

Abstract
Inflammation induced by circulating immunoglobulin G–immune complexes (ICs) characterizes many immune-mediated diseases. In this work, the molecular requirements for the deposition of circulating ICs and subsequent acute leukocyte recruitment in mice were elucidated. We show that after intravenous injection, preformed soluble ICs are rapidly deposited in the postcapillary venules of the cremaster microcirculation, secondary to increased vascular permeability. This deposition is dependent on complement C1q. IC deposition is associated with leukocyte recruitment. Leukocyte rolling, which is mediated by P-selectin in the exteriorized cremaster muscle, is not further increased in response to ICs. In contrast, leukocyte rolling velocity is significantly decreased and leukocyte adhesion is significantly increased in the presence of ICs. The IC-mediated slow leukocyte rolling velocity and subsequent adhesion and emigration are dependent on Fcγ receptors (FcγRs), particularly FcγRIII, with complement C3 and C5 having no detectable role. These studies suggest a regulatory mechanism of IC deposition and leukocyte trafficking in IC-mediated inflammation requiring C1q and FcγRs in sequential, noninteracting roles.
Keywords: antigen–antibody complex, Fc receptors, animal models, complement 1q, microscopy
Introduction
Circulating immune complexes (ICs) are produced continuously in response to infection, tissue injury, and immune reactions to foreign antigens (Ags). Most ICs are of little pathologic significance because they are rapidly cleared by hepatic and splenic phagocytes (1, 2). This cycle of IC formation and clearance is an important component of acquired immunity, ensuring removal or processing of Ags. However, excessive IC accumulation occurs in several immune-mediated diseases in human patients (3, 4). In particular, IgG-containing ICs are responsible for the pathogenesis of systemic lupus erythematosus, rheumatoid arthritis, vasculitis, and glomerulonephritis (2–4). The factors influencing IC deposition have mostly been determined for the glomerulus, which has a unique fenestrated vascular bed that passively traps circulating ICs (2). Mechanisms leading to deposition of circulating ICs in other vascular beds have not been fully elucidated. Studies in the skin of human patients with circulating ICs (5, 6) suggest that altered vascular permeability might be an important contributing factor. Evidence that activated complement components may also be required for IC deposition has also been provided. C1q, which initiates the classical complement pathway by attaching to ICs, is required for IC binding to endothelial cells (ECs) in vitro (7, 8). Bound C1q ultimately activates C3 to C3b, which could facilitate IC deposition by binding to cell surfaces (1, 9). Also, C5, which is activated by C3b-based convertases, appears to be important for IgG–IC deposition in a murine model of rheumatoid arthritis (10).
IC deposition initiates a complex cascade of events, involving adhesion molecules, FcγRs, and complement, which culminate in the recruitment and activation of PMN and macrophages. In non-IC models of inflammation, selectins support leukocyte rolling, a prerequisite for adhesion mediated by leukocyte-specific integrins (CD18) interacting with Ig superfamily members on ECs (11). In the context of IC-mediated diseases, P-selectin was required for leukocyte rolling in an OVA-induced, IgE-mediated hypersensitivity reaction and an IgG-mediated reverse Arthus reaction in murine cremaster muscles (12–14). Importantly, ICs bind to FcγRs and activate complement, and both are critical for leukocyte accumulation and subsequent clinical disease in many murine models of IC-mediated disease (10, 15). However, the relative importance of the FcγR and complement pathway in the pathogenesis of these diseases is contentious (16, 17) and due to their complexity, the exact mechanisms of leukocyte recruitment have not been fully elucidated. Several possibilities exist. IC-mediated activation of FcγRs on mast cells or macrophages may release inflammatory mediators (e.g., histamine) that up-regulate adhesion molecules (18). Leukocyte recruitment may also occur through direct interaction of PMN FcγRs with ICs. Specifically, FcγRIII (a low affinity FcγR) promoted the capture of PMN on IC-coated surfaces under physiological flow in vitro (19). Complement activation may recruit leukocytes through chemokine release and/or up-regulation of EC adhesion molecules (20).
Here we report on a model of circulating IgG-ICs amenable to intravital microscopy (IVM), designed to mimic human immune-mediated diseases characterized by high levels of circulating ICs that access tissues through the vasculature. Mice injected i.v. with soluble preformed ICs developed IC deposits in postcapillary venules, not arterioles and capillaries, within minutes of manipulations, which increased vascular permeability. IC deposition was primarily dependent on C1q. IC deposition recruited PMN by decreasing their P-selectin–dependent rolling velocity and promoting adherence and subsequent emigration into extravascular tissue. The IC-induced alterations in rolling velocity were dependent on FcγRs, whereas adhesion and emigration were specifically dependent on PMN FcγRIII. Our results define a novel in vivo role for C1q in initiating deposition of circulating ICs and for leukocyte FcγRs in mediating subsequent recruitment, thus suggesting that complement and FcγRs have sequential, but noninteracting, roles in acute IC-mediated inflammation.
Materials and Methods
Mice.
7–16-wk-old male mice were used. P-selection-KO (−/−) and WT mice (21) were backcrossed nine generations to C57BL/6J. C1q (C1q−/−; reference 22), C3 (C3−/−; provided by M. Carroll, Harvard Medical School, Boston, MA; reference 23), and platelet-activating receptor−/− mice (provided by S. Ishii and T. Shimizu, University of Tokyo Bunkyo-ku, Tokyo, Japan; reference 24), backcrossed 10 generations to C57Bl/6, were also used with age- and strain-matched controls.
Mice lacking the Fcγ chain and FcγRII, i.e., lacking all three FcγRs, on a mixed background (FcγR−/−) and controls (B6/129S6F1) were from Taconic. FcγRIII (FcγRIII−/−), C5 (C5−/−), and two strains of mast cell (Sl/Sld and W/Wv) KO mice with controls were from The Jackson Laboratory. Mice were housed in a virus antibody (Ab)-free facility at Longwood Medical Research Center, Brigham and Women's Hospital. Experiments were conducted under a protocol approved by the institutional animal care and use committee.
Preparation and Administration of ICs.
Soluble ICs at four to six times Ag excess were formed using BSA (sterile low endotoxin solution; ICN Biomedicals) and polyclonal rabbit anti-BSA Ab (Sigma-Aldrich), and were injected i.v. The amount of rabbit IgG injected was 90–165 μg, as determined by ELISA. Injections of BSA alone or PBS served as controls. ICs were also produced from DNP-conjugated chicken Ig (Biosearch Technologies) and goat anti-DNP Ab (Biogenesis). Deposition was evaluated as described below. All remaining Abs were from Jackson ImmunoResearch Laboratories unless otherwise stated.
Visualization of IC Deposits.
To detect IC deposits, ICs were labeled in vitro or in vivo with secondary fluorescent Abs as described below. The cremaster muscle vasculature was visualized in real-time using IVM, and then the muscle was harvested as a whole mount and examined with a fluorescent microscope. ICs were labeled in vitro with fluorescent-labeled anti–rabbit IgG (1:10) for 20 min, and then were injected i.v. Whole IgG and F(ab′)2 fragments of FITC-donkey anti–rabbit IgG, Cy3− or FITC-donkey, FITC-mouse (Sigma-Aldrich), and FITC-goat (Vector Laboratories) anti–rabbit IgGs were used with equivalent results. The polyclonal rabbit anti-BSA Ab was also directly FITC conjugated (FluoReporter Fluorescein-EX Kit; Pierce Chemical Co.) and ICs thus created were deposited similarly to ICs labeled with secondary Abs. To show that deposition was specific for ICs, we used the following controls: BSA and specific secondary Ab (FITC-donkey anti–rabbit IgG), ICs and nonspecific secondary Ab (FITC-donkey anti–goat IgG), FITC-BSA (Molecular Probes) in PBS, and specific secondary Ab alone in PBS. To detect deposited unlabeled ICs in vivo, unlabeled ICs or BSA was injected i.v., followed by 2 × 108 fluorescent (yellow-green) 1-μm polystyrene microspheres (Polysciences, Inc.) coupled to goat anti–rabbit IgG, given in two boluses at 10-min intervals via the femoral artery. 80 μg Cy3-donkey anti–rabbit IgG was then given i.v. via the retroorbital plexus. Microsphere accumulation was quantified in whole mounts and expressed as numbers per area of harvested muscle.
For detection of ICs by immunohistochemistry (IHC), frozen OCT-embedded, 6-μm cryostat tissue sections were stained for rabbit IgG using standard methods and a streptavidin-peroxidase system (ABC Elite; Vector Laboratories) with amino-ethyl carbazide substrate. IHC for C1q, C3, and platelets was performed using rat anti–murine C1q, C3d (1:20; Cell Science), or CD41 (GPIIb, 1:50; BD Biosciences).
IVM.
Leukocyte recruitment in cremaster muscle venules was evaluated in mice within 1 h of a single i.v. injection of unlabeled ICs or controls. Mice were anesthetized with 90 mg/kg ketamine, 18 mg/kg xylazine, and 0.24 mg/kg atropine. The cremaster was prepared as described previously (25), which took 15–20 min. The mouse was transferred to saline immersion intravital microscope (Mikron Instruments). Fluorescent signals were generated by a xenon lamp, video synchronized with a stroboscope (model 315-T; Colorado Video), and detected using a silicon-intensified camera (Hamamatsu Phototonics). Signals were transferred through an online image processor (Argus 20; Hamamatsu Phototonics) to a video recorder and color monitor (Sony). Transilluminated light was captured by a CCD camera (Sony) that was linked to the video recorder. Data from mice were captured on S-VHS tapes (Maxell) and played back through the Argus 20 for data analysis.
