findings—derived by combining viral sequence data and unbiased, functional T cell analyses that examine every potential HCV epitope after homologous challenge—indicate that the absence of CD4+ T cell help, coupled with strong selective pressure exerted by wild-type–specific CTLs, favor the emergence of immune escape (11) and indicate a patient profile associated with chronicity that might benefit from early antiviral therapy. Elucidation of the mechanisms which underlie the early failure to develop HCV-specific CD4+ T cells and their instructive signals during memory CD8+ T cell differentiation has implications for vaccine strategies that are designed to induce protective immunity to this common disease (22).
MATERIALS AND METHODS
The study protocol was approved by the Human Research Committee and Institutional Review Board of Oregon Health and Science University.
Synthetic peptides.
A total of 750 overlapping (15-mers) peptides spanning the entire polyprotein (Fig. S1) of HCV-1a (accession no. M62321) was synthesized at NMI Laboratories.
ELISPOT assay.
IFN-γ production was detected using an established ELISPOT protocol (23).
Flow cytometry.
Flow cytometric analysis using a fluorescence activated cells sorter FACSCalibur (Becton Dickinson) was performed on the CD8+ T cell population to determine purity and on the CD4+ others to determine the percentage of CD3+CD4+ T cells in the population. Data analysis was performed using CellQuest and FlowJo software. Fluorescent-conjugated anti–human antibodies for T cell subsets, activation, memory, and differentiation markers (BD Biosciences and Caltag) were used. Soluble HLA-A2 tetramers containing core amino acids 132–140 (DLMGYIPLV), NS3 1073–1081 (CINGVWCTV), NS3 1406–1415 (KLVALGINAV), and NS5 2594–2602 (ALYDVVTKL) were synthesized (Beckman Coulter and NIAID tetramer facility) and used to screen responses longitudinally.
In vitro proliferation of CFSE-labeled T cells
107 cells were stained in 0.1% BSA in PBS with 1 μM CFSE (Molecular Probes); PBMCs were cultured with 10 μg/ml each of Core 131, NS3 1073, NS3 1406, and NS5 2594 epitope peptides and 0.5 ng/ml IL-2 (on days 0 and 3).
Isolation, amplification, and sequencing of HCV genomes.
The plasma of both patients were collected by centrifugation in plasma preparation tubes and frozen immediately at −80°C. RNA was isolated with the QIAamp Viral RNA Mini Kit (QIAGEN) according to the manufacturer's instructions. The RNA was reverse transcribed using random oligonucleotide primers and M-MLV Reverse Transcriptase (Fisher Scientific) for 60 min at 37°C. HCV sequences, including nt 93–8566 (reference sequence AF009606), were amplified using nested RT-PCR in four overlapping segments using the primers and conditions that are described in Table S2 (available at http://www.jem.org/cgi/content/full/jem.20042284/DC1). The sequence of the dominant quasispecies was obtained by sequencing both strands of each amplicon directly using primers averaging 300 nt apart, and the full sequence was assembled with the Vector NTI software package.
Statistical analysis.
Results were graphed and analyzed using GraphPad Prism and JMP (SAS Institute) statistical package.
Online supplemental material.
Figs. S1–S3 and Tables S1 and S2 demonstrate the whole genome sequencing and T cell mapping approach. Enumeration and phenotype of HCV-specific, HLA-A2 epitope responses and proliferative responses after stimulation with wild-type and mutant peptides are shown. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20042284/DC1.
Acknowledgments
The authors thank R. Leistikow and J. Grotzke for technical assistance and I. Williams of the Centers for Disease Control and Prevention for providing the donor specimen.
This research was supported by National Institutes of Health grants RO1 DK060590 (to H.R. Rosen) and DK60345 (to J.E. Tavis).
The authors have no conflicting financial interests.
