

Abstract
Circulating murine monocytes comprise two largely exclusive subpopulations that are responsible for seeding normal tissues (Gr-1−/CCR2−/CX3CR1high) or responding to sites of inflammation (Gr-1+/CCR2+/CX3CR1lo). Gr-1+ monocytes are recruited to the site of infection during the early stages of immune response to the intracellular pathogen Toxoplasma gondii. A murine model of toxoplasmosis was thus used to examine the importance of Gr-1+ monocytes in the control of disseminated parasitic infection in vivo. The recruitment of Gr-1+ monocytes was intimately associated with the ability to suppress early parasite replication at the site of inoculation. Infection of CCR2−/− and MCP-1−/− mice with typically nonlethal, low doses of T. gondii resulted in the abrogated recruitment of Gr-1+ monocytes. The failure to recruit Gr-1+ monocytes resulted in greatly enhanced mortality despite the induction of normal Th1 cell responses leading to high levels of IL-12, TNF-α, and IFN-γ. The profound susceptibility of CCR2−/− mice establishes Gr-1+ monocytes as necessary effector cells in the resistance to acute toxoplasmosis and suggests that the CCR2-dependent recruitment of Gr-1+ monocytes may be an important general mechanism for resistance to intracellular pathogens.
Monocytes recruited to the site of an infection potentially bring critical contributions to the immune control of foreign invaders. On arrival, they may differentiate into macrophages that act as effectors or into DCs that efficiently ignite the adaptive immune response (1). Markers for human monocyte subpopulations were described more than a decade ago (2), but markers for corresponding murine subsets have been identified only recently. Circulating murine monocytes comprise two distinct subpopulations that serve very different roles (3). Although Gr-1−/CCR2−/CX3CR1high monocytes seed normal tissues, their counterparts (Gr-1+/CCR2+/CX3CR1lo) represent “inflammatory” monocytes that home to sites of inflammation (3–5). Comparable subsets that exist in humans are known as CD16+/CX3CR1high/CCR2− and CD14+/CX3CR1lo/CCR2+, although their precise roles are less well delineated (3).
Gr-1+ monocytes express the CCR2 chemokine receptor and respond primarily to the ligand MCP-1 (CCL2), as well as MCP-3 (CCL7) and MCP-5 (CCL12; references 6, 7). MCP-1 is produced by a variety of cells in response to inflammation and plays a critical role in the recruitment of monocytes (8). CCR2−/− mice have profound defects in monocyte recruitment though constitutive trafficking remains unaffected, which makes it possible to dissect the role of monocyte recruitment during inflammation (9). Recently, a unique population of TNF/inducible nitric oxide synthase–producing (Tip)–DCs was found to be absent from the spleens of CCR2−/− mice during infection with Listeria monocytogenes (10). Tip-DCs are CCR2+ and contribute to the early control of L. monocytogenes, which suggests that the recruitment of monocytes via CCR2 plays a role in the early control of infection with other intracellular pathogens.
The resistance to an acute challenge with a Toxoplasma gondii infection in a mouse requires the rapid induction of a robust Th1 cell–polarized cell-mediated immune response that leads to the curtailment of early parasite replication (11). Resistance to both the acute and chronic stages of infection with T. gondii hinges primarily on a high level production of IFN-γ (12, 13). Consistent with this requirement, IL-12−/− (14, 15), IFN-γ−/− (16), and IFN-γR−/− mice (13) have all been shown to be extremely susceptible to acute toxoplasmosis and rapidly succumb to a challenge with nonvirulent T. gondii strains that are nonlethal in normal mice. Toxoplasma is able to invade and replicate in a wide variety of cell types, including professional phagocytes, and IFN-γ responsiveness in both hematopoietic and nonhematopoietic cell lineages is important for resistance (13).
The functions of neutrophils (17–19) and CD8α+ splenic DCs (20–22) in the production of IL-12 in early responses to infection with T. gondii have been documented in previous studies. Macrophages cultured in vitro also produce IL-12 when challenged with T. gondii (23); however, it is uncertain as to what extent this response is important in vivo. Although the early induction of cytokines is a key mediator of resistance to infection, it is less clear if phagocytic cells also play important effector roles in the control of acute toxoplasmosis. A prominent feature of nonlethal infections in mice initiated with low doses of a nonvirulent strain of T. gondii is the early predominance of Gr-1+ macrophages at the site of infection (24). We have previously shown that these Gr-1+ monocytes produce inducible nitric oxide synthase, secrete IL-12, and are able to inhibit parasite growth when challenged in vitro (24), all of which suggest that they contribute in vivo to the control of parasite replication.
To determine the role of inflammatory monocytes in the control of toxoplasmosis, we examined the response to infections in MCP-1−/− and CCR2−/− mice in which monocyte recruitment is selectively impaired. Our results demonstrate an essential, nonredundant requirement for CCR2 and a substantial role for MCP-1 in the recruitment of Gr-1+ inflammatory monocytes. We also find that Gr-1+ monocytes are essential for the local control of parasite replication, which indicates that monocytes play a major effector role in the resistance to toxoplasmosis.
Results
Parasites are a potent stimulus for chemokine induction
Previous experiments have demonstrated a profound influx of CD68+/Gr-1+ macrophages during i.p. infection with T. gondii (24). To identify chemokines that might contribute to monocyte recruitment to the site of infection, we examined the expression of chemokines by RNase protection assay after the low-dose infection of mice (Fig. 1 A). Peritoneal exudate cells from T. gondii–infected mice displayed substantially increased mRNA levels for several of the chemokine genes assayed when compared with peritoneal exudate cells from mock-infected mice. Specifically, MCP-1 (CCL2), MIP-1β (CCL4), and lymphotactin (XCL1) mRNA levels were all observed to be increased by >70-fold (Fig. 1 B), and less substantial increases in the mRNA levels were observed for RANTES, MIP-1α, and MIP-2.
Figure 1.

Chemokines are up-regulated in response to an infection with a nonlethal challenge of T. gondii. (A) Peritoneal cells were collected 72 h after i.p. inoculation of mice with 102 T. gondii (Tg) or media diluent (mock). Harvested total RNA was subjected to RNase protection assay for the indicated chemokine genes. (B) Relative levels of expression during infection for chemokine genes assayed when compared with a mock infection control were determined by phosphor image analysis. Data shown are representative of two independent experiments with similar results.
The increase in MCP-1 mRNA levels after infection was intriguing in light of the primary role that MCP-1 has been shown to play in the recruitment of monocytes/macrophages to inflammation sites (4, 8). We examined the production of MCP-1 in response to an infection by measuring its concentration in peritoneal lavage fluid (Fig. 2 A) over the course of the first 5 d after infection and the i.p. inoculation of 100 parasites. The MCP-1 concentration in peritoneal exudates was observed to increase beginning on day 3, with the first significant (P < 0.01) increase in MCP-1 concentration measured at day 4 after infection. The concentration was observed to subsequently decline.
