

Abstract
Primary infection with varicella-zoster virus (VZV) causes the characteristic syndrome of varicella, or chickenpox. Experiments in severe combined immunodeficiency mice with human skin grafts (SCIDhu mice) indicate that VZV infection of T cells can mediate transfer of infectious virus to skin. VZV-infected T cells reached epithelial sites of replication within 24 h after entering the circulation. Memory CD4+ T cells were the predominant population recovered from skin in SCIDhu mice given uninfected or infected mononuclear cells, suggesting that immune surveillance by memory T cells may facilitate VZV transfer. The increased susceptibility of memory T cells to VZV infection may further enhance their role in VZV pathogenesis. During VZV skin infection, viral gene products down-regulated interferon-α to permit focal replication, whereas adjacent epidermal cells mounted a potent interferon-α response against cell–cell spread. Interleukin-1α, although activated in VZV-infected cells, did not trigger expression of endothelial adhesion molecules, thereby avoiding early recruitment of inflammatory cells. The prolonged varicella incubation period appears to represent the time required for VZV to overcome antiviral responses of epidermal cells and generate vesicles at the skin surface. Modulation of VZV replication by cutaneous innate immunity may avoid an incapacitating infection of the host that would limit opportunities for VZV transmission.
Keywords: T cell immune surveillance, viral pathogenesis, inflammation, immune evasion, interferon-α
Introduction
Varicella-zoster virus (VZV) is an alphaherpesvirus that causes varicella (chickenpox), establishes latency in sensory ganglia, and reactivates as zoster. Primary VZV infection begins with respiratory mucosal inoculation, but the characteristic chickenpox rash does not appear for 10–21 d (1). Understanding initial events during human herpesvirus infections is challenging because animal models are often not available, or infection in other species may not fully reproduce the characteristics of human disease. In the absence of experimental data, it has been suggested that the progression of VZV infection during the incubation period of varicella may resemble the stages of infection associated with the pathogenesis of mousepox (2). According to this hypothesis, VZV infects mononuclear cells in regional LNs, causing a primary viremia that carries virus to reticuloendothelial organs, such as the liver. Detection of VZV-infected mononuclear cells in peripheral blood at the end of the incubation period is considered to represent a secondary viremia, resulting from viral amplification in reticuloendothelial tissues. VZV transport to skin has been attributed to this late secondary viremia and is presumed to be followed by the rapid appearance of the classical vesicular skin rash.
VZV infects primary human T cells in vitro and exhibits tropism for T cells in thymus/liver xenografts in our severe combined immunodeficiency (SCID) mouse model in vivo (3–5). In previous experiments, we found that VZV preferentially infected activated memory CD4+ T cells in vitro, which are abundant in tonsillar tissues adjacent to the respiratory epithelial sites of VZV inoculation (3–5). The purpose of these experiments was to use SCID mice with human skin xenografts (SCIDhu) to examine whether VZV-infected tonsil T cells have the capacity to deliver infectious virus from the venous circulation and cause the development of cutaneous lesions in vivo. Since we found that VZV was transferred to skin, the phenotypes of immune cells recovered from skin xenografts in mice given uninfected and VZV-infected cells were compared. Memory CD4+ T cells predominated under both conditions, suggesting that these cells may be important for VZV transfer because memory CD4+ T cells are inherently more likely to traffic to skin. The contribution of memory CD4+ T cells to VZV pathogenesis may be further enhanced by the 3:1 increase in VZV infectivity for this subpopulation compared with naïve CD4+ T cells (5). Our investigations further indicate that the pathogenesis of VZV infection during the incubation period is modulated by potent innate immune responses, especially IFN-α, mounted by epidermal cells within their differentiated tissue microenvironment in vivo. These observations suggest that viral replication in reticuloendothelial organs, although not excluded, is not necessary to account for the 10–21-d incubation period of varicella. Whereas VZV spreads rapidly from cell to cell and causes cytolysis within 24–48 h in vitro, our experiments suggest that innate epidermal immune mechanisms may prevent an incapacitating infection of the host that would otherwise limit opportunities for VZV transmission to other susceptible individuals.
Materials and Methods
VZV Infection of SCIDhu Mice.
Skin xenografts were made in homozygous CB-17scid/scid mice using human fetal tissue obtained according to federal and state regulations; animal use was approved by the Stanford University Administrative Panel on Laboratory Animal Care. Human T cells were isolated from tonsil tissue, obtained according to an approved protocol, and infected by coculture of 5–10 × 106 T cells with VZV-infected primary human lung (HEL) cells (5). Infected T cells were injected into SCIDhu mice with skin xenografts by tail vein; xenografts were harvested at intervals after T cell transfer and tested for infectious virus yields and by immunohistologic analysis (5). In other experiments, skin xenografts were inoculated directly with VZV-infected HEL cells (10 μl) (4). To recover immune cells from skin xenografts, tissues were minced, expressed through a 190-μm-pore-size sieve (Sigma-Aldrich), and centrifuged in Ficoll-Hypaque medium (Promega, Inc.) (6). Total numbers of immune cells were counted, and phenotypes were assessed by flow cytometry using anti-CD3 (cone S4.1, IgG2a), anti-CD4 (clone 3.5, mIgG2a), anti-CD19 (clone SJ25-C1, mIgG1), anti-CD45RA (clone MEM56, mIgG2b) (Caltag Inc.), and anti–cutaneous lymphocyte antigen (CLA) (HECA-452, rat IgM; BD Biosciences). Data for 50,000 events was collected for each marker.
Anti–IFN-α/β Receptor Antibody Blocking.