Endogenous leukocytes were labeled i.v. with 2 mg/ml rhodamine 6G-chloride (Molecular Probes) and detected with the silicon-intensified camera. Five to eight venules were analyzed over 20 min. During analysis, the muscle was bathed in warmed lactated ringer's solution, with 185 μM NaHCO3. Centerline blood flow velocity was measured using a Doppler flow velocimeter (Texas A&M University Health Science Center). The mouse was then injected i.v. with FITC-dextran (10 mg/ml, 160 kD; Sigma-Aldrich) and venule size was measured with the Argus 20. At the completion of microscopy, blood was collected into EDTA for total and differential leukocyte counts.
Analysis of IVM Data.
Leukocyte rolling flux was defined as the number of leukocytes rolling past a perpendicular point in a vessel over 60 s. This was converted to the leukocyte rolling flux fraction as described previously (26). The mean blood flow velocity (Vb) was calculated as centerline velocity/1.6 and wall shear rate as 8 (Vb/venule diameter) (13). Leukocyte rolling velocities were measured by tracking single leukocytes (20/venule) frame by frame and calculating distance moved per unit time (micrometer/second). Adherent leukocytes were defined as cells remaining stationary for 30 s and were expressed per mm2 of venule.
For all mice, analysis was performed in venules ranging from 13 to 66 μm, with Vb of 0.6 to 7.3 mm/s and shear rates of 155 to 2,014 s−1. There were no significant differences (not depicted) in these parameters or leukocyte counts comparing KO and WT mice or treatment groups (mice given ICs, BSA, or PBS), unless otherwise noted.
Analysis of Vascular Permeability.
Permeability changes were evaluated using colloidal carbon, which is retained by leaky vessels and can be assessed microscopically (27, 28), or FITC-dextran. Mice were given labeled ICs, followed 15 min later by colloidal carbon i.v. (200 μl of a 1:4 dilution; Sanford-Higgins). Mice were then killed before or after cremaster exteriorization. To increase EC barrier function, mice were given 5 mg/kg rolipram and 1 mg/kg forskolin (R/F; both from Sigma-Aldrich) i.p. 20 min before IC or 30 min before colloidal carbon injection. Deposition was scored by independent observers. To assess effects of vasoactive mediators, mice were injected i.v. with labeled ICs, followed within 15 min by PBS, 37 μg histamine (Sigma-Aldrich), or 100 ng of vascular endothelial growth factor (VEGF; PeproTech) i.p. Mice were then given colloidal carbon before being killed. H1 receptor antagonists, 30 mg/kg diphenhydramine (Sigma-Aldrich), and 20 mg/kg pyrilamine (Sigma-Aldrich) were given i.p. 15 min before IC injection. All harvested muscles from killed mice were pressed onto poly-l-lysine–coated microscope slides, mounted with Fluorosave (Calbiochem), and colloidal carbon and IC deposits were visualized using phase contrast and fluorescent microscopy, respectively. To evaluate permeability “real-time” during IVM, leakage of FITC-dextran into extravascular tissue (a gross indicator of permeability; reference 29) was assessed at the end of the leukocyte recruitment experiments and was subjectively graded (1 to 4+).
Detection of Leukocytes in Tissues.
Emigrated leukocytes (numbers/mm2) were counted under transillumination in a 0.25-mm2 field of extravascular tissue around each analyzed venule, at the end of the experiment. Extravascular leukocytes were also counted in Giemsa-stained cremaster whole mounts (26). Plastic-embedded sections of cremaster were stained for PMN with chloroacetate-esterase. In addition, differential counts (100 cells) were performed on extravascular and intravascular leukocytes in Giemsa-stained whole mounts and expressed as percentages.
Detection of Adhesion Molecules in Cremaster Muscle Vessels and Circulating PMN.
IHC for E-selectin, vascular cell adhesion molecule (VCAM)-1, and intercellular adhesion molecule (ICAM)-1 was performed on frozen sections, as described above using rat anti–murine E-selectin (BD Biosciences) or VCAM-1 (Cymbus Biotechnology) and Armenian hamster anti–murine ICAM-1 (BD Biosciences).
Blood was obtained from mice 1 h after IC or BSA injection. Standard FACS analysis was performed using the following anti–murine Abs (BD Biosciences) with the following suitable isotype controls: PE anti–Ly6-G (Gr-1) for PMN, FITC anti–Mac-1, and FITC anti–L-selectin. Gr-1+ cells were gated and FITC+ cells were identified on histogram plots.
Peripheral Blood Counts and Platelet Purification and Labeling.
Blood was collected from the retroorbital plexus into EDTA. Platelet counts were performed with a Unopette and hemocytometer (Fisher Scientific). Total leukocyte counts were determined with a Coulter counter (Coulter Electronics). Differential counts (100 cells) were performed on Wright's stained blood smears.
For platelet purification, blood was obtained from WT mice into 25 μM heparin (Sigma-Aldrich), centrifuged at 200 g, washed with modified Tyrode's buffer (with 1 μM prostaglandin E1; Sigma-Aldrich), and then labeled with 0.25 μg/ml calcein acetoxymethyl (Molecular Probes). Labeled platelets were injected i.v. at 108 cells/mouse 15 min before ICs. IVM was performed 1 h later.
ELISA for Measurement of Rabbit IgG and C3.
Concentrations of rabbit IgG in ICs and C3 in sera were determined using a capture ELISA (30). Capture and detection Abs were unconjugated or horseradish peroxidase conjugated with donkey anti–rabbit IgG or goat anti–murine C3 (Cappel; ICN Biomedicals), respectively. Serial sample dilutions were compared with standards (rabbit IgG [Pierce Chemical Co.] or C3 standard [Calbiochem]), using regression analysis of OD (450 nm) versus concentration. In the C3 assay, sera from C3−/− mice had ODs below background, confirming assay specificity.
Statistical Analysis.
Parametric data were represented as mean ± SEM, whereas nonparametric data were represented by medians. With parametric data, multiple groups were compared by analysis of variance. Comparisons between two groups and analysis of variance differences were further evaluated with a nonpaired Student's t test with a Bonferroni adjustment. For nonparametric data, multiple groups were compared with a Kruskal-Wallis test, followed by a Mann-Whitney U-test (31). A paired t test was used to compare total leukocyte and differential counts, platelet counts, and C3 levels in mice before and after IC or BSA injection. Significance was set at P < 0.05.
Online Supplemental Material.
Video 1 illustrates the granular deposition of fluorescently labeled ICs occurring in venules of exteriorized cremasters of anesthetized WT mice. Granule deposits do not extend into capillaries and arterioles, which are diffusely stained. Video 2 illustrates slow leukocyte rolling velocity and several adherent leukocytes after IC injection in WT mice. Leukocytes roll fast and none are adherent in FcγR-KO mice. Videos 1 and 2 are available at http://www.jem.org/cgi/content/full/jem.20040501/DC1.
Results
Characterization of Our In Vivo Model of Circulating ICs.
To examine whether i.v. injection of preformed ICs induced systemic inflammation, we measured C3 levels, leukocyte and platelet counts, and adhesion molecule expression on ECs and PMN 1 h after injection of ICs or BSA into WT mice. Serum C3 and total and differential leukocyte counts did not change after IC injection. Platelet counts decreased by 32% after IC compared with BSA injection (Table I), which might be due to IC-induced platelet aggregation (1, 32). Adhesion molecule expression (E-selectin, ICAM-1, and VCAM-1) was not altered in cremaster vessels and circulating PMN were not detectably activated (no Mac-1 up-regulation or L-selectin shedding) after ICs (not depicted). These results indicate that heterologous ICs do not induce a systemic inflammatory response.
Table I.
Changes in Serum C3 Levels and Peripheral Leukocyte, Neutrophil, and Platelet Counts in Mice 1 h after Injection of BSA/Anti-BSA ICs (n = 12) or BSA Alone (n = 8)
| ICs
|
BSA
|
|||
|---|---|---|---|---|
| Pre | Post | Pre | Post | |
| C3 (μg/ml) |
294 ± 41 | 289 ± 50 | 209 ± 28 | 296 ± 41 |
| Leukocyte (×106/ml) |
6.9 ± 0.5 | 7.9 ± 0.9 | 6.1 ± 1.1 | 6.1 ± 0.8 |
| Neutrophils (×106/ml) |
1.1 ± 0.2a | 3.8 ± 0.3 | 0.9 ± 0.1a | 3.3 ± 0.5 |
| Platelets (×109/ml) |
1.33 ± 0.05a , b | 0.90 ± 0.03 | 1.35 ± 0.04 | 1.22 ± 0.06 |
Note that the increased neutrophil count was not specific to ICs because a similar increase was observed after injection of BSA alone. These increases might be the result of adrenaline- (stress) or anesthesia-induced demargination of leukocytes after i.v. injection of BSA or ICs in anesthetized mice.
Significant difference (P < 0.05) comparing pre- and postinjection values within each group of mice.
Significant difference (P < 0.05) comparing the difference in values (pre- minus postinjection) between mice given ICs and BSA.
Alterations in Vascular Permeability Are Associated with IC Deposition in Tissues.