Figure 2.
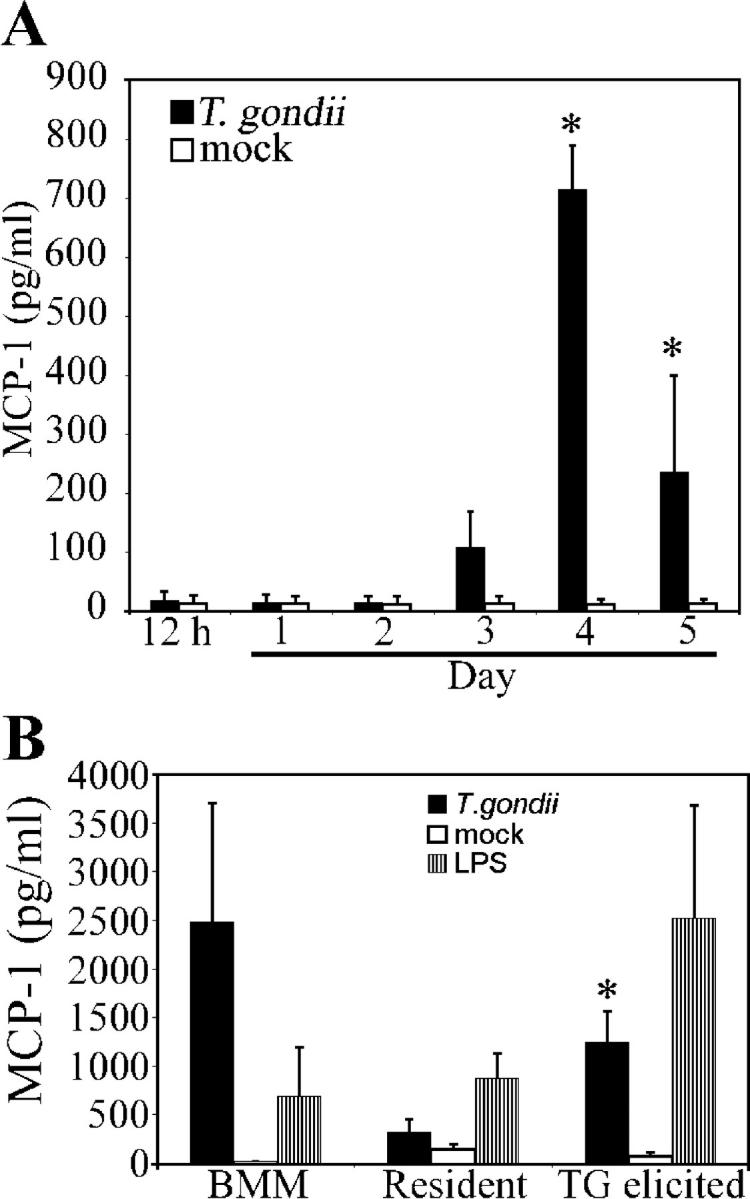
Kinetics of MCP-1 release in peritoneum. (A) C57BL/6J mice were i.p. infected with 102 parasites of T. gondii. The concentration of MCP-1 present in peritoneal lavage fluid was determined by ELISA at the times indicated after infection (*, significantly different from mock infection; P < 0.01). (B) Naive BMM, resident peritoneal macrophages, or thioglycolate (TG)-elicited macrophages were challenged in vitro with live parasites (1:1, cells/parasites). MCP-1 concentrations in supernatant media following 24 h of infection were determined by ELISA (*, significantly different from resident peritoneal macrophages; P < 0.05). Data shown are means ± SE for two independent experiments (using data from three individual mice per time point in A).
MCP-1 could be produced by resident cells or by influxing proinflammatory monocytes. Therefore, we evaluated the potential for these subpopulations to produce MCP-1. Naive bone marrow–derived macrophages (BMM), resident peritoneal macrophages, or thioglycolate-elicited inflammatory macrophages were challenged in vitro with live T. gondii, and MCP-1 production was measured by performing ELISA on samples of supernatant culture media collected after 24 h. We found that all cell types tested, including naive BMM and resident tissue macrophages, produced MCP-1 in response to a T. gondii infection. Thioglycolate-elicited inflammatory macrophages generated significantly higher MCP-1 concentrations (P < 0.05) than resident peritoneal macrophages, which suggests that cells residing in the peritoneum release MCP-1 in response to an infectious challenge, and influxing cells (possibly migrating in response to the early, low levels of MCP-1) further amplify the recruitment signal on their arrival.
MCP-1 and CCR2 are essential for resistance to acute toxoplasmosis
To examine the functional consequences of the high-level induction of MCP-1, we challenged MCP-1−/−, CCR2−/−, and wild-type C57BL/6J mice with doses of T. gondii that are normally nonlethal (Fig. 3). In contrast to wild-type background control mice, which demonstrated high levels of survival at either dose (100% survival at 102 parasites and 80% survival at 103 parasites), both strains of knockout mice displayed a lack of resistance to the acute stage of toxoplasmosis. CCR2−/− mice were profoundly susceptible to acute toxoplasmosis (a dose of 103 parasites was uniformly lethal). Even the lowest dose, 102 parasites, resulted in 90% mortality with 80% of infected mice succumbing by day 15 after infection. MCP-1−/− mice were also more susceptible than wild-type control mice at both inoculum levels tested. Although ∼90% of MCP-1−/− mice survived a challenge with 102 parasites (compared with 100% survival for wild-type C57BL/6J mice), a challenge with 103 parasites resulted in 50% mortality for MCP-1−/− mice (20% mortality for the wild type). Disease progression in MCP-1−/− mice appeared qualitatively different from CCR2−/− mice; mice succumbed at later time points after infection, and some mice displayed signs of neurological abnormality (altered gait or loss of balance) before death at these later time points.
Figure 3.

Survival of CCR2−/− and MCP-1−/− mice after infection with T. gondii. Female CCR2−/−, MCP-1−/−, and wild-type C57BL/6J (WT) mice were infected i.p. with doses of 102 and 103 tachyzoites, and their survival was monitored for 30 d. Data are expressed as a cumulative percentage of two experiments (four to five mice per group).