Immediately after direct inoculation of bilateral skin xenografts with VZV-infected HEL cells, animals were given mouse mAb to human IFN-α/β receptor β chain (IFNα/βRβ) (PBL Biomedical Laboratories) by i.p. injection (25 μg). Administration of anti–IFN-α/β receptor antibody was maintained by s.c. delivery (50 μg) through an Alzet osmotic pump (model 1007D; 0.5 μl/h, 7 d) (Alzet, Cupertino). Skin xenografts were harvested after 7 d.
VZV Lesion Biopsies.
Punch biopsies were obtained from patients with varicella (n = 3) or zoster (n = 2) according to an approved protocol. Specimens were collected between 36–96 h after rash onset, embedded in OCT compound (TissueTec; Miles Laboratories), frozen in isopentane, and stored at −70°C.
Immunohistochemistry.
Formalin-fixed, paraffin-embedded skin sections (5 μm) were deparaffinized, rehydrated, and treated with antigen retrieval reagent (Vector Laboratories). Frozen sections were fixed in 2% paraformaldehyde, treated with 1% Triton X-100 (vol/vol) and 1% sodium citrate (wt/vol), washed in TBS followed by biotin blocking (Vector Laboratories), and treated with 3% hydrogen peroxidase. Primary antibodies included: anti-CD3ɛ (Ab-1, rabbit polyclonal), anti-CD4 (clone KT69-7, mIgG1), anti-CD8 (rabbit polyclonal), anti-CD20 (Ab-1, clone L26, mIgG2aκ), anti-CD45RA (Ab-1, clone 111-1C5, mIgG1κ), and anti-CD31/PECAM (clone JC/70A, mIgG1κ) (Lab Vision); anti-CLA (HECA-452, rat IgM) (BD Biosciences); anti-CD45RO (clone OPD4, mIgG1), anti–E-selectin (clone 1.2B6, mIgG1κ), anti–intercellular adhesion molecule (ICAM)-1 (CD54, clone 6.5B5, mIgG1κ), and anti–vascular adhesion molecule (VCAM)-1 (clone 1.4C2, mIgG1κ) (DAKO); anti–IFN-α (rabbit polyclonal) (PBL Biomedical Laboratories); and anti–IL-1α (clone c-18, goat polyclonal), anti–TNF-α (clone 1E8-G6, mIgG1), and anti-pStat1 (Tyr705, rabbit polyclonal) (Santa Cruz Biotechnology, Inc.). Anti-CCR4 (clone 1G1, mIgG1) mAb was a gift from David Andrew (Millennium Inc., Boston, MA). VZV proteins were detected with human polyclonal anti-VZV IgG or mouse mAb to gE (gpI, mIgG2b; Chemicon). Staining was done with biotinylated secondary antibody and HRP-conjugated streptavidin (Lab Vision). Signals were developed with DAB chromagen (brown) or Vector VIP (purple) and counterstained with hematoxylin (for DAB) or methyl green (for Vector VIP) (Vector Laboratories).
Results
Transfer of VZV to Skin.
VZV-infected tonsil T cells were given i.v. to SCID mice with human skin xenografts that were shown to be vascularized by human CD31+ endothelial cells (Fig. 1 A). 15–30% of CD3+ T cells in the inoculum expressed VZV glycoproteins (Fig. 1 B). CD3+ T cells were detected within the epidermis, dermis, and around hair follicles within 24 h (Fig. 1 C, left). VZV was recovered from skin within 7 d in three experiments. Progression to characteristic VZV lesions, with epidermal thickening, cellular degeneration, and keratin disruption, occurred in 10–21 d (Fig. 1 C, middle). Localized VZV lesions destroyed basement membranes and extended through the epithelial surfaces (Fig. 1 C, right).
Figure 1.

Transfer of VZV by CD3+ T cells. Column-purified CD3+ T cells from tonsils were cocultured with VZV-infected HEL cells for 48 h before i.v. transfusion into SCID mice with human skin xenografts. Skin implants were harvested at various times after T cell transfer, snapped frozen, and sectioned. (A) Human fetal skin xenografts differentiated in SCID mice at 4 wk expressed human CD31 on the endothelial cells (magnification: 100×; insert, 200×). (B) 15–30% of tonsillar T cells used to inject SCIDhu mice expressed VZV glycoproteins as shown in a representative flow cytometric analysis. (C) CD3+ T cells were detected around hair follicles and along basement membranes 24 h after i.v. injection (left, arrows; insert, rabbit IgG control); VZV-induced cytopathology was shown by hematoxylin and eosin (H & E) stain 21 d after T cell transfer compared with uninfected control (middle) and by anti-VZV IgG (brown, top right; bottom right, VZV nonimmune IgG control) (magnification: left, 400×; middle and right, 100×).
Since T cells were infected with VZV in vitro by coculture, some VZV-infected HEL cells might have remained within the T cell inoculum after Ficoll gradient centrifugation and could have been a source of VZV transfer to skin xenografts. Therefore, mice were injected with a high inoculum of VZV-infected HEL cells (5.0 × 106 cells). Infected HEL cells did not transfer VZV to skin.