References
- 1.Alter, M.J., 2002. Prevention of spread of hepatitis C. Hepatology. 36:S93–98. [DOI] [PubMed] [Google Scholar]
- 2.Gordon, S.C. 2003. New insights into acute hepatitis C. Gastroenterology. 125:253–256. [DOI] [PubMed] [Google Scholar]
- 3.Farci, P., A. Shimoda, A. Coiana, G. Diaz, G. Peddis, J.C. Melpolder, A. Strazzera, D.Y. Chien, S.J. Munoz, A. Balestrieri, et al. 2000. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 288:339–344. [DOI] [PubMed] [Google Scholar]
- 4.Cooper, S., A.L. Erickson, E.J. Adams, J. Kansopon, A.J. Weiner, D.Y. Chien, M. Houghton, P. Parham, and C.M. Walker. 1999. Analysis of a successful immune response against hepatitis C virus. Immunity. 10:439–449. [DOI] [PubMed] [Google Scholar]
- 5.Shoukry, N.H., A. Grakoui, M. Houghton, D.Y. Chien, J. Ghrayeb, K.A. Reimann, and C.M. Walker. 2003. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 197:1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thimme, R., D. Oldach, K.M. Chang, C. Steiger, S.C. Ray, and F.V. Chisari. 2001. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 194:1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lechner, F., D.K. Wong, P.R. Dunbar, R. Chapman, R.T. Chung, P. Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B.D. Walker. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 191:1499–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gruner, N.H., T.J. Gerlach, M.C. Jung, H.M. Diepolder, C.A. Schirren, W.W. Schraut, R. Hoffman, R. Zachoval, T. Santantonio, M. Cucciarini, et al. 2000. Association of hepatitis C virus-specific CD8+ T cells with viral clearance in acute hepatitis C. J. Infect. Dis. 181:1528–1536. [DOI] [PubMed] [Google Scholar]
- 9.Erickson A.L., Y. Kimura, S. Igarashi, J. Eichelberger, M. Houghton, J. Sidney, D. McKinney, A. Sette, A.L. Hughes, and C.M. Walker. 2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity. 15:883–895. [DOI] [PubMed] [Google Scholar]
- 10.Weiner, A., A.L. Erickson, J. Kansopon, K. Crawford, E. Muchmore, A.L. Hughes, M. Houghton, and C.M. Walker. 1995. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc. Natl. Acad. Sci. USA. 92:2755–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grakoui, A., N.H. Shoukry, D.J. Woollard, J.H. Han, H.L. Hanson, J. Ghrayeb, K.K. Murthy, C.M. Rice, and C.M. Walker. 2003. HCV persistence and immune evasion in the absence of memory T cell help. Science. 302:659–662. [DOI] [PubMed] [Google Scholar]
- 12.Tsai, S.L., Y.M. Chen, M.H. Chen, C.Y. Huang, I.S. Sheen, C.T. Yeh, J.H. Huang, G.C. Kuo, and Y.F. Liaw. 1998. Hepatitis C virus variants circumventing cytotoxic T lymphocyte activity as a mechanism of chronicity. Gastroenterology. 115:954–965. [DOI] [PubMed] [Google Scholar]
- 13.Timm, J., G.M. Lauer, D.G. Kavanagh, I. Sheridan, A.Y. Kim, M. Lucas, T. Pillay, K. Ouchi, L.L. Reyor, J.S. Zur Wiesch, et al. 2004. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 200:1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wedemeyer, H., X.S. He, M. Nascimbeni, A.R. Davis, H.B. Greenberg, J.H. Hoofnagle, T.J. Liang, H. Alter, and B. Rehermann. 2002. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 169:3447–3458. [DOI] [PubMed] [Google Scholar]
- 15.Lucas, M., A.L. Vargas-Cuero, G.M. Lauer, E. Barnes, C.B. Willberg, N. Semmo, B.D. Walker, R. Phillips, and P. Klenerman. 2004. Pervasive influence of hepatitis C virus on the phenotype of antiviral CD8+ T cells. J. Immunol. 172:1744–1753. [DOI] [PubMed] [Google Scholar]
- 16.Altman, J.D., and M.B. Feinberg. 2004. HIV escape: there and back again. Nat. Med. 10:229–230. [DOI] [PubMed] [Google Scholar]
- 17.Klenerman, P., S. Rowland-Jones, S. McAdam, J. Edwards, S. Daenke, D. Lalloo, B. Koppe, W. Rosenberg, D. Boyd, and A. Edwards. 1994. Cytotoxic T-cell activity antagonized by naturally occurring HIV-1 Gag variants. Nature. 369:403–407. [DOI] [PubMed] [Google Scholar]
- 18.Takaki, A., M. Wiese, G. Maerten, and E. Depla. 2000. Cellular immune responses persist, humoral response responses decrease two decades after recovery from a single source outbreak of hepatitis C. Nat. Med. 6:578–582. [DOI] [PubMed] [Google Scholar]
- 19.Bertoletti A., A. Costanzo, F.V. Chisari, M. Levrero, M. Artini, A. Sette, A. Penna, T. Giuberti, F. Fiaccadori, and C. Ferrari C. 1994. Cytotoxic T lymphocyte response to a wild type hepatitis B virus epitope in patients chronically infected by variant viruses carrying substitutions within the epitope. J. Exp. Med. 180:933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham, T.N., S.A. MacParland, P.M. Mulrooney, H. Cooksley, N.V. Naoumov, and T.I. Michalak. 2004. Hepatitis C virus persistence after spontaneous or treatment-induced resolution of hepatitis C. J. Virol. 78:5867–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klenerman, P., and R.M. Zinkernagel. 1998. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature. 394:482–485. [DOI] [PubMed] [Google Scholar]
- 22.Kaech, S.M., and R. Ahmed. 2003. CD8 T cells remember with a little help. Science. 11:263–265. [DOI] [PubMed] [Google Scholar]
- 23.Wertheimer, A.M., C. Miner, D.M. Lewinsohn, A.W. Sasaki, E. Kaufman, and H.R. Rosen. 2003. Novel CD4+ and CD8+ T-cell determinants within the NS3 protein in subjects with spontaneously resolved HCV infection. Hepatology. 37:577–589. [DOI] [PubMed] [Google Scholar]
- 24.Wedemeyer H., E. Mizukoshi, A.R. Davis, J.R. Bennink, and B. Rehermann. 2001. Cross-reactivity between hepatitis C virus and influenza A virus determinant-specific cytotoxic T cells. J. Virol. 75:11392–11400. [DOI] [PMC free article] [PubMed] [Google Scholar]