Failure to recruit inflammatory monocytes/macrophages to the site of infection
We next wanted to evaluate the necessity of MCP-1 and CCR2 for the recruitment of inflammatory monocytes/macrophages to the peritoneum during the early stages of infection. We used FACS analysis to examine the percentage of Gr-1+/CD68+ cells in the peritoneum of MCP-1−/−, CCR2−/−, and wild-type C57BL/6J mice (Fig. 4). On day 4 after infection (i.p.) with 102 T. gondii, CCR2−/− mice were found to have significantly (P < 0.05) decreased numbers of Gr-1+/CD68+ cells present in the peritoneum (Fig. 4 and Table I). MCP-1−/− mice also failed to recruit wild-type levels of inflammatory monocytes/macrophages. However, on average they suffered a less severe recruitment defect than CCR2−/− mice (Fig. 4 and Table I).
Figure 4.

Recruitment of inflammatory monocytes (Gr-1+/CD68+) is abrogated in CCR2−/− mice, but less so in MCP-1−/− mice. Peritoneal cells from (A) wild-type C57BL/6J (WT), (B) MCP-1−/−, and (C) CCR2−/− mice were harvested 4 d after i.p. inoculation of a dose of 102 T. gondii and analyzed by flow cytometry. Results shown are representative of individual mice. Numbers represent the percentage of gated cells present within the quadrant. The experiment was repeated twice with similar results, using a total of six mice per genotype.
Table I.
Percentage representation of peritoneal populations
| Mouse genotype | |||
|---|---|---|---|
| Population | C57BL/6J | MCP-1− / − | CCR2− / − |
| %a | % | %b | |
| Gr-1+/CD68+ | 28.3 ± 2.7 | 22.8 ± 5.5 | 7.6 ± 0.1 |
| CD68+ | 11.9 ± 2.8 | 5.3 ± 6.0 | 8.1 ± 1.2 |
| Gr-1+ | 4.0 ± 0.6 | 5.1 ± 0.5 | 4.7 ± 0.7 |
Percentages were determined by FACS analysis on day 4 after infection.
Data are means ± SE from two experiments (n = 6 mice).
Significantly different from other genotypes (P < 0.05).
IFN-γ production is essentially normal in CCR2−/− and MCP-1−/− mice
We analyzed the pattern of cytokine production in response to a T. gondii infection in MCP-1−/−, CCR2−/−, and wild-type C57BL/6J mice by measuring the concentrations of cytokines in the peritoneal exudate and serum (Fig. 5) at 4, 7, and 10 d after mice were infected (i.p.) with 102 T. gondii. Interestingly, for the cytokines measured (IL-4, TNF-γ, IL-12p70, and IFN-γ), MCP-1−/− and CCR2−/− mice were capable of generating concentrations comparable to those produced by wild-type C57BL/6J mice at all time points analyzed. Similarly, although the knockout strains of mice generated lower serum concentrations of IL-12p70 at days 4 and 7 after infection, the only concentration found to be significantly lower than wild-type levels (P < 0.05) was the serum level of IL-12p70 in CCR2−/− mice at day 7 after infection. Moreover, no significant deviation from wild-type levels in the serum concentration of IFN-γ, TNF-α, or IL-4 of MCP-1−/− and CCR2−/− mice was identified at any of the time points analyzed, which indicates that the difference in serum IL-12p70 concentration in CCR2−/− mice at day 7 does not result in the systemic underproduction of IFN-γ. Splenocytes harvested from infected CCR2−/− and MCP-1−/− mice (days 7 and 10) and stimulated with low concentrations (5 μg/ml) of soluble Toxoplasma Ag produced more IFN-γ than splenocytes from infected wild-type C57BL/6J mice (unpublished data). This further indicates that the CCR2−/− and MCP-1−/− mice are competent producers of IFN-γ.
Figure 5.

CCR2−/− and MCP-1−/− mice produce high levels of IFN-γ. Peritoneal lavage fluid and serum were collected on days 4, 7, and 10 after infection from C57BL/6J (WT), MCP-1−/−, and CCR2−/− mice that had received a dose of 102 T. gondii (i.p.). Concentrations of IL-12p70, IFN-γ, TNF-α, and IL-4 were determined by ELISA. Data shown are means ± SE from two independent experiments (n = 3 individual mice per genotype) at each time point.
Control of parasite numbers
The lack of resistance to acute toxoplasmosis by mice possessing a targeted deletion of a necessary immune response component is typically accompanied by the failure to control replication of the parasite (e.g., IFN-γ−/− and LRG-47−/− mice). To determine if a similar phenotype was observed in mice in which monocyte recruitment was impaired, parasites present in the peritoneal exudate of infected MCP-1−/−, CCR2−/−, and wild-type C57BL/6J mice were enumerated by plaque assay. Considerably greater numbers of parasites were found in the peritoneal contents of CCR2−/− mice (Fig. 6). CCR2−/− mice were found to have a more profound defect in their ability to suppress parasite numbers than MCP-1−/− mice. This result was also confirmed by real-time PCR assays (unpublished data).
Figure 6.

CCR2−/− and MCP-1−/− mice fail to control early parasite replication, allowing systemic dissemination. CCR2−/−, MCP-1−/−, and wild-type C57BL/6J mice were i.p. infected with a dose of 102 T. gondii. Parasites were enumerated by performing plaque assays using recovered intraperitoneal contents at the indicated times after infection. Data shown are means ± SD (from two mice per genotype) from a representative experiment of two with highly similar outcomes (*, significantly different from WT; P < 0.05).
Neuropathology in MCP-1−/− mice
To explore the possibility that MCP-1−/− mice, which perish at later time points (day 19–30 after infection), succumb to neuropathology, we examined sections of brain tissue from infected MCP-1−/− and wild-type C57BL/6J mice in a blinded fashion. The pathological features observed were characteristic of a T. gondii infection, with three distinguishable grades of severity present in the brain tissues examined. The least severe pattern of pathology found (Fig. 7 B) was a multifocal meningoencephalitis with minimal perivascular cuffing or neuronal degeneration. The intermediate grade of severity (Fig. 7 C) manifested as a diffuse meningoencephalitis and occasionally noted neuronal degeneration. The most severe grade (Fig. 7 D) was assigned to sections that displayed one or more foci of necrosis and a marked loss of parenchyma in addition to the features of the intermediate grade. The composite severity score of independently examined sections revealed that sections from MCP-1−/− mice were significantly more likely (P < 0.05) to display a more severe grade of pathology (Table II). In mice with intermediate or severe grade inflammation, encephalitis was accompanied by meningitis and perivascular infiltration of mononuclear cells (Fig. 7, E and F).
Figure 7.