After i.v. injection of VZV-infected T cells, memory CD45RO+ T cells were common in skin xenografts, whereas no CD45RA+ T cells were detected in >20 serial skin sections. To further investigate these observations, patterns of migration of uninfected mononuclear cells into skin xenografts were analyzed in animals given 5.0 × 106 cells by tail vein infusion. Prior to transfer, the percentages of cells that expressed CD3, CD19, or CD4/CD45RA were 88, 8, and 31%, respectively. The total number of CD3 T cells recovered from skin xenografts (n = 8) was 0.1–1.0% of the initial i.v. inoculum. When the phenotypes of cells recovered from skin xenografts at 48 h after transfer were examined, 90–96% were CD4+ and 97–100% were memory CD4+ T cells, whereas only 0.3–0.5% expressed CD19 (Table I). Whereas the initial percentage of naïve T cells was 31%, only 0.7–3% of CD4+ T cells recovered from the xenografts were CD45RA+. These observations suggested that memory CD4+ T cells predominated in skin xenografts after the inoculation of VZV-infected mononuclear cells because this subpopulation of immune cells had enhanced capacity to traffic to skin independently of VZV infection. The percentage of CLA+ CD4+ T cells recovered from skin was also enriched from 0.7% to 1.5–3.6% after transfer of uninfected mononuclear cells but remained a relatively low frequency subpopulation, as it was in the experiments in which VZV-infected mononuclear cells were transferred (Tables I and II). Based on the expected survival of 5–10% of T cells after adoptive transfer (7), ∼20% infection of T cells in the inoculum and the recovery of 0.1–1.0% of T cells from skin xenografts, we estimate that <50–500 VZV-infected T cells reach the skin xenografts in this model.
Table I.
Total Numbers of CD3 T Cells and Phenotypes of Uninfected Mononuclear Cells Recovered from Skin Xenografts after i.v. Transfer to SCIDhu Mice
| Animala
|
CD3+
T cells |
CD4+
|
Naive CD4
|
Memory CD4
|
CLA+CD4+
|
CD19+
|
|---|---|---|---|---|---|---|
|
|
|
%
|
%
|
%
|
%
|
%
|
| 1 | 1.4 × 103 | 92 | 3.0 | 97 | 1.5 | 0.3 |
| 2 | 3.8 × 103 | 96 | 1.0 | 99 | 3.3 | 0.3 |
| 3 | 4.0 × 103 | 90 | 0 | 100 | 3.6 | 0.3 |
| 4 | 3.6 × 104 | 92 | 0.7 | 99.3 | 2.0 | 0.5 |
Data represent means for two xenografts from each animal; n = 8 skin xenografts.
Table II.
Identification of Mononuclear Cell Populations in VZV-infected Skin Specimens
| Type of skin
|
||
|---|---|---|
| Surface markers expressed by infiltrating cells |
VZV-infected skin xenografts |
VZV patient skin biopsies |
| CD20 | – | + |
| CD3 | + | + |
| CD4 | + | + |
| CD8 | + | + |
| CD45RA | – | + |
| CD45RO | + | + |
| CLA | +, <1% | + |
| CCR4 | – | + |
Results represent cell phenotypes observed in at least three frozen sections from the same skin xenograft or VZV patient lesion biopsy specimen upon testing with each of the markers listed. –, not detected; +, abundantly present.
IL-1α and IFN-α Expression in Uninfected and VZV-infected Skin Xenografts.
Since VZV causes rapid cytolysis of cultured cells in vitro (1), the slow replication of VZV in vivo suggested possible innate immune modulation. IL-1α was expressed constitutively in the cytoplasm of epidermal cells in uninfected skin. In VZV-infected xenografts, IL-1α expression persisted and underwent nuclear translocation within infected cells (Fig. 2, middle), although retaining its cytoplasmic localization in uninfected cells within the epidermal tissues (Fig. 2, right). TNF-α was not detected in uninfected or VZV-infected skin xenografts (not depicted).
Figure 2.

IL-1α expression in VZV-infected and uninfected epidermal cells. In this representative figure, formalin-fixed, paraffin-embedded sections of VZV-infected skin xenografts were stained with anti-VZV IgG, detected with DAB chromagen (brown) as shown in the left panel (magnification, 100×). Serial sections of these samples were stained with anti–IL-1α and detected with DAB as illustrated in the middle and right panels (magnification, 630×). IL-1α expression was analyzed in cells expressing VZV proteins as shown within the area outlined in the left panel and in neighboring uninfected epidermal cells in the same section. IL-1α was activated and translocated to the nuclei of VZV infected cells (middle) but showed a diffuse cytoplasmic stain in neighboring uninfected cells (right).
Like IL-α, IFN-α was expressed constitutively in uninfected skin cells. In VZV-infected skin xenografts, IFN-α was down-regulated consistently within VZV infected cells, whereas it was expressed prominently in neighboring uninfected epidermal cells (Fig. 3 A). Phosphorylation of Stat 1 protein was examined to confirm this evidence of inhibition of the IFN-α pathway in VZV-infected cells and its activation in uninfected cells. IFN-α ligation to IFN-α receptors induces tyrosine phosphorylation of Stat1 by JAK kinases (8). IFN-α is inhibited if Stat1 does not undergo phosphorylation and nuclear translocation. Nuclear, phosphorylated Stat1 was detected in uninfected epidermal cells near VZV-infected cells but not in cells expressing VZV proteins (Fig. 3 B). Although IFN-α was expressed constitutively, Stat1 was not phosphorylated and remained cytoplasmic in uninfected skin xenografts (Fig. 3 B). These observations indicated that a VZV gene product(s) blocks Stat1 activation in order to inhibit IFN-α production in foci of infected skin cells in vivo.
Figure 3.