Previous studies have shown that ICs only deposit in venules after vascular permeability is induced (33, 34). Indeed, mice killed 1 h after prelabeled ICs were given i.v. showed no alterations in permeability (as assessed by colloidal carbon deposition) or IC accumulation (Fig. 1, A and B). In contrast, surgical exteriorization of the cremaster, a step required for IVM, resulted in colloidal carbon and IC deposition (Fig. 1, C and D). When ICs were injected immediately after exteriorization, deposits formed rapidly within 1 min of injection. The surgical-induced permeability and IC deposition were recapitulated by administering permeability mediators, histamine or VEGF, after ICs (Fig. 1, E and F). IC deposition occurred in postcapillary venules, but not arterioles or capillaries, as assessed by IVM (Fig. 1, G and H, and Video 1, available at http://www.jem.org/cgi/content/full/jem.20040501/DC1). This was confirmed by IHC staining for ICs in frozen muscle sections (not depicted). DNP/anti-DNP ICs also deposited, indicating that deposition was independent of IC composition (not depicted). Permeability was increased (not depicted), but IC deposits were not seen in exteriorized muscles of anesthetized mice given BSA (Fig. 1 I). Also, vascular leakage of FITC-dextran was observed in the exteriorized cremaster of BSA- or IC-injected mice (Fig. 1 J), confirming that the surgical manipulation increases permeability.
Figure 1.
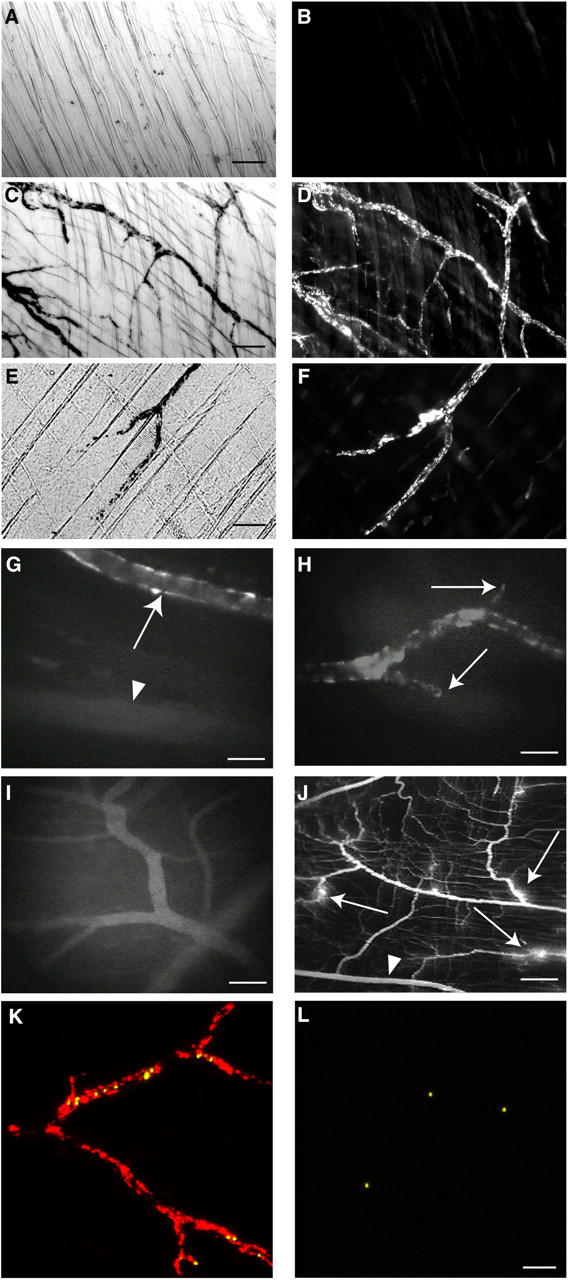
ICs deposit in cremaster muscle venules after induction of vascular permeability and are accessible to circulating leukocytes. (A–F) Deposits of colloidal carbon and ICs (BSA/anti-BSA) were visualized using phase contrast and fluorescent microscopy, respectively, in cremaster whole mounts of mice given prelabeled (Cy-3 anti–rabbit IgG) ICs followed by colloidal carbon i.v. (n = 3 per experiment). (A and B) Mice were killed after colloidal carbon and IC injection and their cremasters were harvested. No deposits of colloidal carbon (A) or ICs (B) were seen (bar, 100 μm). (C and D) The cremaster was exteriorized after colloidal carbon and IC injection, and then harvested. Granular deposits of colloidal carbon (C) and ICs (D) delineated venules (bar, 50 μm). (E and F) Histamine or VEGF (not depicted) was given i.p. between colloidal carbon and IC injections. Mice were then killed and their cremasters were harvested. Colloidal carbon (E) and ICs (F) deposited, mimicking that after cremaster exteriorization (bar, 50 μm). (G–I) Mice were given FITC-prelabeled ICs (G and H) or BSA and FITC anti–rabbit IgG (I). Their cremasters were then exteriorized under anesthesia and visualized with IVM. (G) IC deposits occurred in postcapillary venules (arrows), but not arterioles (arrowhead; bar, 35 μm). (H) Granular IC deposits delineated venules, but did not extend into capillaries (arrows; bar, 35 μm). (I) Diffuse vascular staining was seen in the muscles of mice given BSA and FITC secondary Ab (bar, 70 μm). (J) FITC-dextran, given i.v., diffused into the extravascular space around venules (arrows), but not arterioles (arrowhead) of exteriorized cremasters of mice given ICs (H), PBS, or BSA (not depicted), indicating surgical-induced permeability (bar, 240 μm). (K and L) Unlabeled ICs (K) or BSA (L) were injected i.v. after cremaster exteriorization in anesthetized mice. 1 μm secondary Ab-coupled microspheres were then given, followed by Cy3-labeled secondary Ab. More microspheres (yellow) accumulated in venules after IC than BSA injection. Cy3-Ab (red) identified IC deposits only in vessels of IC-injected mice (bar, 50 μm). See Video 1.
To examine if IC deposition increased with time, we exteriorized cremasters 4, 8, and 24 h after IC injection. The amount of IC deposits decreased with time, with none detectable at 24 h, likely reflecting IC clearance (not depicted). The granular appearance of IC deposits the decrease in platelet counts after IC injection, and reports that platelets accumulate in venules during Arthus reactions (35) prompted us to examine whether IC deposits contained platelets. However, calcein-labeled platelets were not observed at the venule wall with IVM, which was confirmed by IHC (not depicted).
Although permeability changes after surgical exteriorization could be due to histamine, PAF, and mast cells (18), this permeability and IC deposition were not blocked by inhibiting histamine (H1 receptor antagonists), nor was it decreased in mast cell– (Sl/Sld, W/Wv) or platelet-activating receptor–deficient mice (not depicted). However, treatment of mice with R/F, which elevate cAMP and enhance EC barrier function (36), decreased permeability and IC deposition (IC deposition scores: control: 4.0 ± 0.0; R/F: 2.2 ± 0.3; P < 0.05; n = 5). In summary, our data strongly indicate that vascular permeability promotes IC deposition.
IC Deposits Are Accessible to Circulating Leukocytes.
To address whether IC deposits are accessible to circulating leukocytes, we determined if previously administered unlabeled ICs could be detected in vivo by secondary Abs given alone or coupled to 1 μm microspheres. The microsphere approach has been used previously to probe adhesion molecule expression on EC surfaces (37). Microspheres accumulated within 1 min of injection in venules of mice given ICs. Furthermore, Cy3-labeled secondary Ab, given after the microspheres, delineated IC deposits in vessels of mice given ICs (Fig. 1 K), but not BSA (Fig. 1 L). Importantly, significantly more microspheres accumulated in vessels of mice given ICs (5.6 ± 1.3/mm2) than BSA (1.7 ± 0.2/mm2; n = 4; P = 0.03). More ICs were labeled by soluble versus microsphere-coupled secondary Ab. This was attributed to dislodging of microspheres by high shear rates, rigidity of microspheres (which limits access to ICs between ECs), and that more secondary Ab was given alone (80 μg) than as microsphere conjugates (2.6 μg Ab/2 × 108 microspheres). Together, our data indicate that ICs deposited in cremaster venules after exteriorization are directly accessible to macromolecules (IgG) and insoluble particles (microspheres) and thus, by extension, to circulating leukocytes.
IC Deposition Is Dependent on C1q, but Does Not Require C3 or C5.
We examined if C1q and C3 were associated with IC deposits in exteriorized cremaster tissue by IHC. In mice given ICs, C1q colocalized, in a punctuate fashion, with rabbit IgG within vessels, whereas neither were detected in mice given BSA (Fig. 2 A), suggesting that preformed ICs activate and incorporate C1q. In contrast, C3 was identified diffusely in vessels of WT mice (but was absent in C3−/− mice) given ICs or BSA (not depicted), which might be due to continuous low level “Ag-independent” complement activation in vivo (38).
Figure 2.