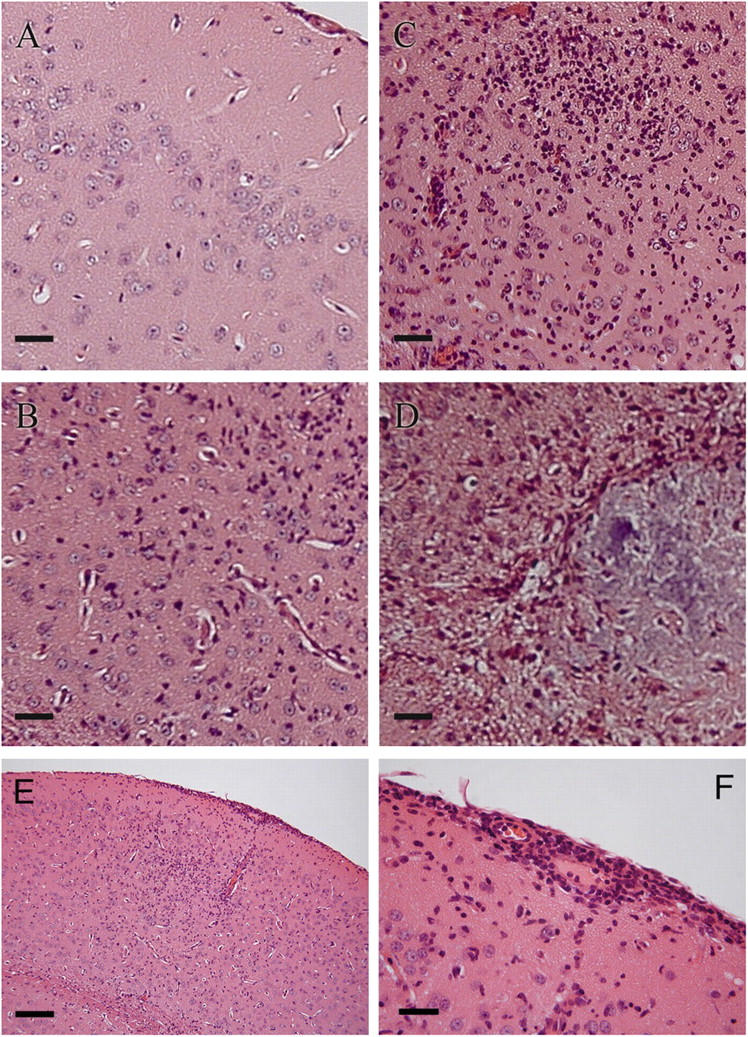
Patterns of CNS inflammation observed during T. gondii infection in MCP-1−/− mice. (A–D) Hematoxylin and eosin–stained sections from brains harvested at day 35 after infection from MCP-1−/− and wild-type C57BL/6J mice infected i.p. with a dose of 102 T. gondii were examined and graded for the severity of inflammation in a blinded fashion. Representative images of the different grades of neuropathology that were observed. (A) Normal control (uninfected); (B) least severe (+); (C) intermediate (++); and (D) most severe (+++) are shown. (E) Example of encephalitis and meningitis with the infiltration of mononuclear cells. (F) Enlarged region of the meninges showing infiltration and perivascular infiltration of mononuclear cells. Similar pathology was found in a second experiment. Bars: (A–D and F) 5 μm; (E) 20 μm.
Table II.
MCP-1−/− mice suffer more severe neuropathology
Sections were assigned a severity grade in a blind fashion. Data represent five nonserial sections per mouse and four mice per genotype.
Distinguishing characteristics of severity grades are as follows: least severe (+), multifocal meningoencephalitis with minimal perivascular cuffing; intermediate (++), diffuse meningoencephalitis with more extensive perivascular cuffing; and most severe (+++), diffuse meningoencephalitis with cuffing the same as in (++), with necrotic foci, and loss of parenchyma.
Significantly more likely to be found to have more severe pathology (Mann-Whitney U test; P < 0.05).
Number of sections assigned to designated grade.
Discussion
Resistance to acute toxoplasmosis in the mouse model is associated with the ability to suppress parasite replication through a variety of IFN-γ–dependent static and cidal mechanisms (11). Effective control requires the early production of IL-12 that may be produced by neutrophils (19, 25, 26), DCs (21), or macrophages (23). Because Gr-1+/CD68+ macrophages are the predominant myeloid population found in the peritoneum early after infection with a nonvirulent strain of T. gondii (24), we examined their recruitment during the early stages of infection with T. gondii. The contribution of Gr-1+ monocytes was delineated by examining infections in mice lacking the major ligand and receptor involved in inflammatory monocyte recruitment. CCR2−/− and MCP-1−/− mice were found to be extremely susceptible to acute toxoplasmosis and were unable to survive typically nonlethal doses of a nonvirulent strain of T. gondii. Increased susceptibility of CCR2−/− or MCP-1−/− mice occurred despite the induction of normally high levels of Th1 cell cytokines, which indicates that in the absence of recruitment of Gr-1+ monocyte cells, these cytokines are not able to control infection. Our results identify an essential requirement for CCR2, as well as a contributory role for MCP-1, in the recruitment of Gr-1+ monocytes during infection, and they document an important in vivo role for Gr-1+ monocytes as effector cells in resistance to acute toxoplasmosis.
T. gondii is capable of infecting a wide range of cell types in vivo, including professional phagocytes (11). Although it is clear that the induction of IL-12 and IFN-γ is essential for controlling acute infection, the effector cells that are responsible for the control of parasite replication are uncertain. Macrophages (27, 28) and nonphagocytic cells (29) can be activated to inhibit or kill the parasites in vitro. Additionally, both hematopoietic and nonhematopoietic cells must express IFN-γ receptors in vivo to adequately control parasite replication (13). However, the relative importance of specific cell types to the control of infection in vivo remains uncertain.
Extensive focus has been placed on the contributions made to early responses to T. gondii infection by neutrophils in the peritoneum (17, 19) and splenic CD8α+ DCs (20–22). Both populations produce IL-12 in response to T. gondii exposure in an MyD88-dependent manner (30, 31), and this response is thought to be important for the induction of IFN-γ, an essential mediator of resistance (12). CCR5 knockout mice develop more severe chronic infections with T. gondii even though they are not impaired in controlling acute infections, which suggests that IL-12 induction via CCR5 positive CD8α+ DCs is primarily important for controlling the later stages of infection (20).
Previous studies have shown that human and murine neutrophils produce IL-12 in response to T. gondii (25, 26); however, this pathway also does not appear to be essential for the control of acute toxoplasmosis in the mouse model. Challenging CXCR2−/− mice with T. gondii results in more cysts being present in the brain of chronically infected mice even though they do not appear to be susceptible to increased mortality during the acute phase (32). It is likely that this phenotype accurately reflects the role of neutrophils as CXCR2−/− mice suffer an important neutrophil recruitment defect, but maintain the normal recruitment of monocytes (32). Several previous studies have used RB6-8C5 mAb to deplete Gr-1+ populations in mice in order to assess the contribution of neutrophils in host defense against T. gondii (18, 19, 33). In light of the recent description of Gr-1+ monocytes and their capacity to specifically home to sites of inflammation (3–5), defects that occur after RB6-8C5 mAb treatment may reflect the combined roles of Gr-1+ monocytes and neutrophils.