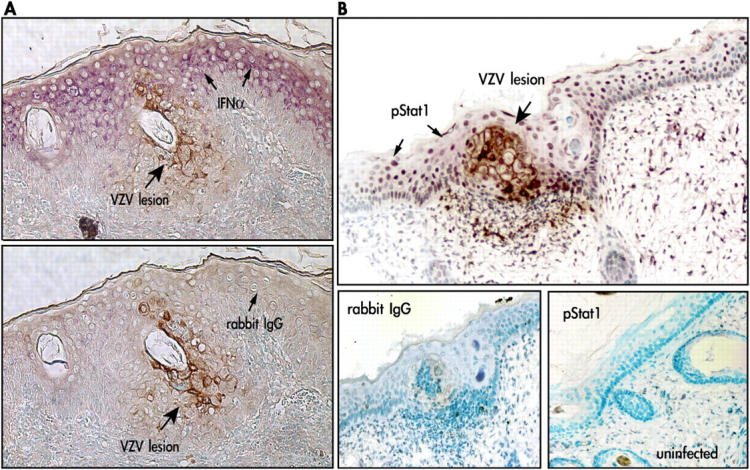
IFN-α expression and Stat1 phosphorylation in VZV-infected and uninfected epidermal cells. Formalin- fixed, paraffin-embedded sections of VZV-infected skin xenografts were pretreated and double stained with anti-VZV IgG, detected with DAB (brown), and anti–IFN-α or anti-pStat1 antibody detected with Vector VIP (purple). Sections are shown at magnification 200×. (A) Infected cells were identified by VZV protein (brown) expression. In double labeled skin sections, IFN-α (purple) expression was prominent in adjacent uninfected cells but not in VZV-infected cells (top) compared with sections stained with rabbit IgG as a control for IFN-α (bottom). (B) In double labeled sections, phosphorylated Stat1 (purple) was up-regulated in adjacent uninfected cells but absent in VZV-infected cells (top); pStat1 was not detected in uninfected skin (bottom right) compared with rabbit IgG control stain (bottom left).
Inhibition of Type I IFN Signaling.
To document the relevance of these observations about IFN-α and Stat1 phosphorylation to VZV pathogenesis, skin xenografts in five mice (n = 10 xenografts) were inoculated with VZV and mice were given neutralizing antibody against the IFN-α/β receptor, which blocks type I IFN activity (9). Four skin xenografts were harvested from mice in the antibody-treated and control groups 7 d after VZV inoculation for viral titrations. The titer of infectious VZV was 10-fold higher in skin from antibody-treated animals (Fig. 4 E). VZV was recovered from all xenografts from mice given anti–IFN-α/β receptor antibody compared with three of four xenografts from controls. In the other xenografts examined by immunohistochemistry, diminished IFN signaling was shown to result in substantially larger cutaneous lesions (Fig. 4, C and D). Stat1 was activated and localized to nuclei of neighboring uninfected cells in control animals (Fig. 4 A). Phosphorylated Stat1 was detected in some cells, primarily at the margins of VZV lesions, in mice given anti–IFN-α/β receptor (Fig. 4 B).
Figure 4.

Blocking of IFN-α signaling enhanced VZV replication in skin. Skin xenografts were infected with VZV by direct inoculation in mice treated with anti-IFNα/βRβ antibody or controls and harvested at day 7 after infection. Half of the skin implant was fixed in 4% paraformaldehyde for histological analysis, and the other half was disassociated into cell suspension to measure virus titers by infectious focus assay. (A and B) Shown are representative results from skin sections examined at 7 d (large arrow, VZV lesion); phosphorylated Stat1 (purple; small arrow) was translocated to nuclei in adjacent cells but not within VZV lesions (magnification, 200×). (C and D) Skin lesions shown by staining with anti-VZV IgG (purple) were larger at day 7 in animals given anti-IFNα/βRβ antibody (right) than controls (left) (magnification, 100×). (E) Mean VZV titers in four skin xenografts from two mice were 10-fold higher in anti-IFNα/βRβ antibody-treated animals compared with controls (P < 0.05, unpaired t test).
Migration of Mononuclear Cells into VZV-infected Skin.
In these experiments, one of two bilateral skin xenografts was inoculated with VZV-infected HEL cells and the other with uninfected fibroblasts. 10 d later, uninfected mononuclear cells were injected i.v., and skin xenografts were harvested after 24–48 h. Mononuclear cells within VZV-infected skin xenografts expressed CD3, whereas no CD20+ B cells were detected in three experiments (Table II). CD4+ and CD8+ T cells were distributed uniformly with no evidence of increased numbers near VZV lesions. Infiltrating cells were exclusively CD45RO+ T cells (Table II). T cells expressing CLA were detected at low frequencies (<1%), but CCR4+ T cells were not identified. Total numbers of T cells recovered from VZV-infected skin were slightly higher than corresponding uninfected xenografts, but surface marker profiles were comparable by flow cytometry. The relative percentages of subpopulations were as observed when uninfected mononuclear cells were transferred (Table I). Thus, T cell migration into skin was not influenced by VZV protein expression or cellular proteins, IL-1α or IFN-α, in the absence of other signals.
The analysis of lesion biopsies from VZV-infected patients showed many infiltrating mononuclear cells, most of which expressed CD4 or CD8; a few CD20+ B cells and IFN-α–expressing cells, probably macrophages, were also detected (Table II). CD45RO+ memory T cells, naive CD45RA+ T cells, and CLA+ and CCR4+ T cells were present in comparable numbers (Table II). The differences in cell profiles between VZV lesion biopsies and VZV-infected skin xenografts after adoptive transfer suggested that recruitment and/or retention of skin homing T cells, naïve T cells, B cells, and macrophages required host immune-mediated signals.
Expression of Endothelial Cell Adhesion Molecules.