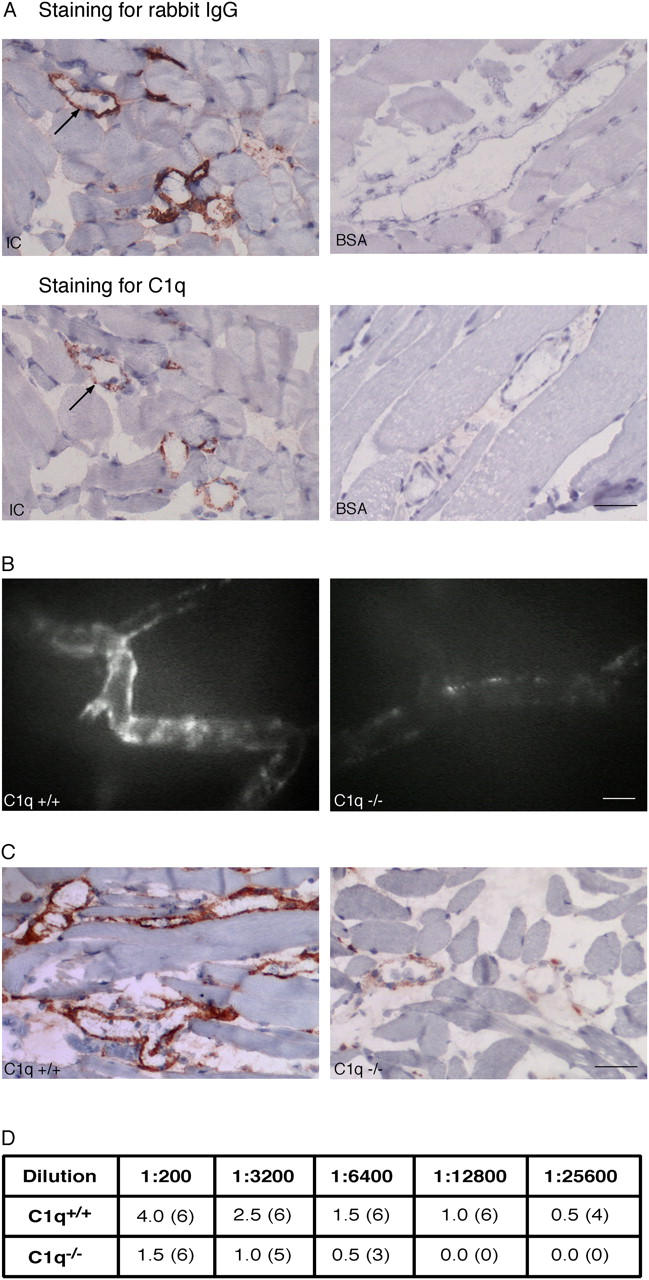
Deposition of ICs is dependent on C1q. (A) C1q colocalizes with rabbit IgG (see arrow for orientation) in venules of WT mice given ICs (left), but staining for both was absent in WT mice given BSA (right; bar, 41 μm). C1q staining was absent in C1q−/− mice (not depicted). (B) Decreased staining of labeled ICs is observed in venules of C1q−/− (right) compared with WT mice (left) under IVM (bar, 35 μm). (C) Decreased staining for rabbit IgG is seen in C1q−/− (right, grade 1+) compared with WT mice (left, grade 4+) with IHC (bar, 50 μm). (D) To semiquantify the deposited rabbit IgG with IHC, the anti–rabbit Ab was serially diluted. Staining was stronger in WT compared with C1q−/− mice at each dilution and disappeared from C1q−/− mice at twofold lower dilutions than WT mice. Numbers of mice with detectable vascular staining is shown in parentheses.
Next, we examined whether C1q, C3, or C5 was required for IC deposition in our model, using relevant KO mice with WT controls. There were no differences in IC deposition in cremaster venules of C3−/− or C5−/− compared with WT mice (n = 6). However, IC deposition was significantly decreased in C1q−/− compared with WT mice (Fig. 2, B–D), despite similar increases in permeability (not depicted). This suggests that C1q, but not C3 or C5, is largely responsible for IC deposition in this model.
In certain microvessels ECs express FcγRs, which could directly bind ICs (39). However, IC deposition was unaltered in mice lacking all three FcγRs (FcγR−/−; n = 6, not depicted), suggesting that FcγRs are not required for deposition of circulating ICs.
IC Deposition Triggers Leukocyte Recruitment That Is Partly Dependent on P-Selectin.
Next, leukocyte recruitment in cremaster venules was examined by IVM after i.v. injection of ICs, BSA, or PBS. Leukocyte rolling flux fractions (i.e., numbers of cells initiating vessel wall contact), velocity of rolling leukocytes (slow rolling facilitates adhesion; reference 40), and adhesion and emigration were measured. Leukocyte rolling flux fractions in WT mice given ICs, BSA, or PBS were similar (Fig. 3 A). However, rolling velocity was significantly (P < 0.05) slower in mice given ICs (median: 21 μm/s) compared with BSA (median, 55 μm/s) or PBS (median, 53 μm/s; n = 200–260 leukocytes/group). Furthermore, leukocyte adhesion was significantly increased after IC, compared with BSA or PBS, injection (Fig. 3 B). Thus, ICs do not affect numbers of rolling leukocytes, but significantly slow rolling velocity, which may favor adhesion, by prolonging transit times (26). Most of the interacting leukocytes were PMN (Fig. 3 C), which comprised 69 ± 2% of intravascular leukocytes on whole mounts.
Figure 3.
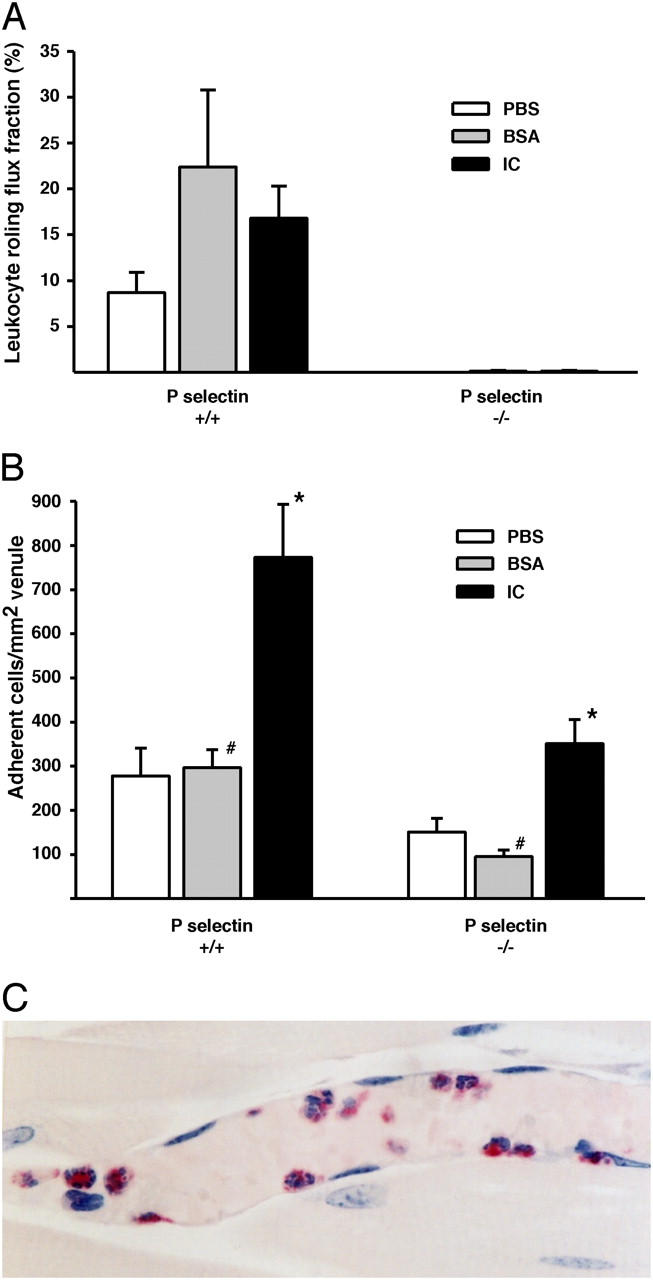
Leukocyte rolling flux fractions and adhesion in WT and P-selectin−/− mice. (A) Leukocyte rolling flux fractions (%) were not significantly different (P > 0.05) after IC, BSA, or PBS injection in WT mice. No rolling was observed in P-selectin−/− mice after any treatment. Hemodynamic parameters were similar between all groups of mice (not depicted). n = 4–6. (B) Numbers of adherent leukocytes were significantly (P < 0.05) increased in WT and P-selectin−/− after IC compared with BSA and PBS injection. Adhesion was proportionally decreased in P-selectin−/− compared with WT mice with all treatments (bars with the same symbols are significantly different; P < 0.05). (C) Chloroacetate-esterase stain for PMN in plastic-embedded section of a cremaster of a WT mouse injected with ICs. Most of the interacting cells are PMN (muscle, red). An original magnification of 500.
Leukocyte rolling in the exteriorized mesentery (21) and cremaster (41) is mediated by P-selectin, which might be constitutively expressed and/or up-regulated after surgery (12, 42). Therefore, we examined rolling and adhesion in P-selectin−/− and WT mice in response to ICs or controls (BSA or PBS). IC deposition and permeability changes were similar in WT and P-selectin−/− mice (not depicted). Rolling was largely abolished in P-selectin−/− mice (Fig. 3 A), regardless of treatment. This was associated with decreased adhesion compared with WT mice, which was comparable among treatments (IC: 55%; BSA: 46%; PBS: 68%). These findings suggest that P-selectin is not up-regulated in response to ICs and that P-selectin–mediated rolling facilitates adhesion regardless of the applied stimulus. Importantly, adhesion was significantly increased in P-selectin−/− mice after ICs compared with controls (Fig. 3 B). Indeed, WT and P-selectin−/− mice showed a similar increase in adhesion (2.7- and 2.8-fold) after IC injection compared with controls. This suggests that ICs may induce adhesion in the absence of P-selectin–mediated rolling.
Leukocyte Adhesion Is Mediated by Fcγ Receptors on Circulating Leukocytes.
FcγR I, II, and III are expressed on murine macrophages, whereas mast cells and PMN constitutively express FcγRII (an inhibitory receptor) and FcγRIII (39). We tested the hypothesis that IC-induced leukocyte recruitment in WT and P-selectin−/− mice in our in vivo model was due to the interaction of circulating leukocyte FcγRs with deposited ICs. Rolling flux fractions following ICs or BSA were similar in WT mice and mice deficient in all three FcgRs (FcγR−/−; not depicted). However, the IC-induced slow leukocyte rolling observed in WT mice was absent in FcγR−/− mice (Video 2, available at http://www.jem.org/cgi/content/full/jem.20040501/DC1, and Fig. 4 A). Also, leukocyte adhesion was significantly reduced in IC-treated FcγR−/− mice (Video 2) and was similar to that in BSA-treated mice (Fig. 5 A). These results suggest a critical role for FcγRs in IC-induced slow leukocyte rolling and adhesion.