Importantly, low-dose intraperitoneal infections with nonvirulent strains of T. gondii, a model that simulates dissemination from natural oral infection, fail to recruit substantial numbers of neutrophils or DCs to this site of primary infection (24). Rather, the primary cell type recruited during this low-dose infection is CD68+/Gr-1+ monocytes that are capable of controlling the proliferation of the parasite (24). Our present study demonstrates that this population of Gr-1+ monocytes is required to control acute toxoplasmosis in the mouse. In the absence of Gr-1+ monocyte recruitment to the site of infection, mice succumbed rapidly to toxoplasmosis characterized by increased burdens of parasites both locally and, later, systemically. Previous experiments have demonstrated that lethality in the mouse model is related to the rapid attainment of the high tissue burdens of parasites (34). CD68+/Gr-1+ monocytes recruited during acute toxoplasmosis also make IL-12 (24); however, this role appears to be secondary as, in their absence, mice develop nearly identical levels of IFN-γ in the peritoneum and systemically. Collectively, these findings indicate that Gr-1+ monocytes are essential effector cells that act primarily downstream of IL-12–IFN-γ induction to control the initial replication of T. gondii in the mouse model. Because an absence of MCP-1 or CCR2 has not been found to notably alter the recruitment of neutrophils (4, 8, 9), the susceptibility of MCP-1−/− and CCR2−/− mice to toxoplasmosis can be specifically attributed to the recruitment of Gr-1+ monocytes. In contrast, the other major subset of murine monocytes (Gr-1−/CCR2−/CX3CR1high) has previously been shown not to play an essential role in the control of toxoplasmosis based on infections done in CX3CR1−/− mice (35).
The substantially higher numbers of parasites present on day 7 after infection in the peritoneal contents of infected MCP-1−/− and CCR2−/− mice compared with wild-type mice clearly indicate a failure to control early parasite replication at the initial site of infection. The observed differences in parasite number (∼7- and ∼2.5-fold more parasites in CCR2−/− and MCP-1−/− mice, respectively, compared with wild-type mice) are similar to those recently reported for highly susceptible STAT1−/− mice (36). The enhanced parasite numbers in CCR2−/− and MCP-1−/− mice also mimic what is seen with virulent strains of the parasite in wild-type mice (14). Collectively, these data suggest that the early control of parasite proliferation is a key determinant of survival that is provided by Gr-1+ monocytes which home to sites of infection primarily in response to MCP-1. Thus, even though nonhematopoetic cells are important in controlling T. gondii replication (13), they are evidently not sufficient to protect animals against lethal infections in the absence of Gr-1+ monocyte recruitment. This requirement may simply reflect the efficient toxoplasmicidal responses of Gr-1+ monocytes, which are recruited in high numbers, produce nitric oxide, and are able to inhibit parasite replication (24).
In other infectious challenge models, CCR2−/− mice have demonstrated a delay in the development of the IFN-γ responses or an imbalance in Th1 versus Th2 cell responses, which contribute to the increased susceptibility to Mycobacterium, Leishmania, and Cryptococcus (37–39). In this study, serum IL-12 concentrations in both CCR2−/− and MCP-1−/− mice were found to lag behind those found in wild-type mice during the first 7 d after infection (Fig. 6). IL-12 production by macrophages in response to nonvirulent strains of T. gondii has only recently been appreciated (23), and it is possible that Gr-1+ monocytes also play a role in vivo in the early induction of IL-12. Although CCR2−/− and MCP-1−/− mice initially produced less IL-12, they developed similar levels of IFN-γ by day 4 after infection, which indicates that other cell types are sufficient for the ignition of this essential axis of resistance to T. gondii.
Tip-DCs have previously been shown to play an important role in resistance to Listeria in the mouse model (10). There are clear phenotypic differences between Tip-DCs and the inflammatory monocytes described here, despite the fact that both rely on MCP-1 and CCR2 for trafficking. Notably, Tip-DCs express DC markers (DEC-205, CD11c) but not macrophage markers such as F4/80; this is the opposite of the phenotype of T. gondii–induced inflammatory Gr-1+ monocytes described here. The control of listeriosis also critically depends on TNF-α supplied by Tip-DCs (10). In contrast, TNF-α responses were not diminished in CCR2−/− mice challenged with T. gondii; this cytokine is important in resistance to chronic infection but is not essential for the control of acute toxoplasmosis (40). Nonetheless, the Tip-DCs and Gr-1+ monocytes described here may represent different manifestations of a common lineage of proinflammatory monocytes that are capable of differentiating into DCs. Indeed, previous studies have shown that Gr-1+ monocytes are capable of differentiating into DCs in vitro and in vivo (3). Hence, Gr-1+ monocytes may also play a vital role in sampling inflammatory sites before differentiating into DCs and migrating from periphery to central lymphatic tissues, where they may also be important in the control of infection.
Although MCP-1−/− mice did not succumb to T. gondii with the rapidity displayed by CCR2−/− mice, their susceptibility to acute toxoplasmosis was considerably enhanced relative to wild-type mice when challenged with higher doses of T. gondii (Fig. 3). CCR2 is the primary receptor for MCP-1 (41), but mice produce two other known CCR2 ligands, the monocyte chemotactic proteins MCP-3 and MCP-5 (42, 43), which might serve to partially compensate for the lack of MCP-1. Consistent with this hypothesis, MCP-1−/− mice were found to recruit fewer Gr-1+ monocytes to the peritoneum after i.p. challenge with T. gondii than wild-type mice. In agreement with such a model, brain tissues from MCP-1−/− mice challenged with a normally nonlethal dose of T. gondii (Fig. 3) were considerably more likely to display severe neuropathology than tissues from chronically infected wild-type mice. Strikingly, the most severe degree of neuropathology, marked by the presence of necrotic foci with a gross loss of brain parenchyma, was never observed in tissues from wild-type mice. Studies of experimental autoimmune encephalomyelitis severity in MCP-1−/− have highlighted a critical role for MCP-1 in the recruitment of macrophages to the tissues of the central nervous system (CNS; reference 44). By extension, tachyzoites of T. gondii that escape initial control may disseminate into the CNS and replicate with impunity in the absence of the MCP-1–mediated recruitment of monocytes. The infection of human fibroblasts and endothelial cells also results in the induction of MCP-1 (45), raising the possibility that the recruitment of inflammatory monocytes is important in the control of human toxoplasmosis, which frequently involves encephalitis in immunocompromised patients.