Since skin homing of memory T cells is regulated by adhesion molecules and chemoattractant receptors (10), adhesion molecule expression was examined to explain differences between T cell subpopulations detected in biopsies of VZV lesions and VZV-infected skin xenografts. Expression of E-selectin, ICAM-1, or VCAM-1 by endothelial cells was not detected in VZV-infected skin xenografts, indicating that VZV replication, which is associated with the expression of all kinetic classes of viral proteins, and altered IL-1α and IFN-α expression alone did not induce adhesion molecules (Fig. 5, left). In contrast, all adhesion molecules were expressed extensively in VZV lesion biopsies when host cellular immunity was intact (Fig. 5, right).
Figure 5.
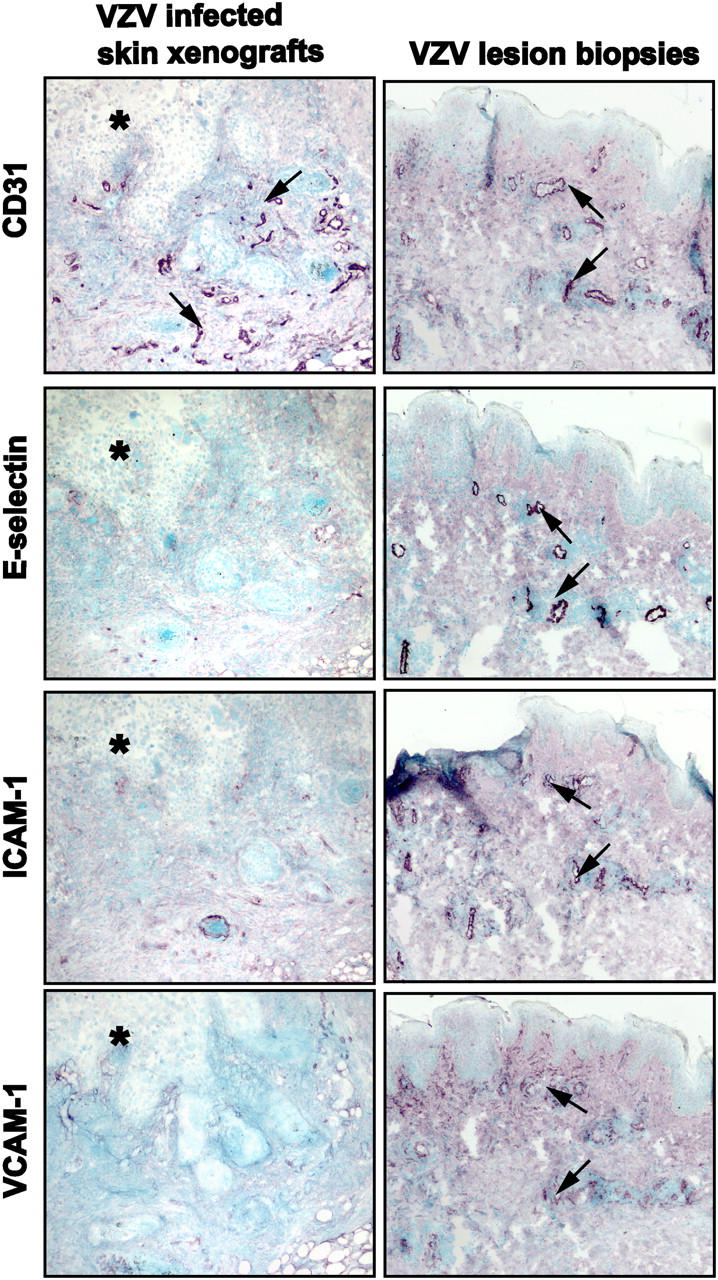
Expression of adhesion molecules by cutaneous endothelial cells in VZV-infected skin xenografts and biopsies of VZV skin lesions. Serial frozen sections of VZV-infected skin xenografts (left) or lesion biopsies from patients with varicella or herpes zoster (right) were stained with antibodies against adhesion molecules expressed by endothelial cells. Signals were detected with Vector VIP (purple) and counterstained with methyl green. Representative results from one of three VZV-infected skin xenografts and one of five VZV lesion biopsies are shown for expression of CD31, E-selectin, ICAM-1, and VCAM-1. E-selectin, ICAM-1, and VCAM-1 were highly expressed by CD31-positive dermal endothelial cells in patient lesion biopsies but were not detected in VZV infected skin xenografts (asterisk, VZV skin lesion). Magnification, 100×.
Discussion
Although the signs and symptoms of infections caused by human herpesviruses are well documented, much less is known about critical events in pathogenesis that precede the onset of the clinical illness. These experiments using the SCIDhu mouse model of VZV pathogenesis provide evidence that infectious VZV can be transferred by T cells through the circulation to sites of replication in skin, resulting in the formation of typical cutaneous lesions. The observations suggest that VZV tropism for T cells, which has been shown in vitro and in T cell xenografts in SCIDhu mice, contributes to the pathogenesis of primary VZV infection in vivo (3–5). The adoptive transfer of uninfected and VZV-infected human mononuclear cells was associated with enrichment of memory CD4+ T cells within human skin xenografts in the SCIDhu mouse model. Therefore, we suggest that VZV tropism for T cells permits the virus to take advantage of the usual pathways for T cell migration through skin during immune surveillance. Whereas naïve T cells enter LNs through the high endothelial venules, exit through efferent lymphatics, and return to the blood, memory T cells traffic through skin and other tissues. In our experiments, the percentage of naïve CD4+ T cells recovered from skin xenografts was only 0.7–3% compared with ∼30% in the infused population. In previous work, we reported that VZV preferentially infects memory T cells compared with naïve CD4+ T cells at a ratio of 3:1 in vitro (5). Lymphoid tissues of the upper respiratory tract, including tonsils and adenoids, contain activated, memory T cells, which may become infected when VZV infects respiratory mucosa. Together, these observations suggest that two factors, which are the intrinsic propensity of memory CD4+ T cells to migrate into skin tissue and the enhanced susceptibility of memory CD4+ T cells to VZV infection, may combine to create an important role for memory CD4+ T cells in VZV pathogenesis. Thus, memory CD4+ T cells that become infected at sites of respiratory epithelial inoculation may be more likely to deliver VZV to epidermal cells and initiate the cutaneous phase of infection than other circulating cells of the immune system, such as naïve CD4+ T cells. As a clinical correlate, VZV dissemination to skin may be modulated in young children, who have few memory T cells, whereas adults with varicella who have >75% memory CD4+ T cells usually develop many cutaneous lesions (1).