Figure 4.
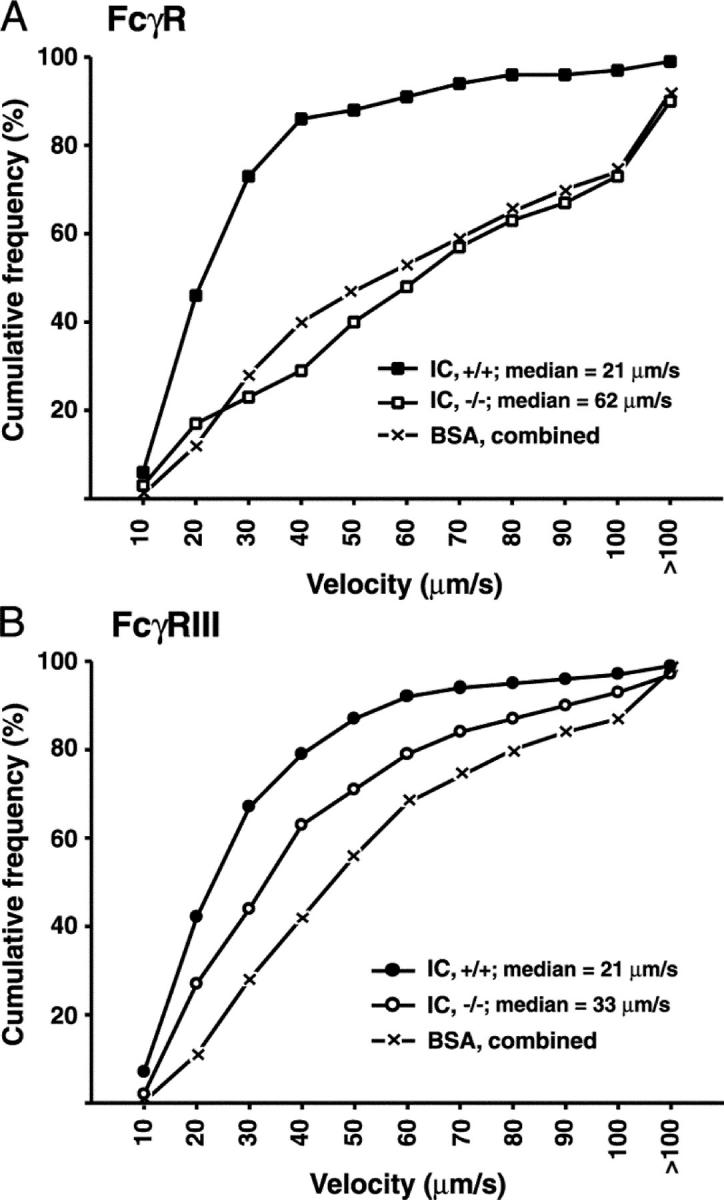
Role of FcγRs in IC-induced slow leukocyte rolling velocity. Cumulative frequency distribution curves of leukocyte rolling velocities in venules of (A) FcγR−/− (−/−), (B) FcγRIII−/− (−/−), and WT cohorts (+/+) 1 h after ICs or BSA (n = 292–420 leukocytes/group). ICs significantly slowed rolling velocity in WT mice. In both genotypes, median rolling velocities after BSA were similar (P > 0.05). Therefore, results were combined. (A) After ICs, rolling velocities were slower (P < 0.05) in WT compared with FcγR−/− mice, which were similar to BSA controls. (B) After ICs, rolling velocities were slower (P < 0.05) in WT than in FcγRIII−/− mice, which in turn were slower (P < 0.05) than BSA controls.
Figure 5.
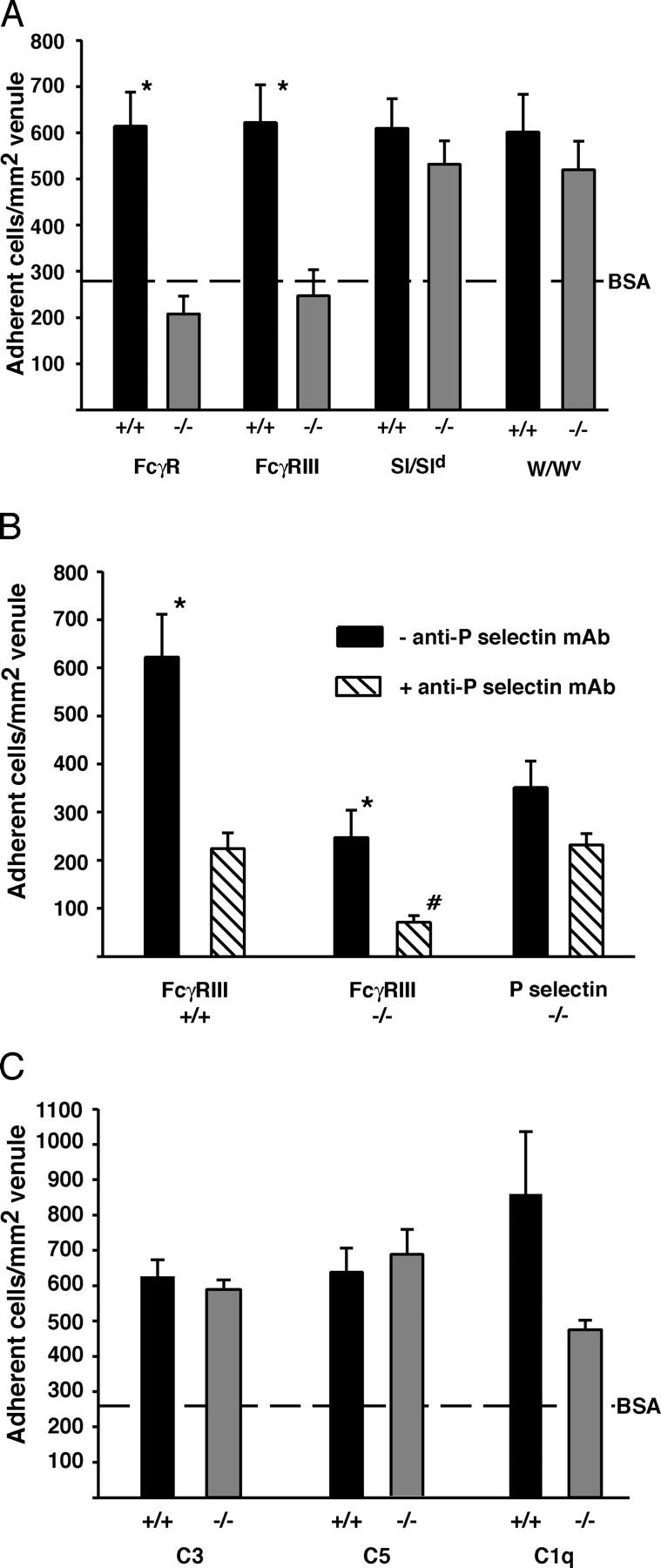
Role of FcγRs, mast cells, and complement in IC-induced leukocyte adhesion. Because adhesion was similar in genotypes given BSA alone, values were combined and the mean is represented as a line. (A) Adhesion was decreased in FcγR−/− and FcγRIII−/− compared with WT mice (*, P < 0.05) and was similar to BSA controls. Adhesion was similar in mast cell KO and WT mice, all of which had significantly more adherent leukocytes compared with BSA controls (297 ± 33 cells/mm2). (B) Adhesion was decreased (*, P < 0.05) in WT and FcγRIII−/− mice given a functional blocking anti–P-selectin mAb (hatched bars) compared with mice given ICs alone (black bars). Adhesion was unaffected in P-selectin−/− mice given the mAb. The mAb reduced adhesion (#, P < 0.05) in FcγRIII−/− compared with WT or P-selectin−/− mice. (C) Adhesion was similar in C3−/− or C5−/−, but was decreased (P = 0.056) in C1q−/− compared with WT mice. All mice had significantly more adherent leukocytes after ICs than BSA (260 ± 56 cells/mm2). See Video 2.
Next, IVM was performed in mice lacking only FcγRIII. Again, rolling flux fractions were similar between treated WT and FcγRIII−/− mice (not depicted). IC-induced slow rolling was only partly FcγRIII dependent (Fig. 4 B), suggesting additional FcγRs, absent in FcγR−/− mice, contribute to IC-induced slow rolling. However, IC-induced adhesion was decreased in FcγRIII−/− mice to similar levels as FcγR−/− mice (Fig. 5 A; P > 0.05). Thus IC-induced adhesion is critically dependent on FcγRIII, whereas slow rolling requires additional FcγRs.
The FcγR-dependent slow rolling and adhesion after ICs support a primary role for leukocyte FcγRs in IC-induced leukocyte recruitment. However, ICs can bind directly to FcγRs on mast cells, releasing factors that could promote PMN recruitment (18). Thus, we examined recruitment in Sl/Sld and W/Wv mast cell KO mice. In both strains, rolling flux fractions, shown earlier to be P-selectin dependent, were significantly reduced (not depicted), whereas IC-induced adhesion was mildly, but not significantly, decreased compared with WT controls (Fig. 5 A). This suggests that mast cells may mediate some P-selectin–dependent rolling, but are not required for FcγR-dependent adhesion in this model.
P-Selectin and FcγRIII Have Distinct, but Overlapping, Roles in IC-mediated Leukocyte Adhesion.