The importance of other chemokines and their receptors for resistance to acute toxoplasmosis has been studied previously by others (46, 47), though these studies focused primarily on the recruitment of T cells. Besides its role in the recruitment of CD8α+ DCs, CCR5 has been shown to mediate the recruitment of TGF-β–secreting CD8+ intraepithelial lymphocytes to sites of intestinal inflammation in mice orally infected with T. gondii (46). The antibody depletion of an interferon-induced 10-kD protein (IP-10), a chemoattractant that signals through the chemokine receptor CXCR3 that is expressed on activated Th1 cells, resulted in extreme susceptibility in mice orally infected with a nonvirulent strain of T. gondii (47). The depressed recruitment (and subsequent expansion) of antigen-specific CD8+ T cells to the spleen in antibody-depleted mice was implicated in the failure to control parasite numbers. Because CD8+ T cells contribute to host defense via the production of IFN-γ, but their cytolytic capability is less critical (48), it is plausible that the inhibited recruitment leads to lower systemic levels of IFN-γ, which depresses the activity of IFN-γ–dependent resistance mechanisms. CCR1−/− mice fail to control parasite numbers in many tissues after oral infection with a nonvirulent strain of T. gondii and rapidly succumb to the infection (49). Notably, in humans both CCR1 and CXCR2 are expressed on CD14+CD16− monocytes that correspond to the murine subset of Gr-1+ inflammatory monocytes (3). However, given the expression of CCR1 on multiple cell types (neutrophils, monocytes, lymphocytes, and eosinophils), CCR1−/− mice likely suffer recruitment defects for multiple cell types.
Our studies demonstrate that the successful recruitment of Gr-1+ monocytes via CCR2 and MCP-1 is intimately associated with the ability of mice to resist acute toxoplasmosis. CCR2−/− and MCP-1−/− mice displayed defective recruitment of Gr-1+ monocytes and failed to control parasite numbers, despite producing normal levels of IFN-γ. Our findings on toxoplasmosis, combined with previous studies on other microorganisms (37–39) argue for a critical role for Gr-1+ monocytes as primary effector cells in resistance to pathogens.
Materials and Methods
Experimental animals.
C57BL/6J and MCP-1−/− mice were obtained from the Jackson Laboratory. CD1 mice were obtained from Charles River Breeding Laboratories. CCR2−/− mice were generated by W.A. Kuziel. MCP-1−/− and CCR2−/− mice represent the 10th generation backcrossed with a C57BL/6 background. Knockout mice were obtained as breeding pairs, with all subsequent breeding and husbandry of animals undertaken under specific pathogen-free conditions at the Washington University School of Medicine. All animal work was conducted in accordance with the Washington University School of Medicine Animal Studies Committee.
Parasite culture and preparations.
Strain PTG (50841; American Type Culture Collection), a cloned line of the ME49 strain of T. gondii, was maintained by serial 2-d passage of tachyzoites in human foreskin fibroblast monolayers as described previously (23). Parasites were routinely tested for Mycoplasma contamination using a GenProbe kit (Fisher Scientific) and remained negative throughout the experiments.
RNase protection assay of chemokine mRNAs.
Total RNA was isolated using TRIzol (Invitrogen). RNase protection assays (BD Biosciences) were performed with 5′-α-[32P]UTP (GE Healthcare), according to the kit manufacturer's directions, using 15 μg of total RNA per sample and an mCK-5 probe set supplied by the manufacturer. Phosphor imaging and data analysis of the resulting gels were accomplished using an FLA-5000 phosphor imager and software supplied by the manufacturer (Fujifilm). For quantification, signal intensity values for individual chemokines were normalized against the value of a housekeeping gene for the respective treatment group.
Harvest of peritoneal exudates.
Immediately after being killed, mice were peritoneally lavaged with 5 ml of ice-cold HBSS supplemented with 10 mM Hepes and 0.1 mM EGTA. For the recovery of cellular contents, recovered lavage fluid was centrifuged at 200 g for 10 min at 4°C. For the analysis of chemokine levels in the peritoneum, recovered lavage fluid was centrifuged at 400 g for 10 min at 4°C, and the clarified supernatant stored at −70°C until analyzed by ELISA.
Macrophage culture.
Resident peritoneal and thioglycolate-elicited macrophages were obtained from C57BL/6J mice. Thioglycolate elicitation was accomplished via i.p. inoculation of 1.5 ml of 3% thioglycolate (Edge Biologicals) 4 d before harvest. Cells were plated in 96-well plates, 2 × 105 per well, in DMEM supplemented with 2% FBS. After incubating for 2 h at 37°C, nonadherent cells were removed by washing the wells three times and replacing the medium with complete DMEM. BMM were obtained as previously described (23).
ELISA measurement of chemokine levels.
MCP-1, TNF-α, IL-12p70, IFN-γ, and IL-4 protein concentrations were determined using an OptEIA kit (BD Biosciences) according to the manufacturer's directions.
FACS analysis.
After treatment with erythrocyte lysis solution (BD Biosciences) and repeat washing, harvested cells were suspended in staining buffer (BD Biosciences), seeded in 96-well plates (106 cells/100 μl), and pretreated with BD FcBlock (clone 2.4G2; BD Biosciences) for 10 min at 4°C. Staining with PE conjugated anti-Gr1 (clone RB6-8C5; BD Biosciences) for 1 h at 4°C and washing preceded fixation, permeabilization (BD Cytofix/Cytoperm; BD Biosciences), and subsequent staining with FITC conjugated anti-CD68 (clone FA-11; Serotec) for 1 h at 4°C. Cells (≥5 × 104) were analyzed using a FACScan flow cytometer (BD Immunocytometry Systems). Data were analyzed using Cell Quest software (BD Immunocytometry Systems).
Parasite enumeration.
Total peritoneal contents were harvested via peritoneal lavage as described in the Harvest of peritoneal exudates section. Aliquots of recovered fluid were immediately (without centrifugation) added to confluent monolayers of human foreskin fibroblast in 96-well culture plates. Cultures were initiated in quadruplicate with independent serial dilutions performed. After 5–7 d of undisturbed culture, plaques were counted to determine the initial number of parasites present in the inoculum.
Histopathology.
Tissues were fixed in 4% neutral buffered formaldehyde and embedded in paraffin wax. 5-μm sections were hematoxylin and eosin or periodic acid Schiff stained according to standard procedures.
Statistics.
The comparison of means was performed using a one-way analysis of variance (F-statistic) to establish whether significant differences were present. Pairwise comparisons between means were performed using the least significant difference test (based on a Student's t test), allowing for the estimation of p-values, when the F-statistic indicated that significant differences existed (50). Analysis of the assignment of neuropathology severity scores was performed using the nonparametric Mann-Whitney U test using Minitab v.12.21 software.