Memory T cells that express CLA or CCR4 are targeted to skin through interactions with adhesion molecules and chemoattracts when inflammation is triggered (11). We detected a two- to fivefold enrichment of CLA+ CD4+ T cells in skin when SCIDhu mice were given uninfected human mononuclear cells, but the percentage remained ≤3.6 of CD4+ T cells. We did not demonstrate any increase in the numbers of CLA+ or CCR4+ CD4 T cells in skin after the adoptive transfer of VZV-infected T cells. These observations suggest that VZV pathogenesis does not depend on a selective infection of memory CD4+ T cell subpopulations that express specific skin homing markers. Since the epidermis should be in its resting state during the initial transfer of VZV to skin by infected T cells, trafficking of CLA+ or CCR4+ T cells should not be selectively involved in the initial phase of VZV pathogenesis. A second clinical correlate is that skin trauma, such as sunburn acquired during the incubation period, is associated with a much more severe varicella rash. Under such conditions, the twofold increase in infectivity of VZV for CLA+ T cells that we observed in vitro, combined with the increased migration of these cells to damaged skin, could facilitate more efficient transfer of VZV to the epidermis. The down-regulation of MHC class I molecules that we have observed on VZV-infected T cells may also contribute to successful VZV transport to skin (12).
VZV transmission to other susceptible individuals requires enough replication to create lesions that contain infectious virus at the skin surface (Fig. 6). However, cell–cell spread in vivo at the rate characteristic of VZV replication in human fibroblasts in vitro would be life threatening to the host and would therefore diminish opportunities for person-to-person transmission. Examining VZV lesion formation in parallel with innate antiviral responses in skin xenografts in vivo showed IFN-α down-regulation in VZV-infected cells and no induction of adhesion molecules on capillary endothelial cells. These conditions favor the pathogen during the early phase of skin infection. At the same time, IFN-α production by epidermal cells adjacent to VZV-infected cells appears to provide an innate barrier against aggressive spread of VZV from cell to cell in skin.
Figure 6.

A model of the pathogenesis of primary VZV infection. T cells within the local lymphoid tissue of the respiratory tract may become infected by transfer of VZV from its initial site of inoculation in respiratory epithelial cells. T cells may then transport the virus to the skin immediately and release infectious VZV. The remainder of the 10–21-d incubation period appears to be the interval required for VZV to overcome the innate IFN-α response in enough epidermal cells to create the typical vesicular lesions containing cell-free virus at the skin surface. The signaling of enhanced IFN-α production in adjacent skin cells may prevent a rapid, uncontrolled cell–cell spread of VZV. Secondary “crops” of varicella lesions may result when T cells traffic through early stage cutaneous lesions become infected and produce a secondary viremia. Intact host immune responses appear to be required to trigger up-regulation of adhesion molecules, facilitating the clearance of VZV by adaptive immunity.
Our analysis of IFN-α and IL-1α in VZV-infected and uninfected skin xenografts revealed a subtle interaction between VZV and epidermal cell cytokine responses. Type I interferons, IFN-α, and to a lesser extent, IFN-β, are the predominant mediators of innate antiviral activity. Constitutive IFN-α/β expression occurs in some cells or tissues, allowing an immediate antiviral response (13). IFN-α/β activation is induced by the Jak/Stat pathway when ligands bind to IFN-α/β receptors and stimulate transcription of the IFN regulatory factors (IRFs) (8). IRF3 and IRF7 are crucial for maximal expression of IFN-α/β (14, 15). Constitutive IFN-α/β signaling is thought to sustain IRF7 expression and enhance IFN-α/β production in a positive feedback loop in response to viral infection (16). Most viruses have mechanisms to inhibit synthesis of IFNs or evade downstream antiviral effects in order to allow viral spread before adaptive immunity develops. HSV, a human alphaherpesvirus closely related to VZV, has several such mechanisms (17).
We found that IFN-α was expressed constitutively in the epidermis, independently of Stat1 activation in uninfected skin xenografts. After VZV infection, IFN-α expression was down-regulated in VZV-infected cells concurrently with inhibition of Stat1 phosphorylation. In contrast, Stat1 was phosphorylated and translocated to nuclei, accompanied by IFN-α up-regulation in neighboring, uninfected cells. The capacity of VZV to block IFN-α in some cells enables the virus to replicate, spread from cell to cell, and create focal cutaneous lesions, whereas IFN-α activation protects the adjacent cells. Blocking the IFN-α/β receptor enhanced VZV replication and lesion formation in vivo, showing that these patterns of IFN-α expression and Jak/Stat signaling have biological importance for regulating VZV pathogenesis. Experiments are in progress to identify VZV proteins that have the capacity to disrupt the IFN-α pathway. Overall, the innate IFN-α response appears to modulate VZV growth, providing a substantial barrier against cell–cell spread in skin during the prolonged varicella incubation period. Our observations suggest that VZV gradually overcomes this barrier in order to form polykaryocytes, eventually creating a vesicular lesion that penetrates the skin surface (Fig. 6).