To address the possibility that P-selectin–mediated rolling facilitates FcγRIII-dependent adhesion, we determined whether adhesion would be abolished if FcγRIII−/− mice are rendered P-selectin deficient. Thus, FcγRIII−/− and WT mice were given 20 μg of a functional blocking mAb against P-selectin. P-selectin−/− mice were given the same mAb as controls. The mAb abolished rolling in WT and FcγRIII−/− mice, but did not affect IC deposition or permeability (not depicted). In WT mice, the mAb decreased adhesion to values similar to that in P-selectin−/− mice given ICs alone or with the mAb (Fig. 5 B), thus confirming Ab specificity. Adhesion was significantly decreased in FcγRIII−/− mice compared with WT or P-selectin−/− mice given ICs and the mAb (Fig. 5 B). Indeed, there was a proportional decrease in adhesion in WT (64%) and FcγRIII−/− mice (71%) given ICs and mAb compared with those given ICs alone, suggesting that the reduced adhesion in these mice is due to the abolition of P-selectin–mediated rolling. The residual adhesion in FcγRIII−/− given ICs and the mAb was similar to that in P-selectin−/− mice given PBS or BSA alone (refer to Fig. 3 A).
Role of Complement in Leukocyte Adhesion.
Complement, particularly C5a, is critical for IC-induced inflammation in most models of IC disease (10, 16). However adhesion was not significantly decreased in C5 or C3 KO mice (Fig. 5 C). There was a trend toward decreased adhesion in C1q−/− mice (by 45%; P = 0.056) compared with WT mice (Fig. 5 C), whereas rolling flux fractions and IC-induced slow rolling were unaltered (not depicted). The reduced adhesion was likely a direct consequence of fewer IC deposits (Fig. 2). These results indicate that C1q is likely not required for PMN adhesion, but indirectly affects recruitment due to its role in IC deposition in this model.
Leukocytes Emigrate into Extravascular Tissue in an FcgRIII-dependent Manner after IC Injection.
Because leukocyte adhesion is a prerequisite to emigration (26), we examined whether leukocytes emigrated into the extravascular tissue after ICs or BSA. This was done using IVM and the data was confirmed on cremaster whole mounts only in sets of mice where a significant decrease in emigration was observed by IVM. Significantly, more leukocytes were observed in extravascular tissue after ICs compared with BSA (Fig. 6). Importantly, although a deficiency in mast cells or complement (C1q, C3, or C5) had no significant effect on IC-induced emigration (not depicted), numbers of emigrated leukocytes were significantly decreased in FcγR−/− and FcγRIII−/− mice compared with WT, and were similar to BSA controls (Fig. 6). These results suggest that IC deposition promotes leukocyte emigration in an FcγRIII-dependent manner. The numbers of emigrated leukocytes in IC-injected WT mice were comparable to that observed 4 h after intrascrotal injection of IL-1β (43, 44).
Figure 6.

Leukocyte emigration in response to IC injection is mediated by FcγRIII. Numbers of emigrated leukocytes under transillumination were significantly increased in WT mice (black bars) given ICs rather than BSA (14 ± 4 cells/mm2). Emigration was decreased after ICs in FcγR−/− and FcγRIII−/− (gray bars) compared with WT (black bars; *, P < 0.05), and was similar to BSA controls (n = 4–7). These observations were confirmed in Giemsa-stained whole mounts (n = 3). Counts are higher with this method because stained cells are easier to visualize.
Discussion
The mechanisms by which circulating ICs deposit and recruit PMN are largely unclear. Here, we present a model of acute, limited exposure to ICs, coupled to real-time analysis, which allowed us to delineate the earliest events in deposition of circulating ICs and PMN recruitment. We show that circulating ICs do not up-regulate adhesion molecules in the cremaster vasculature or activate PMN. We provide evidence that collectively suggests that C1q-bearing ICs deposit in cremaster venules in response to changes in vascular permeability, with C1q being required for deposition. The deposited ICs are accessible to circulating PMN and act as a nidus for their recruitment. Recruitment may occur by direct interaction of PMN low affinity FcγRs (FcγRII and III) with the deposited ICs and/or PMN rolling on P-selectin may interact with IC deposits via their low affinity FcγRs (FcγRII and III). This interaction slows their rolling velocity and promotes firm adhesion via FcγRIII. PMN then migrate into extravascular tissue, with ICs possibly acting as chemotaxins (Fig. 7). Recruited PMN may facilitate clearance of the IC deposits before returning to the circulation as described previously in rats (45). Alternatively, persistent deposition of C1q-containing ICs within vessels, due to chronic exposure to circulating ICs, may cause IC translocation and retention in extravascular tissue. This could result in prolonged FcγR/IC-mediated recruitment and subsequent induction of tissue injury and disease by activating ECs (46, 47).
Figure 7.

Proposed model for leukocyte recruitment to deposited ICs. Soluble ICs deposit in postcapillary venules, secondary to increased vascular permeability. Deposition is facilitated by C1q. (A) PMN tether via P-selectin. (B) Rolling PMN encounter deposited ICs, which slows their rolling velocity, which is critically dependent on FcγRs. (C) Subsequent adhesion is mediated primarily through FcγRIII. In the absence of P-selectin, direct interactions of PMN FcγRIII with deposited ICs may cause adhesion. (D) Lumen-bound ICs and/or ICs diffused extravascularly promote PMN emigration into the extravascular tissue, an FcγRIII-dependent process.
Our studies show that altered vascular permeability induced by histamine, VEGF, or surgical manipulation is accompanied by IC deposition in postcapillary venules. This pattern of deposition is consistent with established observations that permeability occurs principally in small veins and postcapillary venules (27, 48). Similar venular-specific IC deposition occurs after local histamine injection in human patients with vasculitis (5, 6), in Arthus reactions (35, 49), and in a hypersensitivity reaction in guinea pig cheek pouches (29). The surgical-induced permeability and IC deposition was attenuated by cAMP-elevating agents, which increase EC barrier function (36), providing further evidence that permeability promotes IC deposition. The correlation between IC deposition and permeability changes in our and other studies (5, 6, 33, 34) suggests that human patients with circulating soluble ICs could be predisposed to localized IC-mediated inflammation if permeability is altered (e.g., secondary to surgery, blunt trauma, or drugs). Thus, vasoactive factors may have a causative or perpetuating role in diseases associated with circulating ICs.
Although C1q is required for IgG–IC binding to human umbilical vein ECs in vitro (7), this study is the first demonstration that C1q-bound ICs localize to the vessel wall and that C1q is required for permeability-induced IC deposition in vivo. Administration of ICs i.v. causes rapid complement activation through the classical pathway (34), which is triggered when C1q binds to the Fc region of adjacent IgG molecules in ICs (50). Several mechanisms could promote deposition of C1q-containing ICs within the vessel wall. In vitro studies suggest the existence of a putative C1q receptor on ECs (7, 8), although a bona fide receptor has not yet been identified (43, 51). In vivo, we propose that vascular permeability factors may induce these receptors, thus allowing binding of C1q-containing ICs. It is also probable that C1q binds to extracellular matrix molecules exposed through permeability changes because C1q interacts with laminin and fibronectin (52, 53). In addition, C1q could modify the lattice structure of ICs (1), which could facilitate IC deposition. Finally, the residual IC deposition in C1q−/− mice might be due to ICs binding directly to vitronectin or fibronectin (53, 54).
IC-induced complement activation produces chemotaxins (C3a and C5a; reference 20) and is critical for leukocyte recruitment in various murine models of IC-mediated disease (10, 17). Despite this, C3 and C5 were not required for recruitment in our model. This might be due to the relative absence of C3 activation after injection of a single dose of ICs, despite initial binding of C1q. It is likely that injected ICs rapidly bind and activate C1q, but further C activation is down-regulated by inhibitors (9, 55), thus preventing systemic inflammatory effects. C3 and C5 activation is likely pronounced during chronic IC exposure and may have a direct or indirect (via increased adhesion receptor expression) role in leukocyte accumulation under these conditions.
Our data indicate a critical role for FcγRIII in mediating leukocyte adhesion in response to ICs in vivo. This data is in concert with previous results showing that neutrophils bind via FcγRIII to ICs under physiological flow in vitro (19). Notably, IC-induced adhesion occurred in the absence of increased rolling flux fractions, indicating that the IC-induced adhesion is not the result of increased numbers of rolling cells. However, maximal interaction with ICs appears to require P-selectin and FcγRs, suggesting that rolling may facilitate the FcγR-mediated adhesion to ICs. Rolling in this model is likely due to constitutive P-selectin expression (12) or mast cell–mediated up-regulation with cremaster exteriorization (42). Our results suggest that FcγRs, particularly FcγRIII, might be solely responsible for leukocyte recruitment to ICs in vascular beds lacking constitutive P-selectin expression, e.g., the glomerulus (56). Indeed, mice lacking FcγRs have reduced glomerular PMN accumulation in acute nephritis models (19, 57).
Although ICs did not alter rolling flux fractions, the velocity of P-selectin–mediated rolling was significantly slowed in response to ICs, and this was FcγR dependent. The currently held paradigm in non-IC models of inflammation is that slow rolling and long transit times, rather than flux fractions, are critical for efficient leukocyte adhesion (40). The fact that FcγRIII alone was not responsible for the FcγR-dependent, IC-induced slow rolling velocity suggests that FcγRII or a new γ chain–containing FcγR (58) is required for this step. Adhesion in response to ICs was critically dependent on FcγRIII, likely because FcγRIII transmits the activating signals for adhesion in murine PMN (59, 60). These data provide a possible mechanism for the reduced inflammation observed in FcγRIII−/− mice in several models of IC-mediated disease (10, 19, 60). FcγRs are also expressed by mast cells (39), yet they played a minimal role in early PMN recruitment after circulating IC deposition. However, once ICs diffuse into extravascular tissue, it is likely that tissue mast cells or macrophages have an important role in the unfolding inflammatory response.