Acknowledgments
We thank Julie Suetterlin for excellent technical assistance and Drs. Robert Schreiber, Daniel Goldberg, and Stephen Beverley for helpful discussions.
L.D. Sibley was supported by a grant from the National Institutes of Health (AI 36629). P.M. Robben was supported by a grant from the National Institutes of General Medical Sciences Training (5 T32 07200). L.D. Sibley is the recipient of a Scholar Award in Molecular Parasitology from the Burroughs Wellcome Fund.
The authors have no conflicting financial interests.
Abbreviations used: BMM, BM-derived macrophages; CNS, central nervous system; Tip-DC, TNF/inducible nitric oxide synthase–producing DC.
W.A. Kuziel's present address is Autoimmunne and Inflammatory Diseases, Protein Design Laboratories, Inc., Fremont, CA 94555.
References
- 1.Muller, W.A., and G.J. Randolph. 1999. Migration of leukocytes across endothelium and beyond: molecules involved in the transmigration and fate of monocytes. J. Leukoc. Biol. 66:698–704. [DOI] [PubMed] [Google Scholar]
- 2.Passlick, B., D. Flieger, and H.W. Ziegler-Heitbrock. 1989. Identification and characterization of a novel monocytes subpopulation in human peripheral blood. Blood. 74:2527–2534. [PubMed] [Google Scholar]
- 3.Geissmann, F., S. Jung, and D.R. Littman. 2003. Blood monocytes consist of two principle subsets with distinct migratory properties. Immunity. 19:71–82. [DOI] [PubMed] [Google Scholar]
- 4.Henderson, R.B., J.A.R. Hobbs, M. Mathies, and N. Hogg. 2003. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. 102:328–335. [DOI] [PubMed] [Google Scholar]
- 5.Taylor, P.R., and S. Gordon. 2003. Pattern recognition receptors and differentiation antigens define murine myeloid cell heterogeneity ex vivo. Eur. J. Immunol. 33:2090–2097. [DOI] [PubMed] [Google Scholar]
- 6.Luther, S.A., and J.G. Cyster. 2001. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2:102–122. [DOI] [PubMed] [Google Scholar]
- 7.Homey, B., A. Muller, and A. Zlotnik. 2002. Chemokines: agents for the immunotherapy of cancer? Nat. Rev. Immunol. 2:175–184. [DOI] [PubMed] [Google Scholar]
- 8.Lu, B., B.J. Rutledge, L. Gu, J. Fiorillo, N.W. Lukacs, S.L. Kunkel, R. North, C. Gerard, and B.J. Rollins. 1998. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1–deficient mice. J. Exp. Med. 187:601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuziel, W.A., S.J. Morgan, T.C. Dawson, S. Griffin, O. Smithies, K. Ley, and N. Maeda. 1997. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. USA. 94:12053–12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serbina, N.V., T.P. Salazar-Mather, C.A. Biron, W.A. Kuziel, and E.G.P. Am. 2003. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 19:59–70. [DOI] [PubMed] [Google Scholar]
- 11.Hunter, C.A., and G. Reichmann. 2001. Immunology of Toxoplasma infection. Toxoplasmosis. A Comprehensive Clinical Guide. D.H. Joynson and T.J. Wreghitt, editors. Cambridge University Press, New York. 43–57.
- 12.Suzuki, Y., M.A. Orellana, R.D. Schreiber, and J.S. Remington. 1988. Interferon-γ: the major mediator of resistance against Toxoplasma gondii. Science. 240:516–518. [DOI] [PubMed] [Google Scholar]
- 13.Yap, G.S., and A. Sher. 1999. Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-γ– and tumor necrosis factor (TNF)-α–dependent host resistance to the intracellular pathogen, Toxoplasma gondii. J. Exp. Med. 189:1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan, I.A., T. Matsuura, and L.H. Kasper. 1994. Interleukin-12 enhances murine survival against acute toxoplasmosis. Infect. Immun. 62:1639–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gazzinelli, R.T., S. Heiny, T.A. Wynn, S. Wolf, and A. Sher. 1993. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon γ by an intracellular parasite and induces resistance in T-cell-deficient mice. Proc. Natl. Acad. Sci. USA. 90:6115–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scharton-Kersten, T.M., T.A. Wynn, E.Y. Denkers, S. Bala, E. Grunvald, S. Hieny, R.T. Gazzinelli, and A. Sher. 1996. In the absence of endogenous IFN-γ mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol. 157:4045–4054. [PubMed] [Google Scholar]
- 17.Bennouna, S., S.K. Bliss, T.J. Curiel, and E.Y. Denkers. 2003. Cross-talk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J. Immunol. 171:6052–6058. [DOI] [PubMed] [Google Scholar]
- 18.Bliss, S.K., L.C. Gavrilescu, A. Alcaraz, and E.Y. Denkers. 2001. Neutrophil depletion during Toxoplasma gondii infection leads to impaired immunity and lethal systemic pathology. Infect. Immun. 69:4898–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bliss, S.K., B.A. Butcher, and E.Y. Denkers. 2000. Rapid recruitment of neutrophils containing prestored IL-12 during microbial infection. J. Immunol. 165:4515–4521. [DOI] [PubMed] [Google Scholar]
- 20.Aliberti, J., C.R. Sousa, M. Schito, S. Hieny, T. Wells, G.B. Huffnagle, and A. Sher. 2000. CCR5 provides a signal for microbial induced production of IL-12 by CD8-α dendritic cells+. Nat. Immunol. 1:83–87. [DOI] [PubMed] [Google Scholar]
- 21.Sousa, C.R., S. Hieny, T. Scharton-Kersten, D. Jankovic, H. Charest, R.N. Germain, and A. Sher. 1997. In vivo microbial stimulation induces rapid CD40 ligand–independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 186:1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sousa, C.R., G. Yap, O. Schulz, N. Rogers, M. Schito, J. Aliberti, S. Hieny, and A. Sher. 1999. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 11:637–647. [DOI] [PubMed] [Google Scholar]
- 23.Robben, P.M., D.G. Mordue, S.M. Truscott, K. Takeda, S. Akira, and L.D. Sibley. 2004. Induction of IL-12 by Toxoplasma gondii depends on the parasite genotype. J. Immunol. 172:3686–3694. [DOI] [PubMed] [Google Scholar]
- 24.Mordue, D.G., and L.D. Sibley. 2003. A novel population of Gr-1+ activated macrophages induced during acute toxoplasmosis. J. Leukoc. Biol. 74:1015–1025. [DOI] [PubMed] [Google Scholar]