IL-1α is synthesized constitutively and stored in suprabasal layers of the epidermis as a 31-kD precursor molecule, pre–IL-1α (18, 19). When cells die, pre–IL-1α is released and cleaved into a 17-kD IL-1α molecule by extracellular proteases. IL-1α can also be released by the calcium-dependent cysteine proteases called calpains, which are ubiquitously but weakly expressed in all cell types (20). Mature IL-1α displaces the IL-1 receptor antagonist and binds to IL-1αR. Internalization and accumulation of the IL-1α–IL-1 receptor complex in the nucleus induces genes involved in skin cell proliferation and other inflammatory changes (21, 22). We found that IL-1α was expressed constitutively in skin but remained in the cytoplasm of uninfected cells adjacent to and distant from VZV lesions. In contrast, IL-1α accumulated in nuclei of VZV-infected cells. This pattern suggests that IL-1α is activated directly or indirectly by VZV replication within epidermal cells. However, IL-1α is apparently not released, or its release into the local skin microenvironment fails to trigger IL-1α activation or enhanced IL-1α production by neighboring cells.
Expression of cellular adhesion molecules on endothelial cells increases the efficiency of T cell trafficking. Their expression can be regulated by inflammatory cytokines such as IL-1α, TNF-α, and IFN-γ, which were produced by cells infected with some viral pathogens (23–26). In our experiments, VZV-infected T cells migrated into skin and released infectious virus for entry into and replication in epidermal and dermal cells without preexisting expression of E-selectin, ICAM-1, or VCAM-1 on endothelial cells, and without eliciting the expression of these inflammatory proteins. Although IL-1α was activated in VZV-infected cells, it remained cytoplasmic in uninfected cells and did not have the capacity to induce adhesion molecules on endothelial cells in skin xenografts. VZV replication did not induce TNF-α production by skin cells. In contrast, widespread expression of E-selectin, ICAM-1, and VCAM-1 by endothelial cells was observed in biopsies of clinical VZV lesions. CLA+ T cells were enriched in lesion biopsies compared with VZV-infected skin xenografts, which is consistent with a requirement for host immune mechanisms to up-regulate adhesion molecules. This comparative analysis indicated that endothelial cell activation was not stimulated by VZV gene products alone, despite their extensive expression during lytic infection of epidermal and dermal cells, but required host immune mechanisms.
Infectious VZV can be recovered from peripheral blood mononuclear cells during the last few days of the incubation period of varicella in the healthy host and for 2–3 d after the rash appears. We speculate that this secondary viremia results from infection of migrating T cells by virus produced in lesions that are not yet visible at the skin surface (Fig. 6). This hypothesis is suggested by our finding that many T cells entered VZV-infected skin xenografts after adoptive transfer. Thus, viral amplification could result from progressive VZV infection in skin without requiring a phase of replication in reticuloendothelial tissues during the incubation period. Our experiments do not exclude some VZV replication in liver or other reticuloendothelial organs, which might contribute to pathogenesis by increasing the viral load. However, our data indicate that the interval required for VZV to overcome innate immune barriers in skin is sufficient to account for the prolonged incubation period. Continued VZV replication in skin would also be predicted to sustain the viremia associated with malignant varicella, which leads to pneumonia, hepatitis, and encephalitis in immunocompromised children who fail to develop VZV-specific adaptive immunity (1). Further, the transfer of VZV from infected epidermal cells into migratory T cells may also explain how VZV causes viremia and dissemination in high-risk patients with herpes zoster.
Based on these SCIDhu mouse experiments, we offer a new paradigm of primary VZV infection which suggests new directions for the analysis of events in VZV pathogenesis. According to this model, infectious VZV is transferred to skin shortly after inoculation of the respiratory mucosa. Although other cell types may be involved, memory CD4+ T cells play a prominent role in this process because of their intrinsic pattern of skin trafficking and their increased susceptibility to VZV infection. VZV gene products modulate innate epidermal cell defenses, down-regulating IFN-α to ensure localized replication in skin. Epidermal cell infection appears to occur without activating endothelial cell adhesion molecules, thereby delaying the invasion of antiviral inflammatory cells at the early stage of cutaneous lesion formation. At the same time, uninfected epidermal cells are signaled to mount a potent innate IFN-α response, which counters an overly aggressive cell–cell spread in skin (Fig. 6). This model helps to explain how VZV replication may be restricted by the host even though VZV-specific T cell immunity is not detected until after the appearance of vesicular skin lesions (1). Acquisition of T cells that recognize VZV proteins correlates with cessation of skin replication, which we suggest is necessary to stop continued, secondary T cell viremia. A benign infectious process during the initial phase of VZV pathogenesis and a circumscribed clinical illness provides the optimal strategy for VZV to be sustained within the human population (27).
Acknowledgments
We thank Ms. Carolyn Brandt for her assistance with patient biopsies.
This work was supported by National Institutes of Health grants AI20459 and CA49605, and a postdoctoral fellowship to Dr. Ku from the VZV Research Foundation.
The authors have no conflicting financial interests.
H. Ito's present address is Department of Dermatology, Jikei University School of Medicine, 3-19-18 Nishishinbashi Minato, Tokyo, Japan.
Abbreviations used in this paper: CLA, cutaneous lymphocyte antigen; HEL, human lung; ICAM, intercellular adhesion molecule; IRF, IFN regulatory factor; SCID, severe combined immunodeficiency; SCIDhu, SCID with human skin grafts; VACM, vascular adhesion molecule; VZV, varicella-zoster virus.