Finally, IC promoted leukocyte emigration that was entirely FcγRIII dependent, suggesting that ICs might be chemotactic, as reported in vitro (61). ICs may promote leukocyte emigration in an analogous fashion to surface-embedded chemokines, which can induce transendothelial migration under shear conditions without the presence of a chemotactic gradient (62). The increased extravasation may also be a direct consequence of IC-induced leukocyte adhesion. Emigration (as measured under transillumination) was reduced in P-selectin−/− mice (WT: 64 ± 33 cells/mm2; P-selectin−/−: 20 ± 4 cells/mm2; P = 0.22), which paralleled the decreased adhesion in these mice. Thus, although adherence may promote emigration in our model, a chemotactic effect of ICs may also be operative and our results do not distinguish between these possibilities.
In summary, we have developed a model of acute IC-induced inflammation that allows visualization of the earliest events contributing to IC deposition and subsequent PMN accumulation. This model has relevance for human patients with diseases associated with high concentrations of circulating ICs, e.g., systemic lupus erythematosus, and is distinguished from other acute IC models, e.g., the reverse Arthus reaction, in which ICs are primarily generated in extravascular tissues (14, 49). Here, we show that ICs deposit within vessels in response to increased permeability and trigger rapid PMN accumulation by slowing PMN rolling velocity and promoting adhesion. IC deposition is dependent on C1q activation and this might be one of the earliest events permitting IC deposition in vessels in patients with IC-mediated diseases. The lack of an apparent role for C3 and C5 in IC deposition suggests that further complement activation is not required for this step. FcγRs, particularly FcγRIII, are required for several aspects of the subsequent IC-mediated PMN recruitment, namely slowing rolling velocity and mediating adhesion and emigration. Together, these data suggest sequential, noninteracting roles for complement and FcγRs in this model of circulating IC-mediated inflammation.
Acknowledgments
The authors would like to thank Guiyeng-Chen (Center for Blood Research, Harvard Medical School, Boston, MA), Yaw-Chyn Lim, Xavier Cullere, and Daniel Friend (Brigham and Women's Hospital, Boston, MA) for technical and computer assistance. We also gratefully acknowledge Dr. Michael Carroll for providing the C3−/− mice, Drs. Satoshi Ishii and Takao Shimizu for the platelet-activating receptor−/− mice, and Drs. Guido Majno, Isabelle Joris (both from University of Massachusetts, Worcester, MA), and Janice Nagy (Beth Israel Deaconness Medical Center, Harvard Medical School, Boston, MA) for helpful advice with the colloidal carbon technique.
This work was supported by the National Institutes of Health grants HL65095, DK51643, and HL036028 (to T.N. Mayadas), and National Research Service Award HL-67570 (to T. Stokol).
The authors have no conflicting financial interests.
Abbreviations used in this paper: Ab, antibody; Ag, antigen; EC, endothelial cell; IC, immune complex; ICAM, intercellular adhesion molecule; IHC, immunohistochemistry; IVM, intravital microscopy; R/F, rolipram and forskolin; VCAM, vascular cell adhesion molecule; VEGF, vascular endothelial growth factor.
References
- 1.Schifferli, J.A., and R.P. Taylor. 1989. Physiological and pathological aspects of circulating immune complexes. Kidney Int. 35:993–1003. [DOI] [PubMed] [Google Scholar]
- 2.Abrass, C.K. 2001. Mechanisms of immune complex formation and deposition in renal stuctures. Immunologic Renal Disease. E.G. Neilson and W.G. Couser, editors. Lippincott Williams & Wilkins, Philadelphia. 277–295.
- 3.Firestein, G.S. 2003. Evolving concepts of rheumatoid arthritis. Nature. 423:356–361. [DOI] [PubMed] [Google Scholar]
- 4.Lamprecht, P., A. Gause, and W.L. Gross. 1999. Cryoglobulinemic vasculitis. Arthritis Rheum. 42:2507–2516. [DOI] [PubMed] [Google Scholar]
- 5.Braverman, I.M., and A. Yen. 1975. Demonstration of immune complexes in spontaneous and histamine-induced lesions and in normal skin of patients with leukocytoclastic vasculitis. J. Invest. Dermatol. 64:105–112. [DOI] [PubMed] [Google Scholar]
- 6.Wolff, H.H., W. Maciejewski, R. Scherer, and O. Braun-Falco. 1978. Immunoelectronmicroscopic examination of early lesions in histamine induced immune complex vasculitis in man. Br. J. Dermatol. 99:13–24. [DOI] [PubMed] [Google Scholar]
- 7.Daha, M.R., A.M. Miltenburg, P.S. Hiemstra, N. Klar-Mohamad, L.A. Van Es, and V.W. Van Hinsbergh. 1988. The complement subcomponent C1q mediates binding of immune complexes and aggregates to endothelial cells in vitro. Eur. J. Immunol. 18:783–787. [DOI] [PubMed] [Google Scholar]
- 8.Beynon, H.L.C., K.A. Davies, D.O. Haskard, and M.J. Walport. 1994. Erythrocyte complement receptor type I and interactions between immune complexes, neutrophils, and endothelium. J. Immunol. 153:3160–3167. [PubMed] [Google Scholar]
- 9.Walport, M. 2001. Complement: first of two parts. N. Engl. J. Med. 344:1058–1066. [DOI] [PubMed] [Google Scholar]
- 10.Ji, H., K. Ohmura, U. Mahmood, D.M. Lee, F.M. Hofhuis, S.A. Boackle, K. Takahashi, V.M. Holers, M. Walport, C. Gerard, et al. 2002. Arthritis critically dependent on innate immune system players. Immunity. 16:157–168. [DOI] [PubMed] [Google Scholar]
- 11.Kubes, P. 2002. The complexities of leukocyte recruitment. Semin. Immunol. 14:65–72. [DOI] [PubMed] [Google Scholar]
- 12.Hickey, M.J., S. Kanwar, D.M. McCafferty, D.N. Granger, M.J. Eppihimer, and P. Kubes. 1999. Varying roles of E-selectin and P-selectin in different microvascular beds in response to antigen. J. Immunol. 162:1137–1143. [PubMed] [Google Scholar]
- 13.Kanwar, S., D.C. Bullard, M.J. Hickey, C.W. Smith, A.L. Beaudet, B.A. Wolitzky, and P. Kubes. 1997. The association between α4-integrin, P-selectin, and E-selectin in an allergic model of inflammation. J. Exp. Med. 185:1077–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norman, M.U., N.C. Van De Velde, J.R. Timoshanko, A. Issekutz, and M.J. Hickey. 2003. Overlapping roles of endothelial selectins and vascular cell adhesion molecule-1 in immune complex-induced leukocyte recruitment in the cremasteric microvasculature. Am. J. Pathol. 163:1491–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayadas, T.N., A. Rosenkranz, and R.S. Cotran. 1999. Glomerular inflammation: use of genetically deficient mice to elucidate the roles of leukocyte adhesion molecules and Fcγ receptors. Curr. Opin. Nephrol. Hypertens. 8:293–298. [DOI] [PubMed] [Google Scholar]
- 16.Kohl, J., and J.E. Gessner. 1999. On the role of complement and Fc gamma-receptors in the Arthus reaction. Mol. Immunol. 36:893–903. [DOI] [PubMed] [Google Scholar]
- 17.Shushakova, N., J. Skokowa, J. Schulman, U. Baumann, J. Zwirner, R.E. Schmidt, and J.E. Gessner. 2002. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J. Clin. Invest. 110:1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robbie-Ryan, M., and M. Brown. 2002. The role of mast cells in allergy and autoimmunity. Curr. Opin. Immunol. 14:728–733. [DOI] [PubMed] [Google Scholar]
- 19.Coxon, A., X. Cullere, S. Knight, S. Sethi, M.W. Wakelin, G. Stavrakis, F.W. Luscinskas, and T.N. Mayadas. 2001. Fc gamma RIII mediates neutrophil recruitment to immune complexes. A mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 14:693–704. [DOI] [PubMed] [Google Scholar]
- 20.Ember, J.A., M.A. Jagels, and T.E. Hugli. 1998. Characterization of complement anaphylatoxins and their biological responses. The Human Complement System in Health and Disease. J.E. Volanakis, editor. Marcel Dekker Inc, New York. 241–284.
- 21.Mayadas, T.N., R.C. Johnson, H. Rayburn, R.O. Hynes, and D.D. Wagner. 1993. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. 74:541–554. [DOI] [PubMed] [Google Scholar]
- 22.Botto, M., C. Dell'Agnola, A.E. Bygrave, E.M. Thompson, H.T. Cook, F. Petry, M. Loos, P.P. Pandolfi, and M.J. Walport. 1998. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 19:56–59. [DOI] [PubMed] [Google Scholar]
- 23.Wessels, M.R., P. Butko, M. Ma, H.B. Warren, A.L. Lage, and M.C. Carroll. 1995. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA. 92:11490–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishii, S., T. Kuwaki, T. Nagase, K. Maki, F. Tashiro, S. Sunaga, W.H. Cao, K. Kume, Y. Fukuchi, K. Ikuta, et al. 1998. Impaired anaphylactic responses with intact sensitivity to endotoxin in mice lacking a platelet-activating factor receptor. J. Exp. Med. 187:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coxon, A., P. Rieu, F.J. Barkalow, S. Askari, A.H. Sharpe, U.H. von Andrian, M.A. Arnaout, and T.N. Mayadas. 1996. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 5:653–666. [DOI] [PubMed] [Google Scholar]
- 26.Jung, U., K.E. Norman, K. Scharffetter-Kochanek, A.L. Beaudet, and K. Ley. 1998. Transit time of leukocytes rolling through venules controls cytokine-induced inflammatory cell recruitment in vivo. J. Clin. Invest. 102:1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majno, G., G.E. Palade, and G.I. Schoefl. 1961. Studies on inflammation. II. The site of action of histamine and serotonin along the vascular tree: a topographic study. J Biophys. Biochem. Cytol. 11:607–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humphrey, D.M., L.M. McManus, D.J. Hanahan, and R.N. Pinckard. 1984. Morphologic basis of increased vascular permeability induced by acetyl glyceryl ether phosphorylcholine. Lab. Invest. 50:16–25. [PubMed] [Google Scholar]
- 29.Smedegard, G., J. Bjork, and K.E. Arfors. 1985. An intravital microscopy model for studies of immune complex induced inflammation at the microvascular level. Int. J. Tissue React. 7:55–60. [PubMed] [Google Scholar]
- 30.Taktak, Y.S., and B. Stenning. 1992. Solid phase enzyme immunoassays for the quantification of serum amyloid P (SAP) and complement component 3 (C3) proteins in acute-phase mouse sera. Horm. Metab. Res. 24:371–374. [DOI] [PubMed] [Google Scholar]
- 31.Dawson-Saunders, B., and R.G. Trapp. 1990. Comparing three or more means. Basic and Clinical Biostatistics. B. Dawson-Saunders and R.G. Trapp, editors. Appleton and Lange, Norwalk, CO. 124–141.