- 25.Bliss, S.K., A.J. Marshall, Y. Zhang, and E.Y. Denkers. 1999. Human polymorphonuclear leukocytes produce IL-12, TNF-α, and the chemokines macrophage-inflammatory protein-1α and -1β in response to Toxoplasma gondii antigens. J. Immunol. 162:7369–7375. [PubMed] [Google Scholar]
- 26.Bliss, S.K., Y. Zhang, and E.Y. Denkers. 1999. Murine neutrophil stimulation by Toxoplasma gondii antigen drives high level production of IFN-γ-independent IL-12. J. Immunol. 163:2081–2088. [PubMed] [Google Scholar]
- 27.Adams, L.B., J.B. Hibbs, R.R. Taintor, and J.L. Krahenbuhl. 1990. Microbiostatic effect of murine-activated macrophages for Toxoplasma gondii: role for synthesis of inorganic nitrogen oxides from L-arginine. J. Immunol. 144:2725–2729. [PubMed] [Google Scholar]
- 28.Collazo, C.M., G.S. Yap, G.D. Sempowski, K.C. Lusby, L. Tessarollo, G.F. Woude, A. Sher, and G.A. Taylor. 2001. Inactivation of LRG-47 and IRG-47 reveals a family of interferon γ–inducible genes with essential, pathogen-specific roles in resistance to infection. J. Exp. Med. 194:181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfefferkorn, E.R. 1984. Interferon-γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host to degrade tryptophan. Proc. Natl. Acad. Sci. USA. 81:908–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Rio, L., B.A. Butcher, S. Bennouna, S. Hieny, A. Sher, and E.Y. Denkers. 2004. Toxoplasma gondii triggers myeloid differentiation factor 88-dependent IL-12 and chemokine ligand 2 (monocyte chemoattractant protein 1) responses using distinct parasite molecules and host receptors. J. Immunol. 172:6954–6960. [DOI] [PubMed] [Google Scholar]
- 31.Scanga, C.A., J. Aliberti, D. Jankovic, F. Tilloy, S. Bennouna, E.Y. Denkers, R. Medzhitov, and A. Sher. 2002. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 168:5997–6001. [DOI] [PubMed] [Google Scholar]
- 32.Del Rio, L., S. Bennouna, J. Salinas, and E.Y. Denkers. 2001. CXCR2 deficiency confers impaired neutrophil recruitment and increased susceptibility during Toxoplasma gondii infection. J. Immunol. 167:6503–6509. [DOI] [PubMed] [Google Scholar]
- 33.Sayles, P.C., and L.L. Johnson. 1996. –97. Exacerbation of toxoplasmosis in neutrophil-depleted mice. Nat. Immun. 15:249–258. [PubMed] [Google Scholar]
- 34.Mordue, D.G., F. Monroy, M. La Regina, C.A. Dinarello, and L.D. Sibley. 2001. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J. Immunol. 167:4574–4584. [DOI] [PubMed] [Google Scholar]
- 35.Jung, S., J. Aliberti, P. Graemmel, M.J. Sunshine, G.W. Kreutzberg, A. Sher, and D.R. Littman. 2000. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20:4106-4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gavrilescu, L.C., B.A. Butcher, L. Del Rio, G.A. Taylor, and E.Y. Denkers. 2004. STAT1 is essential for antimicrobial effector function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infect. Immun. 72:1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters, W., H.M. Scott, H.F. Chambers, J.L. Flynn, I.F. Charo, and J.D. Ernst. 2001. Chemokine receptor 2 serves and early and essential role in resistance to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA. 98:7958–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato, N., W.A. Kuziel, P.C. Melby, R.L. Reddick, V. Kostecki, W. Zhao, N. Maeda, S.K. Ahuja, and S.S. Ahuka. 1999. Defects in the generation of IFN-gamma are overcome to control infection with Leishmania donovani in CC chemokine receptor (CCR) 5-, macrophage inflammatory protein-1 alpha- or CCR2-deficient mice. J. Immunol. 163:5519–5525. [PubMed] [Google Scholar]
- 39.Traynor, T.R., W.A. Kuziel, G.B. Toews, and G.B. Huffnagle. 2000. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J. Immunol. 164:2021–2027. [DOI] [PubMed] [Google Scholar]
- 40.Yap, G.S., T. Scharton-Kersten, H. Charest, and A. Sher. 1998. Decreased resistance of TNF receptor p55- and p75-deficient mice to chronic toxoplasmosis despite normal activation of inducible nitric oxide synthase in vivo. J. Immunol. 160:1340–1345. [PubMed] [Google Scholar]
- 41.Gu, L., S.C. Tseng, and B.J. Rollins. 1999. Monocyte chemoattractant protein-1. Chem. Immunol. 72:7–29. [DOI] [PubMed] [Google Scholar]
- 42.Combiadere, C., S.K. Ahuka, J. Van Damme, H.L. Tiffany, J.L. Gao, and P.M. Murphy. 1995. Monocyte chemoattractant protein-3 is a functional ligand for CC chemokine receptors 1 and 2B. J. Biol. Chem. 270:29671–29675. [DOI] [PubMed] [Google Scholar]
- 43.Sarafi, M.N., E.A. Garcia-Zepeda, J.A. MacLean, I.F. Charo, and A.D. Luster. 1997. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J. Exp. Med. 185:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang, D.R., J. Wang, P. Kivisakk, B.J. Rollins, and R.M. Ransohoff. 2001. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 193:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denney, C.F., L. Eckmann, and S.L. Reed. 1999. Chemokine secretion of human cells in response to Toxoplasma gondii infection. Infect. Immun. 67:1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luangsay, S., L.H. Kasper, N. Rachinel, L.A. Minns, F.J. Mennechet, A. Vandewalle, and D. Buzoni-Gatel. 2003. CCR5 mediates specific migration of Toxoplasma gondii-primed CD8 lymphocytes to inflammatory intestinal epithelial cells. Gastroenterology. 125:491–500. [DOI] [PubMed] [Google Scholar]
- 47.Khan, I.A., J.A. MacLean, F.S. Lee, L. Casciotti, E. DeHaan, J.D. Schwartzman, and A.D. Luster. 2000. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 12:483–494. [DOI] [PubMed] [Google Scholar]
- 48.Yap, G.S., and A. Sher. 1999. Cell-mediated immunity to Toxoplasma gondii: initiation, regulation and effector function. Immunobiology. 201:240–247. [DOI] [PubMed] [Google Scholar]
- 49.Khan, I.A., P.M. Murphy, L. Casciotti, J.D. Schwartzman, J. Collins, J.L. Gao, and G.R. Yeaman. 2001. Mice lacking the chemokine receptor CCR1 show increased susceptibility to Toxoplasma gondii infection. J. Immunol. 166:1930–1937. [DOI] [PubMed] [Google Scholar]
- 50.Kuehl, R.O. 1994. Statistical Principles of Research Design and Analysis. Duxbury Press, Belmont, CA. 686 pp.