References
- 1.Arvin, A. 2001. Chapter 79. Varicella-Zoster Virus. Fields Virology. D. Knipe and P. Howley, editors. Lippincott Williams & Wilkins, Philadelphia, PA. 2731–2767.
- 2.Grose, C. 1981. Variation on a theme by Fenner: the pathogenesis of chickenpox. Pediatrics. 68:735–737. [PubMed] [Google Scholar]
- 3.Soong, W., J. Schultz, A. Patera, M. Sommer, and J. Cohen. 2000. Infection of human T lymphocytes with varicella-zoster virus: an analysis with viral mutants and clinical isolates. J. Virol. 74:1864–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moffat, J.F., M.D. Stein, H. Kaneshima, and A.M. Arvin. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 69:5236–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ku, C.C., J.A. Padilla, C. Grose, E.C. Butcher, and A.M. Arvin. 2002. Tropism of varicella-zoster virus for human tonsillar CD4(+) T lymphocytes that express activation, memory, and skin homing markers. J. Virol. 76:11425–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koelle, D.M., L. Corey, R.L. Burke, R.J. Eisenberg, G.H. Cohen, R. Pichyangkura, and S.J. Triezenberg. 1994. Antigenic specificities of human CD4+ T-cell clones recovered from recurrent genital herpes simplex virus type 2 lesions. J. Virol. 68:2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ku, C.C., J. Kappler, and P. Marrack. 2001. The growth of the very large CD8+ T cell clones in older mice is controlled by cytokines. J. Immunol. 166:2186–2193. [DOI] [PubMed] [Google Scholar]
- 8.Darnell, J.E., Jr., I.M. Kerr, and G.R. Stark. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 264:1415–1421. [DOI] [PubMed] [Google Scholar]
- 9.Colamonici, O.R., and P. Domanski. 1993. Identification of a novel subunit of the type I interferon receptor localized to human chromosome 21. J. Biol. Chem. 268:10895–10899. [PubMed] [Google Scholar]
- 10.Campbell, J.J., and E.C. Butcher. 2000. Chemokines in tissue-specific and microenvironment-specific lymphocyte homing. Curr. Opin. Immunol. 12:336–341. [DOI] [PubMed] [Google Scholar]
- 11.Campbell, D.J., and E.C. Butcher. 2002. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J. Exp. Med. 195:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abendroth, A., I. Lin, B. Slobedman, H. Ploegh, and A.M. Arvin. 2001. Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J. Virol. 75:4878–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gresser, I. 1990. Biologic effects of interferons. J. Invest. Dermatol. 95:66S–71S. [DOI] [PubMed] [Google Scholar]
- 14.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya, M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 13:539–548. [DOI] [PubMed] [Google Scholar]
- 15.Lin, R., Y. Mamane, and J. Hiscott. 2000. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275:34320–34327. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi, T., and A. Takaoka. 2001. A weak signal for strong responses: interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol. 2:378–386. [DOI] [PubMed] [Google Scholar]
- 17.Katze, M.G., Y. He, and M. Gale Jr. 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675–687. [DOI] [PubMed] [Google Scholar]
- 18.Dinarello, C.A. 1996. Biologic basis for interleukin-1 in disease. Blood. 87:2095–2147. [PubMed] [Google Scholar]
- 19.Murphy, J.E., C. Robert, and T.S. Kupper. 2000. Interleukin-1 and cutaneous inflammation: a crucial link between innate and acquired immunity. J. Invest. Dermatol. 114:602–608. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi, Y., K. Yamamoto, T. Saido, H. Kawasaki, J. Oppenheim, and K. Matsushima. 1990. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1α. Proc. Natl. Acad. Sci. USA. 87:5548–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maier, J.A., M. Statuto, and G. Ragnotti. 1994. Endogenous interleukin 1 alpha must be transported to the nucleus to exert its activity in human endothelial cells. Mol. Cell. Biol. 14:1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freedberg, I.M., M. Tomic-Canic, M. Komine, and M. Blumenberg. 2001. Keratins and the keratinocyte activation cycle. J. Invest. Dermatol. 116:633–640. [DOI] [PubMed] [Google Scholar]
- 23.Pati, S., M. Cavrois, H.-G. Guo, J.S. Foulke Jr., J. Kim, R.A. Feldman, and M. Reitz. 2001. Activation of NF-kB by the human herpesvirus 8 chemokine receptor ORF74: evidence for a paracrine model of Kaposi's sarcoma pathogenesis. J. Virol. 75:8660–8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dustin, M.L., R. Rothlein, A.K. Bhan, C.A. Dinarello, and T.A. Springer. 1986. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J. Immunol. 137:245–254. [PubMed] [Google Scholar]
- 25.Leeuwenberg, J.F., T.M. Jeunhomme, and W.A. Buurman. 1989. Induction of an activation antigen on human endothelial cells in vitro. Eur. J. Immunol. 19:715–720. [DOI] [PubMed] [Google Scholar]
- 26.Swerlick, R.A., K.H. Lee, L.J. Li, N.T. Sepp, S.W. Caughman, and T.J. Lawley. 1992. Regulation of vascular cell adhesion molecule 1 on human dermal microvascular endothelial cells. J. Immunol. 149:698–705. [PubMed] [Google Scholar]
- 27.McGeoch, D.J., and A.J. Davison. 1999. The molecular evolutionary history of the herpesviruses. Origin and Evolution of Viruses. E. Domingo, R.G. Webseter, and J.J. Holland, editors. Academic Press, London. 441–466.