- 32.Peerschke, E.I., and B. Ghebrehiwet. 1997. C1q augments platelet activation in response to aggregated Ig. J. Immunol. 159:5594–5598. [PubMed] [Google Scholar]
- 33.Cochrane, C.G. 1963. Studies on the localization of circulating antigen–antibody complexes and other macromolecules in vessels. II. Pathogenetic and pharmacodynamic studies. J. Exp. Med. 118:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cochrane, C.G., and D. Hawkins. 1968. Studies on circulating immune complexes. 3. Factors governing the ability of circulating complexes to localize in blood vessels. J. Exp. Med. 127:137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crawford, J.P., H.Z. Movat, J.O. Minta, and M. Opas. 1985. Acute inflammation induced by immune complexes in the microcirculation. Exp. Mol. Pathol. 42:175–193. [DOI] [PubMed] [Google Scholar]
- 36.Irie, K., E. Fujii, H. Ishida, K. Wada, T. Suganuma, T. Nishikori, T. Yoshioka, and T. Muraki. 2001. Inhibitory effects of cyclic AMP elevating agents on lipopolysaccharide (LPS)-induced microvascular permeability change in mouse skin. Br. J. Pharmacol. 133:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazo, I.B., E.J. Quackenbush, J.B. Lowe, and U.H. von Andrian. 2002. Total body irradiation causes profound changes in endothelial traffic molecules for hematopoietic progenitor cell recruitment to bone marrow. Blood. 99:4182–4191. [DOI] [PubMed] [Google Scholar]
- 38.Manderson, A.P., M.C. Pickering, M. Botto, M.J. Walport, and C.R. Parish. 2001. Continual low-level activation of the classical complement pathway. J. Exp. Med. 194:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gessner, J.E., H. Heiken, A. Tamm, and R.E. Schmidt. 1998. The IgG Fc receptor family. Ann. Hematol. 76:231–248. [DOI] [PubMed] [Google Scholar]
- 40.Ley, K. 2002. Integration of inflammatory signals by rolling neutrophils. Immunol. Rev. 186:8–18. [DOI] [PubMed] [Google Scholar]
- 41.Ley, K., D.C. Bullard, M.L. Arbones, R. Bosse, D. Vestweber, T.F. Tedder, and A.L. Beaudet. 1995. Sequential contribution of L- and P-selectin to leukocyte rolling in vivo. J. Exp. Med. 181:669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kubes, P., and S. Kanwar. 1994. Histamine induces leukocyte rolling in post-capillary venules - A P-selectin-mediated event. J. Immunol. 152:3570–3577. [PubMed] [Google Scholar]
- 43.Norsworthy, P.J., L. Fossati-Jimack, J. Cortes-Hernandez, P.R. Taylor, A.E. Bygrave, R.D. Thompson, S. Nourshargh, M.J. Walport, and M. Botto. 2004. Murine CD93 (C1qRp) contributes to the removal of apoptotic cells in vivo but is not required for C1q-mediated enhancement of phagocytosis. J. Immunol. 172:3406–3414. [DOI] [PubMed] [Google Scholar]
- 44.Young, R.E., R.D. Thompson, and S. Nourshargh. 2002. Divergent mechanisms of action of the inflammatory cytokines interleukin 1-beta and tumour necrosis factor-alpha in mouse cremasteric venules. Br. J. Pharmacol. 137:1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fries, J.W.U., D.L. Mendrick, and H.G. Rennke. 1988. Determinants of immune complex-mediated glomerulonephritis. Kidney Int. 34:333–345. [DOI] [PubMed] [Google Scholar]
- 46.Lozada, C., R.I. Levin, M. Huie, R. Hirschhorn, D. Naime, M. Whitlow, P.A. Recht, B. Golden, and B.N. Cronstein. 1995. Identification of C1q as the heat-labile serum cofactor required for immune complexes to stimulate endothelial expression of the adhesion molecules E-selectin and intercellular and vascular cell adhesion molecules 1. Proc. Natl. Acad. Sci. USA. 92:8378–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao, S., C. Xu, and J.N. Jarvis. 2001. C1q-bearing immune complexes induce IL-8 secretion in human umbilical vein endothelial cells (HUVEC) through tyrosine kinase- and mitogen-activated protein kinase-dependent mechanisms: evidence that the 126 kD phagocytic C1q receptor mediates immune complex activation of HUVEC. Clin. Exp. Immunol. 125:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thurston, G., P. Baluk, and D.M. McDonald. 2000. Determinants of endothelial cell phenotype in venules. Microcirculation. 7:67–80. [PubMed] [Google Scholar]
- 49.Cream, J.J., A.D. Bryceson, and G. Ryder. 1971. Disappearance of immunoglobulin and complement from the Arthus reaction and its relevance to studies of vasculitis in man. Br. J. Dermatol. 84:106–109. [DOI] [PubMed] [Google Scholar]
- 50.Eggleton, P., K.B. Reid, and A.J. Tenner. 1998. C1q-how many functions? How many receptors? Trends Cell Biol. 8:428–431. [DOI] [PubMed] [Google Scholar]
- 51.McGreal, E., and P. Gasque. 2001. Structure-function studies of the receptors for complement C1q. Biochem. Soc. Trans. 30:1010–1014. [DOI] [PubMed] [Google Scholar]
- 52.Bohnsack, J.F., A.J. Tenner, G.W. Laurie, H.K. Kleinman, G.R. Martin, and E.J. Brown. 1985. The C1q subunit of the first component of complement binds to laminin: a mechanism for the deposition and retention of immune complexes in basement membrane. Proc. Natl. Acad. Sci. USA. 82:3824–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rostagno, A.A., G. Gallo, and L.I. Gold. 2002. Binding of polymeric IgG to fibronectin in extracellular matrices: an in vitro paradigm for immune-complex deposition. Mol. Immunol. 38:1101–1111. [DOI] [PubMed] [Google Scholar]
- 54.Maehnss, K., J. Kobarg, W.H. Schmitt, H.P. Hansen, H. Lange, E. Csernok, W.L. Gross, and H. Lemke. 2002. Vitronectin- and fibronectin-containing immune complexes in primary systemic vasculitis. J. Autoimmun. 18:239–250. [DOI] [PubMed] [Google Scholar]
- 55.Liszewski, M.K., T.C. Farries, D.M. Lublin, I.A. Rooney, and J.P. Atkinson. 1996. Control of the complement system. Adv. Immunol. 61:201–283. [DOI] [PubMed] [Google Scholar]
- 56.Rosenkranz, A.R., D.L. Mendrick, R.S. Cotran, and T.N. Mayadas. 1999. P-selectin deficiency exacerbates experimental glomerulonephritis: a protective role for endothelial P-selectin in inflammation. J. Clin. Invest. 103:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tarzi, R.M., K.A. Davies, M.G. Robson, L. Fossati-Jimack, T. Saito, M.J. Walport, and H.T. Cook. 2002. Nephrotoxic nephritis is mediated by Fcgamma receptors on circulating leukocytes and not intrinsic renal cells. Kidney Int. 62:2087–2096. [DOI] [PubMed] [Google Scholar]
- 58.Davis, R.S., Y.H. Wang, H. Kubagawa, and M.D. Cooper. 2001. Identification of a family of Fc receptor homologs with preferential B cell expression. Proc. Natl. Acad. Sci. USA. 98:9772–9777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clynes, R., J.S. Maizes, R. Guinamard, M. Ono, T. Takai, and J.V. Ravetch. 1999. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 189:179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nabbe, K.C., A.B. Blom, A.E. Holthuysen, P. Boross, J. Roth, S. Verbeek, P.L. van Lent, and W.B. van den Berg. 2003. Coordinate expression of activating Fc gamma receptors I and III and inhibiting Fc gamma receptor type II in the determination of joint inflammation and cartilage destruction during immune complex-mediated arthritis. Arthritis Rheum. 48:255–265. [DOI] [PubMed] [Google Scholar]
- 61.Trevani, A.S., P.A. Fontan, G.A. Andonegui, M.A. Isturiz, and J.R. Geffner. 1995. Neutrophil chemotaxis induced by immune complexes. Clin. Immunol. Immunopathol. 74:107–111. [DOI] [PubMed] [Google Scholar]
- 62.Cinamon, G., V. Shinder, and R. Alon. 2001. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat. Immunol. 2:515–522. [DOI] [PubMed] [Google Scholar]